


Unveiling Palmitoyl Thymidine Derivatives as Antimicrobial/Antiviral Inhibitors: Synthesis, Molecular Docking, Dynamic Simulations, ADMET, and Assessment of Protein–Ligand Interactions

 ,
,  , ,
, ,  , ,
, ,  , ,
, ,  and
and 
Abstract

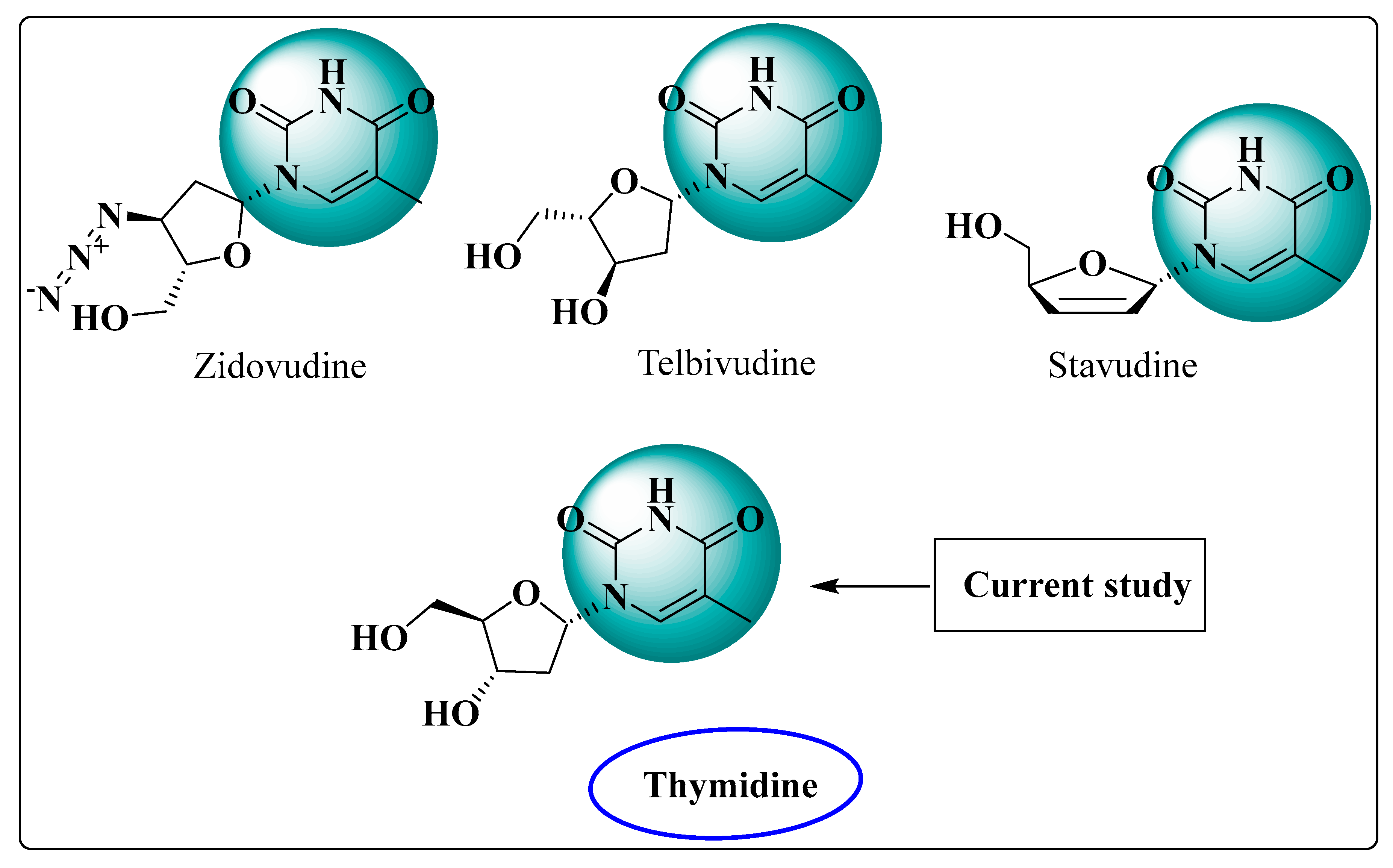
1. Introduction
2. Results and Discussion
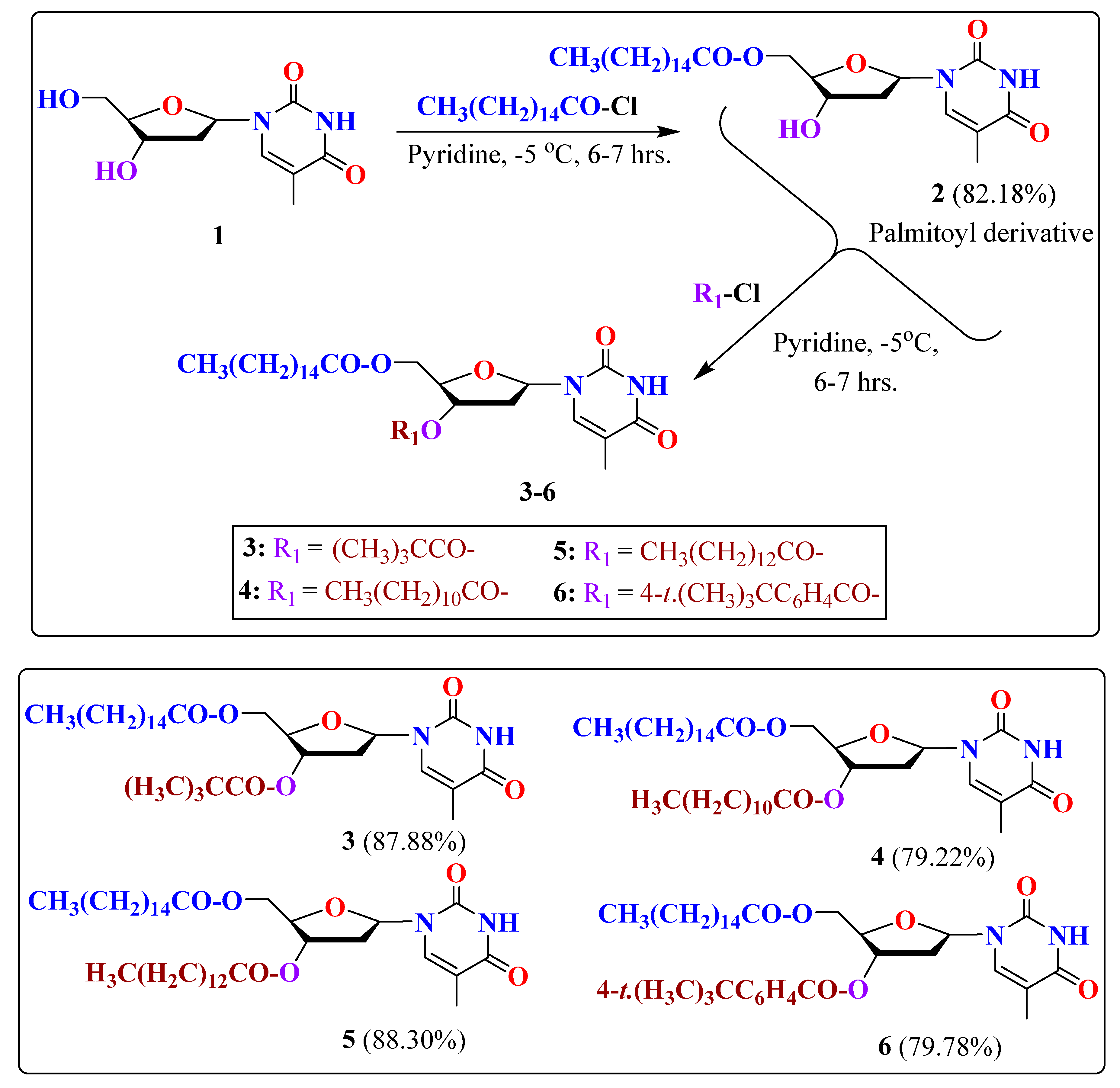
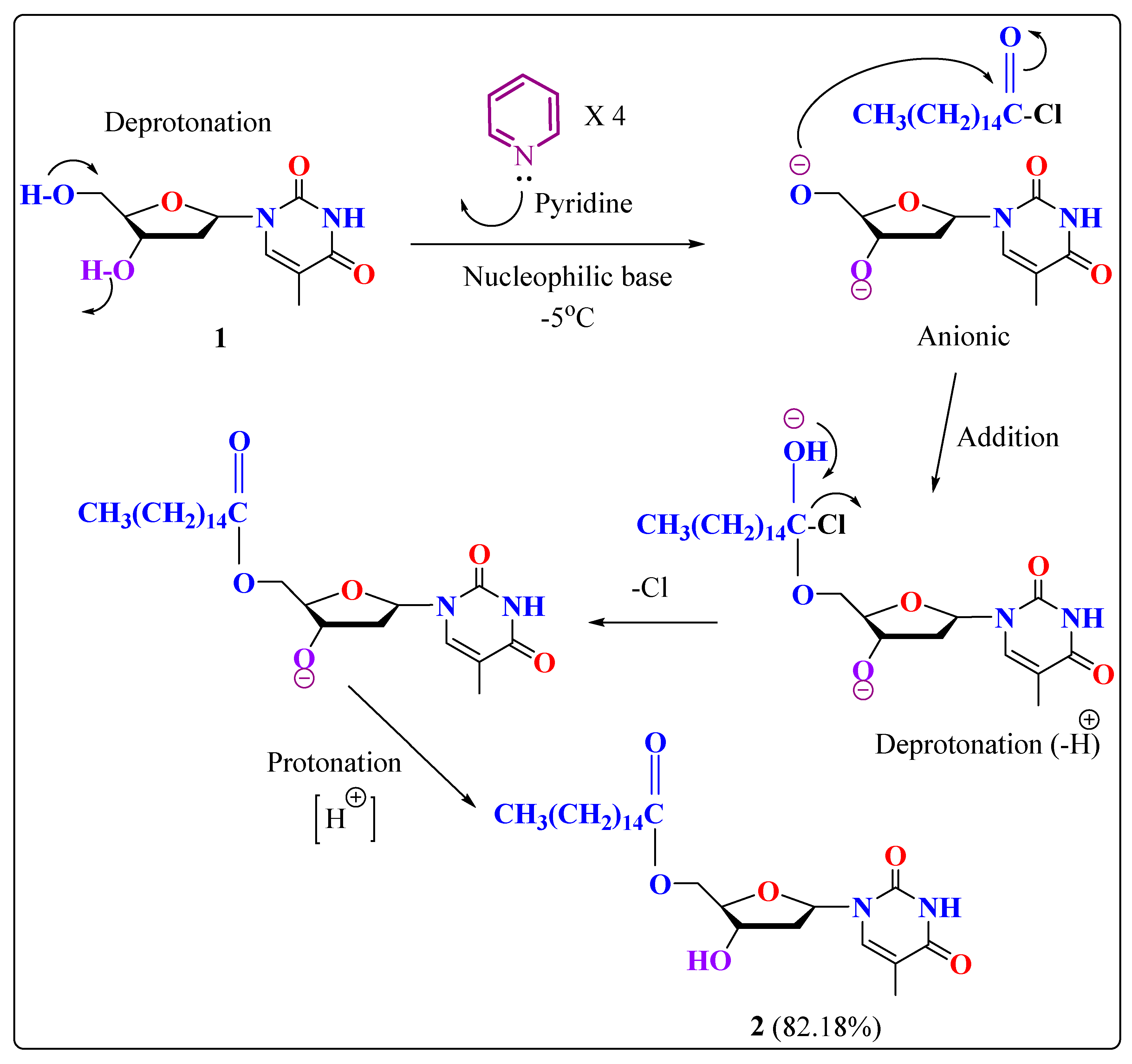
2.1. Chemistry
2.2. Characterization
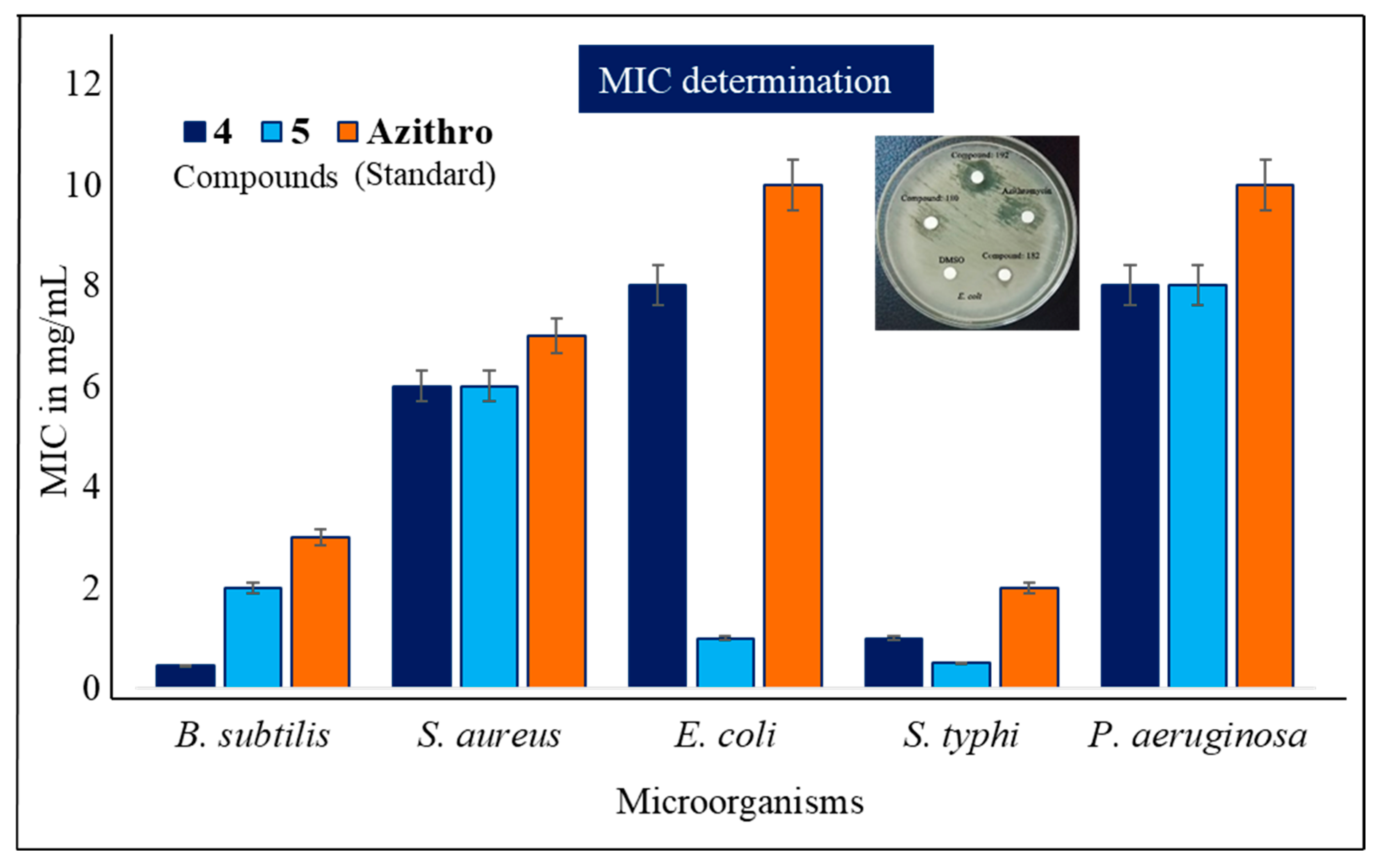
2.3. Antibacterial Potential
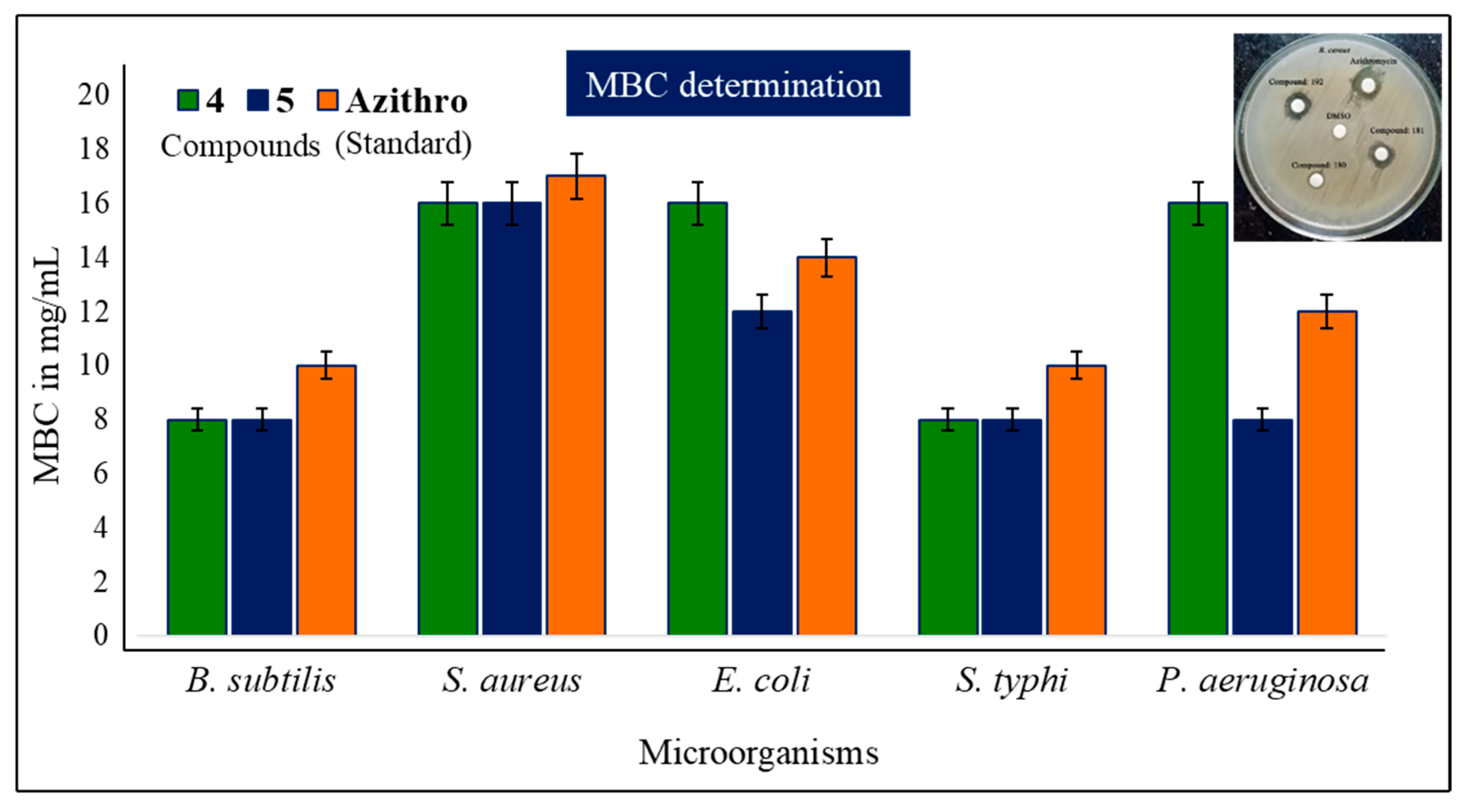
2.4. MIC and MBC Evaluation
