

Target Mapping in Cancer: Ligandable Protein Pockets on 3D OncoPPI Networks

 ,
,  ,
, 
Abstract

1. Introduction
2. Results and Discussion
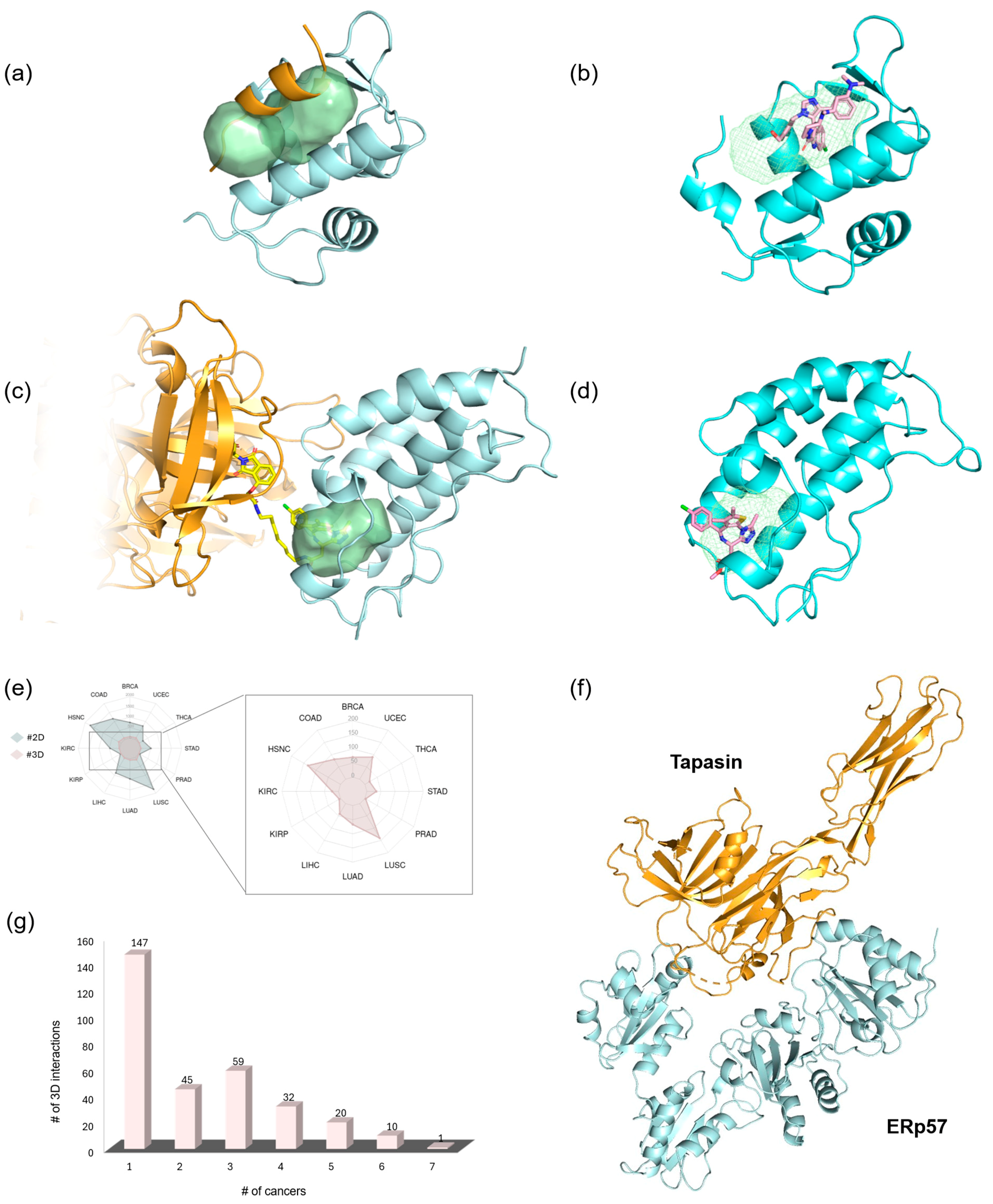
2.1. Detection of Known oncoPPI Inhibitor and PROTAC Binding Pockets
2.2. 3D Dataset of oncoPPIs
2.3. 3D Protein Pocket Mapping: Development of the oncoPPI Pocketome
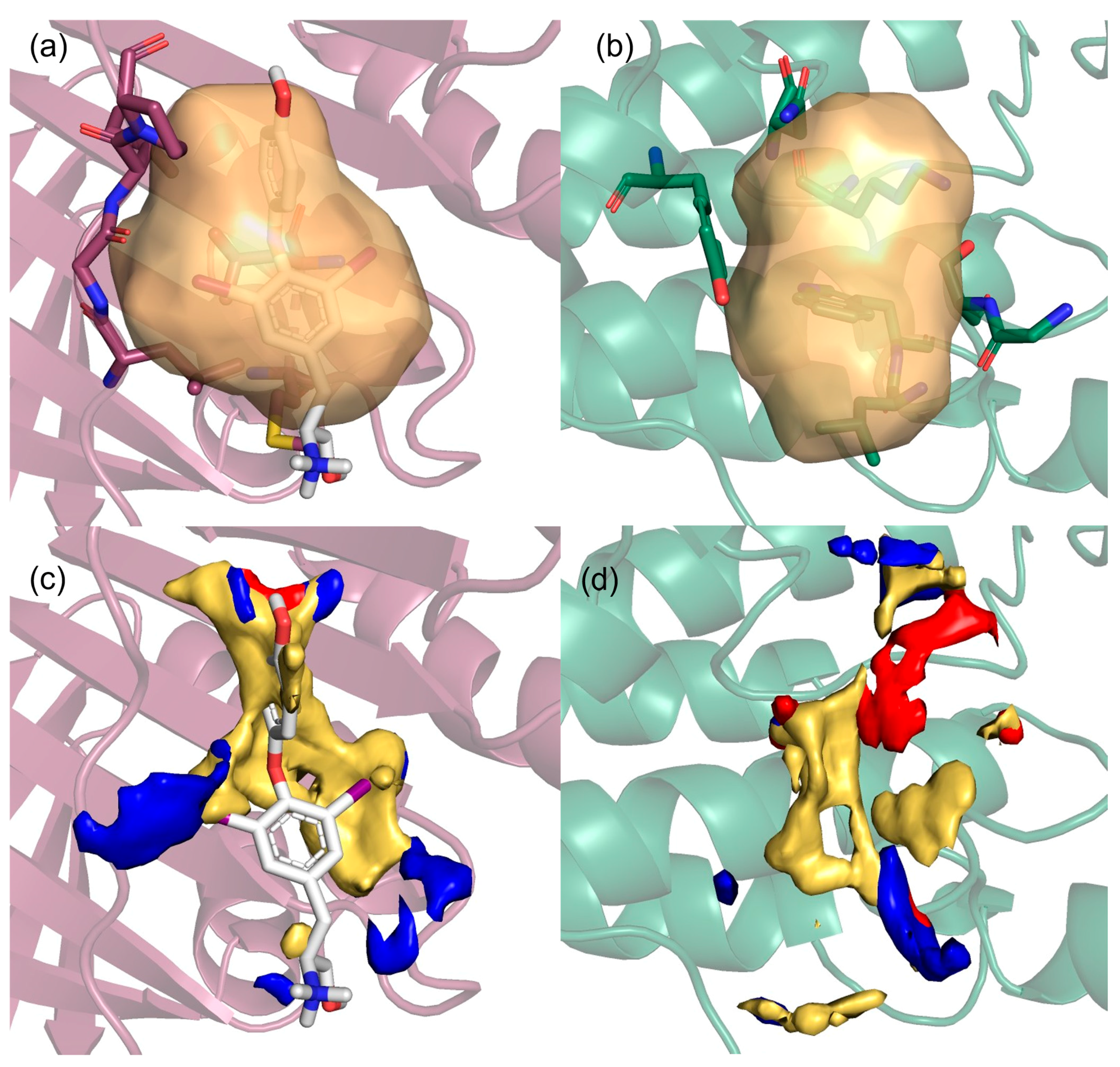
- Detached partners: the search for pockets was carried out on each individual partner separately by splitting the crystallographic complex, so that any pocket at the interaction interface could be revealed for both partners (Figure 2a,b).
