


Synthesis, Biological Evaluation, and In Silico Characterization of Novel Imidazothiadiazole–Chalcone Hybrids as Multi-Target Enzyme Inhibitors


Abstract

1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Activity Studies
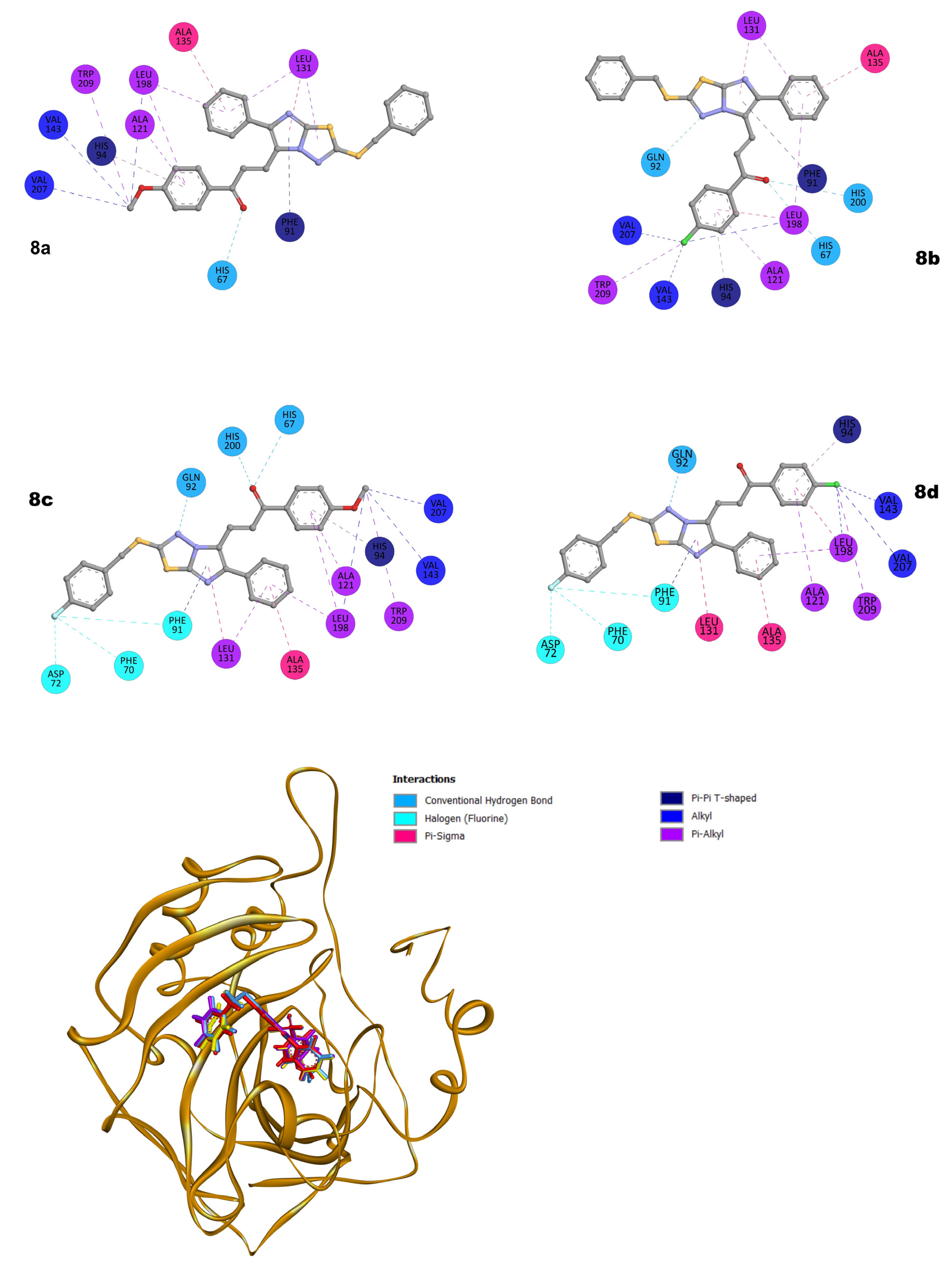
2.3. Molecular Docking Studies
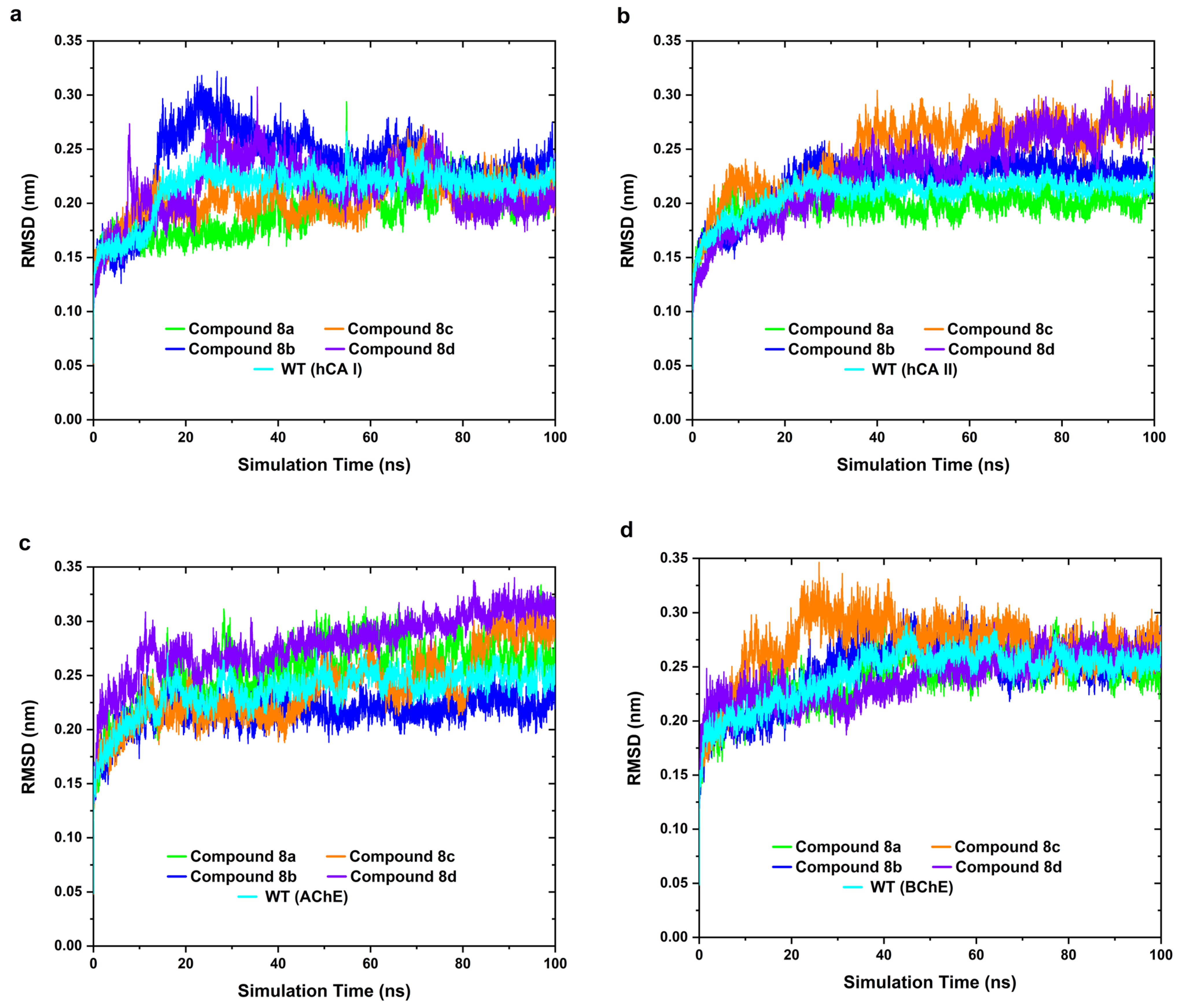
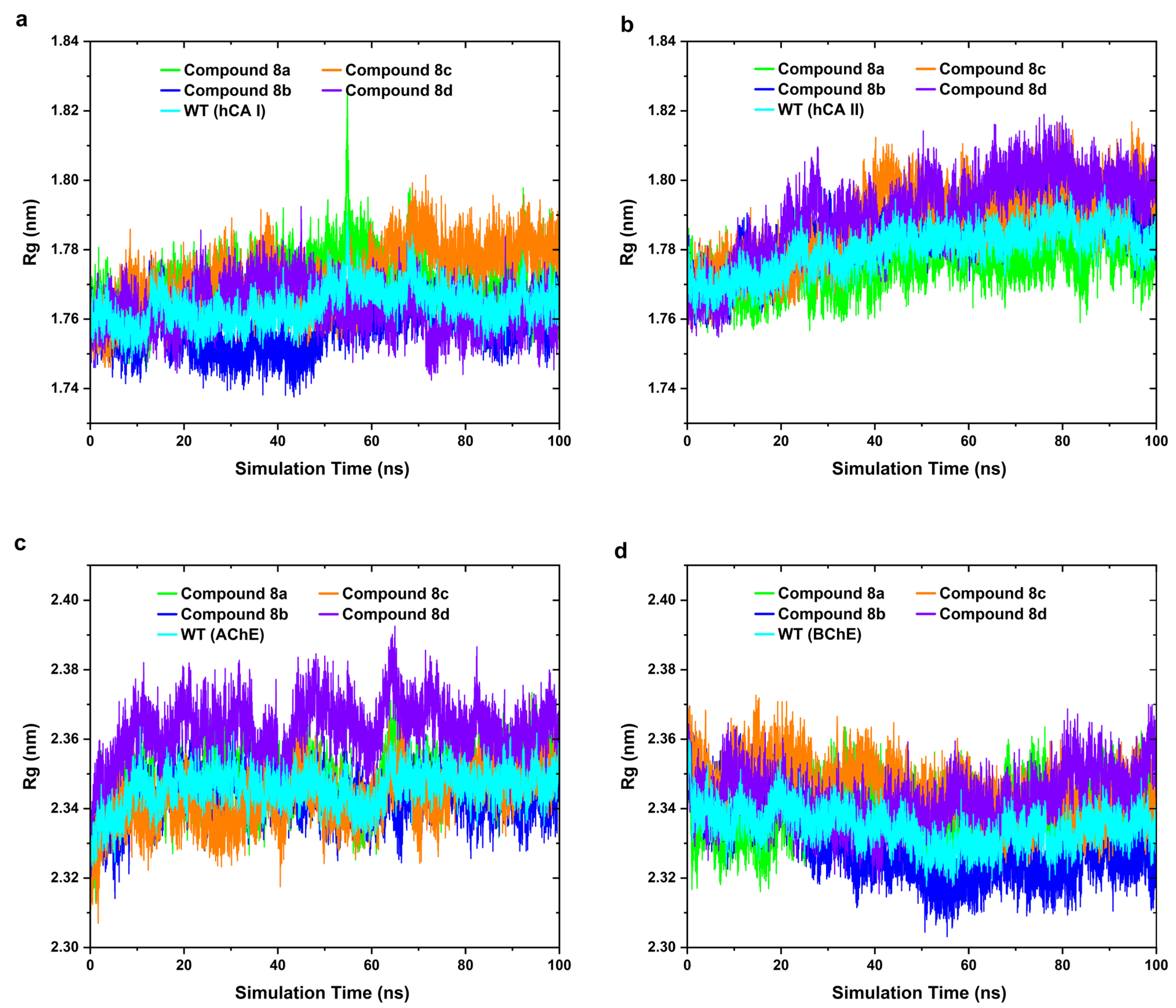
2.4. MD Simulation
2.5. In Silico Pharmacokinetic and Toxicological Evaluation
3. Materials and Methods
3.1. Experimental Synthesis
3.1.1. General Methods
General Synthesis of 2-amino-1,3,4-thiadiazole Derivatives (3a–b)
General Synthesis of 2,6-disubstituted imidazo[2,1-b][1,3,4]thiadiazole Derivatives (5a–d)
General Synthesis of 2-(substituted-benzylthio)-6-phenylimidazo[2,1-b][1,3,4]thiadiazole-5-carbaldehyde Derivatives (6a–b)
- 2-(Benzylthio)-6-phenylimidazo[2,1-b][1,3,4]thiadiazole-5-carbaldehyde (6a)
- 2-((4-Fluorobenzyl)thio)-6-phenylimidazo[2,1-b][1,3,4]thiadiazole-5-carbaldehyde (6b)
General Synthesis of Chalcone Derivatives (8a–d)
- (E)-3-(2-(Benzylthio)-6-phenylimidazo[2,1-b][1,3,4]thiadiazol-5-yl)-1-(4-methoxyphenyl)prop-2-en-1-one (8a)
- (E)-3-(2-(Benzylthio)-6-phenylimidazo[2,1-b][1,3,4]thiadiazol-5-yl)-1-(4-chlorophenyl)prop-2-en-1-one (8b)
- (E)-3-(2-((4-Fluorobenzyl)thio)-6-phenylimidazo[2,1-b][1,3,4]thiadiazol-5-yl)-1-(4-methoxyphenyl)prop-2-en-1-one (8c)
- (E)-1-(4-Chlorophenyl)-3-(2-((4-fluorobenzyl)thio)-6-phenylimidazo[2,1-b][1,3,4]thiadiazol-5-yl)prop-2-en-1-one (8d)
3.2. Biological Activity
3.2.1. Activities of CA I and II Isoenzymes
3.2.2. AChE and BChE Activities
3.2.3. IC50 and Ki Studies for Both Enzymes
3.3. In Silico Studies
3.3.1. Molecular Docking Setups
3.3.2. Sequence Origin and Conservation Analysis
3.3.3. MD Simulations
3.3.4. ADMET Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alvárez-Builla, J.; Barluenga, J. Heterocyclic Compounds: An Introduction. In Modern Heterocyclic Chemistry; Wiley-VCH Verlag GmbH & Co., KGaA: Weinheim, Germany, 2011; pp. 1–9. [Google Scholar]
- Strzemecka, L.; Urbańczyk-Lipkowska, Z. The structure of N-allyl-(5-phenyl-[1,3,4]thiadiazol-2-yl) amine in solution and the solid state studied by the 1H, 13C, 15N NMR spectroscopy and X-ray crystallography. J. Mol. Struct. 2010, 970, 1–13. [Google Scholar] [CrossRef]
- Yi, R.; Liu, S.; Gao, H.; Liang, Z.; Xu, X.; Li, N. Iodine-promoted direct thiolation (selenylation) of imidazole with disulfides (diselenide): A convenient and metal-free protocol for the synthesis of 2-arylthio (seleno) imidazole. Tetrahedron 2020, 76, 130951. [Google Scholar] [CrossRef]
- Alwan, W.S.; Karpoormath, R.; Palkar, M.B.; Patel, H.M.; Rane, R.A.; Shaikh, M.S.; Kajee, A.; Mlisana, K.P. Novel imidazo [2,1-b]-1,3,4-thiadiazoles as promising antifungal agents against clinical isolate of Cryptococcus neoformans. Eur. J. Med. Chem. 2015, 95, 514–525. [Google Scholar] [CrossRef]
