


Structure-Based Design and In-Silico Evaluation of Computationally Proposed Curcumin Derivatives as Potential Inhibitors of the Coronaviral PLpro Enzymes


Abstract

1. Introduction
2. Results and Discussion
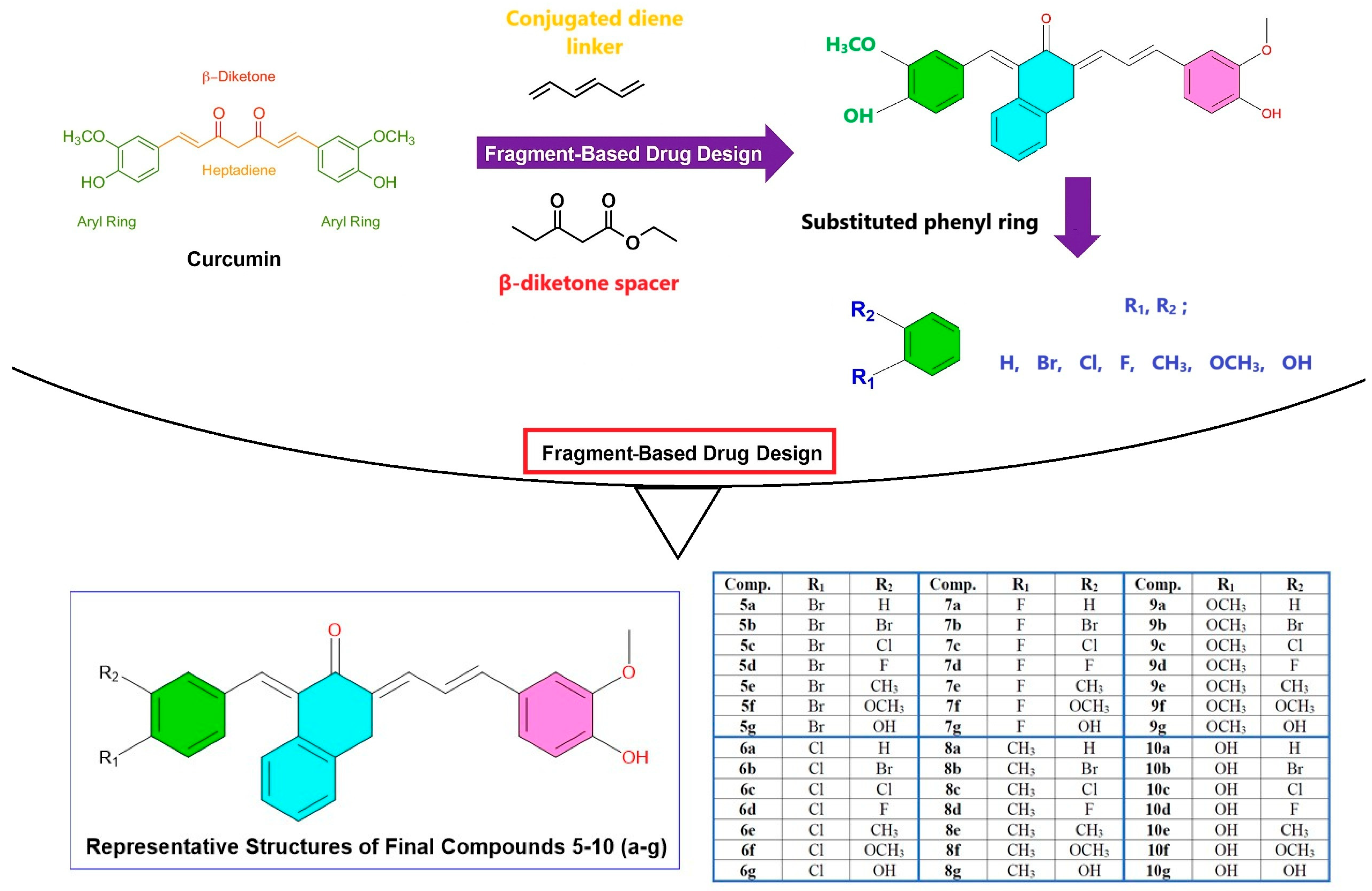
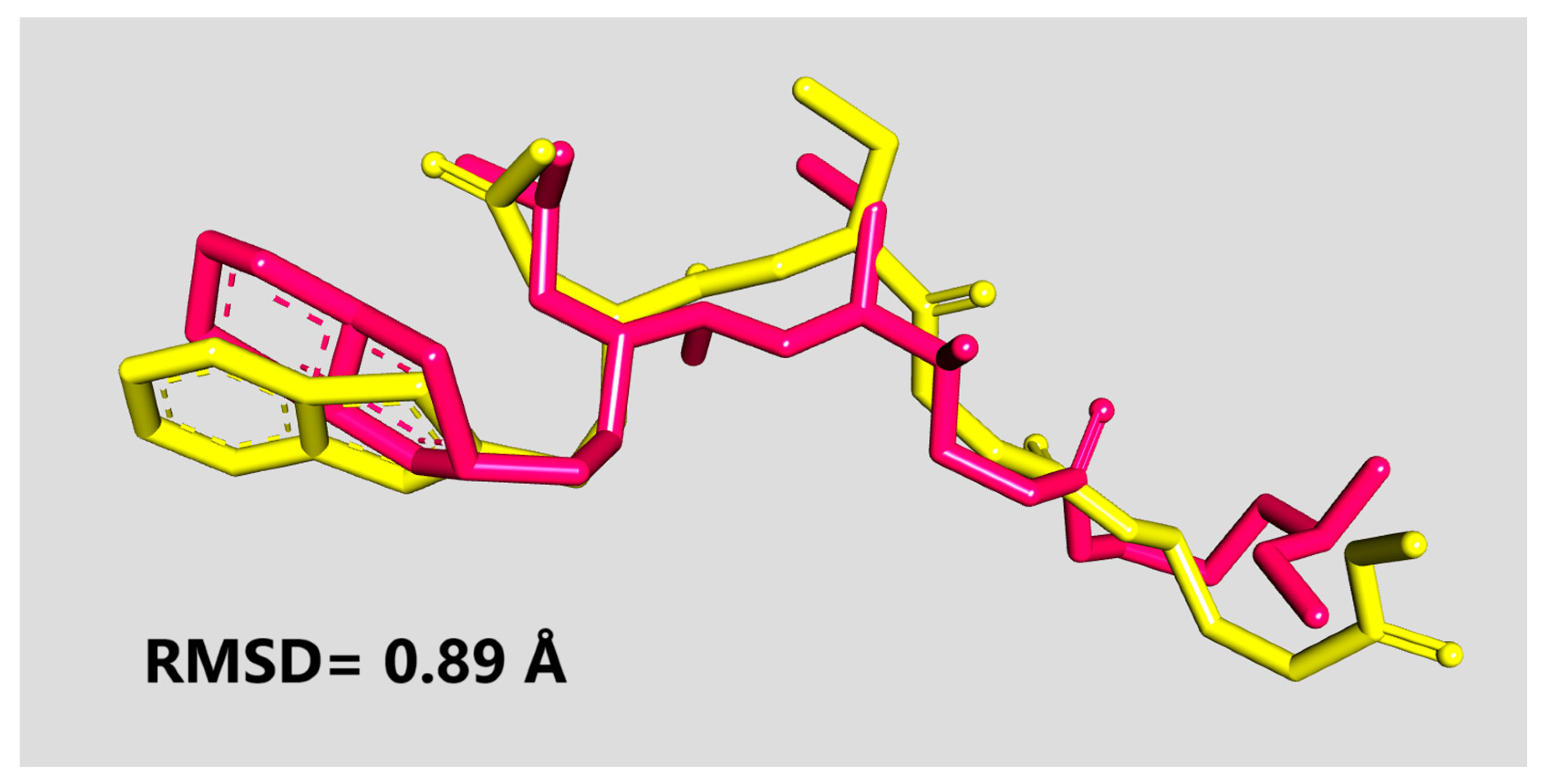
2.1. Design Strategy and Structural Modifications
2.2. ADMET Evolation
2.2.1. Evaluation of Drug-Likeness and Pharmacokinetic Properties of Curcumin Derivatives Based on Ro5
- -
- Molecular Weight (MW) ≤ 500 Da
- -
- Hydrogen Bond Donor Number (HBD) ≤ 5
- -
- Hydrogen Bond Acceptor Number (HBA) ≤ 10
- -
- Lipophilicity (logP) ≤ 5
2.2.2. Absorption and Oral Bioavailability
2.2.3. Distribution Properties and Tissue Penetration Potential
2.2.4. Metabolic Stability and Cytochromes P450 (CYP) Enzyme Interactions
2.2.5. Excretion Profiles and Clearance Characteristics
2.2.6. Toxicological Evaluation: Safety Profile and Risk Assessment
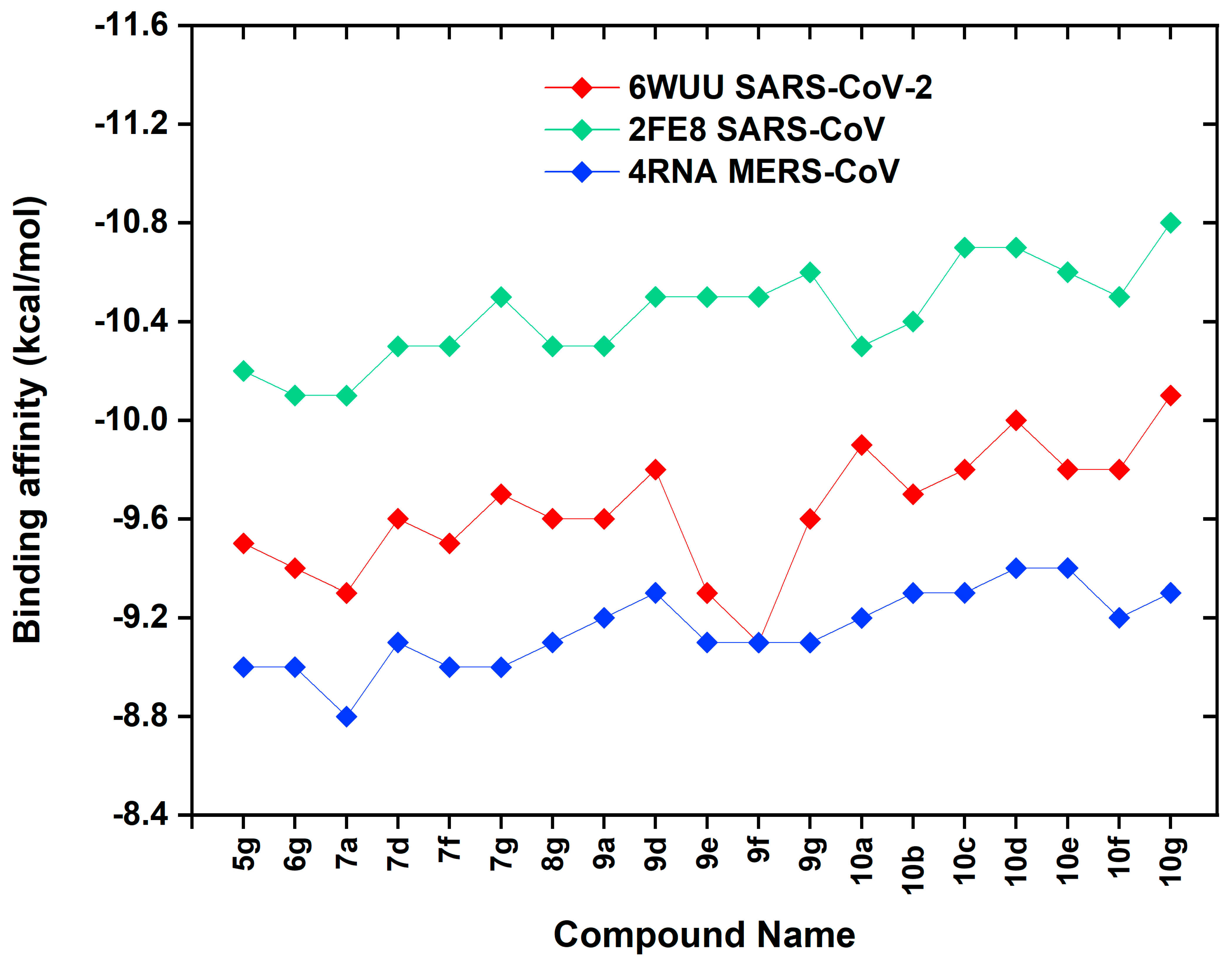
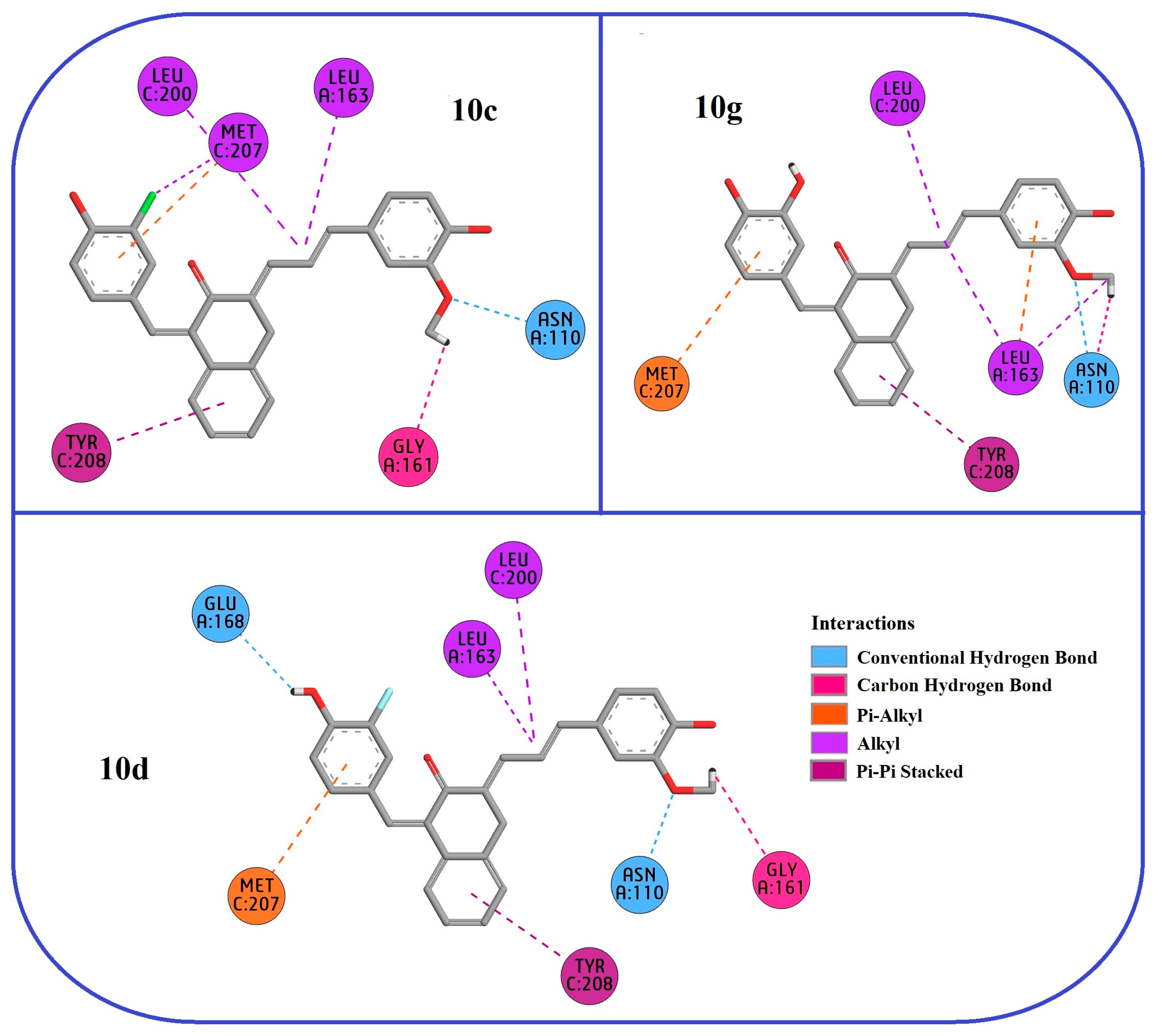
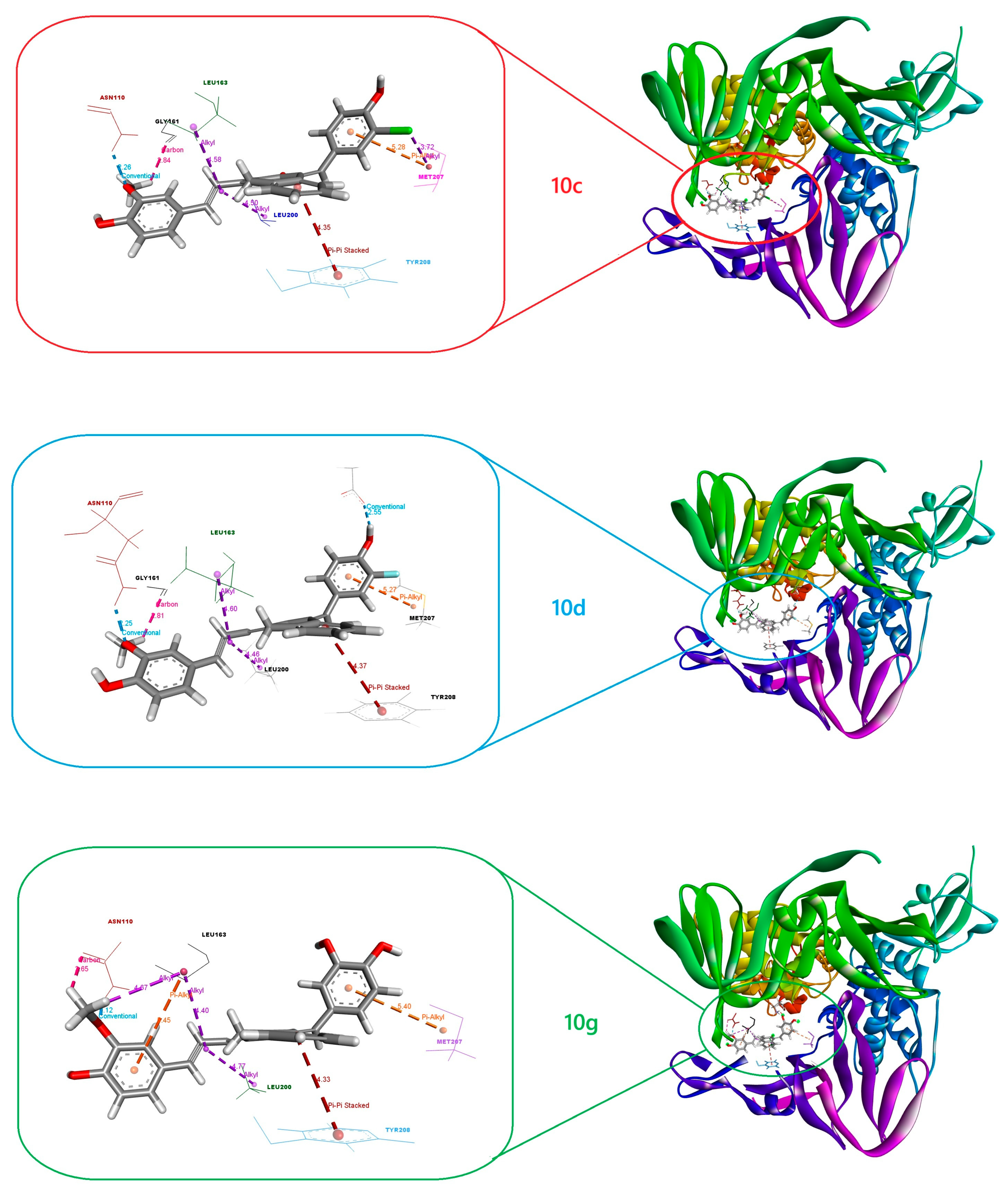
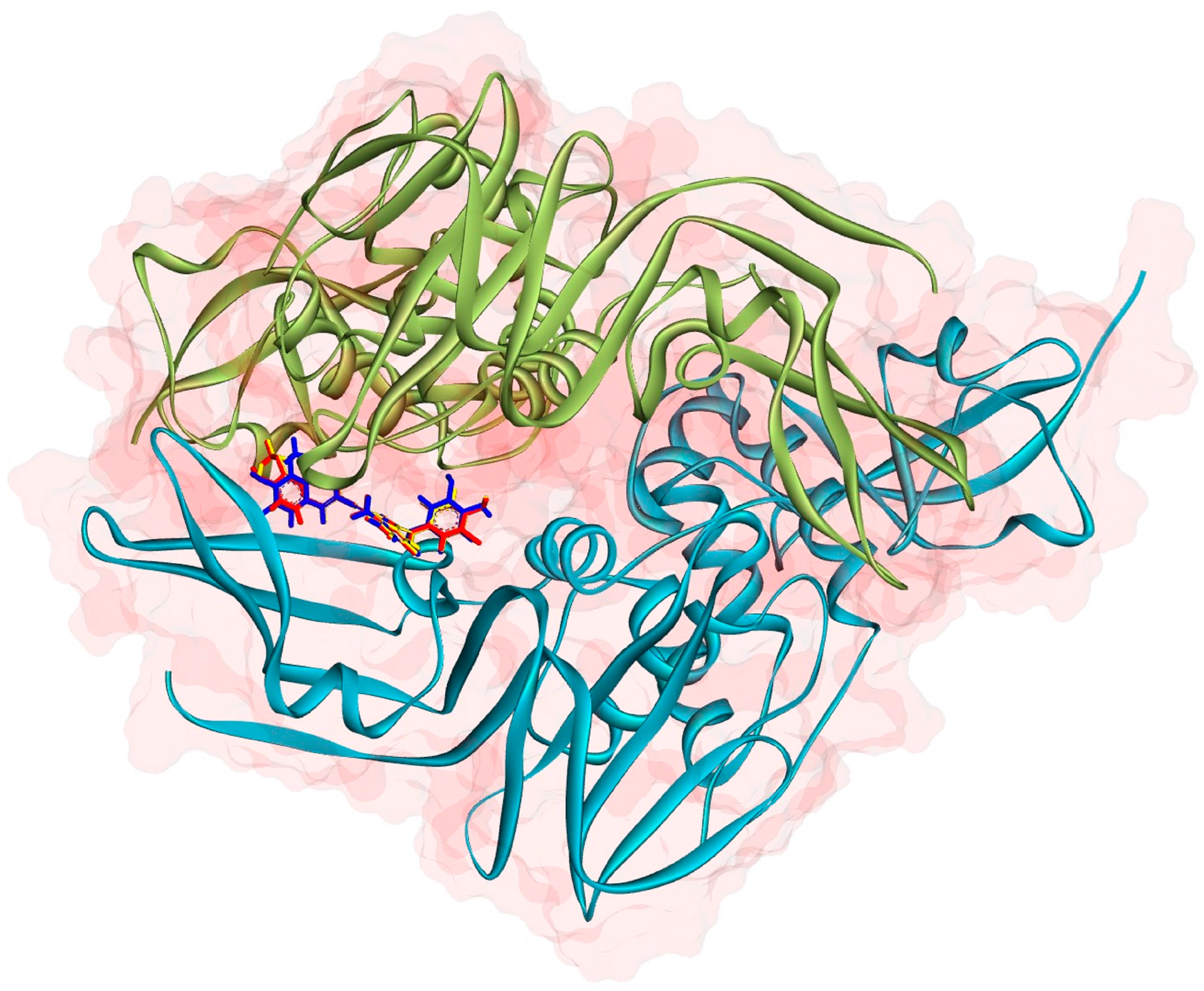
2.3. Molecular Docking
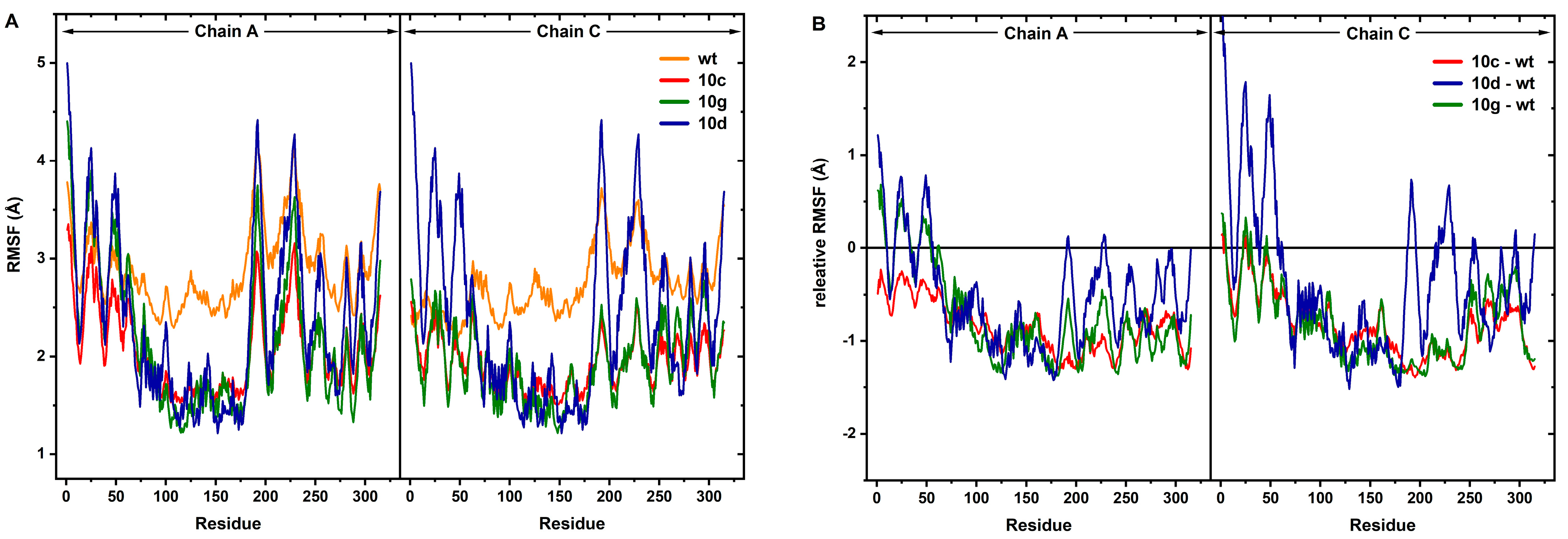
2.4. MD Simulations and MM-PBSA Analysis
3. Materials and Methods
3.1. ADMET Predictions
3.2. Molecular Docking Calculations
3.3. MD Simulation Protocol
3.4. MM/PBSA Free Energy Calculations
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Peiris, J.S.M.; Chu, C.M.; Cheng, V.C.C.; Chan, K.S.; Hung, I.F.N.; Poon, L.L.M.; Law, K.I.; Tang, B.S.F.; Hon, T.Y.W.; Chan, C.S.; et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: A prospective study. Lancet 2003, 361, 1767–1772. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Zaki Ali, M.; van Boheemen, S.; Bestebroer Theo, M.; Osterhaus Albert, D.M.E.; Fouchier Ron, A.M. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Zumla, A.; Chan, J.F.W.; Azhar, E.I.; Hui, D.S.C.; Yuen, K.-Y. Coronaviruses—Drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; De Clercq, E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat. Rev. Drug Discov. 2020, 19, 149–150. [Google Scholar] [CrossRef]
- Shyr, Z.A.; Gorshkov, K.; Chen, C.Z.; Zheng, W. Drug Discovery Strategies for SARS-CoV-2. J. Pharmacol. Exp. Ther. 2020, 375, 127–138. [Google Scholar] [CrossRef]
- Rabie, A.M. The informative nature of the disappeared SARS-CoV-2 genomic sequences: A mini-review with perspectives. Adv. Chemicobiol. Res. 2022, 1, 58–64. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M.; Shahabi, D.; Chapman, M.E.; Mesecar, A.D. Drug Development and Medicinal Chemistry Efforts toward SARS-Coronavirus and Covid-19 Therapeutics. ChemMedChem 2020, 15, 907–932. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Baez-Santos, Y.M.; St John, S.E.; Mesecar, A.D. The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015, 115, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Rut, W.; Lv, Z.; Zmudzinski, M.; Patchett, S.; Nayak, D.; Snipas, S.J.; El Oualid, F.; Huang, T.T.; Bekes, M.; Drag, M.; et al. Activity profiling and structures of inhibitor-bound SARS-CoV-2-PLpro protease provides a framework for anti-COVID-19 drug design. Sci. Adv. 2020, 6, eabd4596. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Barretto, N.; Jukneliene, D.; Ratia, K.; Chen, Z.; Mesecar, A.D.; Baker, S.C. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 2005, 79, 15189–15198. [Google Scholar] [CrossRef] [PubMed]
- Ratia, K.; Saikatendu, K.S.; Santarsiero, B.D.; Barretto, N.; Baker, S.C.; Stevens, R.C.; Mesecar, A.D. Severe acute respiratory syndrome coronavirus papain-like protease: Structure of a viral deubiquitinating enzyme. Proc. Natl. Acad. Sci. USA 2006, 103, 5717–5722. [Google Scholar] [CrossRef]
- van Vliet, V.J.E.; Huynh, N.; Palà, J.; Patel, A.; Singer, A.; Slater, C.; Chung, J.; van Huizen, M.; Teyra, J.; Miersch, S.; et al. Ubiquitin variants potently inhibit SARS-CoV-2 PLpro and viral replication via a novel site distal to the protease active site. PLoS Pathog. 2022, 18, e1011065. [Google Scholar] [CrossRef]