- Complexed partners: in this case, we analyzed the interaction zone of the whole complex in order to identify pockets involved in the bound/unbound equilibrium (Figure 2c).
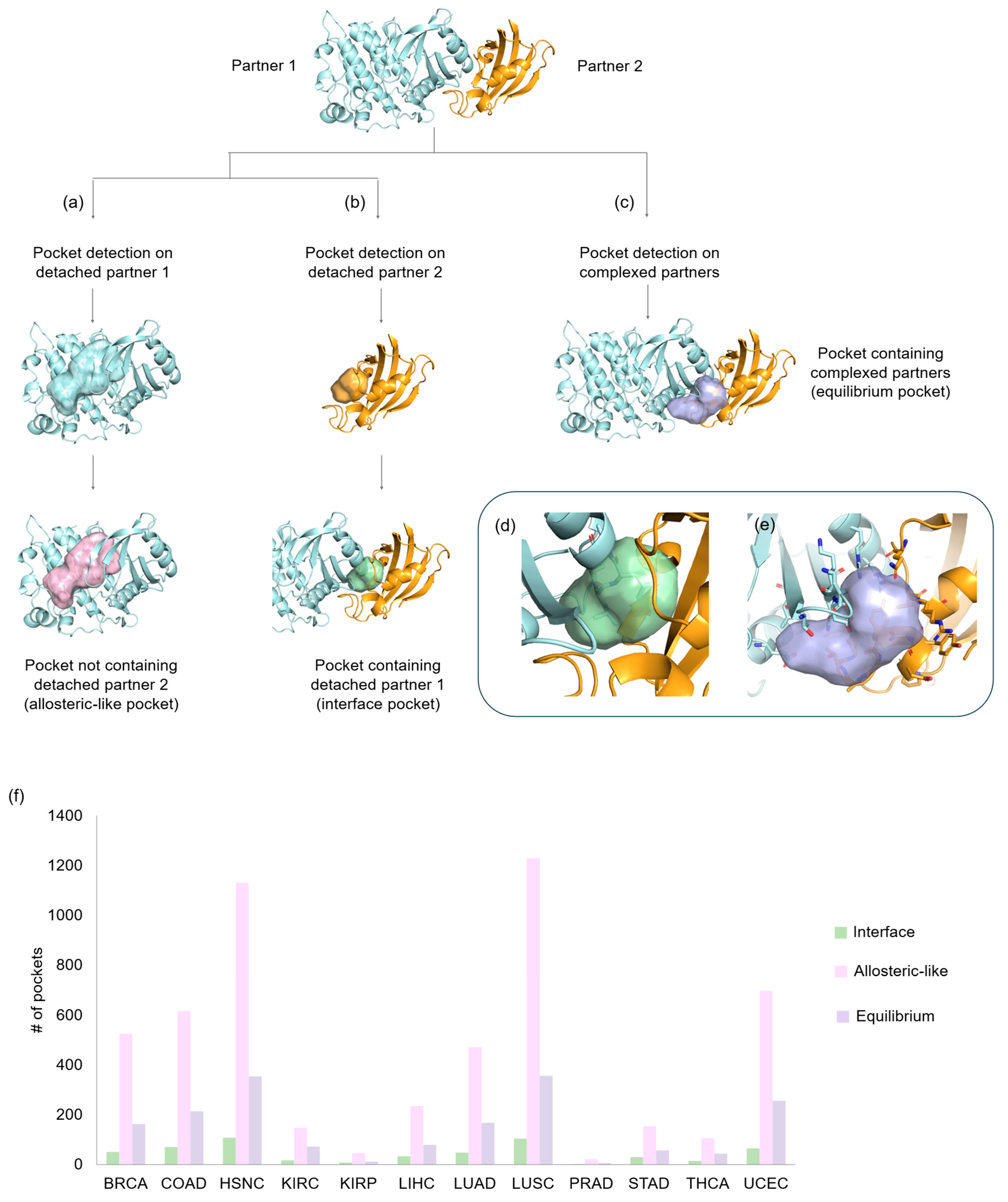
- Interface pockets: these pockets were identified on individual detached partners and are located in regions involved in 3D oncoPPIs. A pocket is classified as an interface pocket if the protein residues of the interacting partner are enclosed within it (Figure 2d).
- Allosteric-like pockets: these pockets are found on individual detached partners and do not correspond to regions directly involved in 3D oncoPPIs.
- Equilibrium pockets: these pockets were computed on the complexed partners and are located in regions defining the interaction interface between the two protein partners. To be classified as an equilibrium pocket, it must consist of residues belonging to both interacting partners (Figure 2e).