- Khazi, I.A.M.; Gadad, A.K.; Lamani, R.S.; Bhongade, B.A. Chemistry of imidazo[2,1-b][1,3,4]thiadiazoles. Tetrahedron 2011, 67, 3289–3316. [Google Scholar] [CrossRef]
- Tahtaci, H.; Karacık, H.; Ece, A.; Er, M.; Şeker, M.G. Design, Synthesis, SAR and Molecular Modeling Studies of Novel Imidazo[2,1-b][1,3,4]Thiadiazole Derivatives as Highly Potent Antimicrobial Agents. Mol. Inform. 2018, 37, 1700083. [Google Scholar] [CrossRef]
- Fascio, M.L.; Errea, M.I.; D’Accorso, N.B. Imidazothiazole and related heterocyclic systems. Synthesis, chemical and biological properties. Eur. J. Med. Chem. 2015, 90, 666–683. [Google Scholar] [CrossRef] [PubMed]
- Jabeen, T.; Aslam, S.; Yaseen, M.; Jawwad Saif, M.; Ahmad, M.; Al-Hussain, S.A.; Zaki, M.E.A. Recent synthetic strategies of medicinally important imidazothiadiazoles. J. Saudi Chem. Soc. 2023, 27, 101679. [Google Scholar] [CrossRef]
- Kumar, R.S.; Praveen, S.; Shridharshini, K.; Maruthamuthu, M.; Mohanapriya, K.; Mythili, A. Biological applications of imidazothiazole scaffolds: A current review. J. Adv. Chem. Sci. 2022, 8, 756–769. [Google Scholar] [CrossRef]
- Cascioferro, S.; Petri, G.L.; Parrino, B.; Carbone, D.; Funel, N.; Bergonzini, C.; Mantini, G.; Dekker, H.; Geerke, D.; Peters, G.J.; et al. Imidazo[2,1-b] [1,3,4]thiadiazoles with antiproliferative activity against primary and gemcitabine-resistant pancreatic cancer cells. Eur. J. Med. Chem. 2020, 189, 112088. [Google Scholar] [CrossRef]
- Narasimha Rao, M.P.; Nagaraju, B.; Kovvuri, J.; Polepalli, S.; Alavala, S.; Vishnuvardhan, M.V.P.S.; Swapna, P.; Nimbarte, V.D.; Lakshmi, J.K.; Jain, N.; et al. Synthesis of imidazo-thiadiazole linked indolinone conjugates and evaluated their microtubule network disrupting and apoptosis inducing ability. Bioorg. Chem. 2018, 76, 420–436. [Google Scholar] [CrossRef]
- Gireesh, T.M.; Kamble, R.R.; Taj, T. Synthesis and antimicrobial and anticancer activity of new of imidazo[2,1-b][1,3,4]thiadiazoles. Pharm. Chem. J. 2011, 45, 313–316. [Google Scholar] [CrossRef]
- Alegaon, S.G.; Alagawadi, K.R. Synthesis, characterization and antimicrobial activity evaluation of new imidazo [2, 1-b][1, 3, 4] thiadiazole derivatives. Eur. J. Chem. 2011, 2, 94–99. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, S.; Liu, Z.J.; Chen, W.; Fu, J.; Zeng, Q.F.; Zhu, H.L. Synthesis and antimicrobical evaluation of a novel class of 1,3,4-thiadiazole: Derivatives bearing 1,2,4-triazolo[1,5-a]pyrimidine moiety. Eur. J. Med. Chem. 2013, 64, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Chandrakantha, B.; Isloor, A.M.; Shetty, P.; Fun, H.K.; Hegde, G. Synthesis and biological evaluation of novel substituted 1,3,4-thiadiazole and 2,6-di aryl substituted imidazo [2,1-b] [1,3,4] thiadiazole derivatives. Eur. J. Med. Chem. 2014, 71, 316–323. [Google Scholar] [CrossRef]