- Gold, I.M.; Reis, N.; Glaser, F.; Glickman, M.H. Coronaviral PLpro proteases and the immunomodulatory roles of conjugated versus free Interferon Stimulated Gene product-15 (ISG15). Semin. Cell Dev. Biol. 2022, 132, 16–26. [Google Scholar] [CrossRef]
- Varghese, A.; Liu, J.; Liu, B.; Guo, W.; Dong, F.; Patterson, T.A.; Hong, H. Analysis of Structures of SARS-CoV-2 Papain-like Protease Bound with Ligands Unveils Structural Features for Inhibiting the Enzyme. Molecules 2025, 30, 491. [Google Scholar] [CrossRef]
- Alici, H.; Tahtaci, H.; Demir, K. Design and various in silico studies of the novel curcumin derivatives as potential candidates against COVID-19 -associated main enzymes. Comput. Biol. Chem. 2022, 98, 107657. [Google Scholar] [CrossRef]
- Teixeira, S.A.; Medeiros, S.R.A.; da Silva Oliveira, G.L.; Acha, B.T.; Pereira-Freire, J.A. Effect of Curcumin on the Process of Neuroinflammation Caused by COVID-19. In Curcumin and Neurodegenerative Diseases: From Traditional to Translational Medicines; Rai, M., Feitosa, C.M., Eds.; Springer Nature Singapore: Singapore, 2023; pp. 293–310. [Google Scholar]
- Farooqui, A.A. (Ed.) Metabolism, Bioavailability, Biochemical Effects of Curcumin in Visceral Organs and the Brain. In Therapeutic Potentials of Curcumin for Alzheimer Disease; Springer International Publishing: Cham, Switzerland, 2016; pp. 113–149. [Google Scholar]
- Prasad, S.; Tyagi, A.K.; Aggarwal, B.B. Recent Developments in Delivery, Bioavailability, Absorption and Metabolism of Curcumin: The Golden Pigment from Golden Spice. Cancer Res. Treat. 2014, 46, 2–18. [Google Scholar] [CrossRef] [PubMed]
- Siviero, A.; Gallo, E.; Maggini, V.; Gori, L.; Mugelli, A.; Firenzuoli, F.; Vannacci, A. Curcumin, a golden spice with a low bioavailability. J. Herb. Med. 2015, 5, 57–70. [Google Scholar] [CrossRef]
- Suresh, K.; Nangia, A. Curcumin: Pharmaceutical solids as a platform to improve solubility and bioavailability. CrystEngComm 2018, 20, 3277–3296. [Google Scholar] [CrossRef]
- Teshima, K.; Takeshi, T.; Zhengmao, Y.; Ken, I.; Takao, M.; Tamotsu, S.; Ayumu, M.; Masato, H.; Mayo, Y.; Komatsu, H. Antiviral activity of curcumin and its analogs selected by an artificial intelligence-supported activity prediction system in SARS-CoV-2-infected VeroE6 cells. Nat. Prod. Res. 2024, 38, 867–872. [Google Scholar] [CrossRef]
- Fibriani, A.; Taharuddin, A.A.P.; Stephanie, R.; Yamahoki, N.; Laurelia, J.; Wisnuwardhani, P.H.; Agustiyanti, D.F.; Angelina, M.; Rubiyana, Y.; Ningrum, R.A.; et al. Curcumin-derived carbon-dots as a potential COVID-19 antiviral drug. Heliyon 2023, 9, e20089. [Google Scholar] [CrossRef]
- Dourado, D.; Freire, D.T.; Pereira, D.T.; Amaral-Machado, L.; Alencar, É.N.; de Barros, A.L.B.; Egito, E.S.T. Will curcumin nanosystems be the next promising antiviral alternatives in COVID-19 treatment trials? Biomed. Pharmacother. 2021, 139, 111578. [Google Scholar] [CrossRef]
- Bormann, M.; Alt, M.; Schipper, L.; van de Sand, L.; Le-Trilling, V.T.; Rink, L.; Heinen, N.; Madel, R.J.; Otte, M.; Wuensch, K.; et al. Turmeric Root and Its Bioactive Ingredient Curcumin Effectively Neutralize SARS-CoV-2 In Vitro. Viruses 2021, 13, 1914. [Google Scholar] [CrossRef] [PubMed]
- Nicoliche, T.; Bartolomeo, C.S.; Lemes, R.M.R.; Pereira, G.C.; Nunes, T.A.; Oliveira, R.B.; Nicastro, A.L.M.; Soares, É.N.; da Cunha Lima, B.F.; Rodrigues, B.M.; et al. Antiviral, anti-inflammatory and antioxidant effects of curcumin and curcuminoids in SH-SY5Y cells infected by SARS-CoV-2. Sci. Rep. 2024, 14, 10696. [Google Scholar] [CrossRef]
- Azarkar, S.; Abedi, M.; Lavasani, A.S.O.; Ammameh, A.H.; Goharipanah, F.; Baloochi, K.; Bakhshi, H.; Jafari, A. Curcumin as a natural potential drug candidate against important zoonotic viruses and prions: A narrative review. Phytother. Res. 2024, 38, 3080–3121. [Google Scholar] [CrossRef]
- Fakih, T.M.; Ritmaleni; Zainul, R.; Muchtaridi, M. Molecular docking-based virtual screening and computational investigations of biomolecules (curcumin analogs) as potential lead inhibitors for SARS-CoV-2 papain-like protease. Pharmacia 2024, 71, 1–19. [Google Scholar] [CrossRef]