2.5. Antifungal Susceptibility
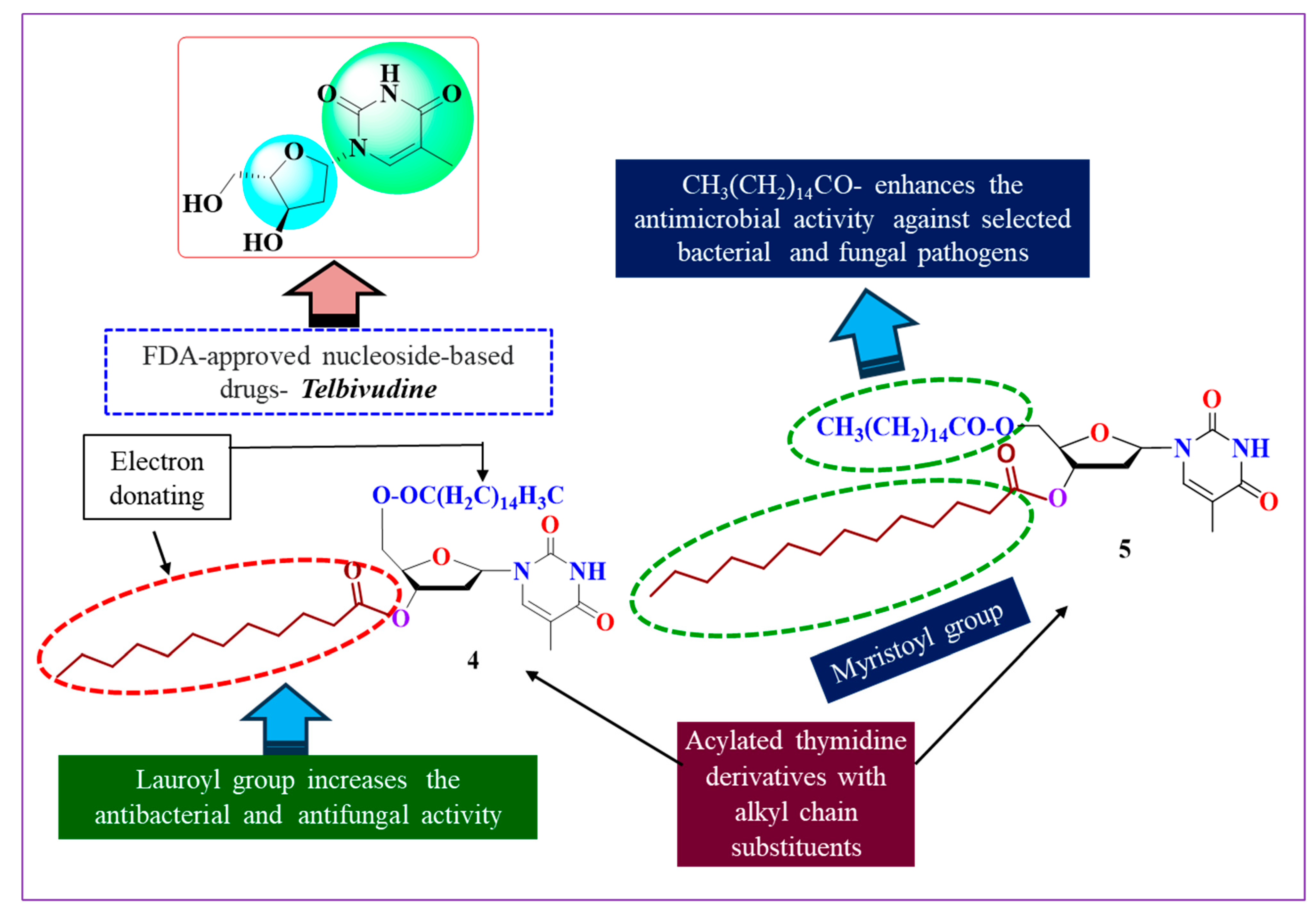
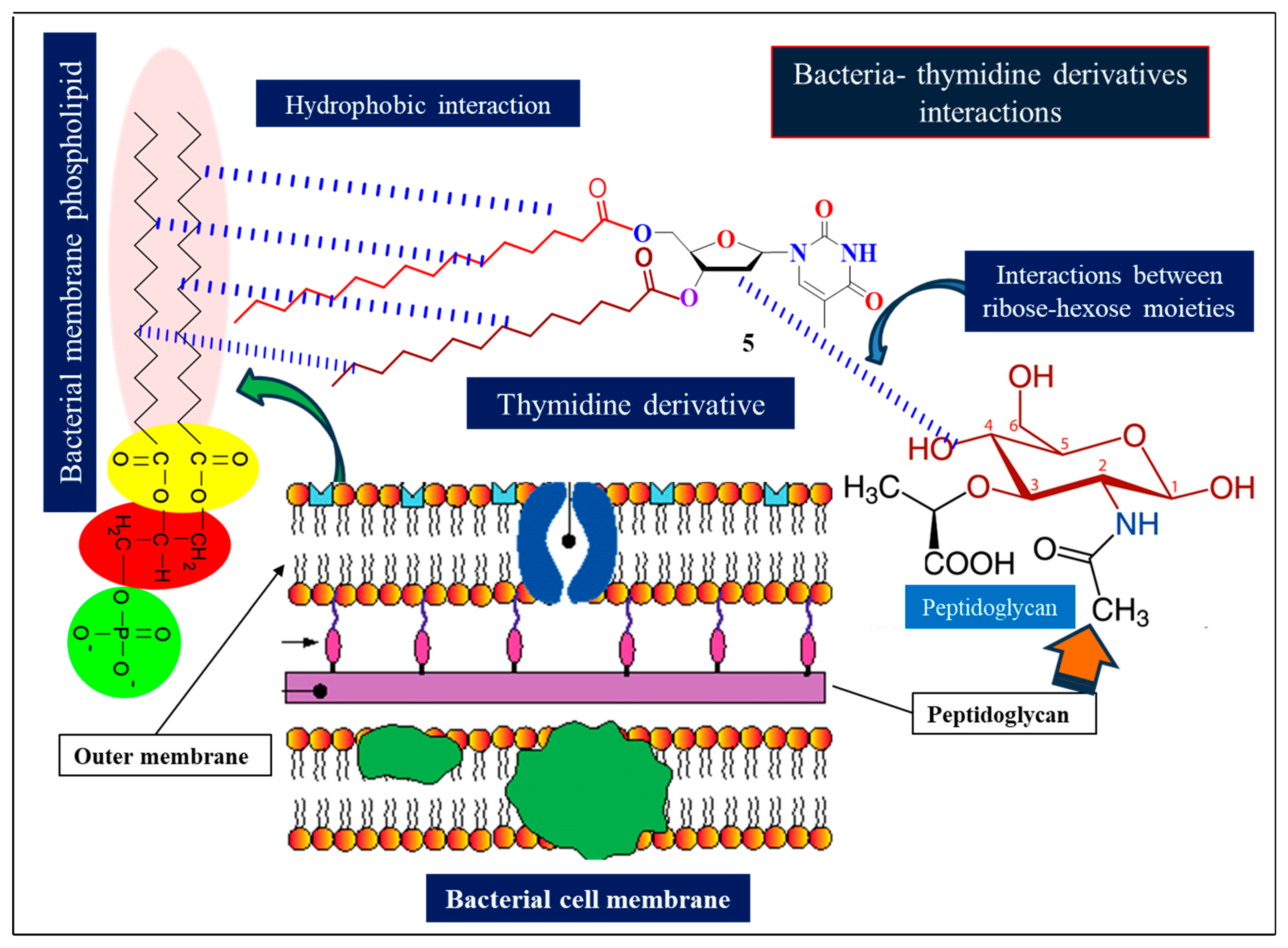
2.6. SAR Assessment
- ✓
- The potential implications for long-chain aliphatic hydrocarbons (lauroyl, myristoyl);
- ✓
- The binding acquaintances are enhanced by the attachment of the palmitoyl-substituted acyl group at position 5′;
- ✓
- By combining the acyl groups that are substituted at position 3′ with the lauroyl and myristoyl groups, the binding affinity is increased;
- ✓
- The presence of pyrimidine and ribose rings is also important for the efficacy of the activity.