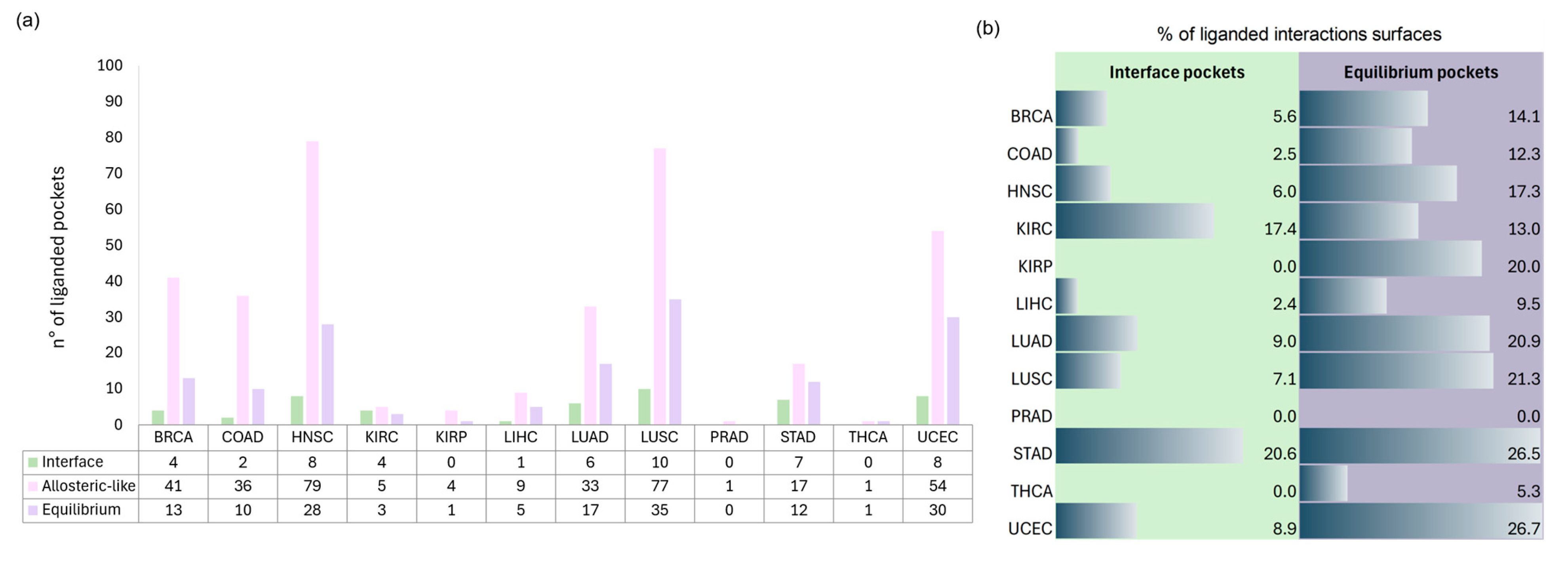
2.3.1. Interface and Allosteric-like Pockets on Detached Partner
Interface Pockets
Allosteric-like Pockets
2.3.2. Equilibrium Pockets on Complexed Partners
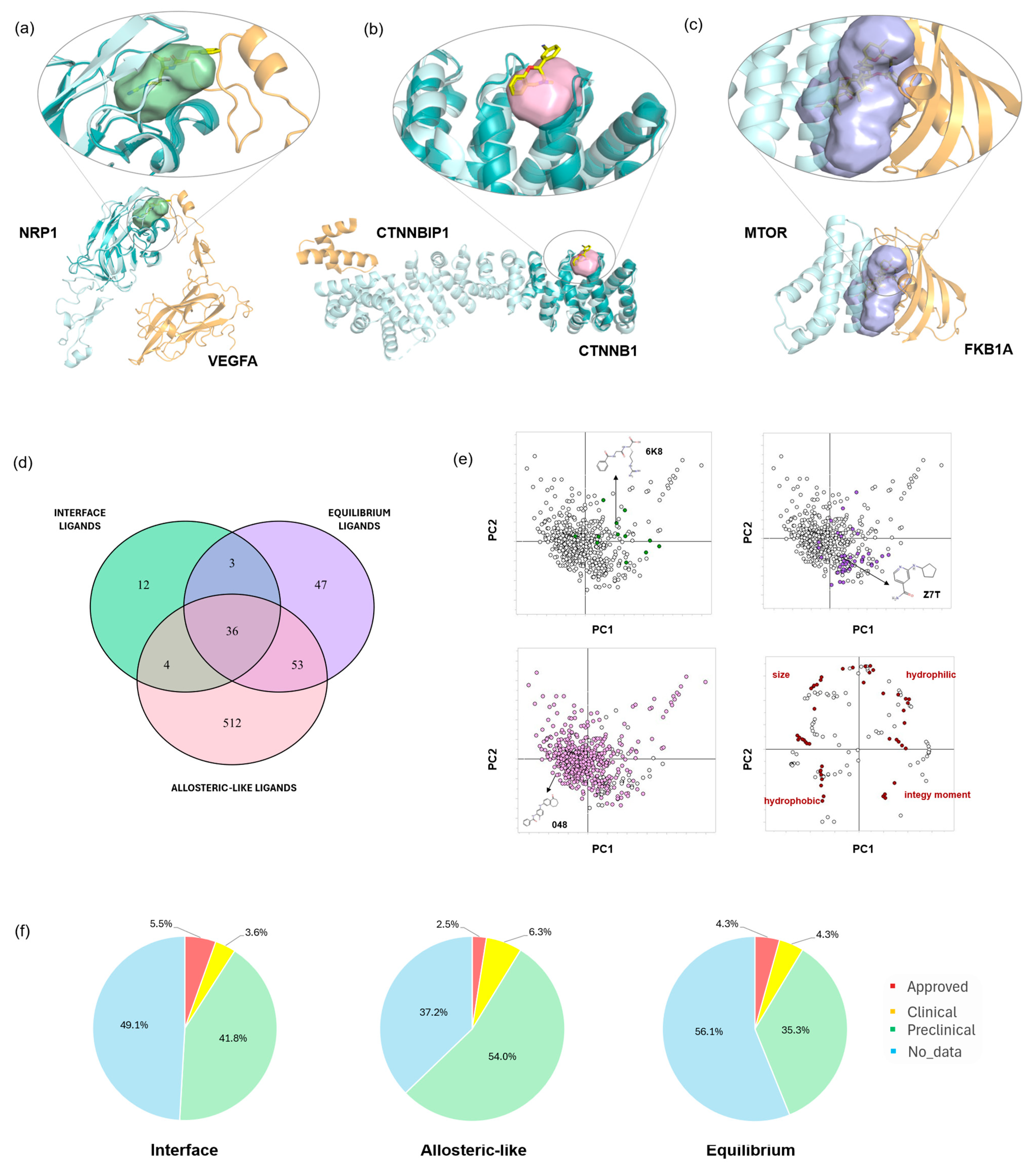
2.4. Ligandable Pockets for 3D oncoPPI Modulators and PROTAC Design
2.5. Geometric and Energetic Anatomy of the 3D oncoPPI Pocketome