- Ferreira, J.C.; Villanueva, A.J.; Al Adem, K.; Fadl, S.; Alzyoud, L.; Ghattas, M.A.; Rabeh, W.M. Identification of novel allosteric sites of SARS-CoV-2 papain-like protease (PLpro) for the development of COVID-19 antivirals. J. Biol. Chem. 2024, 300, 107821. [Google Scholar] [CrossRef] [PubMed]
- Rabie, A.M. RNA: The most attractive target in recent viral diseases. Chem. Biol. Drug Des. 2024, 103, e14404. [Google Scholar] [CrossRef] [PubMed]
- Rabie, A.M.; Khedraoui, M.; Chtita, S. Targeting Conserved Regions of the SARS-CoV-2 Polymerase (RdRp) with Kinase Inhibitors as an Effective New Tactic for Discovering Dual-Action “Antiviral─Antiinflammatory” Drugs against COVID-19. Comput. Biol. Chem. 2025, 108454, in press. [Google Scholar] [CrossRef]
- Rabie, A.M. Future of the current anticoronaviral agents: A viewpoint on the validation for the next COVIDs and pandemics. Biocell 2023, 47, 2133–2139. [Google Scholar] [CrossRef]
- Rabie, A.M.; Yamari, I.; Chtita, S. The isoquinoline derivative “CYNOVID” as a prospective anti-SARS-CoV-2 agent: An expanded investigative computational study. Eur. J. Med. Chem. Rep. 2024, 12, 100214. [Google Scholar] [CrossRef]
- Rabie, A.M. Potent Inhibitory Activities of the Adenosine Analogue Cordycepin on SARS-CoV-2 Replication. ACS Omega 2022, 7, 2960–2969. [Google Scholar] [CrossRef]
- Rabie, A.M.; Abdalla, M. Forodesine and Riboprine Exhibit Strong Anti-SARS-CoV-2 Repurposing Potential: In Silico and In Vitro Studies. ACS Bio Med. Chem. Au 2022, 2, 565–585. [Google Scholar] [CrossRef]
- Rabie, A.M. Teriflunomide: A possible effective drug for the comprehensive treatment of COVID-19. Curr. Res. Pharmacol. Drug Discov. 2021, 2, 100055. [Google Scholar] [CrossRef]
- Eltayb, W.A.; Abdalla, M.; Rabie, A.M. Novel Investigational Anti-SARS-CoV-2 Agent Ensitrelvir “S-217622”: A Very Promising Potential Universal Broad-Spectrum Antiviral at the Therapeutic Frontline of Coronavirus Species. ACS Omega 2023, 8, 5234–5246. [Google Scholar] [CrossRef]
- Rabie, A.M.; Abdalla, M. Evaluation of a series of nucleoside analogs as effective anticoronaviral-2 drugs against the Omicron-B.1.1.529/BA.2 subvariant: A repurposing research study. Med. Chem. Res. 2023, 32, 326–341. [Google Scholar] [CrossRef]
- Rabie, A.M.; Eltayb, W.A. Potent Dual Polymerase/Exonuclease Inhibitory Activities of Antioxidant Aminothiadiazoles Against the COVID-19 Omicron Virus: A Promising In Silico/In Vitro Repositioning Research Study. Mol. Biotechnol. 2024, 66, 592–611. [Google Scholar] [CrossRef] [PubMed]
- Rabie, A.M. Efficacious Preclinical Repurposing of the Nucleoside Analogue Didanosine against COVID-19 Polymerase and Exonuclease. ACS Omega 2022, 7, 21385–21396. [Google Scholar] [CrossRef] [PubMed]
- Rabie, A.M. Potent toxic effects of Taroxaz-104 on the replication of SARS-CoV-2 particles. Chem.-Biol. Interact. 2021, 343, 109480. [Google Scholar] [CrossRef]
- Nandi, S.; Kumar, M.; Saxena, A.K. Repurposing of Drugs and HTS to Combat SARS-CoV-2 Main Protease Utilizing Structure-Based Molecular Docking. Lett. Drug Des. Discov. 2022, 19, 413–427. [Google Scholar] [CrossRef]
- Nandi, S.; Kumar, M.; Saxena, A.K. QSAR of SARS-CoV-2 Main Protease Inhibitors Utilizing Theoretical Molecular Descriptors. Lett. Drug Des. Discov. 2024, 21, 116–132. [Google Scholar] [CrossRef]
- Rabie, A.M. Improved synthesis of the anti-SARS-CoV-2 investigational agent (E)-N-(4-cyanobenzylidene)-6-fluoro-3-hydroxypyrazine-2-carboxamide (cyanorona-20). Rev. Chim. 2022, 73, 69–75. [Google Scholar] [CrossRef]
- Rabie, A.M. Two antioxidant 2,5-disubstituted-1,3,4-oxadiazoles (CoViTris2020 and ChloViD2020): Successful repurposing against COVID-19 as the first potent multitarget anti-SARS-CoV-2 drugs. New J. Chem. 2021, 45, 761–771. [Google Scholar] [CrossRef]
- Rabie, A.M. New Potential Inhibitors of Coronaviral Main Protease (CoV-Mpro): Strychnine Bush, Pineapple, and Ginger could be Natural Enemies of COVID-19. Int. J. New Chem. 2022, 9, 225–237. [Google Scholar]
- Smith, D.A.; Jones, B.C.; Walker, D.K. Design of Drugs Involving the Concepts and Theories of Drug Metabolism and Pharmacokinetics. Med. Res. Rev. 1996, 16, 243–266. [Google Scholar] [CrossRef]