- Ramprasad, J.; Nayak, N.; Dalimba, U.; Yogeeswari, P.; Sriram, D. One-pot synthesis of new triazole—Imidazo[2,1-b][1,3,4]thiadiazole hybrids via click chemistry and evaluation of their antitubercular activity. Bioorg. Med. Chem. Lett. 2015, 25, 4169–4173. [Google Scholar] [CrossRef]
- Taflan, E.; Bayrak, H.; Er, M.; Alpay Karaoğlu, Ş.; Bozdeveci, A. Novel imidazo[2,1-b][1,3,4]thiadiazole (ITD) hybrid compounds: Design, synthesis, efficient antibacterial activity and antioxidant effects. Bioorg. Chem. 2019, 89, 102998. [Google Scholar] [CrossRef]
- Rammohan, A.; Reddy, J.S.; Sravya, G.; Rao, C.N.; Zyryanov, G.V. Chalcone synthesis, properties and medicinal applications: A review. Environ. Chem. Lett. 2020, 18, 433–458. [Google Scholar] [CrossRef]
- Basappa, V.C.; Ramaiah, S.; Penubolu, S.; Kariyappa, A.K. Recent developments on the synthetic and biological applications of chalcones—A review. Biointerface Res. Appl. Chem 2021, 12, 180–195. [Google Scholar]
- Narwal, S.; Devi, B.; Dhanda, T.; Kumar, S.; Tahlan, S. Exploring Chalcone Derivatives: Synthesis and Their Therapeutic Potential. J. Mol. Struct. 2024, 1303, 137554. [Google Scholar] [CrossRef]
- Elkanzi, N.A.A.; Hrichi, H.; Alolayan, R.A.; Derafa, W.; Zahou, F.M.; Bakr, R.B. Synthesis of Chalcones Derivatives and Their Biological Activities: A Review. ACS Omega 2022, 7, 27769–277869. [Google Scholar] [CrossRef]
- Nguyen, P.; Ananthapavan, J.; Tan, E.J.; Crosland, P.; Bowe, S.J.; Gao, L.; Dunstan, D.W.; Moodie, M. Modelling the potential health and economic benefits of reducing population sitting time in Australia. Int. J. Behav. Nutr. Phys. Act. 2022, 19, 28. [Google Scholar] [CrossRef] [PubMed]
- Huo, P.C.; Hu, Q.; Shu, S.; Zhou, Q.H.; He, R.J.; Hou, J.; Guan, X.Q.; Tu, D.Z.; Hou, X.D.; Liu, P.; et al. Design, synthesis and biological evaluation of novel chalcone-like compounds as potent and reversible pancreatic lipase inhibitors. Bioorg. Med. Chem. 2021, 29, 115853. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Boone, C.D.; Bhargav, K.; McKenna, R. Structural annotation of human carbonic anhydrases. J. Enzym. Inhib. Med. Chem. 2013, 28, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Pastorekova, S.; Parkkila, S.; Pastorek, J.; Supuran, C.T. Carbonic anhydrases: Current state of the art, therapeutic applications and future prospects. J. Enzyme Inhib. Med. Chem. 2004, 19, 199–229. [Google Scholar] [CrossRef]
- Mishra, C.B.; Tiwari, M.; Supuran, C.T. Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: Where are we today? Med. Res. Rev. 2020, 40, 2485–2565. [Google Scholar] [CrossRef]