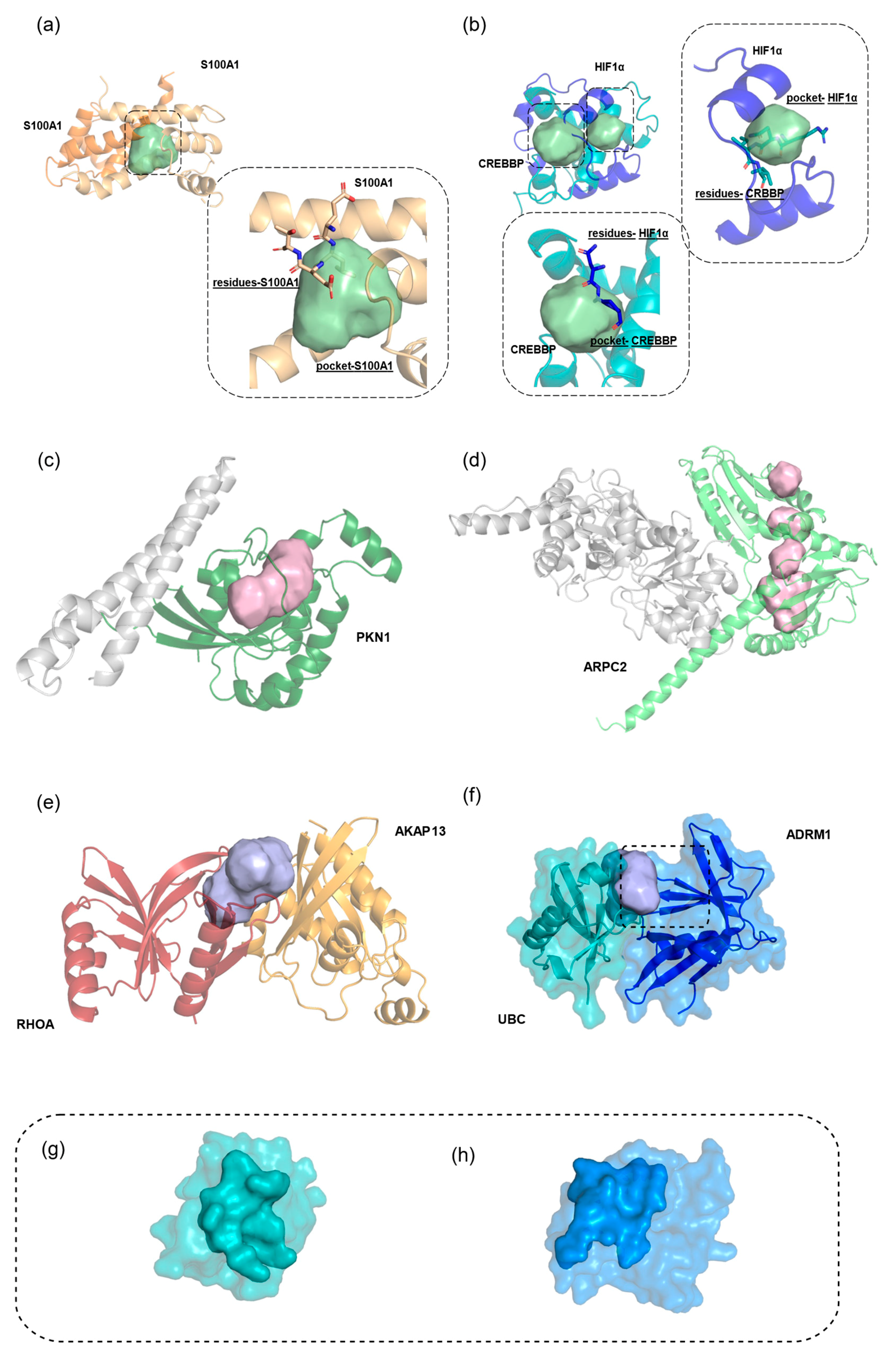
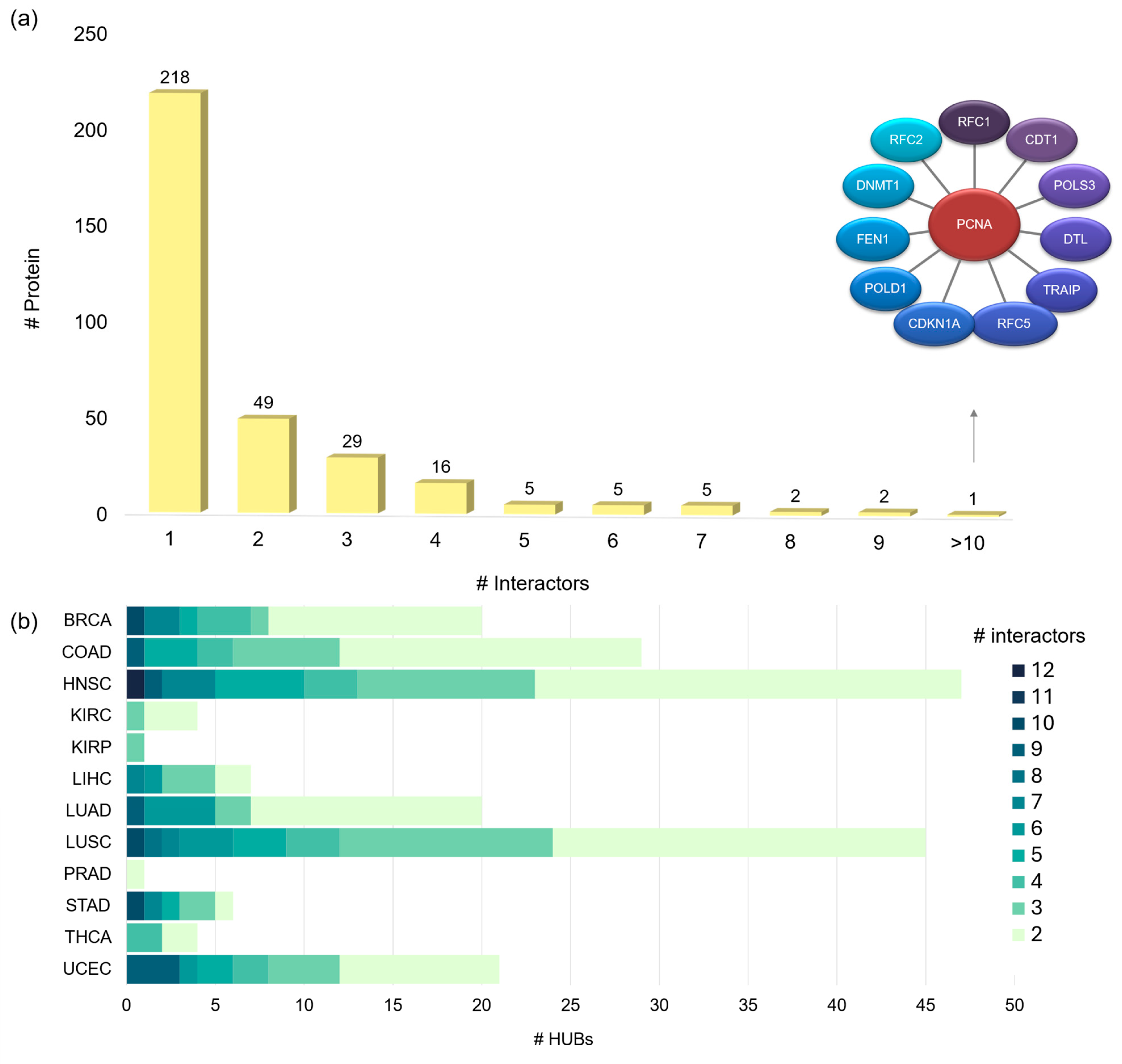
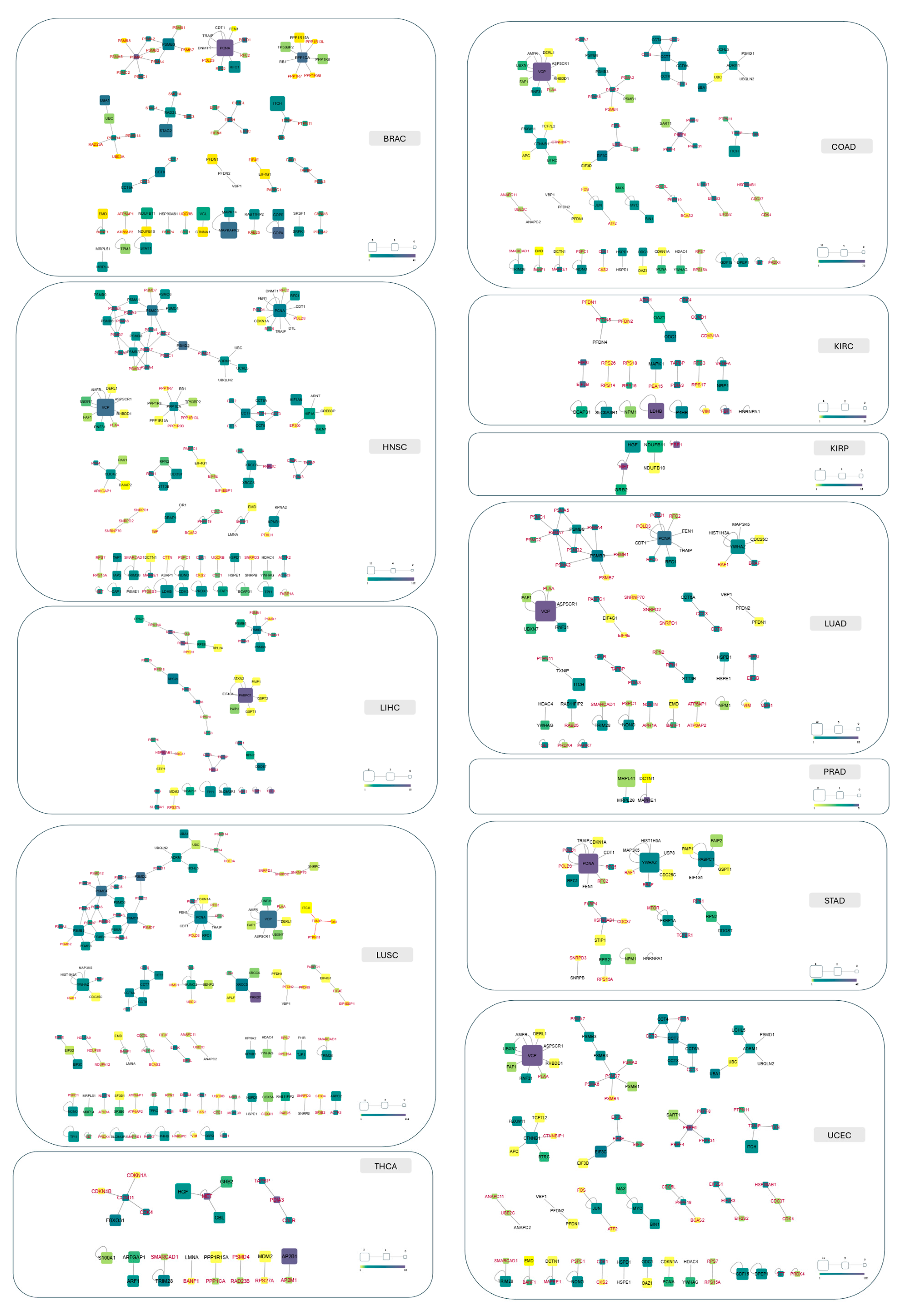
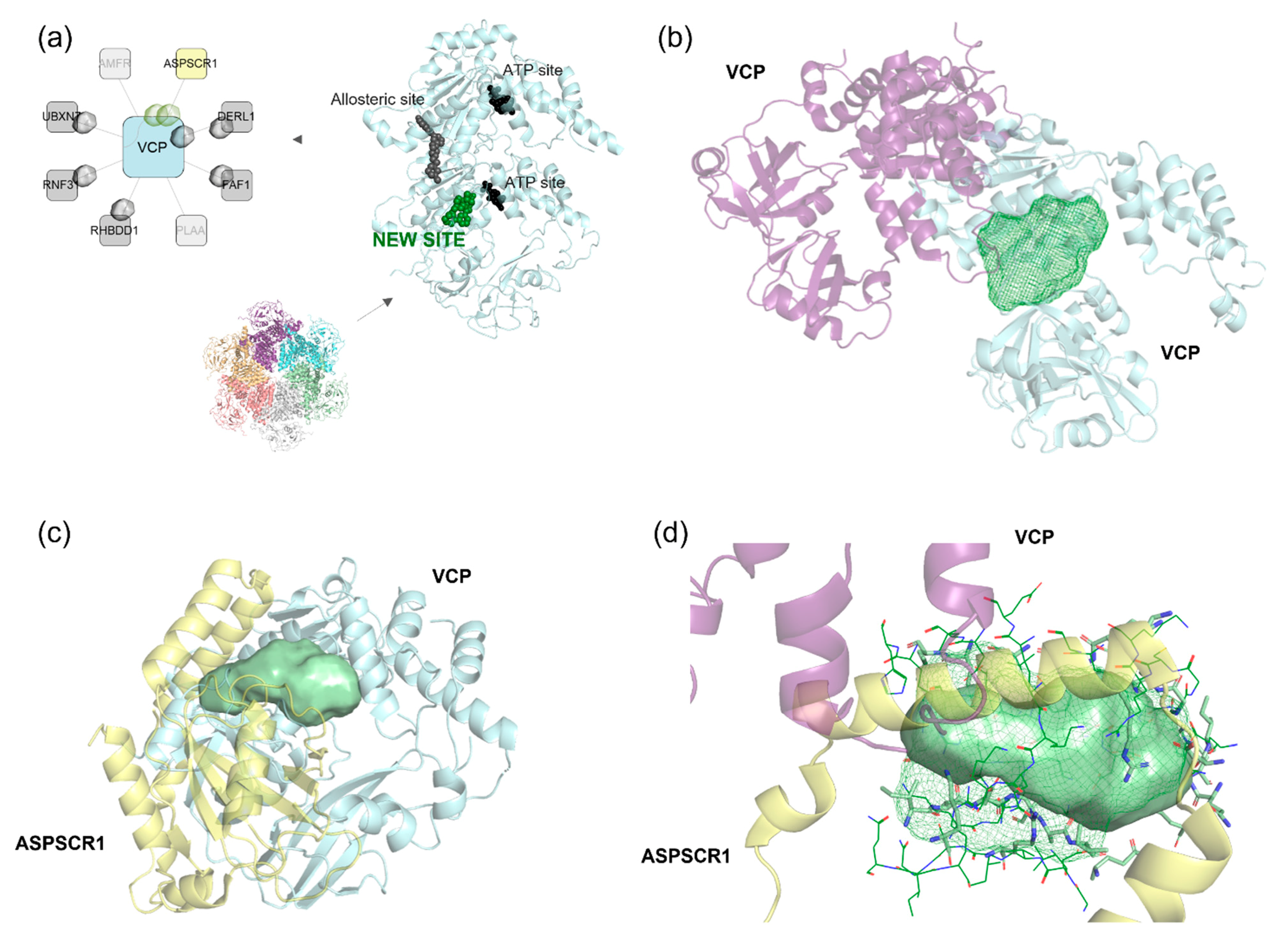
2.6. Hub Proteins and Hub Pockets in 3D oncoPPI Networks
Discovery of a New Ligandable Hub Pocket on VCP
3. Materials and Methods
3.1. Protein Validation Set
3.2. oncoPPI Dataset
3.3. Protein Preparation
3.4. Pockets Detection
- In the validation set, for each protein, the pockets were calculated: (i) on the inhibitor-bound form, (ii) on the protein-bound form, or (iii) on the PROTAC-bound form. In all cases of complexes with inhibitors, protein partners, or PROTACs, any binder was previously extracted to find the cavity involved in the binding.