- Çevik, U.A.; Işik, A.; Karakaya, A. ADMET and Physicochemical Assessments in Drug Design. In Computational Methods for Rational Drug Design; Wiley: Hoboken, NJ, USA, 2025; pp. 123–151. [Google Scholar]
- Pillai, O.; Dhanikula, A.B.; Panchagnula, R. Drug delivery: An odyssey of 100 years. Curr. Opin. Chem. Biol. 2001, 5, 439–446. [Google Scholar] [CrossRef]
- Calkilic, N.M.; Alici, H.; Direkel, S.; Tahtaci, H. Synthesis, Characterization, Theoretical Analyses, and Investigation of Their Biological Activities of Acetovanillone-Derived Novel Benzyl Ethers. Polycycl. Aromat. Comp. 2022, 42, 5671–5695. [Google Scholar] [CrossRef]
- Ozcan, I.; Akkoc, S.; Alici, H.; Capanlar, S.; Sahin, O.; Tahtaci, H. Novel Thioether-Bridged 2,6-Disubstituted and 2,5,6-Trisubstituted Imidazothiadiazole Analogues: Synthesis, Antiproliferative Activity, ADME, and Molecular Docking Studies. Chem. Biodivers. 2023, 20, e202200884. [Google Scholar] [CrossRef]
- Ozcan, I.; Alici, H.; Taslimi, P.; Tahtaci, H. Novel 1,2,4-triazole-derived Schiff base derivatives: Design, synthesis, and multi-enzyme targeting potential for therapeutic applications. Bioorg. Chem. 2025, 157, 108246. [Google Scholar] [CrossRef] [PubMed]
- Jung, W.; Goo, S.; Hwang, T.; Lee, H.; Kim, Y.-K.; Chae, J.-w.; Yun, H.-y.; Jung, S. Absorption Distribution Metabolism Excretion and Toxicity Property Prediction Utilizing a Pre-Trained Natural Language Processing Model and Its Applications in Early-Stage Drug Development. Pharmaceuticals 2024, 17, 382. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- da Silva Lopes, L.; Pereira, S.K.S.; Lima, L.K.F. Pharmacokinetics and Pharmacodynamics of Curcumin. In Curcumin and Neurodegenerative Diseases: From Traditional to Translational Medicines; Rai, M., Feitosa, C.M., Eds.; Springer Nature Singapore: Singapore, 2023; pp. 3–19. [Google Scholar]
- Dei Cas, M.; Ghidoni, R. Dietary Curcumin: Correlation between Bioavailability and Health Potential. Nutrients 2019, 11, 2147. [Google Scholar] [CrossRef]
- Wei, X.; Senanayake, T.H.; Bohling, A.; Vinogradov, S.V. Targeted Nanogel Conjugate for Improved Stability and Cellular Permeability of Curcumin: Synthesis, Pharmacokinetics, and Tumor Growth Inhibition. Mol. Pharm. 2014, 11, 3112–3122. [Google Scholar] [CrossRef]
- Silvestre, F.; Santos, C.; Silva, V.; Ombredane, A.; Pinheiro, W.; Andrade, L.; Garcia, M.; Pacheco, T.; Joanitti, G.; Luz, G.; et al. Pharmacokinetics of Curcumin Delivered by Nanoparticles and the Relationship with Antitumor Efficacy: A Systematic Review. Pharmaceuticals 2023, 16, 943. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Lien, J.-C.; Chen, C.-Y.; Hung, C.-C.; Lin, H.-C. Design, Synthesis and Evaluation of Novel Derivatives of Curcuminoids with Cytotoxicity. Int. J. Mol. Sci. 2021, 22, 12171. [Google Scholar] [CrossRef]
- Gupta, S.C.; Prasad, S.; Kim, J.H.; Patchva, S.; Webb, L.J.; Priyadarsini, I.K.; Aggarwal, B.B. Multitargeting by curcumin as revealed by molecular interaction studies. Nat. Prod. Rep. 2011, 28, 1937–1955. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Zhang, S.; Zheng, X.; Luo, Z.; Chen, H.; Yao, X. Advances in β-Diketocyclisation of Curcumin Derivatives and their Antitumor Activity. Chem. Biodivers. 2024, 21, e202301556. [Google Scholar] [CrossRef] [PubMed]
- Rabie, A.M. Revolutionizing Playing with Skeleton Atoms: Molecular Editing Surgery in Medicinal Chemistry. Mini Rev. Med. Chem. 2025, 25, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Szakács, G.; Váradi, A.; Özvegy-Laczka, C.; Sarkadi, B. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME–Tox). Drug Discov. Today 2008, 13, 379–393. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]
- Li, D.; Edward, H.K.; Guy, T.C. Drug-Like Property Concepts in Pharmaceutical Design. Curr. Pharm. Des. 2009, 15, 2184–2194. [Google Scholar]
- Waring, M.J. Lipophilicity in drug discovery. Expert. Opin. Drug Discov. 2010, 5, 235–248. [Google Scholar] [CrossRef]
- Pelkonen, O.; Boobis, A.R.; Gundert-Remy, U. In vitro prediction of gastrointestinal absorption and bioavailability: An experts’ meeting report. Eur. J. Clin. Pharmacol. 2001, 57, 621–629. [Google Scholar] [CrossRef]