- Cabaleiro-Lago, C.; Lundqvist, M. The Effect of Nanoparticles on the Structure and Enzymatic Activity of Human Carbonic Anhydrase I and II. Molecules 2020, 25, 4405. [Google Scholar] [CrossRef]
- Breton, S. The cellular physiology of carbonic anhydrases. Jop 2001, 2, 159–164. [Google Scholar]
- Patočka, J.; Kuča, K.; Jun, D. Acetylcholinesterase and butyrylcholinesterase–important enzymes of human body. Acta Med. 2004, 47, 215–228. [Google Scholar] [CrossRef]
- Geula, C.; Darvesh, S. Butyrylcholinesterase, cholinergic neurotransmission and the pathology of Alzheimer’s disease. Drugs Today 2004, 40, 711–721. [Google Scholar] [CrossRef]
- Tsim, K.; Soreq, H. Acetylcholinesterase: Old questions and new developments. Front. Mol. Neurosci. 2013, 5, 101. [Google Scholar] [CrossRef]
- Greig, N.H.; Lahiri, D.K.; Sambamurti, K. Butyrylcholinesterase: An important new target in Alzheimer’s disease therapy. Int. Psychogeriatr. 2002, 14 (Suppl. S1), 77–91. [Google Scholar] [CrossRef] [PubMed]
- Pohanka, M. Diagnoses of Pathological States Based on Acetylcholinesterase and Butyrylcholinesterase. Curr. Med. Chem. 2020, 27, 2994–3011. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, I.; Akkoc, S.; Alici, H.; Capanlar, S.; Sahin, O.; Tahtaci, H. Novel Thioether-Bridged 2,6-Disubstituted and 2,5,6-Trisubstituted Imidazothiadiazole Analogues: Synthesis, Antiproliferative Activity, ADME, and Molecular Docking Studies. Chem. Biodivers. 2023, 20, e202200884. [Google Scholar] [CrossRef]
- Mirghani, A.H.; Pehlivanoglu, S.; Alici, H.; Tahtaci, H.; Uysal, S. Synthesis and Characterization of Schiff Bases and Their Ag(I) Complexes Containing 2,5,6-Trisubstituted Imidazothiadiazole Derivatives: Molecular Docking and In Vitro Cytotoxic Effects Against Nonsmall Lung Cancer Cell Line. J. Biochem. Mol. Toxicol. 2025, 39, e70142. [Google Scholar] [CrossRef]
- Dagli, M.; Er, M.; Karakurt, T.; Onaran, A.; Alici, H.; Tahtaci, H. Synthesis, Characterization, Antimicrobial Evaluation, and Computational Investigation of Substituted Imidazo [2,1-b][1,3,4]Thiadiazole Derivatives. ChemistrySelect 2020, 5, 11753–11763. [Google Scholar] [CrossRef]
- Askin, S.; Tahtaci, H.; Türkeş, C.; Demir, Y.; Ece, A.; Akalın Çiftçi, G.; Beydemir, Ş. Design, synthesis, characterization, in vitro and in silico evaluation of novel imidazo [2,1-b][1,3,4]thiadiazoles as highly potent acetylcholinesterase and non-classical carbonic anhydrase inhibitors. Bioorg. Chem. 2021, 113, 105009. [Google Scholar] [CrossRef]
- Tunel, H.; Er, M.; Alici, H.; Onaran, A.; Karakurt, T.; Tahtaci, H. Synthesis, structural characterization, biological activity, and theoretical studies of some novel thioether-bridged 2,6-disubstituted imidazothiadiazole analogues. J. Heterocycl. Chem. 2021, 58, 1321–1343. [Google Scholar] [CrossRef]