- In the 3D oncoPPI datasets, the pockets were calculated on each individual partner separately, by splitting the crystallographic complex (i.e., detached partner) and by considering the entire complex (i.e., complexed partner). In the first case, each individual chain representing the partner protein was used as input for BioGPS to collect its pockets. In the second case, the original crystallographic complex of the two chains was used as input for pocket detection.
3.5. Classification of Pockets in the 3D oncoPPI Dataset
- Interface pockets: these pockets were identified on individual (detached) protein partners and are located in regions involved in PPIs. A pocket is classified as an interface pocket if the fraction of volume of the interacting partner contained within the pocket is greater than 0.
- Allosteric-like pockets: these pockets were identified on individual (detached) protein partners that do not correspond to any regions directly involved in PPIs.
- Equilibrium pockets: these pockets were calculated by considering the whole crystallographic complex and are located in regions that define the interaction interface between the two protein partners. Specifically, for a pocket to be classified as an equilibrium pocket, it must be composed of residues contributed by both interacting partners.
3.6. Ligand-Bound Pockets
3.7. Ligands’ Physicochemical Properties
3.8. Ligands’ Classification
3.9. 3D oncoPPI Network
3.10. Pockets Physicochemical Properties
- ▪
- Globularity: quantifies the degree of sphericity of the pocket. It is equal to 1.0 for perfect spherical objects, whereas it assumes values lower than 1.0 for real spheroidal ones;
- ▪
- Rugosity: indicates the presence of molecular wrinkles or creases on the pocket surface expressed as the ratio of volume to surface. The higher the ratio, the higher the rugosity;
- ▪
- Hydrophobic volume: proportional to the number of points in the DRY field, filtered to include only those with energy lower than −0.5 Kcal/mol;
- ▪
- Hydrophilic volume: proportional to the total number of points in the OH2 field, considering only points with energy below −3.5 Kcal/mol;
- ▪
- Exposed to solvent: describes the surface of the pocket accessible to the solvent and not in contact with protein residues. It is proportional to the external points in the H field that are at least 2.2 Å away from the protein atoms;
- ▪
- Buried volume: measures the volume of points embedded within the protein cavity, calculated by summing the “collisions” of 50 vectors intersecting with the protein surface. Each “collision” adds to the buriedness, and the final value is an average of all the values across all points. The reported value refers to the pocket points with low buried volume.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- OECD. Tackling the Impact of Cancer on Health, the Economy and Society, OECD Health Policy Studies; OECD Publishing: Paris, France, 2024. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small Molecules in Targeted Cancer Therapy: Advances, Challenges, and Future Perspectives. Sig Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Chovanec, M.; Lauritsen, J.; Bandak, M.; Oing, C.; Kier, G.G.; Kreiberg, M.; Rosenvilde, J.; Wagner, T.; Bokemeyer, C.; Daugaard, G. Late Adverse Effects and Quality of Life in Survivors of Testicular Germ Cell Tumour. Nat. Rev. Urol. 2021, 18, 227–245. [Google Scholar] [CrossRef] [PubMed]
- Sharifi Tabar, M.; Francis, H.; Yeo, D.; Bailey, C.G.; Rasko, J.E.J. Mapping Oncogenic Protein Interactions for Precision Medicine. Int. J. Cancer 2022, 151, 7–19. [Google Scholar] [CrossRef]
- Kamburov, A.; Lawrence, M.S.; Polak, P.; Leshchiner, I.; Lage, K.; Golub, T.R.; Lander, E.S.; Getz, G. Comprehensive Assessment of Cancer Missense Mutation Clustering in Protein Structures. Proc. Natl. Acad. Sci. USA 2015, 112, E5486–E5495. [Google Scholar] [CrossRef]
- Li, Z.; Ivanov, A.A.; Su, R.; Gonzalez-Pecchi, V.; Qi, Q.; Liu, S.; Webber, P.; McMillan, E.; Rusnak, L.; Pham, C.; et al. The OncoPPi Network of Cancer-Focused Protein–Protein Interactions to Inform Biological Insights and Therapeutic Strategies. Nat. Commun. 2017, 8, 14356. [Google Scholar] [CrossRef]
- Gulfidan, G.; Turanli, B.; Beklen, H.; Sinha, R.; Arga, K.Y. Pan-Cancer Mapping of Differential Protein-Protein Interactions. Sci. Rep. 2020, 10, 3272. [Google Scholar] [CrossRef]
- Du, Y.; Cai, M.; Xing, X.; Ji, J.; Yang, E.; Wu, J. PINA 3.0: Mining Cancer Interactome. Nucleic Acids Res. 2021, 49, D1351–D1357. [Google Scholar] [CrossRef]
- Lostao, A.; Lim, K.; Pallarés, M.C.; Ptak, A.; Marcuello, C. Recent Advances in Sensing the Inter-Biomolecular Interactions at the Nanoscale—A Comprehensive Review of AFM-Based Force Spectroscopy. Int. J. Biol. Macromol. 2023, 238, 124089. [Google Scholar] [CrossRef]
- Ha, T.; Fei, J.; Schmid, S.; Lee, N.K.; Gonzalez, R.L.; Paul, S.; Yeou, S. Fluorescence Resonance Energy Transfer at the Single-Molecule Level. Nat. Rev. Methods Primers 2024, 4, 21. [Google Scholar] [CrossRef]
- Kar, G.; Gursoy, A.; Keskin, O. Human Cancer Protein-Protein Interaction Network: A Structural Perspective. PLoS Comput. Biol. 2009, 5, e1000601. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Jalal, S.I.; Sledge, G.W.; Meroueh, S.O. Small-Molecule Binding Sites to Explore Protein–Protein Interactions in the Cancer Proteome. Mol. BioSyst. 2016, 12, 3067–3087. [Google Scholar] [CrossRef] [PubMed]
- Trisciuzzi, D.; Villoutreix, B.O.; Siragusa, L.; Baroni, M.; Cruciani, G.; Nicolotti, O. Targeting Protein-Protein Interactions with Low Molecular Weight and Short Peptide Modulators: Insights on Disease Pathways and Starting Points for Drug Discovery. Expert. Opin. Drug Discov. 2023, 18, 737–752. [Google Scholar] [CrossRef] [PubMed]