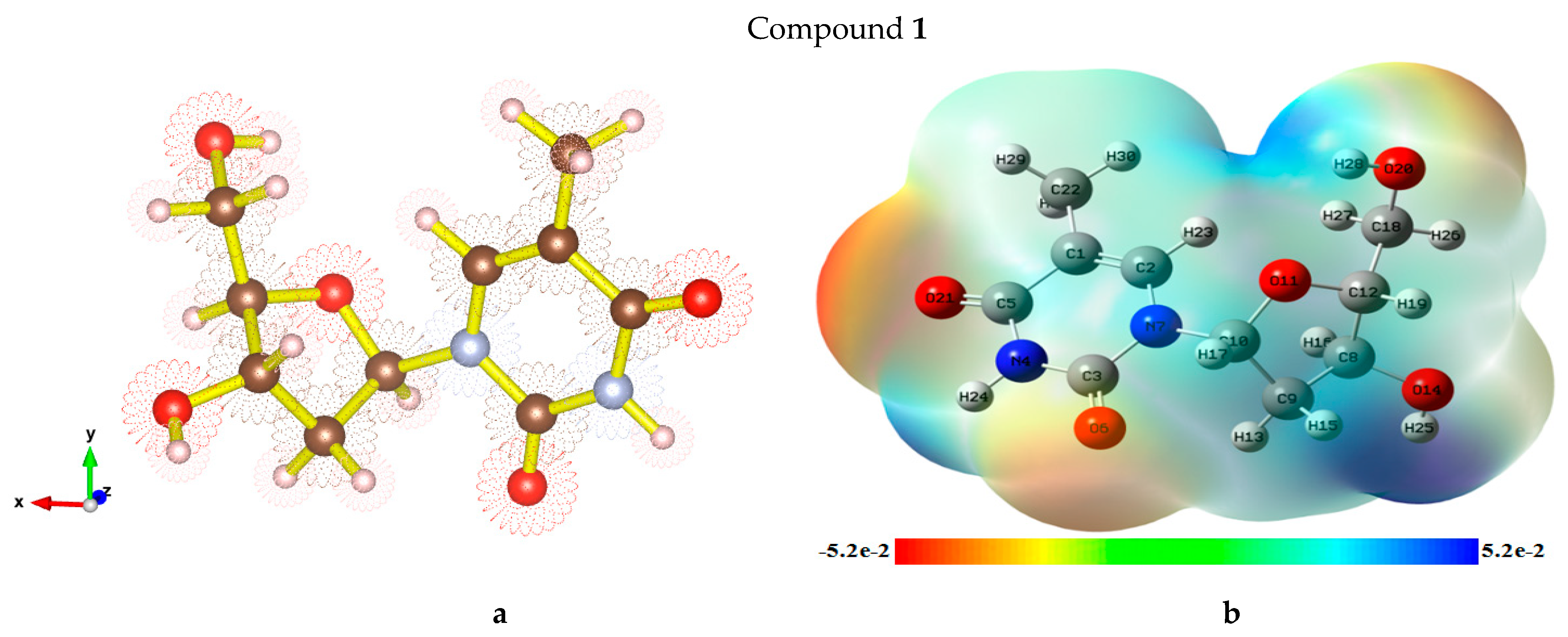
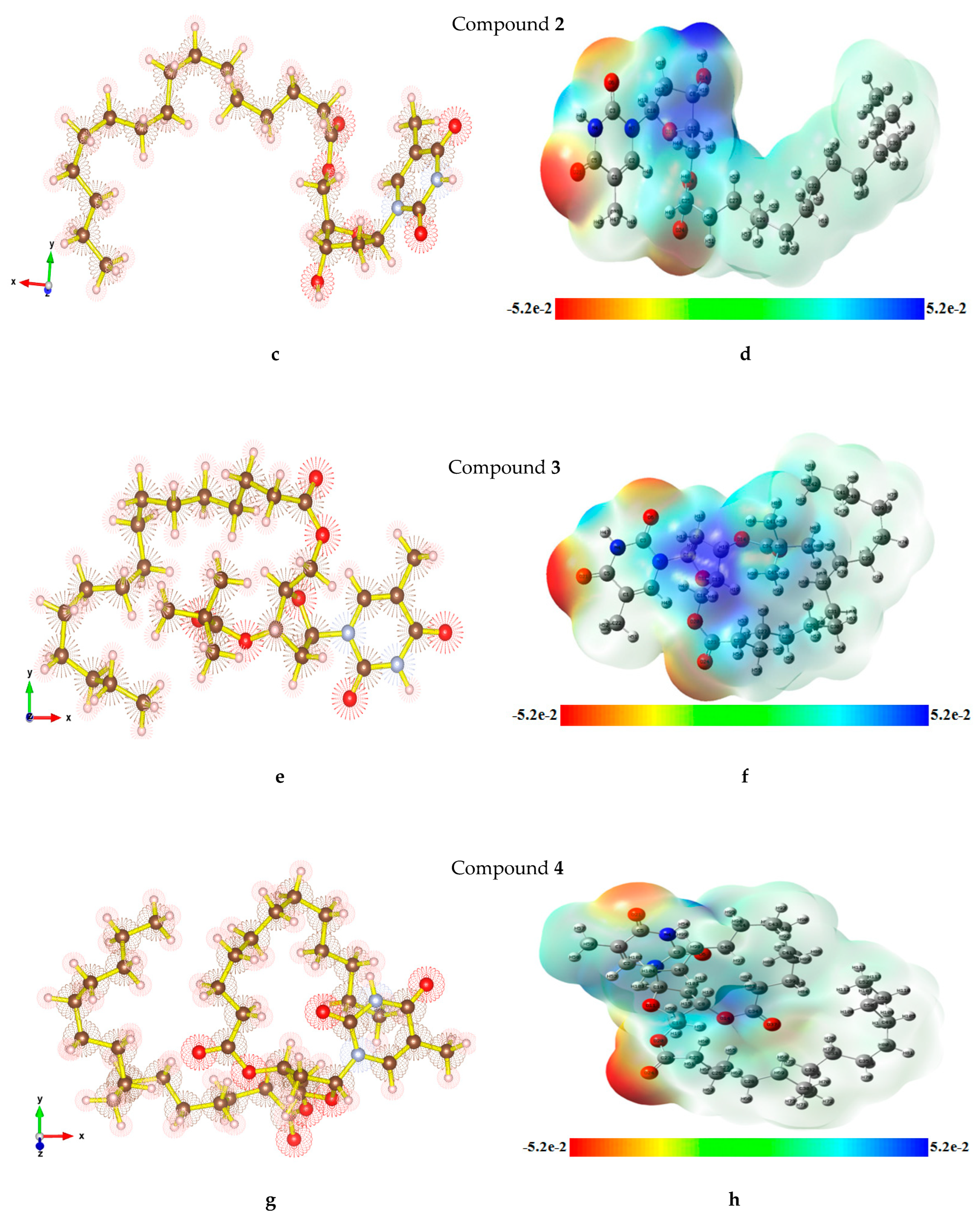
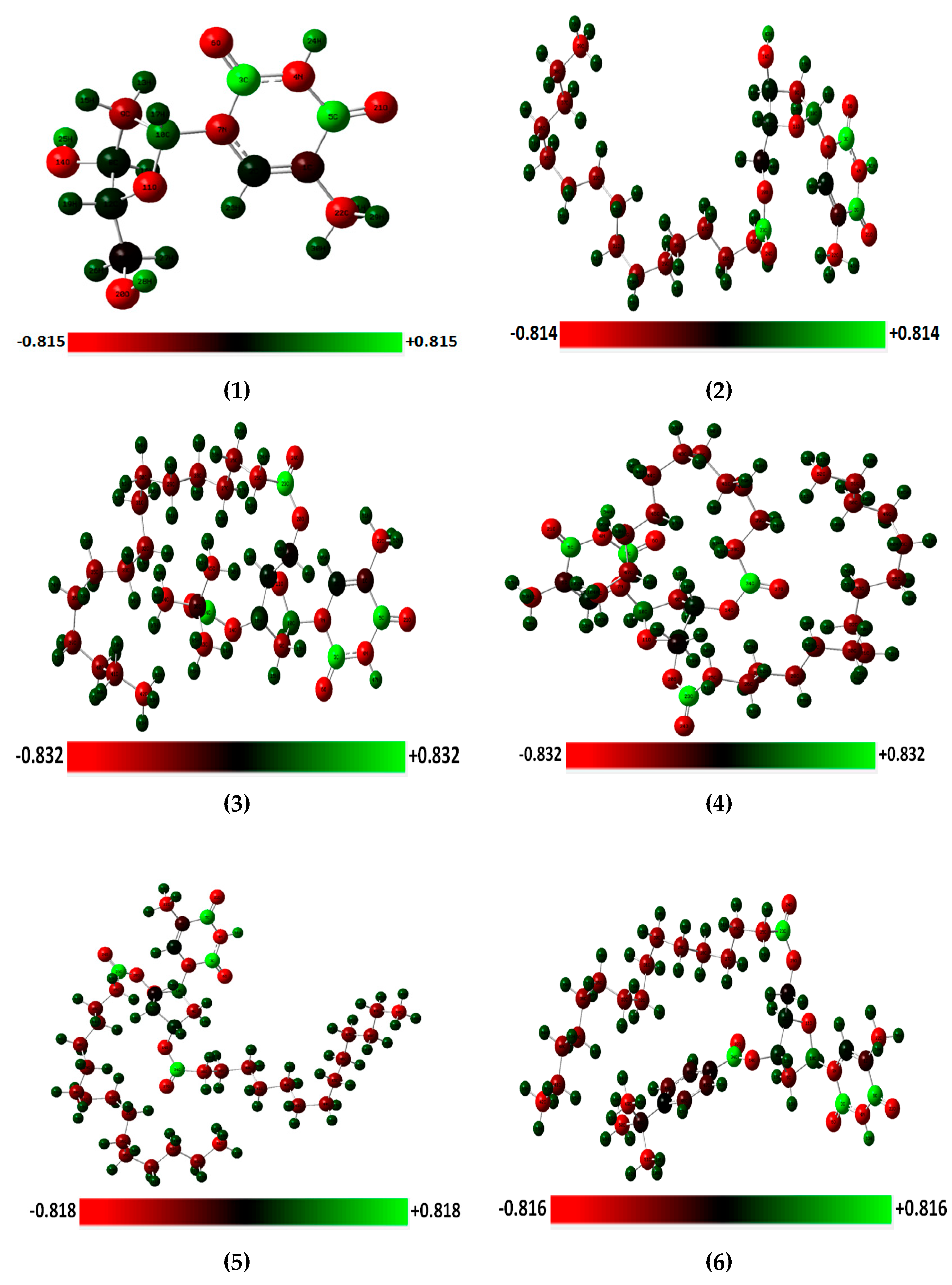
2.7. Optimized Structures and MEP
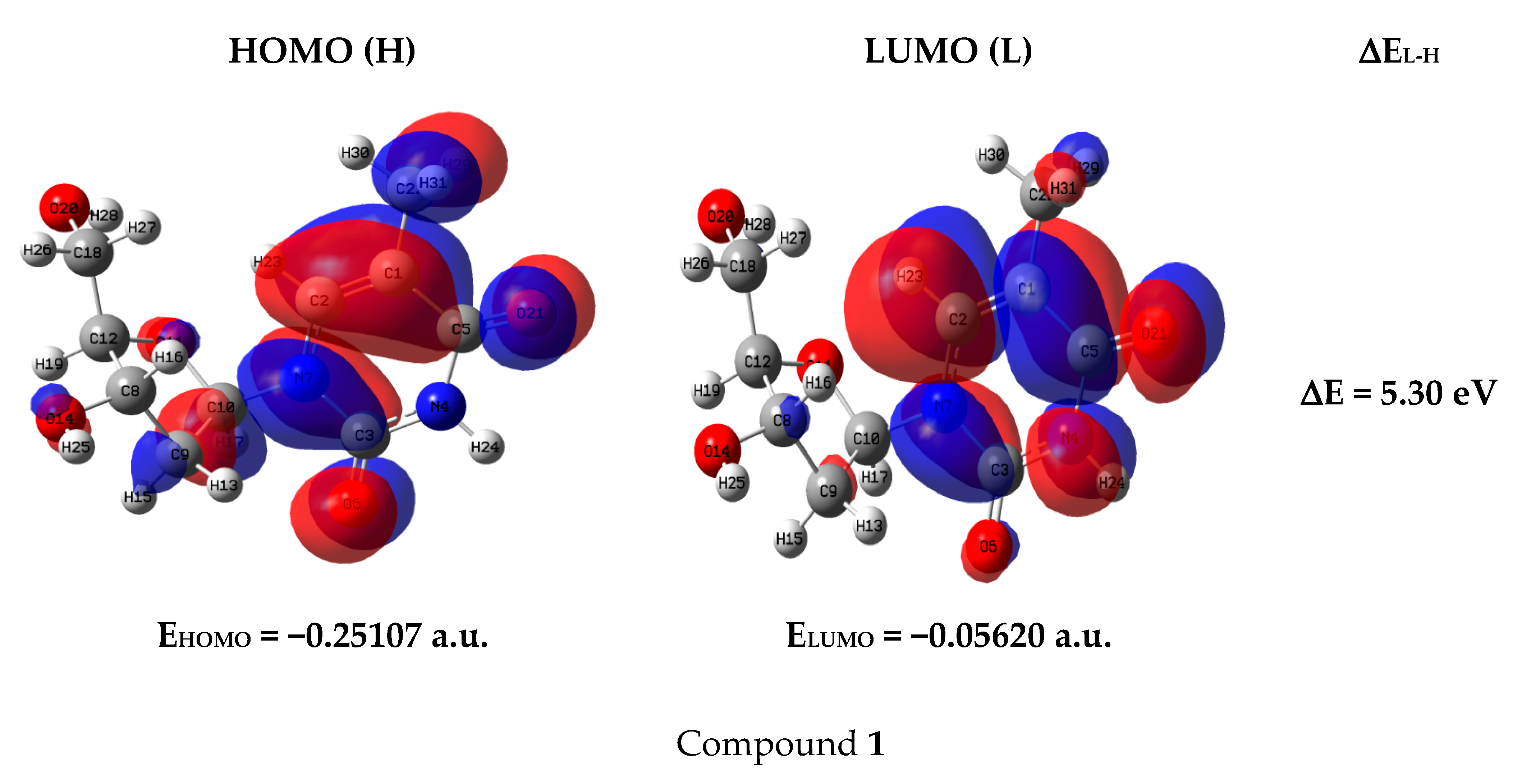
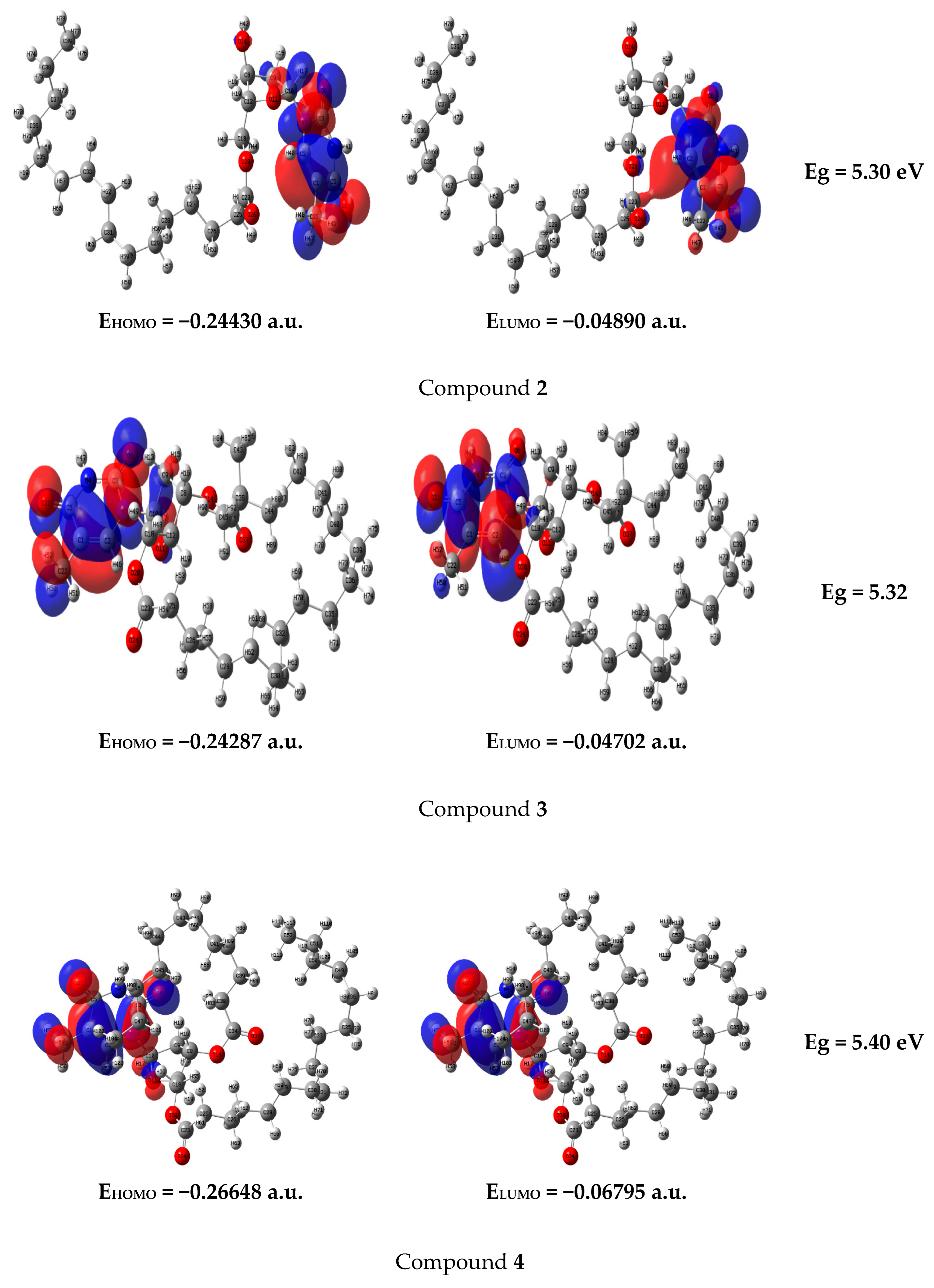
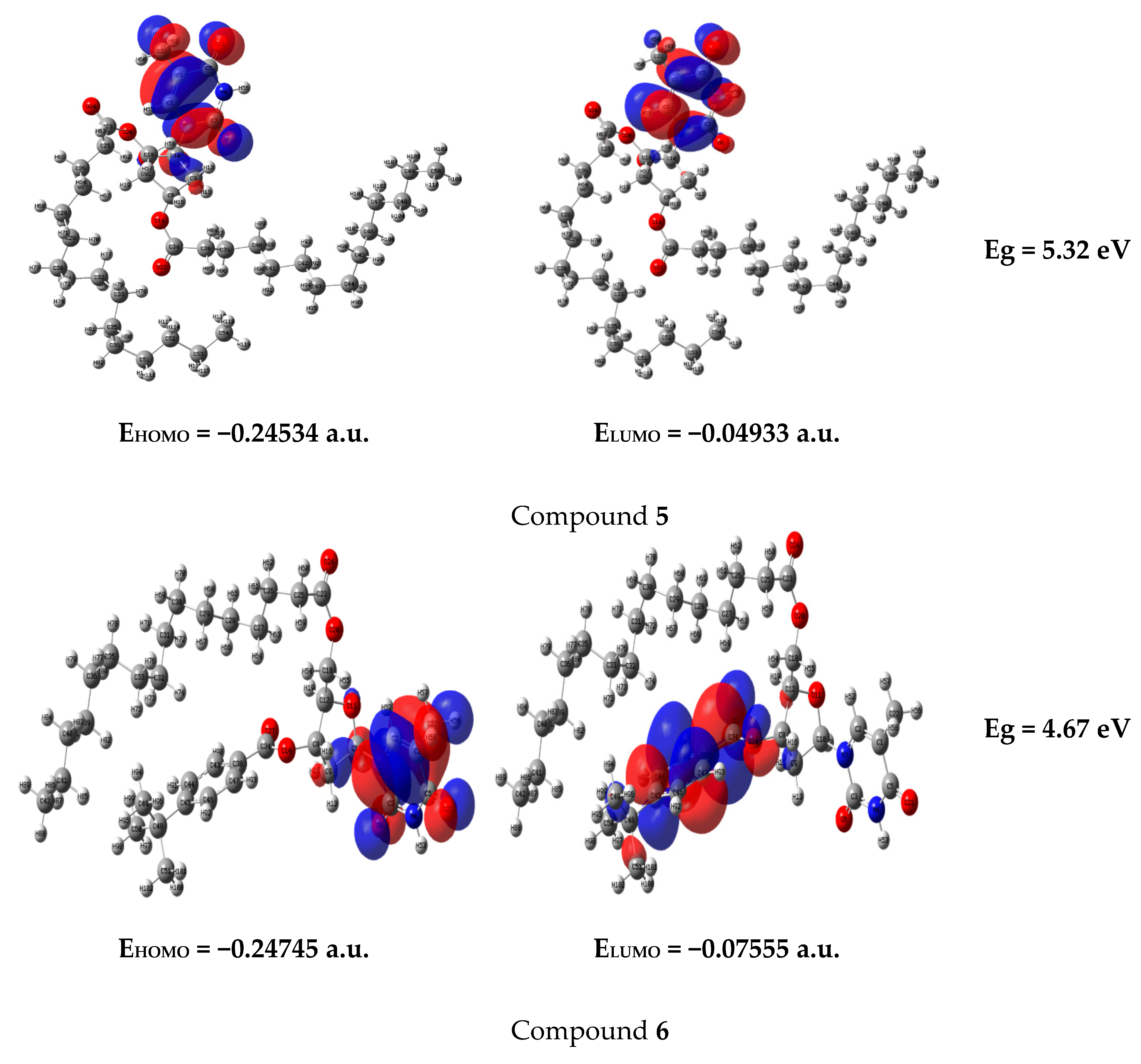
2.8. Frontier Molecular Orbital (FMO)
2.9. Global Reactivity Parameters
2.10. 3D-NBO Charge
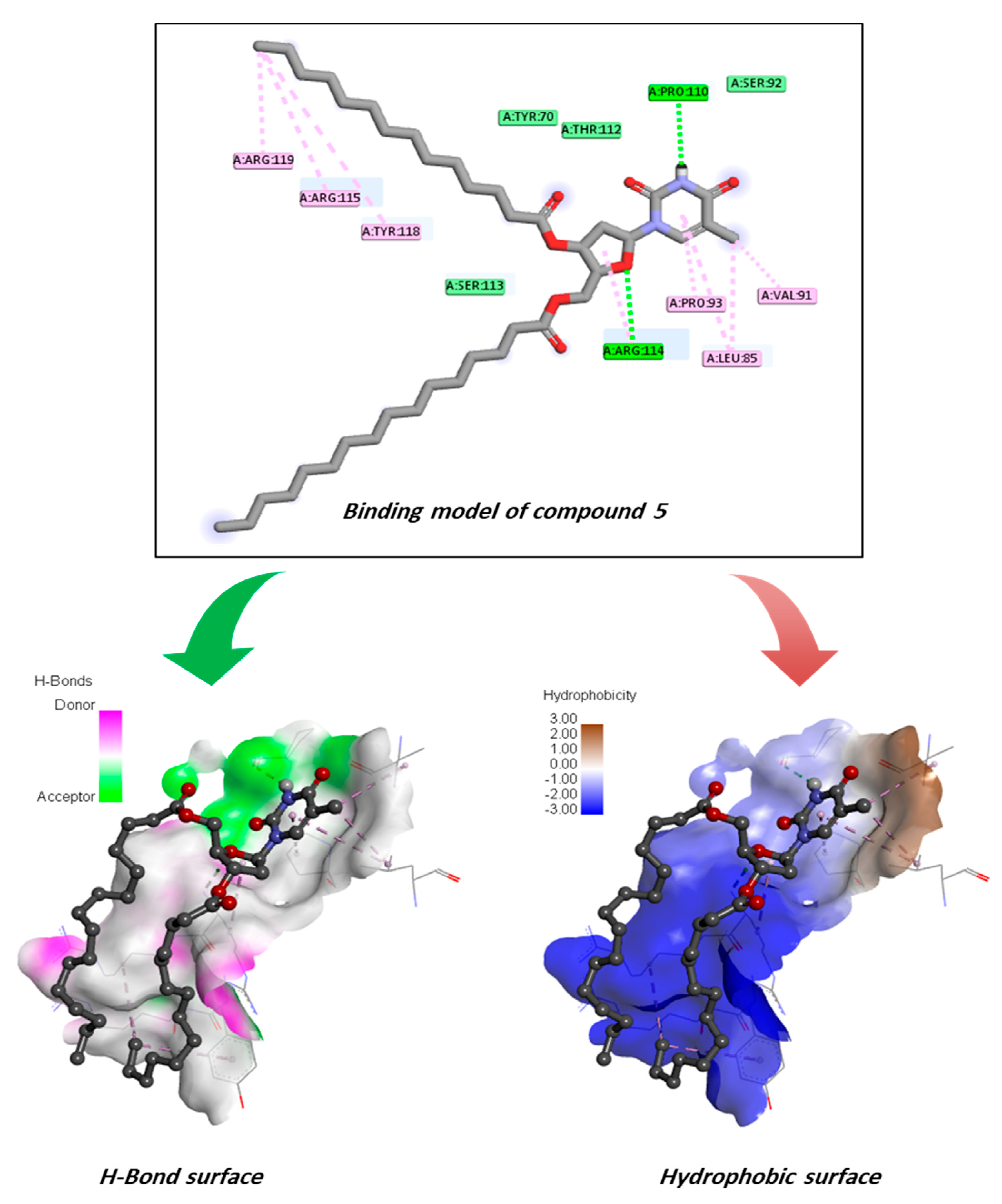
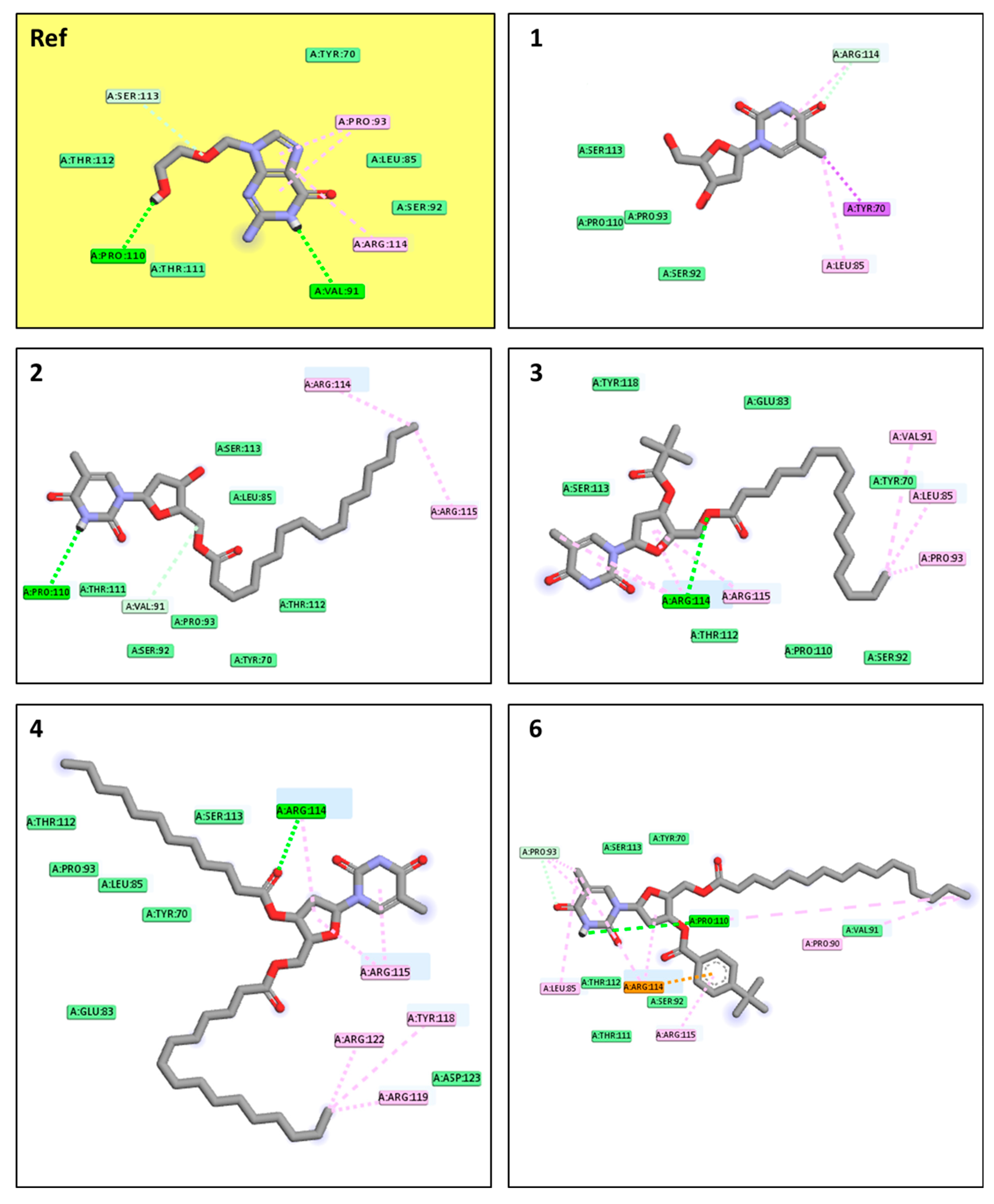
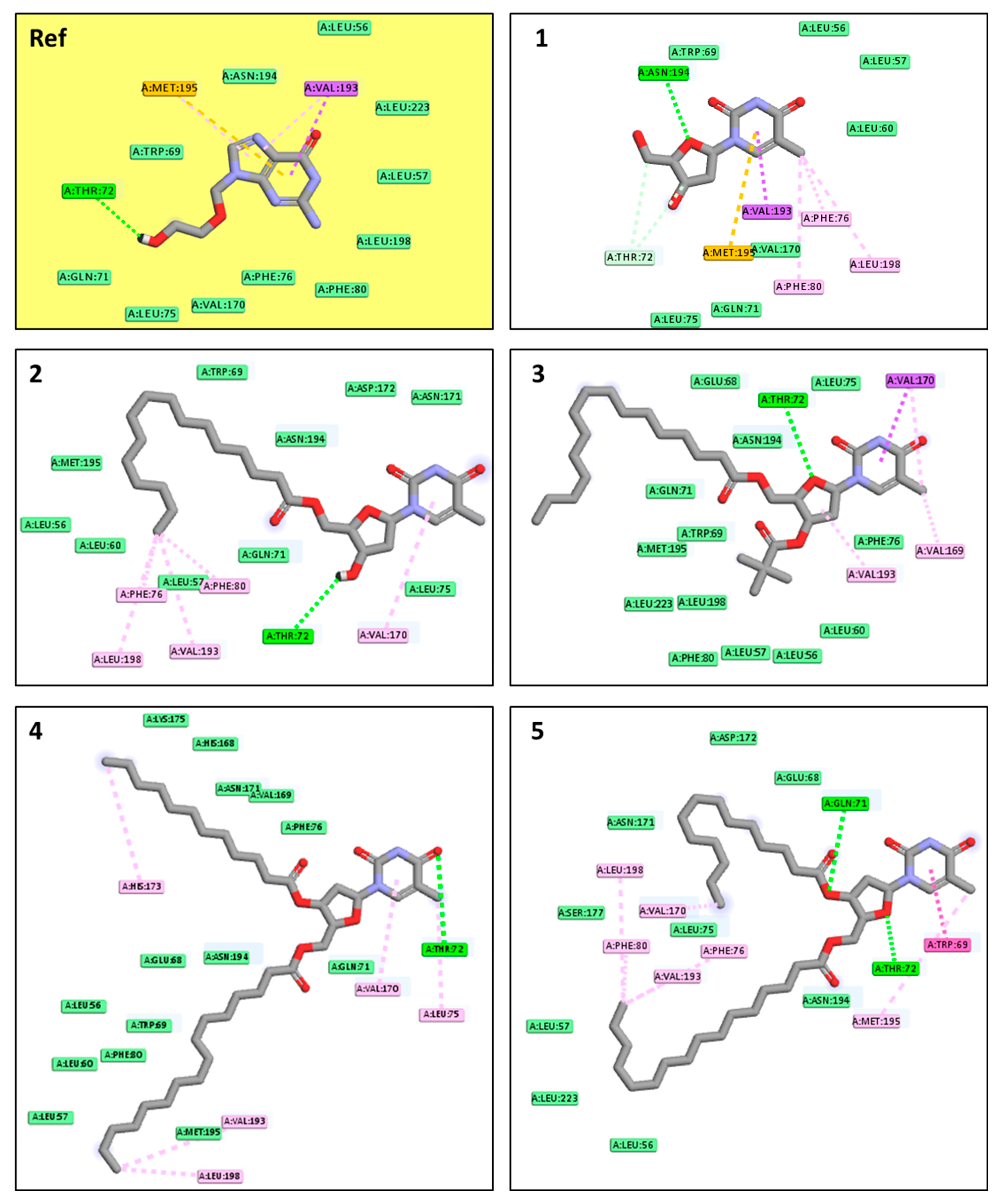
2.11. Molecular Docking Pose and Interaction Analysis of the Monkeypox Virus
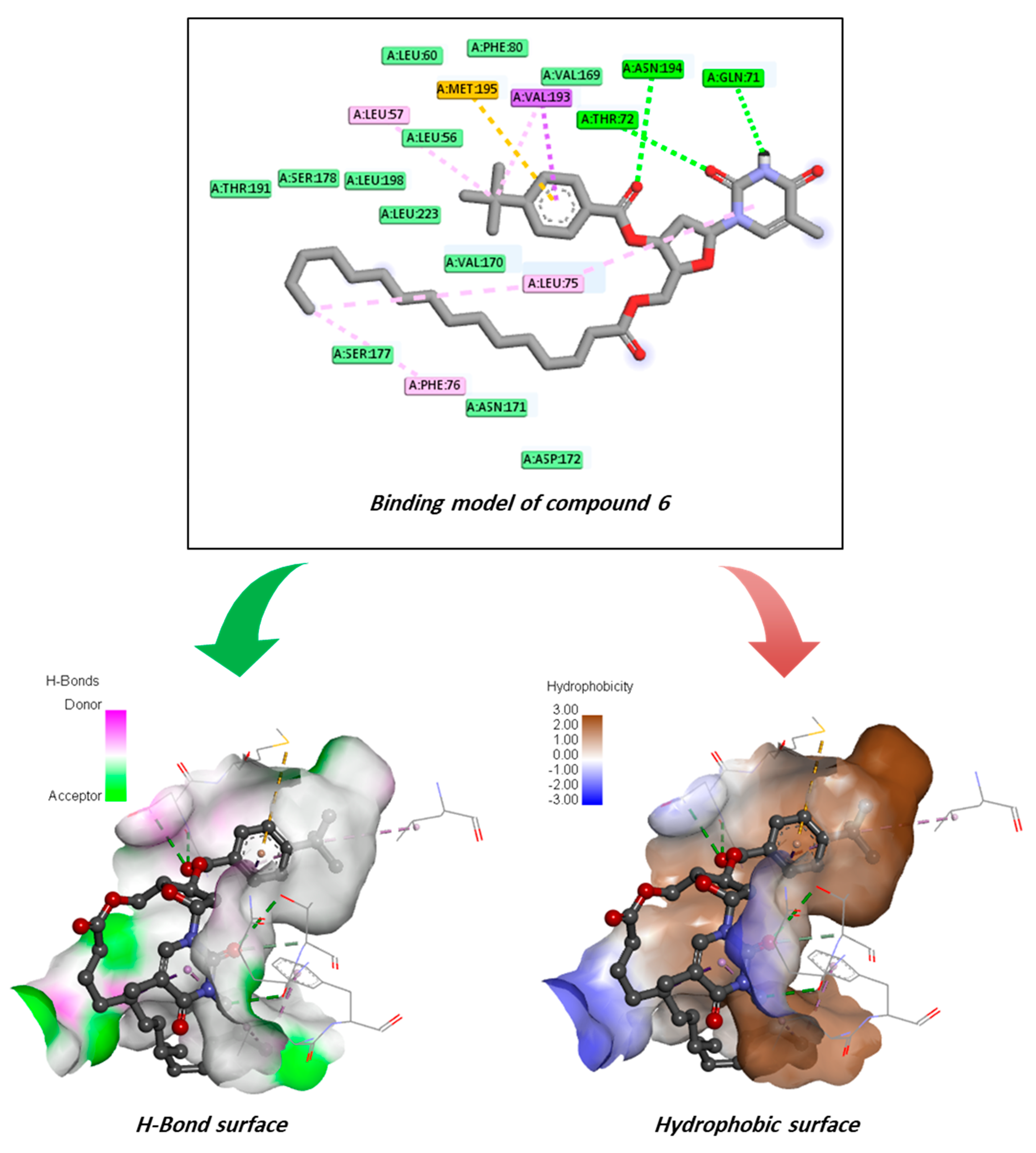
2.12. Molecular Docking Pose and Interaction Analysis of the Marburg Virus
2.13. ADMET Prediction
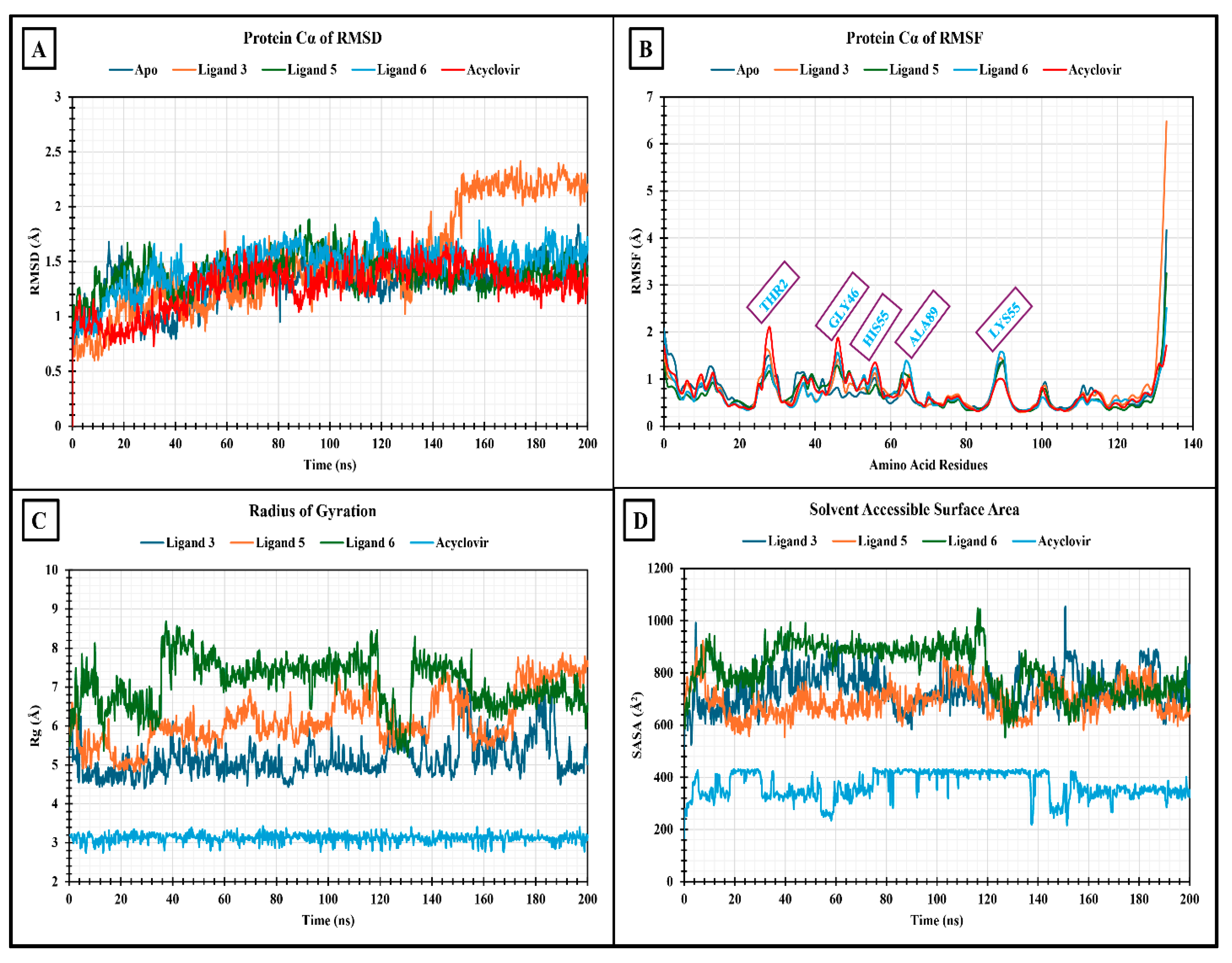
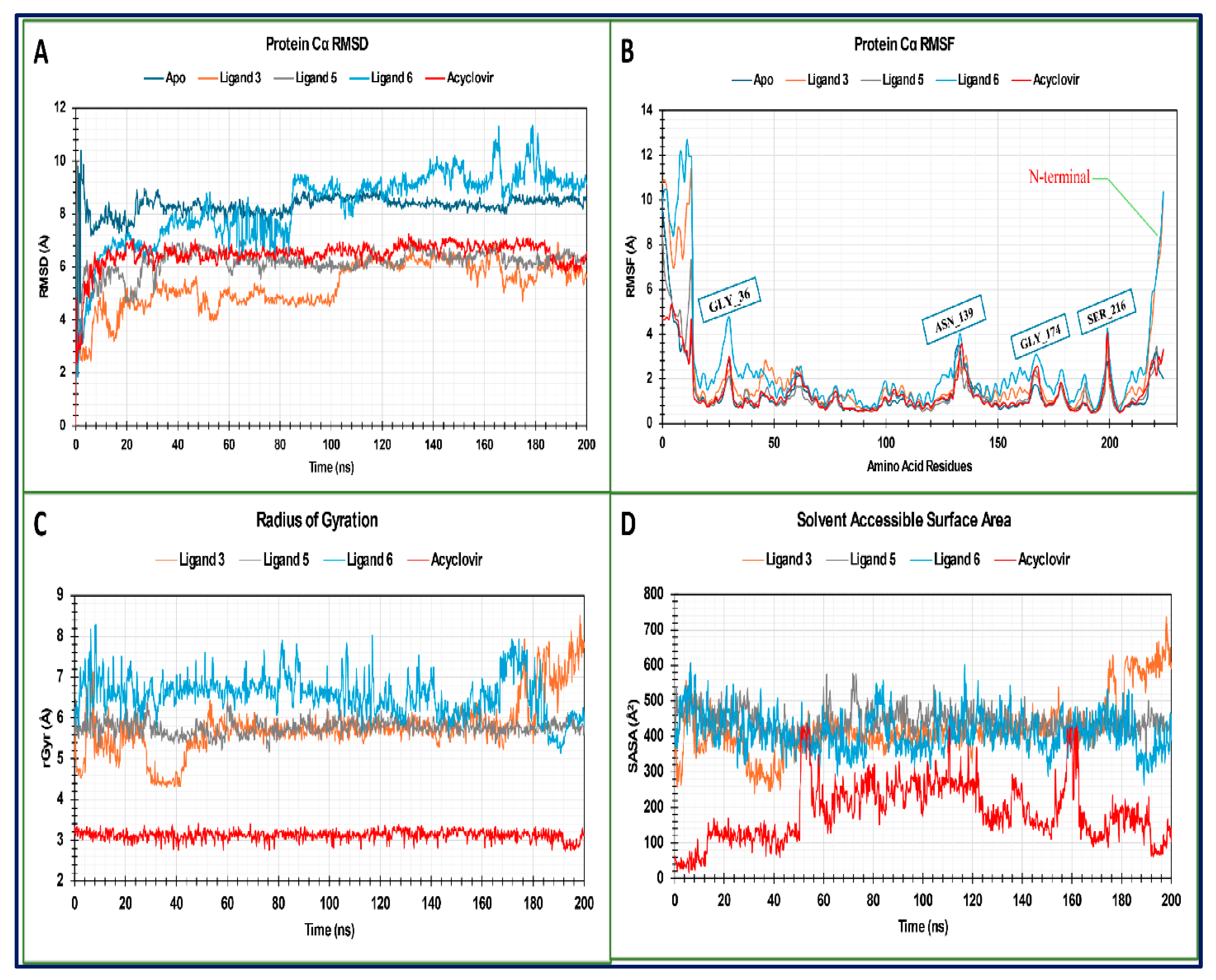
2.14. MD Trajectory Data Analysis of Monkeypox (Mpox) Virus
2.14.1. RMSD Analysis
2.14.2. RMSF Analysis
2.14.3. Radius of Gyration (Rg) Analysis
2.14.4. Solvent Accessible Surface Area (SASA)
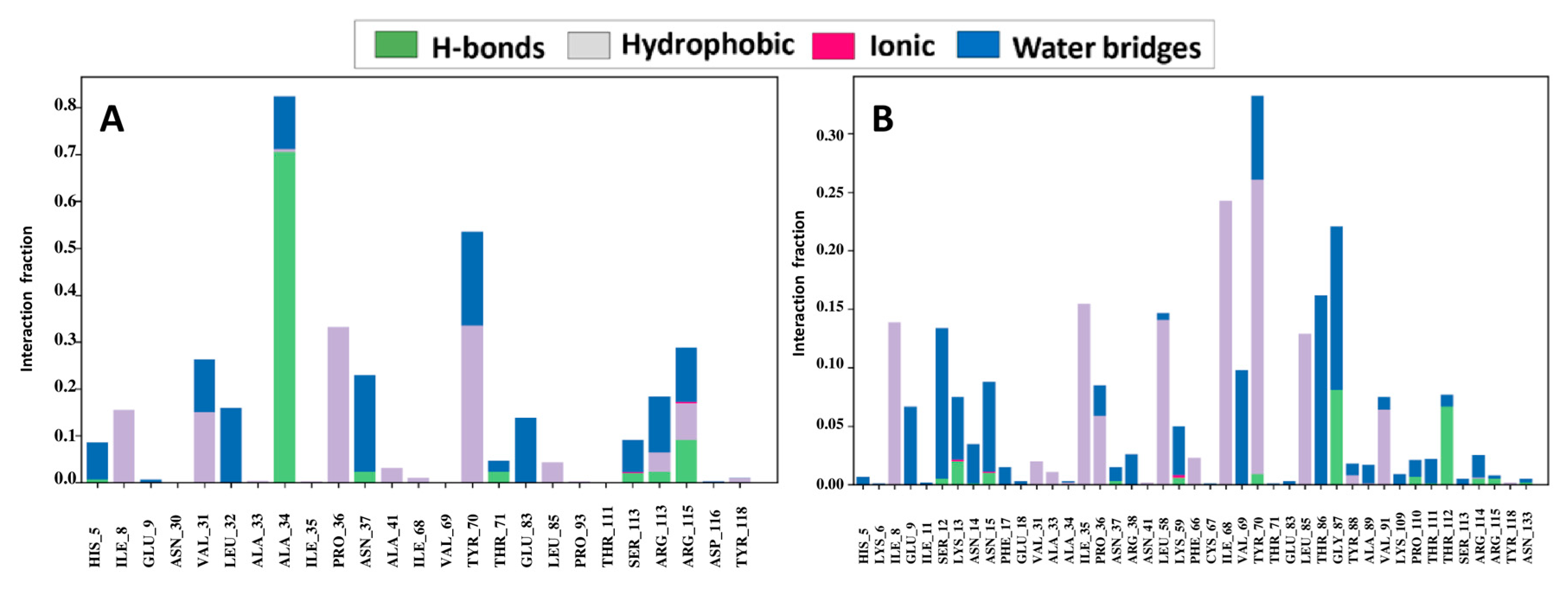
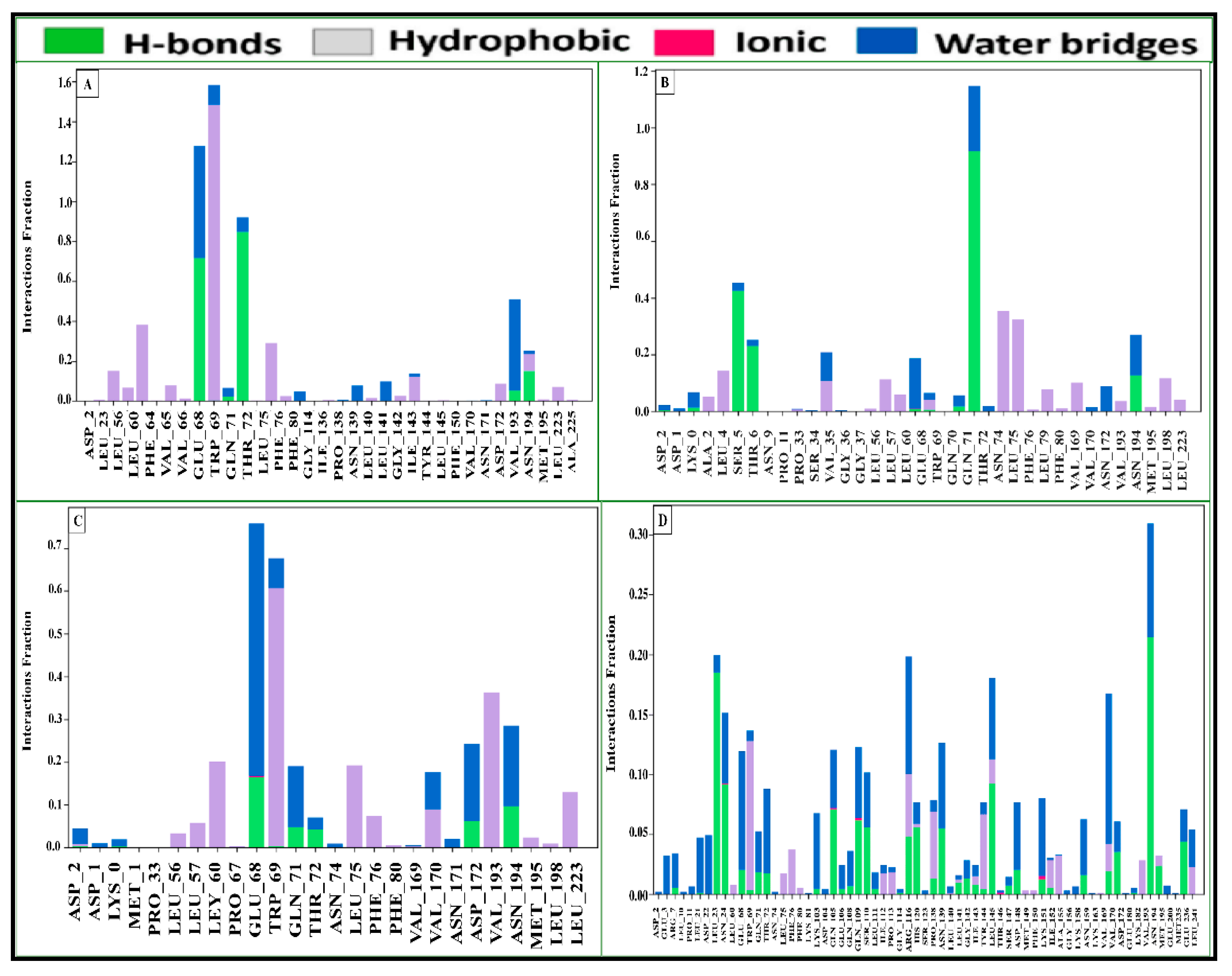
2.15. Assessment of Protein–Ligand Interactions
2.16. MD Trajectory Data Analysis of Marburg Virus
2.16.1. RMSD Analysis
2.16.2. RMSF Analysis
2.16.3. The Radius of Gyration (rGyr) Analysis
2.16.4. Solvent Accessible Surface Area (SASA)
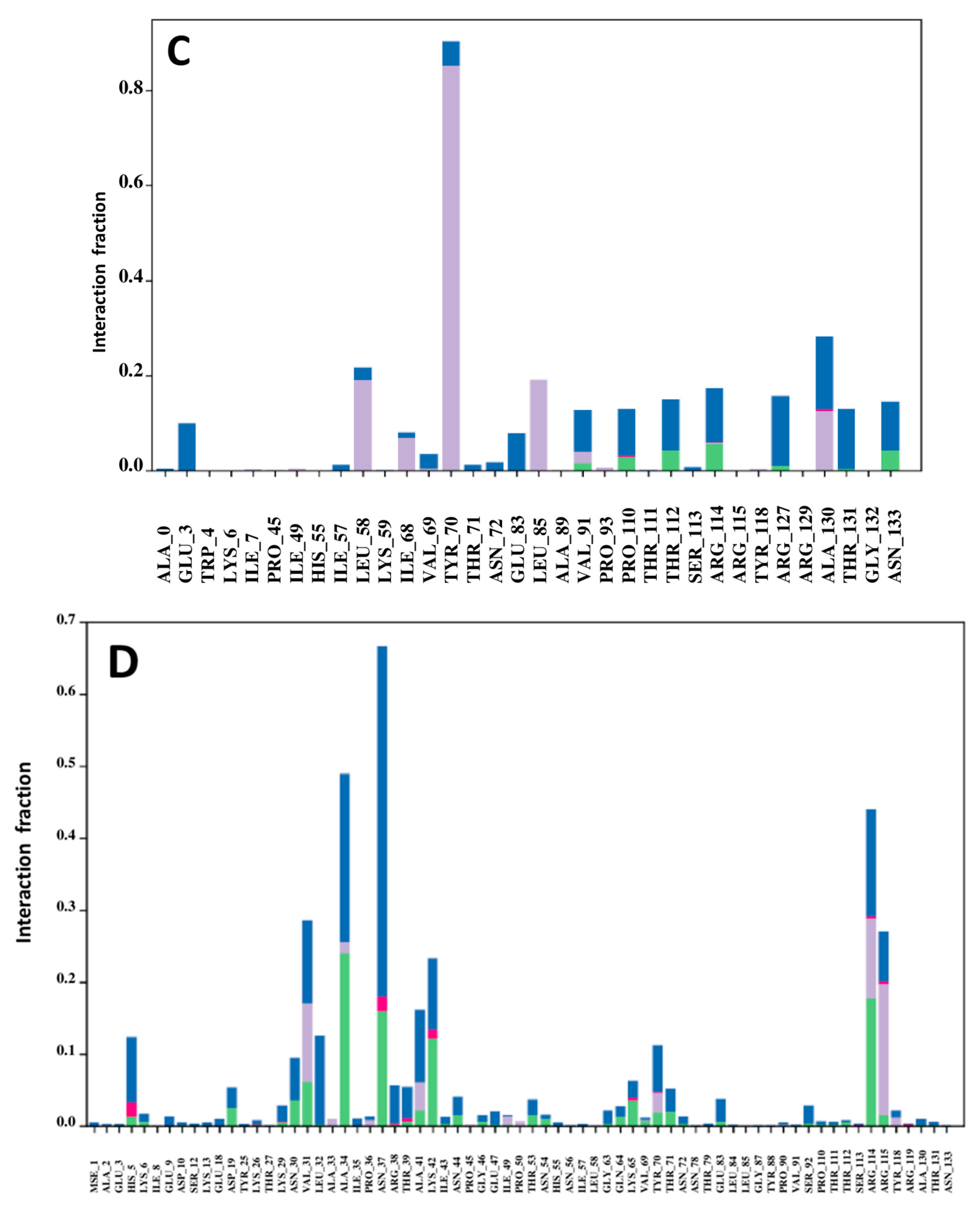
2.17. Protein–Ligand Contact Analysis
3. Materials and Methods
3.1. Materials, Samples, and Equipment
3.2. Synthesis
3.2.1. The 5′-O-(Palmitoyl)thymidine (2)
3.2.2. The General Procedure for the Preparation of Palmitoyl Derivatives (3–6)
5′-O-Palmitoyl-3′-O-(pivaloyl)thymidine (3)
3.2.3. The 3′-O-Lauroyl-5′-O-(palmitoyl)thymidine (4)
3.2.4. The 3′-O-Myristoyl-5′-O-(palmitoyl)thymidine (5)
3.2.5. The 3′-O-(4-t-Butylbenzoyl)-5′-O-(palmitoyl)thymidine (6)
3.3. In Vitro Antibacterial Activity Test
3.4. Determination of the MIC and MBC via the Broth Microdilution Method
3.5. Determination of Mycelial Growth
3.6. Structure–Activity Relationship
3.7. Computational Details
3.8. Protein Selection and Molecular Docking
3.9. ADMET Properties Involving Pharmacokinetic Prediction
3.10. Molecular Dynamics Simulation
4. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADMET | absorption, distribution, metabolism, excretion, and toxicity |
| DFT | density functional theory |
| FMO | frontier molecular orbital |
| HOMO | highest occupied molecular orbital |
| LUMO | lowest unoccupied molecular orbital |
| MBC | minimum bactericidal concentration |
| MD | molecular dynamics |
| MEP | molecular electrostatic potential |
| MIC | minimum inhibitory concentration |
| NBO | natural bond charge |
| PASS | prediction of substance activity spectra |
| SAR | structure–activity relationship |
References
- Li, N.; Smith, T.J.; Zong, M.-H. Biocatalytic transformation of nucleoside derivatives. Biotechnol. Adv. 2010, 28, 348–366. [Google Scholar] [CrossRef] [PubMed]
- Whitley, R.J.; Roizman, B. Herpes simplex virus infections. Lancet 2001, 357, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Staszewski, S.; Morales-Ramirez, J.; Tashima, K.T.; Rachlis, A.; Skiest, D.; Stanford, J.; Stryker, R.; Johnson, P.; Labriola, D.F.; Farina, D. Efavirenz plus zidovudine and lamivudine, efavirenz plus indinavir, and indinavir plus zidovudine and lamivudine in the treatment of HIV-1 infection in adults. N. Engl. J. Med. 1999, 341, 1865–1873. [Google Scholar] [CrossRef] [PubMed]
- Schettler, C. Virus hepatitis of geese 3. Properties of the causal agent. Avian Pathol. 1973, 2, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Mayer, R.J.; Davis, R.B.; Schiffer, C.A.; Berg, D.T.; Powell, B.L.; Schulman, P.; Omura, G.A.; Moore, J.O.; McIntyre, O.R.; Frei, E. Intensive postremission chemotherapy in adults with acute myeloid leukemia. N. Engl. J. Med. 1994, 331, 896–903. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Bulbul, M.Z.; Chowdhury, T.S.; Misbah, M.M.; Ferdous, J.; Dey, S.; Hasan, I.; Fujii, Y.; Ozeki, Y.; Kawsar, S.M. Synthesis of new series of pyrimidine nucleoside derivatives bearing the acyl moieties as potential antimicrobial agents. Pharmacia 2021, 68, 23–34. [Google Scholar] [CrossRef]
- Yuen, G.J.; Weller, S.; Pakes, G.E. A review of the pharmacokinetics of abacavir. Clin. Pharmacokinet. 2008, 47, 351–371. [Google Scholar] [CrossRef]
- Elwell, L.P.; Ferone, R.; Freeman, G.A.; Fyfe, J.A.; Hill, J.A.; Ray, P.H.; Richards, C.A.; Singer, S.C.; Knick, V.B.; Rideout, J.L. Antibacterial activity and mechanism of action of 3′-azido-3′-deoxythymidine (BW A509U). Antimicrob. Agents Chemother. 1987, 31, 274–280. [Google Scholar] [CrossRef]
- Fischer, J.; Ganellin, C.R. Analogue-based drug discovery. Chem. Int. Newsmag. IUPAC 2010, 32, 12–15. [Google Scholar]
- Styrt, B.; Freiman, J.P. Hepatotoxicity of antiviral agents. Gastroenterol. Clin. N. Am. 1995, 24, 839–852. [Google Scholar] [CrossRef]