- Kaya Çavuşoğlu, B.; Sağlık, B.N.; Acar Çevik, U.; Osmaniye, D.; Levent, S.; Özkay, Y.; Kaplancıklı, Z.A. Design, synthesis, biological evaluation, and docking studies of some novel chalcones as selective COX-2 inhibitors. Arch. Pharm. 2021, 354, 2000273. [Google Scholar] [CrossRef]
- Koudad, M.; El Hamouti, C.; Elaatiaoui, A.; Dadou, S.; Oussaid, A.; Abrigach, F.; Pilet, G.; Benchat, N.; Allali, M. Synthesis, crystal structure, antimicrobial activity and docking studies of new imidazothiazole derivatives. J. Iran. Chem. Soc. 2020, 17, 297–306. [Google Scholar] [CrossRef]
- Kamal, A.; Prabhakar, S.; Janaki Ramaiah, M.; Venkat Reddy, P.; Ratna Reddy, C.; Mallareddy, A.; Shankaraiah, N.; Lakshmi Narayan Reddy, T.; Pushpavalli, S.N.C.V.L.; Pal-Bhadra, M. Synthesis and anticancer activity of chalcone-pyrrolobenzodiazepine conjugates linked via 1,2,3-triazole ring side-armed with alkane spacers. Eur. J. Med. Chem. 2011, 46, 3820–3831. [Google Scholar] [CrossRef]
- Abdullah, M.N.; Osw, P.; Hassan, S.A.; Othman, S. Two new cyclohexenone derivatives: Synthesis, DFT estimation, biological activities and molecular docking study. J. Mol. Struct. 2024, 1301, 137361. [Google Scholar] [CrossRef]
- Pereira, R.; Silva, A.M.S.; Ribeiro, D.; Silva, V.L.M.; Fernandes, E. Bis-chalcones: A review of synthetic methodologies and anti-inflammatory effects. Eur. J. Med. Chem. 2023, 252, 115280. [Google Scholar] [CrossRef] [PubMed]
- Mphahlele, M.J.; Agbo, E.N.; Choong, Y.S. Synthesis, Structure, Carbohydrate Enzyme Inhibition, Antioxidant Activity, In Silico Drug-Receptor Interactions and Drug-Like Profiling of the 5-Styryl-2-Aminochalcone Hybrids. Molecules 2021, 26, 2692. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Reddy, V.S.; Santosh, K.; Bharath Kumar, G.; Shaik, A.B.; Mahesh, R.; Chourasiya, S.S.; Sayeed, I.B.; Kotamraju, S. Synthesis of imidazo [2,1-b][1,3,4]thiadiazole–chalcones as apoptosis inducing anticancer agents. MedChemComm 2014, 5, 1718–1723. [Google Scholar] [CrossRef]
- Durmaz, L.; Gülçin, I.; Tüzün, B. Isofraxidin: Antioxidant, Anti-carbonic Anhydrase, Anti-cholinesterase, Anti-diabetic, and in Silico Properties. ChemistrySelect 2023, 8, e202300170. [Google Scholar] [CrossRef]
- Zahedi, N.A.; Mohammadi-Khanaposhtani, M.; Rezaei, P.; Askarzadeh, M.; Alikhani, M.; Adib, M.; Mahdavi, M.; Larijani, B.; Niakan, S.; Tehrani, M.B.; et al. Dual functional cholinesterase and carbonic anhydrase inhibitors for the treatment of Alzheimer’s disease: Design, synthesis, in vitro, and in silico evaluations of coumarin-dihydropyridine derivatives. J. Mol. Struct. 2023, 1276, 134767. [Google Scholar] [CrossRef]
- Zareei, S.; Mohammadi-Khanaposhtani, M.; Adib, M.; Mahdavi, M.; Taslimi, P. Sulfonamide-phosphonate hybrids as new carbonic anhydrase inhibitors: In vitro enzymatic inhibition, molecular modeling, and ADMET prediction. J. Mol. Struct. 2023, 1271, 134114. [Google Scholar] [CrossRef]