- Stillhart, C.; Vučićević, K.; Augustijns, P.; Basit, A.W.; Batchelor, H.; Flanagan, T.R.; Gesquiere, I.; Greupink, R.; Keszthelyi, D.; Koskinen, M.; et al. Impact of gastrointestinal physiology on drug absorption in special populations—An UNGAP review. Eur. J. Pharm. Sci. 2020, 147, 105280. [Google Scholar] [CrossRef]
- O’Hagan, S.; Kell, D.B. The apparent permeabilities of Caco-2 cells to marketed drugs: Magnitude, and independence from both biophysical properties and endogenite similarities. PeerJ 2015, 3, e1405. [Google Scholar] [CrossRef] [PubMed]
- Hilgers, A.R.; Conradi, R.A.; Burton, P.S. Caco-2 cell monolayers as a model for drug transport across the intestinal mucosa. Pharm. Res. 1990, 7, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Taniyama, K.; Aoyama, T.; Watanabe, Y. Evaluation of the Role of P-glycoprotein (P-gp)-Mediated Efflux in the Intestinal Absorption of Common Substrates with Elacridar, a P-gp Inhibitor, in Rats. Eur. J. Drug Metab. Pharmacokinet. 2020, 45, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Kono, Y.; Kawahara, I.; Shinozaki, K.; Nomura, I.; Marutani, H.; Yamamoto, A.; Fujita, T. Characterization of P-Glycoprotein Inhibitors for Evaluating the Effect of P-Glycoprotein on the Intestinal Absorption of Drugs. Pharmaceutics 2021, 13, 388. [Google Scholar] [CrossRef]
- Waters, N.J.; Lombardo, F. Use of the Øie-Tozer Model in Understanding Mechanisms and Determinants of Drug Distribution. Drug Metab. Dispos. 2010, 38, 1159–1165. [Google Scholar] [CrossRef]
- Stepensky, D. Use of unbound volumes of drug distribution in pharmacokinetic calculations. Eur. J. Pharm. Sci. 2011, 42, 91–98. [Google Scholar] [CrossRef]
- Korzekwa, K.; Nagar, S. Drug Distribution Part 2. Predicting Volume of Distribution from Plasma Protein Binding and Membrane Partitioning. Pharm. Res. 2017, 34, 544–551. [Google Scholar] [CrossRef]
- Knights, K.M.; Stresser, D.M.; Miners, J.O.; Crespi, C.L. In Vitro Drug Metabolism Using Liver Microsomes. Curr. Protoc. Pharmacol. 2016, 74, 7.8.1–7.8.24. [Google Scholar] [CrossRef]
- Armani, S.; Geier, A.; Forst, T.; Merle, U.; Alpers, D.H.; Lunnon, M.W. Effect of changes in metabolic enzymes and transporters on drug metabolism in the context of liver disease: Impact on pharmacokinetics and drug–drug interactions. Br. J. Clin. Pharmacol. 2024, 90, 942–958. [Google Scholar] [CrossRef]
- Ajavon, A.D.; Bonate, P.L.; Taft, D.R. Renal excretion of clofarabine: Assessment of dose-linearity and role of renal transport systems on drug excretion. Eur. J. Pharm. Sci. 2010, 40, 209–216. [Google Scholar] [CrossRef]
- Severance, A.C.; Sandoval, P.J.; Wright, S.H. Correlation between Apparent Substrate Affinity and OCT2 Transport Turnover. J. Pharmacol. Exp. Ther. 2017, 362, 405–412. [Google Scholar] [CrossRef]
- Nishizawa, K.; Yoda, N.; Morokado, F.; Komori, H.; Nakanishi, T.; Tamai, I. Changes of drug pharmacokinetics mediated by downregulation of kidney organic cation transporters Mate1 and Oct2 in a rat model of hyperuricemia. PLoS ONE 2019, 14, e0214862. [Google Scholar] [CrossRef] [PubMed]
- Garrido, A.; Lepailleur, A.; Mignani, S.M.; Dallemagne, P.; Rochais, C. hERG toxicity assessment: Useful guidelines for drug design. European J. Med. Chem. 2020, 195, 112290. [Google Scholar] [CrossRef] [PubMed]
- Guy, R.C. Saccharin. In Encyclopedia of Toxicology, 4th ed.; Wexler, P., Ed.; Academic Press: Oxford, UK, 2024; pp. 377–379. [Google Scholar]
- Mortelmans, K.; Zeiger, E. The Ames Salmonella/microsome mutagenicity assay. Mutat. Res.-Fundam. Mol. Mech. Mutagen. 2000, 455, 29–60. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Zhang, X.; Zhang, X.; Wang, X.; Sun, W.; Zhang, Y.; Hu, Z. Unveiling the mechanism of action of a novel natural dual inhibitor of SARS-CoV-2 Mpro and PLpro with molecular dynamics simulations. Nat. Prod. Bioprospect. 2025, 15, 3. [Google Scholar] [CrossRef]