- Machado-Vieira, R.; Salvadore, G.; DiazGranados, N.; Ibrahim, L.; Latov, D.; Wheeler-Castillo, C.; Baumann, J.; Henter, I.D.; Zarate, C.A. New therapeutic targets for mood disorders. Sci. World J. 2010, 10, 713–726. [Google Scholar] [CrossRef]
- Matsumoto, R.; Fujii, Y.; Kawsar, S.M.; Kanaly, R.A.; Yasumitsu, H.; Koide, Y.; Hasan, I.; Iwahara, C.; Ogawa, Y.; Im, C.H. Cytotoxicity and glycan-binding properties of an 18 kDa lectin isolated from the marine sponge Halichondria okadai. Toxins 2012, 4, 323–338. [Google Scholar] [CrossRef]
- Fujii, Y.; Kawsar, S.M.; Matsumoto, R.; Yasumitsu, H.; Ishizaki, N.; Dogasaki, C.; Hosono, M.; Nitta, K.; Hamako, J.; Taei, M. A D-galactose-binding lectin purified from coronate moon turban, Turbo (Lunella) coreensis, with a unique amino acid sequence and the ability to recognize lacto-series glycosphingolipids. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2011, 158, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Sartorius, B.; Gray, A.P.; Weaver, N.D.; Aguilar, G.R.; Swetschinski, L.R.; Ikuta, K.S.; Mestrovic, T.; Chung, E.; Wool, E.E.; Han, C. The burden of bacterial antimicrobial resistance in the WHO African region in 2019: A cross-country systematic analysis. Lancet Glob. Health 2024, 12, e201–e216. [Google Scholar] [CrossRef] [PubMed]
- Ruckert, A.; Lake, S.; Van Katwyk, S.R. Developing a protocol on antimicrobial resistance through WHO’s pandemic treaty will protect lives in future pandemics. Global Health 2024, 20, 10. [Google Scholar] [CrossRef]
- Salam, M.A.; Al-Amin, M.Y.; Salam, M.T.; Pawar, J.S.; Akhter, N.; Rabaan, A.A.; Alqumber, M.A.A. Antimicrobial Resistance: A Growing Serious Threat for Global Public Health. Healthcare 2023, 11, 1946. [Google Scholar] [CrossRef]
- Asghar, A.; Khalid, A.; Baqar, Z.; Hussain, N.; Saleem, M.Z.; Sairash, K.; Rizwan, A. An insights into emerging trends to control the threats of antimicrobial resistance (AMR): An address to public health risks. Arch. Microbiol. 2024, 206, 72. [Google Scholar] [CrossRef]
- Tang, K.W.K.; Millar, B.C.; Moore, J.E. Antimicrobial resistance (AMR). Braz. J. Biomed. Sci. 2023, 80, 11387. [Google Scholar] [CrossRef]
- Murray, C.J.L.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Aguilar, G.R.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Abdallah, E.M.; Alhatlani, B.Y.; de Paula Menezes, R.; Martins, C.H.G. Back to nature: Medicinal plants as promising sources for antibacterial drugs in the post-antibiotic era. Plants 2023, 12, 3077. [Google Scholar] [CrossRef] [PubMed]
- Schitter, G.; Wrodnigg, T.M. Update on carbohydrate-containing antibacterial agents. Expert Opin. Drug Discov. 2009, 4, 315–356. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Dhara, A.K.; Nayak, A.K. Biological macromolecules: Sources, properties, and functions. In Biological Macromolecules; Elsevier: Amsterdam, The Netherlands, 2022; pp. 3–22. [Google Scholar]
- Chowdhury, S.A.; Bhuiyan, M.M.; Ozeki, Y.; Kawsar, S.M. Simple and rapid synthesis of some nucleoside derivatives: Structural and spectral characterization. Curr. Chem. Lett. 2016, 5, 83–92. [Google Scholar] [CrossRef]
- Maowa, J.; Alam, A.; Rana, K.M.; Dey, S.; Hosen, A.; Fujii, Y.; Hasan, I.; Ozeki, Y.; Kawsar, S.M. Synthesis, characterization, synergistic antimicrobial properties and molecular docking of sugar modified uridine derivatives. Ovidius. Univ. Ann. Chem 2021, 32, 6–21. [Google Scholar] [CrossRef]
- Bhuiyan, T.S.; Said, M.A.; Bulbul, M.Z.; Ahmed, S.; Bhat, A.R.; Chalkha, M.; Kawsar, S.M. Synthesis, antimicrobial, and in silico studies of c5′-O-substituted cytidine derivatives: Cinnamoylation leads to improvement of antimicrobial activity. Nucleosides Nucleotides Nucleic Acids 2024, 43, 1472–1510. [Google Scholar] [CrossRef]
- Arifuzzaman, M.; Islam, M.M.; Rahman, M.M.; Rahman, M.A.; Kawsar, S.M. An efficient approach to the synthesis of thymidine derivatives containing various acyl groups: Characterization and antibacterial activities. ACTA Pharm. Sci. 2018, 56, 7–22. [Google Scholar] [CrossRef]
- Alam, A.; Hosen, M.A.; Islam, M.; Ferdous, J.; Fujii, Y.; Ozeki, Y.; Kawsar, S.M. Synthesis, Antibacterial and cytotoxicity assessment of modified uridine molecules. Curr. Adv. Chem. Biochem. 2021, 6, 114–129. [Google Scholar]
- Kawsar, S.M.; Ara, H.A.; Uddin, S.A.; Hossain, M.K.; Chowdhury, S.A.; Sanaullah, A.F.; Manchur, M.A.; Hasan, I.; Ogawa, Y.; Fujii, Y. Chemically modified uridine molecules incorporating acyl residues to enhance antibacterial and cytotoxic activities. Int. J. Org. Chem. 2015, 5, 232. [Google Scholar] [CrossRef]
- Rana, K.M.; Ferdous, J.; Hosen, A.; Kawsar, S.M.A. Ribose moieties acylation and characterization of some cytidine analogs. J. Sib. Fed. University. Chem. 2020, 13, 465–478. [Google Scholar] [CrossRef]