- Gülçin, İ.; Trofimov, B.; Kaya, R.; Taslimi, P.; Sobenina, L.; Schmidt, E.; Petrova, O.; Malysheva, S.; Gusarova, N.; Farzaliyev, V.; et al. Synthesis of nitrogen, phosphorus, selenium and sulfur-containing heterocyclic compounds—Determination of their carbonic anhydrase, acetylcholinesterase, butyrylcholinesterase and α-glycosidase inhibition properties. Bioorg. Chem. 2020, 103, 104171. [Google Scholar] [CrossRef]
- Hashmi, S.; Khan, S.; Shafiq, Z.; Taslimi, P.; Ishaq, M.; Sadeghian, N.; Karaman, H.S.; Akhtar, N.; Islam, M.; Asari, A.; et al. Probing 4-(diethylamino)-salicylaldehyde-based thiosemicarbazones as multi-target directed ligands against cholinesterases, carbonic anhydrases and α-glycosidase enzymes. Bioorg. Chem. 2021, 107, 104554. [Google Scholar] [CrossRef]
- Manasa, K.L.; Pujitha, S.; Sethi, A.; Arifuddin, M.; Alvala, M.; Angeli, A.; Supuran, C.T. Synthesis and Biological Evaluation of Imidazo [2,1-b]Thiazole based Sulfonyl Piperazines as Novel Carbonic Anhydrase II Inhibitors. Metabolites 2020, 10, 136. [Google Scholar] [CrossRef]
- Keçeci Sarıkaya, M.; Yıldırım, Ş.; Kocyigit, U.M.; Ceylan, M.; Yırtıcı, Ü.; Eyüpoğlu, V. Novel Aminothiazole–Chalcone Analogs: Synthesis, Evaluation Acetylcholinesterase Activity, In Silico Analysis. Chem. Biodivers. 2025, 22, e202402777. [Google Scholar] [CrossRef] [PubMed]
- Güzel, E.; Koçyiğit, Ü.M.; Taslimi, P.; Gülçin, İ.; Erkan, S.; Nebioğlu, M.; Arslan, B.S.; Şişman, İ. Phthalocyanine complexes with (4-isopropylbenzyl)oxy substituents: Preparation and evaluation of anti-carbonic anhydrase, anticholinesterase enzymes and molecular docking studies. J. Biomol. Struct. Dyn. 2022, 40, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Taslimi, P.; Işık, M.; Türkan, F.; Durgun, M.; Türkeş, C.; Gülçin, İ.; Beydemir, Ş. Benzenesulfonamide derivatives as potent acetylcholinesterase, α-glycosidase, and glutathione S-transferase inhibitors: Biological evaluation and molecular docking studies. J. Biomol. Struct. Dyn. 2021, 39, 5449–5460. [Google Scholar] [CrossRef]
- Dadou, S.; Altay, A.; Koudad, M.; Türkmenoğlu, B.; Yeniçeri, E.; Çağlar, S.; Allali, M.; Oussaid, A.; Benchat, N.; Karrouchi, K. Design, synthesis, anticancer evaluation and molecular docking studies of new imidazo [2, 1-b] thiazole -based chalcones. Med. Chem. Res. 2022, 31, 1369–1383. [Google Scholar] [CrossRef]
- Er, M.; Ahmadov, F.; Karakurt, T.; Direkel, Ş.; Tahtaci, H. A Novel Class Substituted Imidazo [2,1-b][1,3,4]thiadiazole Derivatives: Synthesis, Characterization, In Vitro Biological Activity, and Potential Inhibitors Design Studies. ChemistrySelect 2019, 4, 14281–14290. [Google Scholar] [CrossRef]