- Pathmanathan, S.; Grozavu, I.; Lyakisheva, A.; Stagljar, I. Drugging the Undruggable Proteins in Cancer: A Systems Biology Approach. Curr. Opin. Chem. Biol. 2022, 66, 102079. [Google Scholar] [CrossRef]
- Kelm, J.M.; Pandey, D.S.; Malin, E.; Kansou, H.; Arora, S.; Kumar, R.; Gavande, N.S. PROTAC’ing Oncoproteins: Targeted Protein Degradation for Cancer Therapy. Mol. Cancer 2023, 22, 62. [Google Scholar] [CrossRef]
- Bai, L.; Zhou, H.; Xu, R.; Zhao, Y.; Chinnaswamy, K.; McEachern, D.; Chen, J.; Yang, C.-Y.; Liu, Z.; Wang, M.; et al. A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo. Cancer Cell 2019, 36, 498–511.e17. [Google Scholar] [CrossRef]
- Lee, G.T.; Nagaya, N.; Desantis, J.; Madura, K.; Sabaawy, H.E.; Kim, W.-J.; Vaz, R.J.; Cruciani, G.; Kim, I.Y. Effects of MTX-23, a Novel PROTAC of Androgen Receptor Splice Variant-7 and Androgen Receptor, on CRPC Resistant to Second-Line Antiandrogen Therapy. Mol. Cancer Ther. 2021, 20, 490–499. [Google Scholar] [CrossRef]
- Chen, Y.; Ning, Y.; Bai, G.; Tong, L.; Zhang, T.; Zhou, J.; Zhang, H.; Xie, H.; Ding, J.; Duan, W. Design, Synthesis, and Biological Evaluation of IRAK4-Targeting PROTACs. ACS Med. Chem. Lett. 2021, 12, 82–87. [Google Scholar] [CrossRef]
- Hanafi, M.; Chen, X.; Neamati, N. Discovery of a Napabucasin PROTAC as an Effective Degrader of the E3 Ligase ZFP91. J. Med. Chem. 2021, 64, 1626–1648. [Google Scholar] [CrossRef]
- Yu, X.; Li, D.; Kottur, J.; Shen, Y.; Kim, H.S.; Park, K.-S.; Tsai, Y.-H.; Gong, W.; Wang, J.; Suzuki, K.; et al. A Selective WDR5 Degrader Inhibits Acute Myeloid Leukemia in Patient-Derived Mouse Models. Sci. Transl. Med. 2021, 13, eabj1578. [Google Scholar] [CrossRef]
- Palomba, T.; Baroni, M.; Cross, S.; Cruciani, G.; Siragusa, L. ELIOT: A Platform to Navigate the E3 Pocketome and Aid the Design of New PROTACs. Chem. Biol. Drug Des. 2023, 101, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Trisciuzzi, D.; Siragusa, L.; Baroni, M.; Autiero, I.; Nicolotti, O.; Cruciani, G. Getting Insights into Structural and Energetic Properties of Reciprocal Peptide–Protein Interactions. J. Chem. Inf. Model. 2022, 62, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Trisciuzzi, D.; Siragusa, L.; Baroni, M.; Cruciani, G.; Nicolotti, O. An Integrated Machine Learning Model To Spot Peptide Binding Pockets in 3D Protein Screening. J. Chem. Inf. Model. 2022, 62, 6812–6824. [Google Scholar] [CrossRef]
- Farias, A.B.; Candiotto, G.; Siragusa, L.; Goracci, L.; Cruciani, G.; Oliveira, E.R.A.; Horta, B.A.C. Targeting Nsp9 as an Anti-SARS-CoV-2 Strategy. New J. Chem. 2021, 45, 522–525. [Google Scholar] [CrossRef]
- Goodford, P.J. A Computational Procedure for Determining Energetically Favorable Binding Sites on Biologically Important Macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar] [CrossRef]
- Siragusa, L.; Cross, S.; Baroni, M.; Goracci, L.; Cruciani, G. BioGPS: Navigating Biological Space to Predict Polypharmacology, off-Targeting, and Selectivity. Proteins 2015, 83, 517–532. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent Advances in the Development of Protein-Protein Interactions Modulators: Mechanisms and Clinical Trials. Signal Transduct. Target. Ther. 2020, 5, 213. [Google Scholar] [CrossRef]
- Weng, G.; Li, D.; Kang, Y.; Hou, T. Integrative Modeling of PROTAC-Mediated Ternary Complexes. J. Med. Chem. 2021, 64, 16271–16281. [Google Scholar] [CrossRef]
- Ignatov, M.; Jindal, A.; Kotelnikov, S.; Beglov, D.; Posternak, G.; Tang, X.; Maisonneuve, P.; Poda, G.; Batey, R.A.; Sicheri, F.; et al. High Accuracy Prediction of PROTAC Complex Structures. J. Am. Chem. Soc. 2023, 145, 7123–7135. [Google Scholar] [CrossRef]
- Chicco, D.; Tötsch, N.; Jurman, G. The Matthews Correlation Coefficient (MCC) Is More Reliable than Balanced Accuracy, Bookmaker Informedness, and Markedness in Two-Class Confusion Matrix Evaluation. BioData Mining 2021, 14, 13. [Google Scholar] [CrossRef]
- Tripathi, B.K.; Hirsh, N.H.; Qian, X.; Durkin, M.E.; Wang, D.; Papageorge, A.G.; Lake, R.; Evrard, Y.A.; Marcus, A.I.; Ramalingam, S.S.; et al. The Pro-Oncogenic Noncanonical Activity of a RAS•GTP:RanGAP1 Complex Facilitates Nuclear Protein Export. Nat. Cancer 2024, 5, 1902–1918. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Liu, H.; Wu, J.; Gao, X.; Hao, J.; Fan, D. Insights into the Role of ERp57 in Cancer. J. Cancer 2021, 12, 2456–2464. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, H.; Cui, X.; Xu, T.; Dong, M.; Li, S.; Li, X. S100A1 Is a Potential Biomarker for Papillary Thyroid Carcinoma Diagnosis and Prognosis. J. Cancer 2021, 12, 5760–5771. [Google Scholar] [CrossRef]
- Wu, D.; Zhang, R.; Zhao, R.; Chen, G.; Cai, Y.; Jin, J. A Novel Function of Novobiocin: Disrupting the Interaction of HIF 1α and P300/CBP through Direct Binding to the HIF1α C-Terminal Activation Domain. PLoS ONE 2013, 8, e62014. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, B.-B.; Wei, W.; Huang, G.-C.; Liu, L.-L.; Chen, W.-W.; Wang, J.; Zhao, X.-Y.; Lu, L.; Fang, R.; et al. ITCH Facilitates Proteasomal Degradation of TXNIP in Hypoxia-Induced Lung Cancer Cells. Thorac. Cancer 2022, 13, 2235–2247. [Google Scholar] [CrossRef]
- Wang, X.; Ge, Y.; Shi, M.; Dai, H.; Liu, W.; Wang, P. Protein Kinase N1 Promotes Proliferation and Invasion of Liver Cancer. Exp. Ther. Med. 2021, 21, 651. [Google Scholar] [CrossRef]
- Diviani, D.; Raimondi, F.; Del Vescovo, C.D.; Dreyer, E.; Reggi, E.; Osman, H.; Ruggieri, L.; Gonano, C.; Cavin, S.; Box, C.L.; et al. Small-Molecule Protein-Protein Interaction Inhibitor of Oncogenic Rho Signaling. Cell Chem. Biol. 2016, 23, 1135–1146. [Google Scholar] [CrossRef]
- Kessler, D.; Mayer, M.; Zahn, S.K.; Zeeb, M.; Wöhrle, S.; Bergner, A.; Bruchhaus, J.; Ciftci, T.; Dahmann, G.; Dettling, M.; et al. Getting a Grip on the Undrugged: Targeting β-Catenin with Fragment-Based Methods. ChemMedChem 2021, 16, 1420–1424. [Google Scholar] [CrossRef]