- Devi, S.R.; Jesmin, S.; Rahman, M.; Manchur, M.A.; Fujii, Y.; Ozeki, Y.; Kawsar, S.M. Microbial efficacy and two step synthesis of uridine derivatives with spectral characterization. ACTA Pharm. Sci. 2019, 57, 47–68. [Google Scholar] [CrossRef]
- Kawsar, S.M.; Islam, M.; Jesmin, S.; Manchur, M.A.; Hasan, I.; Rajia, S. Evaluation of the antimicrobial activity and cytotoxic effect of some uridine derivatives. Int. J. Biosci. 2018, 12, 211–219. [Google Scholar]
- Petersen, E.; Kantele, A.; Koopmans, M.; Asogun, D.; Yinka-Ogunleye, A.; Ihekweazu, C.; Zumla, A. Human monkeypox: Epidemiologic and clinical characteristics, diagnosis, and prevention. Infect. Dis. Clin. N. Am. 2019, 33, 1027. [Google Scholar] [CrossRef] [PubMed]
- McCollum, A.M.; Damon, I.K. Human monkeypox. Clin. Infect. Dis. 2014, 58, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Mitjà, O.; Ogoina, D.; Titanji, B.K.; Galvan, C.; Muyembe, J.-J.; Marks, M.; Orkin, C.M. Monkeypox. Lancet 2023, 401, 60–74. [Google Scholar] [CrossRef]
- Khan, S.H.; Iqbal, R.; Naz, S. A Recent Survey of the Advancements in Deep Learning Techniques for Monkeypox Disease Detection. arXiv 2023, arXiv:2311.10754. [Google Scholar]
- Guinan, M.; Benckendorff, C.; Smith, M.; Miller, G.J. Recent advances in the chemical synthesis and evaluation of anticancer nucleoside analogues. Molecules 2020, 25, 2050. [Google Scholar] [CrossRef]
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef]
- Koszalka, G.W.; Daluge, S.M.; Boyd, F.L. Advances in Nucleoside and Nucleotide Antiviral Therapies. In Annual Reports in Medicinal Chemistry; Elsevier: Amsterdam, The Netherlands, 1998; Volume 33, pp. 163–171. [Google Scholar]
- Kabir, A.K.; Kawsar, S.M.; Bhuiyan, M.M.; Rahman, M.S.; Banu, B. Biological evaluation of some octanoyl derivatives of methyl 4, 6-O-cyclohexylidene-α-D-glucopyranoside. Chittagong Univ. J. Biol. Sci. 2008, 3, 53–64. [Google Scholar] [CrossRef]
- Ahammad, F.; Alam, R.; Mahmud, R.; Akhter, S.; Talukder, E.K.; Tonmoy, A.M.; Fahim, S.; Al-Ghamdi, K.; Samad, A.; Qadri, I. Pharmacoinformatics and molecular dynamics simulation-based phytochemical screening of neem plant (Azadiractha indica) against human cancer by targeting MCM7 protein. Brief. Bioinform. 2021, 22, bbab098. [Google Scholar] [CrossRef]
- El-Demerdash, A.; Al-Karmalawy, A.A.; Abdel-Aziz, T.M.; Elhady, S.S.; Darwish, K.M.; Hassan, A.H. Investigating the structure–activity relationship of marine natural polyketides as promising SARS-CoV-2 main protease inhibitors. RSC Adv. 2021, 11, 31339–31363. [Google Scholar] [CrossRef] [PubMed]
- Tasneem, S.; Ferdous, J.; Bulbul, M.; Misbah, M.; Sujan, D.; Hasan, I.; Kawsar, S. Antimicrobial and Anticancer activities of some partially acylated Thymidine derivatives. J. Bio-Sci. 2021, 29, 11–22. [Google Scholar] [CrossRef]
- Huang, R.-M.; Chen, Y.-N.; Zeng, Z.; Gao, C.-H.; Su, X.; Peng, Y. Marine nucleosides: Structure, bioactivity, synthesis and biosynthesis. Mar. Drugs 2014, 12, 5817–5838. [Google Scholar] [CrossRef] [PubMed]
- Rashad, A.E.; Shamroukh, A.H.; Sayed, H.H.; Awad, S.M.; Abdelwahed, N.A. Some novel thiopyrimidine nucleoside analogs: Synthesis and in vitro antimicrobial evaluation. Synth. Commun. 2011, 41, 652–661. [Google Scholar] [CrossRef]
- Akter, N.; Saha, S.; Hossain, M.A.; Uddin, K.M.; Bhat, A.R.; Ahmed, S.; Kawsar, S.M. Acylated glucopyranosides: FTIR, NMR, FMO, MEP, molecular docking, dynamics simulation, ADMET and antimicrobial activity against bacterial and fungal pathogens. Chem. Phys. Impact 2024, 9, 100700. [Google Scholar] [CrossRef]
- Serpi, M.; Ferrari, V.; Pertusati, F. Nucleoside derived antibiotics to fight microbial drug resistance: New utilities for an established class of drugs? J. Med. Chem. 2016, 59, 10343–10382. [Google Scholar] [CrossRef]
- Dmochowska, B.; Pellowska-Januszek, L.; Samaszko-Fiertek, J.; Slusarz, R.; Wakiec, R.; Madaj, J. Efficient synthesis and antifungal investigation of nucleosides’ quaternary ammonium salt derivatives. Turk. J. Chem. 2019, 43, 157–171. [Google Scholar] [CrossRef]
- Alexandrova, L.A.; Jasko, M.V.; Negrya, S.D.; Solyev, P.N.; Shevchenko, O.V.; Solodinin, A.P.; Kolonitskaya, D.P.; Karpenko, I.L.; Efremenkova, O.V.; Glukhova, A.A. Discovery of novel N4-alkylcytidines as promising antimicrobial agents. Eur. J. Med. Chem. 2021, 215, 113212. [Google Scholar] [CrossRef]
- Rusu, A.; Moga, I.-M.; Uncu, L.; Hancu, G. The role of five-membered heterocycles in the molecular structure of antibacterial drugs used in therapy. Pharmaceutics 2023, 15, 2554. [Google Scholar] [CrossRef]
- Jibroo, R.N.; Mustafa, Y.F.; Al-Shakarchi, W. Synthesis and evaluation of linearly fused thiadiazolocoumarins as prospects with broad-spectrum bioactivity. Results Chem. 2024, 7, 101494. [Google Scholar] [CrossRef]