- Verpoorte, J.A.; Mehta, S.; Edsall, J.T. Esterase Activities of Human Carbonic Anhydrases B and C. J. Biol. Chem. 1967, 242, 4221–4229. [Google Scholar] [CrossRef]
- Akkus, M.; Kirici, M.; Poustforoosh, A.; Erdoğan, M.; Tüzün, B. Phenolic Compounds: Investigating Their Anti-Carbonic Anhydrase, Anti-Cholinesterase, Anticancer, Anticholinergic, and Antiepileptic Properties Through Molecular Docking, MM-GBSA, and Dynamics Analyses. Korean J. Chem. Eng. 2025, 42, 1149–1168. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Feather-Stone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Maharramov, A.; Kurbanova, M.; Demir, Y.; Safarova, A.; Huseynov, E.; Sujayev, A.; Alwasel, S.; Gülçin, I. Synthesis, characterization, crystal structure and bioactivities of novel enamine and pyrrole derivatives endowed with acetylcholinesterase, α-glycosidase and human carbonic anhydrase inhibition effects. Org. Commun. 2021, 78, 144–156. [Google Scholar]
- Chakravarty, S.; Kannan, K.K. Drug-Protein Interactions: Refined Structures of Three Sulfonamide Drug Complexes of Human Carbonic Anhydrase I Enzyme. J. Mol. Biol. 1994, 243, 298–309. [Google Scholar] [CrossRef]
- Sippel, K.H.; Robbins, A.H.; Domsic, J.; Genis, C.; Agbandje-McKenna, M.; McKenna, R. High-resolution structure of human carbonic anhydrase II complexed with acetazolamide reveals insights into inhibitor drug design. Struct. Biol. Cryst. Commun. 2009, 65, 992–995. [Google Scholar] [CrossRef] [PubMed]
- Dileep, K.V.; Ihara, K.; Mishima-Tsumagari, C.; Kukimoto-Niino, M.; Yonemochi, M.; Hanada, K.; Shirouzu, M.; Zhang, K.Y.J. Crystal structure of human acetylcholinesterase in complex with tacrine: Implications for drug discovery. Int. J. Biol. Macromol. 2022, 210, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Nachon, F.; Carletti, E.; Ronco, C.; Trovaslet, M.; Nicolet, Y.; Jean, L.; Renard, P.-Y. Crystal structures of human cholinesterases in complex with huprine W and tacrine: Elements of specificity for anti-Alzheimer’s drugs targeting acetyl- and butyryl-cholinesterase. Biochem. J. 2013, 453, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Biovia, D.S. Discovery Studio Visualizer; Dassault Systèmes BIOVIA.: San Diego, CA, USA, 2023. [Google Scholar]
- Consortium, U. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 27852791. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. In Chemical Biology: Methods and Protocols; Hempel, J.E., Williams, C.H., Hong, C.C., Eds.; Springer: New York, NY, USA, 2015; pp. 243–250. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]

 I2 (the second inhibitor);
I2 (the second inhibitor);  I1 (the first inhibitor);
I1 (the first inhibitor);  control (no inhibitor).
I2 (the second inhibitor); I1 (the first inhibitor); control (no inhibitor).
control (no inhibitor).