- Imane, Y.; Lamiae El, B.; Oussama, A.; Mohammed, B.; Mhammed El, K.; Abdelouahid, S.; Samir, C. Integrated Exploration of Pyranocoumarin Derivatives as Synergistic Inhibitors of Dual-target for Mpro and PLpro Proteins of SARS-CoV-2 through Molecular Docking, ADMET Analysis, and Molecular Dynamics Simulation. Curr. Med. Chem. 2024, 31, 1–14. [Google Scholar]
- Valdés-Albuernes, J.L.; Díaz-Pico, E.; Alfaro, S.; Caballero, J. Modeling of noncovalent inhibitors of the papain-like protease (PLpro) from SARS-CoV-2 considering the protein flexibility by using molecular dynamics and cross-docking. Front. Mol. Biosci. 2024, 11, 1374364. [Google Scholar] [CrossRef]
- Metwaly, A.M.; Elkaeed, E.B.; Khalifa, M.M.; Alsfouk, A.A.; Amin, F.G.; Ibrahim, I.M.; Eissa, I.H. Discovery of potential FDA-approved SARS-CoV-2 Papain-like protease inhibitors: A multi-phase in silico approach. J. Chem. Res. 2024, 48, 17475198241298547. [Google Scholar] [CrossRef]
- Bader, S.M.; Calleja, D.J.; Devine, S.M.; Kuchel, N.W.; Lu, B.G.C.; Wu, X.; Birkinshaw, R.W.; Bhandari, R.; Loi, K.; Volpe, R.; et al. A novel PLpro inhibitor improves outcomes in a pre-clinical model of long COVID. Nat. Commun. 2025, 16, 2900. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, w5–w14. [Google Scholar] [CrossRef]
- Braga, R.C.; Alves, V.M.; Silva, M.F.B.; Muratov, E.; Fourches, D.; Lião, L.M.; Tropsha, A.; Andrade, C.H. Pred-hERG: A Novel web-Accessible Computational Tool for Predicting Cardiac Toxicity. Mol. Inform. 2015, 34, 698–701. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lei, H.; Santarsiero, B.D.; Gatuz, J.L.; Cao, S.; Rice, A.J.; Patel, K.; Szypulinski, M.Z.; Ojeda, I.; Ghosh, A.K.; et al. Inhibitor Recognition Specificity of MERS-CoV Papain-like Protease May Differ from That of SARS-CoV. ACS Chem. Biol. 2015, 10, 1456–1465. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Bastos, R.S.; de Aguiar, C.P.O.; Cruz, J.N.; Ramos, R.S.; Kimani, N.M.; de Souza, J.S.N.; Chaves, M.H.; de Freitas, H.F.; Pita, S.S.R.; Santos, C.B.R.d. Rational Approach toward COVID-19′s Main Protease Inhibitors: A Hierarchical Biochemoinformatics Analysis. Int. J. Mol. Sci. 2024, 25, 6715. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Andersen, H.C. Rattle: A “velocity” version of the shake algorithm for molecular dynamics calculations. J. Comput. Phys. 1983, 52, 24–34. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physicochemical and Liphophilicity Parameters | Drug-Likeness Properties | |||||
|---|---|---|---|---|---|---|
| Compound Name | MW (Dalton) | HBA | HBD | logP | Violations of Ro5 Rules | Violations |
| 5a | 472.07 | 3 | 1 | 5.448 | 1 | LOGP > 5.00 |
| 5b | 549.98 | 3 | 1 | 5.815 | 2 | MW > 500, LOGP > 5.00 |
| 5c | 506.03 | 3 | 1 | 5.828 | 2 | MW > 500, LOGP > 5.00 |
| 5d | 490.06 | 4 | 1 | 5.438 | 1 | LOGP > 5.00 |
| 5e | 486.08 | 3 | 1 | 5.758 | 1 | LOGP > 5.00 |
| 5f | 502.08 | 4 | 1 | 5.190 | 2 | MW > 500, LOGP > 5.00 |
| 5g | 488.06 | 4 | 2 | 4.985 | ||
| 6a | 428.12 | 3 | 1 | 5.305 | 1 | |
| 6b | 506.03 | 3 | 1 | 5.781 | 2 | MW > 500, LOGP > 5.00 |
| 6c | 462.08 | 3 | 1 | 5.765 | 1 | LOGP > 5.00 |
| 6d | 446.11 | 3 | 1 | 5.373 | 1 | LOGP > 5.00 |
| 6e | 442.13 | 3 | 1 | 5.724 | 1 | LOGP > 5.00 |
| 6f | 458.13 | 4 | 1 | 5.050 | 1 | LOGP > 5.00 |
| 6g | 444.11 | 4 | 2 | 4.826 | ||
| 7a | 412.15 | 3 | 1 | 4.765 | ||
| 7b | 490.06 | 3 | 1 | 5.345 | 1 | LOGP > 5.00 |
| 7c | 446.11 | 3 | 1 | 5.332 | 1 | LOGP > 5.00 |
| 7d | 430.14 | 3 | 1 | 4.942 | ||
| 7e | 426.16 | 3 | 1 | 5.228 | 1 | LOGP > 5.00 |
| 7f | 442.16 | 4 | 1 | 4.672 | ||
| 7g | 428.14 | 4 | 2 | 4.466 | ||
| 8a | 408.17 | 3 | 1 | 5.081 | 1 | LOGP > 5.00 |
| 8b | 486.08 | 3 | 1 | 5.669 | 1 | LOGP > 5.00 |
| 8c | 442.13 | 3 | 1 | 5.716 | 1 | LOGP > 5.00 |
| 8d | 426.16 | 3 | 1 | 5.257 | 1 | LOGP > 5.00 |
| 8e | 422.19 | 3 | 1 | 5.519 | 1 | LOGP > 5.00 |
| 8f | 438.18 | 4 | 1 | 5.121 | 1 | LOGP > 5.00 |
| 8g | 424.17 | 4 | 2 | 4.627 | ||
| 9a | 424.17 | 4 | 1 | 4.666 | ||
| 9b | 502.08 | 4 | 1 | 5.102 | 2 | MW > 500, LOGP > 5.00 |
| 9c | 458.13 | 4 | 1 | 5.038 | 1 | LOGP > 5.00 |
| 9d | 442.16 | 4 | 1 | 4.629 | ||
| 9e | 438.18 | 4 | 1 | 4.309 | ||
| 9f | 454.18 | 5 | 1 | 4.287 | ||
| 9g | 440.16 | 5 | 2 | 4.143 | ||