- Paudel, A.; Kaneko, K.; Watanabe, A.; Shigeki, M.; Motomu, K.; Hamamoto, H.; Sekimizu, K. Structure–activity relationship study of novel iminothiadiazolo-pyrimidinone antimicrobial agents. J. Antibiot. 2013, 66, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Paracini, N.; Schneck, E.; Imberty, A.; Micciulla, S. Lipopolysaccharides at solid and liquid interfaces: Models for biophysical studies of the gram-negative bacterial outer membrane. Adv. Colloid Interface Sci. 2022, 301, 102603. [Google Scholar] [CrossRef]
- Wright, G.D. Bacterial resistance to antibiotics: Enzymatic degradation and modification. Adv. Drug Deliv. Rev. 2005, 57, 1451–1470. [Google Scholar] [CrossRef]
- Malanovic, N.; Lohner, K. Antimicrobial peptides targeting gram-positive bacteria. Pharmaceuticals 2016, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Judge, V.; Narasimhan, B.; Ahuja, M.; Sriram, D.; Yogeeswari, P.; De Clercq, E.; Pannecouque, C.; Balzarini, J. Synthesis, antimycobacterial, antiviral, antimicrobial activity and QSAR studies of N2-acyl isonicotinic acid hydrazide derivatives. Med. Chem. 2013, 9, 53–76. [Google Scholar] [CrossRef]
- Munia, N.S.; Alanazi, M.M.; El Bakri, Y.; Alanazi, A.S.; Mukhrish, Y.E.; Hasan, I.; Kawsar, S.M. Uridine derivatives: Synthesis, Biological evaluation, and in Silico Studies as Antimicrobial and Anticancer agents. Medicina 2023, 59, 1107. [Google Scholar] [CrossRef] [PubMed]
- Kawsar, S.M.A.; Hosen, M.A.; Ahmad, S.; El Bakri, Y.; Ahmad, S.; Affi, S.T.; Goumri-Said, S. In silico approach for potential antimicrobial agents through antiviral, molecular docking, molecular dynamics, pharmacokinetic and bioactivity predictions of galactopyranoside derivatives. Arab. J. Basic Appl. Sci. 2022, 29, 99–112. [Google Scholar] [CrossRef]
- Li, W.-R.; Xie, X.-B.; Shi, Q.-S.; Zeng, H.-Y.; Ou-Yang, Y.-S.; Chen, Y.-B. Antibacterial activity and mechanism of silver nanoparticles on Escherichia coli. Appl. Microbiol. Biotechnol. 2010, 85, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Sworakowski, J. How accurate are energies of HOMO and LUMO levels in small-molecule organic semiconductors determined from cyclic voltammetry or optical spectroscopy? Synth. Met. 2018, 235, 125–130. [Google Scholar] [CrossRef]
- Janani, S.; Rajagopal, H.; Muthu, S.; Aayisha, S.; Raja, M. Molecular structure, spectroscopic (FT-IR, FT-Raman, NMR), HOMO-LUMO, chemical reactivity, AIM, ELF, LOL and Molecular docking studies on 1-Benzyl-4-(N-Boc-amino) piperidine. J. Mol. Struct. 2021, 1230, 129657. [Google Scholar] [CrossRef]
- Hosen, M.A.; Alam, A.; Islam, M.; Fujii, Y.; Ozeki, Y.; Kawsar, S.A. Geometrical optimization, PASS prediction, molecular docking, and in silico ADMET studies of thymidine derivatives against FimH adhesin of Escherichia coli. Bulg. Chem. Commun. 2021, 53, 327–342. [Google Scholar]
- Gassoumi, B.; Mahmoud, A.A.; Nasr, S.; Karayel, A.; Özkınalı, S.; Castro, M.; Melendez, F.; Mahdouani, M.; Nouar, L.; Madi, F. Revealing the effect of Co/Cu (d7/d9) cationic doping on an electronic acceptor ZnO nanocage surface for the adsorption of citric acid, vinyl alcohol, and sulfamethoxazole ligands: DFT-D3, QTAIM, IGM-NCI, and MD analysis. Mater. Chem. Phys. 2023, 309, 128364. [Google Scholar] [CrossRef]
- Kawsar, S.M.; Hossain, M.A.; Saha, S.; Abdallah, E.M.; Bhat, A.R.; Ahmed, S.; Jamalis, J.; Ozeki, Y. Nucleoside-based drug target with general antimicrobial screening and specific computational studies against SARS-CoV-2 main protease. ChemistrySelect 2024, 9, e202304774. [Google Scholar] [CrossRef]
- Shimizu, A.; Ishizaki, Y.; Horiuchi, S.; Hirose, T.; Matsuda, K.; Sato, H.; Yoshida, J.-i. HOMO–LUMO energy-gap tuning of π-conjugated zwitterions composed of electron-donating anion and electron-accepting cation. J. Org. Chem. 2020, 86, 770–781. [Google Scholar] [CrossRef]
- Mumit, M.A.; Pal, T.K.; Alam, M.A.; Islam, M.A.-A.-A.-A.; Paul, S.; Sheikh, M.C. DFT studies on vibrational and electronic spectra, HOMO–LUMO, MEP, HOMA, NBO and molecular docking analysis of benzyl-3-N-(2, 4, 5-trimethoxyphenylmethylene) hydrazinecarbodithioate. J. Mol. Struct. 2020, 1220, 128715. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Emara, A.A.; Taha, A.; Adly, O.M.; Nabeel, A.I.; Aziz, M.A.; Salah, N. Synthesis, characterization, TD-DFT, molecular docking, biological applications, and solvatochromic studies of some new metal complexes derived from semicarbazone of pyrano [3, 2-c] quinoline-3-carboxaldehyde. Appl. Organomet. Chem. 2023, 37, e7169. [Google Scholar] [CrossRef]
- Besbes, M.; Hamdi, A.; Horchani, M.; Majouli, K.; Ghorbel, M.; Lotfi, S.; Alshammari, A.A.; Jilani, S.; Lajimi, R.H.; Jannet, H.B. Phytochemical Screening, Phytotoxic Effects and In Silico Studies of Zilla Spinosa L. and Farsetia Aegyptia Turra Extracts Growing in Hail Region. J. Soil Sci. Plant Nutr. 2025, 25, 2052–2069. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Shamsuddin, T.; Hosen, M.A.; Alam, M.S.; Emran, T.B.; Kawsar, S.M.A. Uridine derivatives: Antifungal, PASS outcomes, ADME/T, drug-likeliness, molecular docking and binding energy calculations. Med. Sci. Int. Med. J. 2021, 10, 1373–1386. [Google Scholar] [CrossRef]