I2 (the second inhibitor); I1 (the first inhibitor); control (no inhibitor). 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R1 | R2 |
|---|---|---|
| 8a | -H | 4-OMe |
| 8b | -H | 4-Cl |
| 8c | -F | 4-OMe |
| 8d | -F | 4-Cl |
| Compound | IC50 Values (nM) | Ki Values (nM) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| hCA I | r2 | hCA II | r2 | AChE | r2 | BChE | r2 | hCA I | hCA II | AChE | BChE | |
| 8a | 73.05 | 0.983 | 41.07 | 0.947 | 4.36 | 0.945 | 1.37 | 0.937 | 81.24 ± 4.36 | 49.33 ± 5.03 | 3.86 ± 0.20 | 1.01 ± 0.08 |
| 8b | 64.37 | 0.936 | 49.32 | 0.924 | 6.77 | 0.929 | 1.95 | 0.902 | 78.01 ± 5.48 | 52.45 ± 6.24 | 6.01 ± 0.35 | 1.70 ± 0.10 |
| 8c | 42.16 | 0.989 | 30.21 | 0.983 | 13.04 | 0.993 | 2.03 | 0.915 | 48.36 ± 6.98 | 36.08 ± 5.36 | 11.35 ± 1.02 | 1.56 ± 0.06 |
| 8d | 39.14 | 0.961 | 34.08 | 0.940 | 9.30 | 0.946 | 1.98 | 0.938 | 45.13 ± 3.91 | 39.57 ± 4.91 | 7.98 ± 0.60 | 1.78 ± 0.12 |
| AZA * | 129.03 | 0.907 | 102.55 | 0.936 | - | - | - | - | 145.73 ± 8.03 | 112.63 ± 6.87 | - | - |
| TAC * | - | - | - | - | 20.34 | 0.968 | 4.35 | 0.905 | - | - | 14.27 ± 2.03 | 3.57 ± 0.35 |
| Compound | Docking Score (AChE) [kcal/mol] | IC50 (AChE) [μM] | Docking Score (BChE) [kcal/mol] | IC50 (BChE) [μM] | Docking Score (hCA I) [kcal/mol] | IC50 (hCA I) [μM] | Docking Score (hCA II) [kcal/mol] | IC50 (hCA II) [μM] |
|---|---|---|---|---|---|---|---|---|
| 8a | −10.2 | 2.8 | −9.5 | 5.6 | −9.1 | 2.41 ± 0.11 | −9.3 | 3.18 ± 0.15 |
| 8b | −9.8 | 4.1 | −8.9 | 7.3 | −8.9 | 3.05 ± 0.13 | −9.2 | 3.97 ± 0.20 |
| 8c | −10.0 | 3.5 | −9.7 | 6.0 | −9.0 | 2.87 ± 0.10 | −9.1 | 3.45 ± 0.17 |
| 8d | −9.6 | 5.0 | −9.1 | 7.8 | −8.7 | 3.40 ± 0.16 | −9.0 | 4.12 ± 0.21 |
| Characteristic | Compound 8a | Compound 8b | Compound 8c | Compound 8d |
|---|---|---|---|---|
| Intestinal absorption (%) | 87.03 | 85.52 | 86.50 | 84.99 |
| BBB permeability (logBB) | 0.079 | 0.383 | 0.202 | 0.506 |
| CNS permeability (logPS) | –1.76 | –0.95 | –1.83 | –0.98 |
| AMES toxicity | No | Yes | No | Yes |
| hERG I inhibition | Yes | No | No | No |
| Hepatotoxicity | No | No | No | No |
| CYP3A4 substrate/inhibitor | Yes/Yes | Yes/Yes | Yes/Yes | Yes/Yes |
| P-gp substrate/inhibitor | Yes/Yes | Yes/Yes | Yes/Yes | Yes/Yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alici, H.; Topuz, S.; Demir, K.; Taslimi, P.; Tahtaci, H. Synthesis, Biological Evaluation, and In Silico Characterization of Novel Imidazothiadiazole–Chalcone Hybrids as Multi-Target Enzyme Inhibitors. Pharmaceuticals 2025, 18, 962. https://doi.org/10.3390/ph18070962
Alici H, Topuz S, Demir K, Taslimi P, Tahtaci H. Synthesis, Biological Evaluation, and In Silico Characterization of Novel Imidazothiadiazole–Chalcone Hybrids as Multi-Target Enzyme Inhibitors. Pharmaceuticals. 2025; 18(7):962. https://doi.org/10.3390/ph18070962
Chicago/Turabian StyleAlici, Hakan, Senol Topuz, Kadir Demir, Parham Taslimi, and Hakan Tahtaci. 2025. "Synthesis, Biological Evaluation, and In Silico Characterization of Novel Imidazothiadiazole–Chalcone Hybrids as Multi-Target Enzyme Inhibitors" Pharmaceuticals 18, no. 7: 962. https://doi.org/10.3390/ph18070962
APA StyleAlici, H., Topuz, S., Demir, K., Taslimi, P., & Tahtaci, H. (2025). Synthesis, Biological Evaluation, and In Silico Characterization of Novel Imidazothiadiazole–Chalcone Hybrids as Multi-Target Enzyme Inhibitors. Pharmaceuticals, 18(7), 962. https://doi.org/10.3390/ph18070962