| 10a | 410.15 | 4 | 2 | 4.283 | ||
| 10b | 488.06 | 4 | 2 | 4.890 | ||
| 10c | 444.11 | 4 | 2 | 4.824 | ||
| 10d | 428.14 | 4 | 2 | 4.452 | ||
| 10e | 424.17 | 4 | 2 | 4.640 | ||
| 10f | 440.16 | 5 | 2 | 4.163 | ||
| 10g | 426.15 | 5 | 3 | 3.899 | ||
| Curcumin | 368.38 | 6 | 2 | 1.47 | ||
| Remdesivir | 602.58 | 12 | 4 | 0.18 | 2 | MW > 500, HBA > 10 |
| Lopinavir | 628.80 | 5 | 4 | 2.93 | 1 | MW > 500 |
| Compound | Intestinal Absorption (% Absorbed) | Caco-2 Permeability (log Papp) | P-gp Substrate | P-gp I Inhibitor | P-gp II Inhibitor | Skin Permeability (log Kp) | Water Solubility (log mol/L) |
|---|---|---|---|---|---|---|---|
| 5g | 93.098 | 0.515 | Yes | Yes | Yes | −2.742 | −4.683 |
| 6g | 93.166 | 0.523 | Yes | Yes | Yes | −2.742 | −4.669 |
| 7a | 95.753 | 1.273 | Yes | Yes | Yes | −2.734 | −5.391 |
| 7d | 96.110 | 1.296 | No | Yes | Yes | −2.735 | −4.75 |
| 7f | 97.097 | 1.258 | No | Yes | Yes | −2.735 | −4.811 |
| 7g | 94.131 | 1.049 | Yes | Yes | Yes | −2.739 | −4.359 |
| 8g | 94.624 | 0.573 | Yes | Yes | Yes | −2.743 | −4.629 |
| 9a | 94.310 | 1.203 | Yes | Yes | Yes | −2.736 | −6.301 |
| 9d | 94.849 | 1.259 | Yes | Yes | Yes | −2.735 | −6.291 |
| 9e | 96.177 | 1.206 | Yes | Yes | Yes | −2.738 | −6.232 |
| 9f | 96.108 | 0.474 | Yes | Yes | Yes | −2.734 | −6.336 |
| 9g | 92.807 | 0.505 | Yes | Yes | Yes | −2.738 | −5.21 |
| 10a | 91.302 | 0.533 | Yes | Yes | Yes | −2.736 | −4.492 |
| 10b | 89.716 | 0.440 | Yes | Yes | Yes | −2.737 | −5.13 |
| 10c | 89.783 | 0.448 | Yes | Yes | Yes | −2.737 | −5.122 |
| 10d | 90.526 | 0.487 | Yes | Yes | Yes | −2.737 | −5.043 |
| 10e | 92.590 | 0.451 | Yes | Yes | Yes | −2.741 | −5.464 |
| 10f | 91.207 | 0.514 | Yes | Yes | Yes | −2.739 | −5.258 |
| 10g | 93.677 | 0.679 | Yes | Yes | Yes | −2.737 | −4.048 |
| Curcumin | 82.190 | −0.093 | Yes | Yes | Yes | −2.764 | −4.01 |
| Compound | VDss (log L/kg) | Fraction Unbound (Fu) | BBB Permeability (log BB) | CNS Permeability (log PS) |
|---|---|---|---|---|
| 5g | −0.708 | 0.000 | −0.475 | −1.528 |
| 6g | −0.717 | 0.000 | −0.474 | −1.551 |
| 7a | −0.611 | 0.038 | −0.241 | −1.300 |
| 7d | −0.839 | 0.073 | −0.224 | −1.319 |
| 7f | −0.786 | 0.042 | −0.376 | −1.699 |
| 7g | −0.926 | 0.044 | −0.441 | −1.707 |
| 8g | −0.696 | 0.000 | −0.461 | −1.591 |
| 9a | −0.374 | 0.032 | −0.291 | −1.653 |
| 9d | −0.643 | 0.040 | −0.277 | −1.662 |
| 9e | −0.259 | 0.035 | −0.446 | −1.543 |
| 9f | −0.606 | 0.031 | −0.432 | −1.788 |
| 9g | −0.999 | 0.000 | +0.231 | −1.761 |
| 10a | −0.879 | 0.000 | −0.279 | −1.568 |
| 10b | −0.805 | 0.000 | −0.296 | −1.442 |
| 10c | −0.814 | 0.000 | −0.295 | −1.465 |
| 10d | −0.994 | 0.000 | −0.254 | −1.596 |
| 10e | −0.662 | 0.000 | −0.501 | −1.525 |
| 10f | −0.882 | 0.000 | −0.080 | −1.721 |
| 10g | −1.054 | 0.000 | −0.749 | −1.910 |
| Curcumin | −0.215 | 0.000 | −0.562 | −2.990 |
| Compound | CYP2D6 Substrate | CYP3A4 Substrate | CYP3A4 Inhibitor | CYP1A2 Inhibitor | CYP2C19 Inhibitor | CYP2C9 Inhibitor |
|---|---|---|---|---|---|---|
| 5g | No | Yes | No | No | Yes | Yes |
| 6g | No | Yes | No | No | Yes | Yes |
| 7a | No | Yes | No | No | Yes | Yes |
| 7d | No | Yes | Yes | Yes | Yes | Yes |
| 7f | No | Yes | Yes | No | Yes | Yes |
| 7g | No | Yes | Yes | No | Yes | Yes |
| 8g | No | Yes | No | No | Yes | Yes |
| 9a | No | Yes | No | No | Yes | No |
| 9d | No | Yes | No | No | Yes | Yes |
| 9e | No | Yes | No | No | Yes | No |
| 9f | No | Yes | Yes | No | Yes | Yes |
| 9g | No | Yes | Yes | No | Yes | Yes |
| 10a | No | Yes | No | No | Yes | Yes |
| 10b | No | Yes | No | No | Yes | Yes |
| 10c | No | Yes | No | No | Yes | Yes |
| 10d | No | Yes | Yes | No | Yes | Yes |
| 10e | No | Yes | No | No | Yes | No |
| 10f | No | Yes | No | No | Yes | Yes |
| 10g | No | Yes | No | No | Yes | Yes |
| Curcumin | No | Yes | Yes | No | No | No |
| Compound | Total Clearance (log mL/min/kg) | Renal OCT2 Substrate |
|---|---|---|
| 5g | −0.219 | No |
| 6g | −0.039 | No |
| 7a | +0.091 | No |
| 7d | −0.002 | No |
| 7f | +0.117 | No |
| 7g | +0.050 | No |
| 8g | +0.183 | No |
| 9a | +0.194 | No |
| 9d | +0.050 | No |
| 9e | +0.183 | No |
| 9f | +0.187 | No |