- Angelis, I.D.; Turco, L. Caco-2 cells as a model for intestinal absorption. Curr. Protoc. Toxicol. 2011, 47, 20.6.1–20.6.15. [Google Scholar] [CrossRef]
- Manikandan, P.; Nagini, S. Cytochrome P450 structure, function and clinical significance: A review. Curr. Drug Targets 2018, 19, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Kawsar, S.M.A.; Hosen, M.A.; Chowdhury, T.S.; Rana, K.M.; Fujii, Y.; Ozeki, Y. Thermochemical, PASS, Molecular Docking, Drug-Likeness and In Silico ADMET Prediction of Cytidine Derivatives against HIV-1 Reverse Transcriptase. Rev. De Chimie 2021, 72, 159–178. [Google Scholar] [CrossRef]
- Stampfer, H.G.; Gabb, G.M.; Dimmitt, S.B. Why maximum tolerated dose? Br. J. Clin. Pharmacol. 2019, 85, 2213–2217. [Google Scholar] [CrossRef] [PubMed]
- Siddiquee, N.H.; Hossain, M.I.; Talukder, M.E.K.; Nirob, S.A.A.; Shourav, M.; Jahan, I.; Tamanna, U.H.A.; Das, P.; Akter, R.; Hasan, M. In-silico identification of novel natural drug leads against the Ebola virus VP40 protein: A promising approach for developing new antiviral therapeutics. Inform. Med. Unlocked 2024, 45, 101458. [Google Scholar] [CrossRef]
- Siddiquee, N.H.; Tanni, A.A.; Sarker, N.; Sourav, S.H.; Islam, L.; Mili, M.A.; Akter, F.; Roy, S.C.; Abdullah-Al-Mamun, M.; Malek, S. Insights into novel inhibitors intending HCMV protease a computational molecular modelling investigation for antiviral drug repurposing. Inform. Med. Unlocked 2024, 48, 101522. [Google Scholar] [CrossRef]
- Ali, S.; Khan, F.I.; Mohammad, T.; Lan, D.; Hassan, M.I.; Wang, Y. Identification and evaluation of inhibitors of lipase from Malassezia restricta using virtual high-throughput screening and molecular dynamics studies. Int. J. Mol. Sci. 2019, 20, 884. [Google Scholar] [CrossRef]
- Al-Mijalli, S.H.; Mrabti, H.N.; Elbouzidi, A.; Ashmawy, N.S.; Batbat, A.; Abdallah, E.M.; Laaboudi, W.; Aladhadh, M.; Alshabrmi, F.M.; Alnasser, S.M. Thymus serpyllum L. Essential Oil: Phytochemistry and in Vitro and in Silico Screening of Its Antimicrobial, Antioxidant and Anti-Inflammatory Properties. Phyton 2025, 94, 209–227. [Google Scholar] [CrossRef]
- Matlou, T.D.; Matotoka, M.M.; Mnisi, T.J.; Masoko, P. Biological Activities of Leonotis ocymifolia (Burm. f.) and Its Antibacterial Activities Against ESKAPE Pathogens. Antibiotics 2025, 14, 238. [Google Scholar] [CrossRef]
- Yavuz, D.Ö.; Dinler, H.; Morca, A.U. Determination of Antifungal Activities on Some Plant Extracts on Alternaria alternata. Turk. J. Agric. Food Sci. Technol. 2024, 12, 500–506. [Google Scholar]
- Nyembe, P.; Ntombela, T.; Makatini, M. Review: Structure-Activity Relationship of Antimicrobial Peptoids. Pharmaceutics 2023, 15, 1506. [Google Scholar] [CrossRef]
- Caricato, M.; Frisch, M.J.; Hiscocks, J.; Frisch, M.J. Gaussian 09: IOps Reference; Gaussian: Wallingford, CT, USA, 2009. [Google Scholar]
- Adjir, K.; Sekkal-Rahal, M.; Springborg, M. DFT evaluation of structural, electronic and variation properties for complex carbohydrates with biological interest. J. Biomol. Struct. Dyn. 2023, 41, 5981–5989. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.E.; Trucks, G.; Schlegel, H.B.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Minasov, G.; Inniss, N.L.; Shuvalova, L.; Anderson, W.F.; Satchell, K.J. Structure of the Monkeypox virus profilin-like protein A42R reveals potential functional differences from cellular profilins. Struct. Biol. Cryst. Commun. 2022, 78, 371–377. [Google Scholar] [CrossRef]
- Zhang, A.P.; Bornholdt, Z.A.; Abelson, D.M.; Saphire, E.O. Crystal structure of Marburg virus VP24. J. Virol. 2014, 88, 5859–5863. [Google Scholar] [CrossRef] [PubMed]
- Siddiquee, N.H.; Malek, S.; Tanni, A.A.; Mitu, I.J.; Arpa, S.H.; Hasan, M.R.; Shammi, S.E.J.; Chakma, C.; Mahinur, M.; Wajed, S. Unveiling the antiviral activity of 2′, 3, 5, 7-Tetrahydroxyflavanone as potential inhibitor of chikungunya virus envelope glycoprotein. Inform. Med. Unlocked 2024, 47, 101486. [Google Scholar] [CrossRef]
- Asseri, A.H.; Alam, M.J.; Alzahrani, F.; Khames, A.; Pathan, M.T.; Abourehab, M.A.; Hosawi, S.; Ahmed, R.; Sultana, S.A.; Alam, N.F. Toward the identification of natural antiviral drug candidates against merkel cell polyomavirus: Computational drug design approaches. Pharmaceuticals 2022, 15, 501. [Google Scholar] [CrossRef]
- Hassan, S.A.; Aziz, D.M.; Abdullah, M.N.; Bhat, A.R.; Dongre, R.S.; Hadda, T.B.; Almalki, F.A.; Kawsar, S.M.; Rahiman, A.K.; Ahmed, S. In vitro and in vivo evaluation of the antimicrobial, antioxidant, cytotoxic, hemolytic activities and in silico POM/DFT/DNA-binding and pharmacokinetic analyses of new sulfonamide bearing thiazolidin-4-ones. J. Biomol. Struct. Dyn. 2024, 42, 3747–3763. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diameter of Inhibition Zone (mm) | |||||