- Tortorella, S.; Carosati, E.; Sorbi, G.; Bocci, G.; Cross, S.; Cruciani, G.; Storchi, L. Combining Machine Learning and Quantum Mechanics Yields More Chemically Aware Molecular Descriptors for Medicinal Chemistry Applications. J. Comput. Chem. 2021, 42, 2068–2078. [Google Scholar] [CrossRef]
- Knox, C.; Wilson, M.; Klinger, C.M.; Franklin, M.; Oler, E.; Wilson, A.; Pon, A.; Cox, J.; Chin, N.E.L.; Strawbridge, S.A.; et al. DrugBank 6.0: The DrugBank Knowledgebase for 2024. Nucleic Acids Res. 2024, 52, D1265–D1275. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards Direct Deposition of Bioassay Data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Kikuchi, S.; Hishiki, A.; Shao, Y.; Heath, R.; Evison, B.J.; Actis, M.; Canman, C.E.; Hashimoto, H.; Fujii, N. A Small Molecule Inhibitor of Monoubiquitinated Proliferating Cell Nuclear Antigen (PCNA) Inhibits Repair of Interstrand DNA Cross-Link, Enhances DNA Double Strand Break, and Sensitizes Cancer Cells to Cisplatin. J. Biol. Chem. 2014, 289, 7109–7120. [Google Scholar] [CrossRef] [PubMed]
- Mingione, V.R.; Paung, Y.; Outhwaite, I.R.; Seeliger, M.A. Allosteric Regulation and Inhibition of Protein Kinases. Biochem. Soc. Trans. 2023, 51, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Nithya, C.; Kiran, M.; Nagarajaram, H.A. Dissection of Hubs and Bottlenecks in a Protein-Protein Interaction Network. Comput. Biol. Chem. 2023, 102, 107802. [Google Scholar] [CrossRef]
- Hudock, N.L.; Mani, K.; Khunsriraksakul, C.; Walter, V.; Nekhlyudov, L.; Wang, M.; Lehrer, E.J.; Hudock, M.R.; Liu, D.J.; Spratt, D.E.; et al. Future Trends in Incidence and Long-Term Survival of Metastatic Cancer in the United States. Commun. Med. 2023, 3, 76. [Google Scholar] [CrossRef]
- Kilgas, S.; Ramadan, K. Inhibitors of the ATPase P97/VCP: From Basic Research to Clinical Applications. Cell Chem. Biol. 2023, 30, 3–21. [Google Scholar] [CrossRef]
- Arumughan, A.; Roske, Y.; Barth, C.; Forero, L.L.; Bravo-Rodriguez, K.; Redel, A.; Kostova, S.; McShane, E.; Opitz, R.; Faelber, K.; et al. Quantitative Interaction Mapping Reveals an Extended UBX Domain in ASPL That Disrupts Functional P97 Hexamers. Nat. Commun. 2016, 7, 13047. [Google Scholar] [CrossRef]
- Tsai, C.-J.; Ma, B.; Nussinov, R. Protein–Protein Interaction Networks: How Can a Hub Protein Bind so Many Different Partners? Trends Biochem. Sci. 2009, 34, 594–600. [Google Scholar] [CrossRef]
- Basse, M.-J.; Betzi, S.; Morelli, X.; Roche, P. 2P2Idb v2: Update of a Structural Database Dedicated to Orthosteric Modulation of Protein-Protein Interactions. Database 2016, 2016, baw007. [Google Scholar] [CrossRef]
- The UniProt Consortium UniProt: The Universal Protein Knowledgebase in 2025. Nucleic Acids Res. 2025, 53, D609–D617. [CrossRef]
- Mosca, R.; Céol, A.; Aloy, P. Interactome3D: Adding Structural Details to Protein Networks. Nat. Methods 2013, 10, 47–53. [Google Scholar] [CrossRef]
- Schrödinger, L.; DeLano, W. PyMOL. 2020. Available online: http://www.pymol.org/pymol (accessed on 1 June 2025).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2024; Available online: https://www.r-project.org/ (accessed on 1 June 2025).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interface pockets | ||
| n° X-ray ligands | Liganded partner protein | Interacting partner protein |
| 16 | Transforming protein RhoA | Rho GTPase-activating protein 1 |
| 11 | 14-3-3 protein zeta/delta | Mitogen-activated protein kinase kinase kinase 5 |
| 9 | Proteasome subunit beta type-6 | Proteasome subunit beta type-4 |
| 6 | Neuropilin-1 | Vascular endothelial growth factor A |
| 4 | Proliferating cell nuclear antigen | DNA polymerase delta subunit 3 |
| Allosteric-like pockets | ||
| n° X-ray ligands | Liganded partner protein | |
| 177 | Mitogen-activated protein kinase 14 | - |
| 126 | Mitogen-activated protein kinase 1 | - |
| 60 | Serine/threonine-protein kinase B-raf | - |
| 43 | Fructose-1,6-bisphosphatase 1 | - |
| 35 | Peptidyl-prolyl cis-trans isomerase FKBP1A | - |
| Equilibrium pockets | ||
| n° X-ray ligands | Liganded partner protein 1 | Liganded partner protein 2 |
| 27 | Transforming protein RhoA | Rho guanine nucleotide exchange factor 12 |
| 23 | Serine/threonine-protein kinase PAK 4 | Cell division control protein 42 homolog |
| 22 | Proteasome subunit beta type-1 | Proteasome subunit beta type-8 |
| 10 | Transforming protein RhoA | Rho GTPase-activating protein 1 |
| 10 | Proteasome subunit beta type-4 | Proteasome subunit beta type-6 |
| Rugosity | Globularity | Hydrophilic Volume | Hydrophobic Volume | Buried Volume | Exposition to Solvent | |
|---|---|---|---|---|---|---|
| Ligand-bound interface PCNA | 2.213 | 0.820 | 52.312 | 46.406 | 645.559 | 159.188 |
| Allosteric-like BRAF | 2.159 | 0.851 | 52.734 | 32.062 | 552.502 | 187.312 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trisciuzzi, D.; Nicolotti, O.; Cruciani, G.; Menna, G.; Siragusa, L. Target Mapping in Cancer: Ligandable Protein Pockets on 3D OncoPPI Networks. Pharmaceuticals 2025, 18, 958. https://doi.org/10.3390/ph18070958
Trisciuzzi D, Nicolotti O, Cruciani G, Menna G, Siragusa L. Target Mapping in Cancer: Ligandable Protein Pockets on 3D OncoPPI Networks. Pharmaceuticals. 2025; 18(7):958. https://doi.org/10.3390/ph18070958
Chicago/Turabian StyleTrisciuzzi, Daniela, Orazio Nicolotti, Gabriele Cruciani, Gabriele Menna, and Lydia Siragusa. 2025. "Target Mapping in Cancer: Ligandable Protein Pockets on 3D OncoPPI Networks" Pharmaceuticals 18, no. 7: 958. https://doi.org/10.3390/ph18070958
APA StyleTrisciuzzi, D., Nicolotti, O., Cruciani, G., Menna, G., & Siragusa, L. (2025). Target Mapping in Cancer: Ligandable Protein Pockets on 3D OncoPPI Networks. Pharmaceuticals, 18(7), 958. https://doi.org/10.3390/ph18070958