|---|---|---|---|---|---|
| Entry | B. subtilis (+ve) | B. cereus (+ve) | E. coli (−ve) | S. typhi (−ve) | P. aeruginosa (−ve) |
| 1 | NI | NI | NI | NI | NI |
| 2 | NI | NI | 7.84 ± 0.1 | NI | NI |
| 3 | NI | NI | 7.75 ± 0.2 | NI | NI |
| 4 | * 11.00 ± 0.1 | * 17.00 ± 0.4 | * 20.75 ± 0.2 | 9.75 ± 0.2 | * 17.75 ± 0.2 |
| 5 | * 10.00 ± 0.2 | * 15.00 ± 0.2 | 9.26 ± 0.2 | 8.13 ± 0.3 | * 14.00 ± 0.1 |
| 6 | NI | NI | NI | NI | NI |
| Azithromycin | 18.25 ± 0.3 ** | 17.75 ± 0.3 ** | ** 17.25 ± 0.1 | ** 18.00 ± 0.2 | ** 18.5 ± 0.3 |
| Entry | % Fungal Mycelial Growth Inhibition in mm (20 μg/μL) | |
|---|---|---|
| Aspergillus niger | Aspergillusflavus | |
| 1 | NI | NI |
| 2 | * 73.72 ± 1.2 | * 77.45 ± 1.0 |
| 3 | * 77.54 ± 1.1 | NI |
| 4 | * 64.40 ± 1.3 | * 74.59 ± 1.2 |
| 5 | 55.61 ± 1.1 | * 61.52 ± 1.0 |
| 6 | NI | NI |
| Nystatin | ** 65.4 ± 1.0 | ** 64.1 ± 1.0 |
| HOMO | LUMO | (eV) | η (eV) | μ (eV) | ω (eV) | ||
|---|---|---|---|---|---|---|---|
| 1 | −6.83 | −1.52 | 4.17 | 2.65 | 0.37 | −4.17 | 3.28 |
| 2 | −6.64 | −1.33 | 3.98 | 2.65 | 0.37 | −3.98 | 2.98 |
| 3 | −6.60 | −1.27 | 3.93 | 2.66 | 0.37 | −3.93 | 2.90 |
| 4 | −7.25 | −1.84 | 4.54 | 2.70 | 0.37 | −4.54 | 3.81 |
| 5 | −6.67 | −1.34 | 4.00 | 2.66 | 0.37 | −4.00 | 3.00 |
| 6 | −6.73 | −2.05 | 4.39 | 2.34 | 0.42 | −4.39 | 4.11 |
| Ligand | Binding Affinity (kcal/mol) | Number of H-Bonds Formed |
|---|---|---|
| 1 | −4.5 | 0 |
| 2 | −4.6 | 1 |
| 3 | −5.1 | 1 |
| 4 | −5.0 | 1 |
| 5 | −6.2 | 2 |
| 6 | −5.7 | 1 |
| Reference (acyclovir) | −4.2 | 2 |
| Ligand | Binding Affinity (kcal/mol) | Number of H-Bonds Formed |
|---|---|---|
| 1 | −4.9 | 1 |
| 2 | −5.3 | 1 |
| 3 | −5.5 | 1 |
| 4 | −5.3 | 1 |
| 5 | −5.6 | 2 |
| 6 | −7.0 | 3 |
| Reference (acyclovir) | −4.3 | 1 |
| 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|
| Water solubility (log mol/L) | −5.061 | −2.892 | −5.4 | −3.597 | −2.892 |
| Caco-2 permeability (log Papp in 10−6 cm/s) | 0.53 | 3.008 | 0.703 | 0.185 | 3.008 |
| Intestinal absorption (% Absorbed) | 68.559 | 69.881 | 77.117 | 71.954 | 69.881 |
| Skin permeability (log Kp) | −2.767 | −2.735 | −2.714 | −2.735 | −2.735 |
| VDss (log L/kg) | −0.732 | 0.011 | −0.738 | −1.203 | 0.011 |
| Fraction unbound (Fu) | 0.137 | 0.381 | 0 | 0.089 | 0.381 |
| BBB permeability (log BB) | −1.283 | −0.245 | −1.254 | −1.784 | −0.245 |
| CNS permeability (log PS) | −3.409 | −1.488 | −2.85 | −3.782 | −1.488 |
| CYP2D6 substrate | No | No | No | No | No |
| CYP3A4 substrate | Yes | No | Yes | Yes | No |
| CYP1A2 inhibitor | No | Yes | No | No | Yes |
| CYP2C19 inhibitor | No | No | No | No | No |
| CYP2C9 inhibitor | No | No | No | No | No |
| CYP2D6 inhibitor | No | No | No | No | No |
| CYP3A4 inhibitor | No | No | No | No | No |
| Renal OCT2 substrate | No | No | No | No | No |
| AMES toxicity | No | Yes | No | No | Yes |
| Max. tolerated dose (log mg/kg/day) | −0.2 | 0.438 | 0.069 | 0.168 | 0.438 |
| Hepatotoxicity | Yes | No | Yes | Yes | No |
| hERG I inhibitor | No | No | No | No | No |
| hERG II inhibitor | No | No | No | No | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawsar, S.M.A.; Al-mijalli, S.H.; Bouzid, G.; Abdallah, E.M.; Siddiquee, N.H.; Hosen, M.A.; Horchani, M.; Ghalla, H.; B. Jannet, H.; Fujii, Y.; et al. Unveiling Palmitoyl Thymidine Derivatives as Antimicrobial/Antiviral Inhibitors: Synthesis, Molecular Docking, Dynamic Simulations, ADMET, and Assessment of Protein–Ligand Interactions. Pharmaceuticals 2025, 18, 806. https://doi.org/10.3390/ph18060806
Kawsar SMA, Al-mijalli SH, Bouzid G, Abdallah EM, Siddiquee NH, Hosen MA, Horchani M, Ghalla H, B. Jannet H, Fujii Y, et al. Unveiling Palmitoyl Thymidine Derivatives as Antimicrobial/Antiviral Inhibitors: Synthesis, Molecular Docking, Dynamic Simulations, ADMET, and Assessment of Protein–Ligand Interactions. Pharmaceuticals. 2025; 18(6):806. https://doi.org/10.3390/ph18060806
Chicago/Turabian StyleKawsar, Sarkar M. A., Samiah Hamad Al-mijalli, Gassoumi Bouzid, Emad M. Abdallah, Noimul H. Siddiquee, Mohammed A. Hosen, Mabrouk Horchani, Houcine Ghalla, Hichem B. Jannet, Yuki Fujii, and et al. 2025. "Unveiling Palmitoyl Thymidine Derivatives as Antimicrobial/Antiviral Inhibitors: Synthesis, Molecular Docking, Dynamic Simulations, ADMET, and Assessment of Protein–Ligand Interactions" Pharmaceuticals 18, no. 6: 806. https://doi.org/10.3390/ph18060806
APA StyleKawsar, S. M. A., Al-mijalli, S. H., Bouzid, G., Abdallah, E. M., Siddiquee, N. H., Hosen, M. A., Horchani, M., Ghalla, H., B. Jannet, H., Fujii, Y., & Ozeki, Y. (2025). Unveiling Palmitoyl Thymidine Derivatives as Antimicrobial/Antiviral Inhibitors: Synthesis, Molecular Docking, Dynamic Simulations, ADMET, and Assessment of Protein–Ligand Interactions. Pharmaceuticals, 18(6), 806. https://doi.org/10.3390/ph18060806