| 9g | +0.131 | No |
| 10a | +0.139 | No |
| 10b | −0.227 | No |
| 10c | −0.047 | No |
| 10d | −0.001 | No |
| 10e | +0.127 | No |
| 10f | +0.136 | No |
| 10g | +0.093 | No |
| Curcumin | −0.002 | No |
| Compound | AMES Toxicity | Max. Tolerated Dose (log g/kg/day) | hERG I Inhibitor | Oral Rat Acute Toxicity (LD50, mol/kg) | Oral Rat Chronic Toxicity (LOALEL) (logmg/kgbw/day) | Hepatotoxicity | T. Pyriformis Toxicity (log µg/L) | Minnow Toxicity (log mM) |
|---|---|---|---|---|---|---|---|---|
| 5g | Yes | 0.052 | No | 2.110 | 1.403 | Yes | 0.293 | −3.283 |
| 6g | Yes | 0.050 | No | 2.106 | 1.430 | Yes | 0.293 | −3.137 |
| 7a | No | 0.258 | No | 2.826 | 1.404 | No | 0.296 | −2.868 |
| 7d | No | 0.366 | No | 2.783 | 2.150 | Yes | 0.291 | −3.714 |
| 7f | No | 0.218 | No | 2.758 | 1.964 | Yes | 0.291 | −3.750 |
| 7g | No | 0.178 | No | 2.016 | 2.265 | Yes | 0.290 | −3.126 |
| 8g | Yes | 0.047 | No | 2.077 | 1.374 | Yes | 0.293 | −2.919 |
| 9a | No | 0.270 | No | 3.107 | 1.502 | Yes | 0.299 | −3.088 |
| 9d | No | 0.238 | No | 2.760 | 1.254 | Yes | 0.293 | −4.342 |
| 9e | Yes | 0.148 | No | 3.004 | 1.423 | Yes | 0.305 | −3.816 |
| 9f | Yes | 0.132 | No | 2.719 | 1.045 | No | 0.293 | −4.572 |
| 9g | Yes | −0.028 | No | 1.967 | 1.325 | Yes | 0.291 | −1.909 |
| 10a | Yes | 0.087 | No | 1.894 | 1.383 | Yes | 0.293 | −1.273 |
| 10b | Yes | 0.065 | No | 1.950 | 1.273 | Yes | 0.294 | −1.832 |
| 10c | Yes | 0.064 | No | 1.948 | 1.301 | Yes | 0.294 | −1.686 |
| 10d | Yes | 0.060 | No | 1.979 | 1.422 | Yes | 0.291 | −1.341 |
| 10e | Yes | 0.108 | No | 2.244 | 1.346 | Yes | 0.299 | −1.005 |
| 10f | Yes | 0.080 | No | 2.033 | 1.448 | No | 0.292 | −1.124 |
| 10g | No | 0.089 | No | 1.853 | 3.834 | No | 0.290 | −2.312 |
| Curcumin | No | 0.081 | No | 1.833 | 0.835 | No | 0.305 | −4.572 |
| Coranavirus Targets | |||
|---|---|---|---|
| Compound Name | 6WUU SARS-CoV-2 | 2FE8 SARS-CoV | 4RNA MERS-CoV |
| 5g | −9.5 | −10.2 | −9.0 |
| 6g | −9.4 | −10.1 | −9.0 |
| 7a | −9.3 | −10.1 | −8.8 |
| 7d | −9.6 | −10.3 | −9.1 |
| 7f | −9.5 | −10.3 | −9.0 |
| 7g | −9.7 | −10.5 | −9.0 |
| 8g | −9.6 | −10.3 | −9.1 |
| 9a | −9.6 | −10.3 | −9.2 |
| 9d | −9.8 | −10.5 | −9.3 |
| 9e | −9.3 | −10.5 | −9.1 |
| 9f | −9.1 | −10.5 | −9.1 |
| 9g | −9.6 | −10.6 | −9.1 |
| 10a | −9.9 | −10.3 | −9.2 |
| 10b | −9.7 | −10.4 | −9.3 |
| 10c | −9.8 | −10.7 | −9.3 |
| 10d | −10.0 | −10.7 | −9.4 |
| 10e | −9.8 | −10.6 | −9.4 |
| 10f | −9.8 | −10.5 | −9.2 |
| 10g | −10.1 | −10.8 | −9.3 |
| Bisdemothoxycurcumin | −7.5 | −7.5 | −7.5 |
| Curcumin | −8.0 | −7.8 | −7.6 |
| Demothoxycurcumin | −7.9 | −8.4 | −7.6 |
| Favipiravir | −5.7 | −6.0 | −5.3 |
| Hydroxychloroquine | −6.7 | −6.9 | −6.1 |
| Lopinavir | −8.7 | −10.1 | −8.7 |
| Remdesivir | −8.8 | −9.0 | −8.1 |
| Warfarin | −8.5 | −8.4 | −7.4 |
| VIR250 | −7.6 | −8.2 | −6.2 |
| Residue | ΔGbind | ΔGvW | ΔGelec | ΔGps | ΔGnps |
|---|---|---|---|---|---|
| 10c | −259.43 | −315.12 | −98.65 | +148.87 | −5.47 |
| 10d | −268.85 | −328.67 | −105.23 | +157.56 | −6.51 |
| 10g | −280.72 | −342.89 | −112.75 | +165.98 | −7.04 |
| 10c (kJ/mol) | 10d (kJ/mol) | 10g (kJ/mol) | ||||
|---|---|---|---|---|---|---|
| Residue | Chain A | Chain C | Chain A | Chain C | Chain A | Chain C |
| Arg82 | 1.11 | 1.75 | 1.30 | 2.98 | 2.45 | 1.60 |
| Lys108 | 2.22 | 1.85 | 1.51 | 1.92 | 1.89 | 2.35 |
| Asn110 | −2.44 | −0.32 | −2.41 | −0.64 | −2.27 | −0.52 |
| Lys157 | 1.95 | 2.32 | 1.10 | 2.21 | 1.45 | 1.68 |
| Gly161 | −2.34 | −0.15 | −2.11 | −0.90 | −2.67 | −1.56 |
| Glu162 | −2.07 | −0.22 | −1.92 | −0.80 | −2.45 | −0.53 |
| Leu163 | −1.98 | −1.04 | −2.12 | −0.98 | −2.75 | −1.00 |
| Asp164 | −2.07 | −0.50 | −2.45 | −0.67 | −2.89 | −0.10 |
| Glu167 | 2.75 | 1.68 | 2.10 | 2.20 | 2.45 | 3.52 |
| Arg183 | 3.05 | 1.85 | 1.21 | 3.10 | 1.75 | 1.50 |
| Thr198 | −0.33 | −2.28 | −0.23 | 1.80 | −0.30 | −2.75 |
| Leu200 | −0.25 | −2.12 | −0.32 | −2.35 | −0.15 | −2.68 |
| Val203 | −0.25 | −1.97 | −0.02 | −2.04 | −0.11 | −2.00 |
| Glu204 | 1.50 | 2.34 | 1.89 | 1.85 | 1.22 | 2.11 |
| Met207 | −0.26 | −3.78 | −0.20 | −4.10 | −0.50 | −4.75 |
| Tyr208 | −0.45 | −3.10 | −0.80 | −3.50 | −0.05 | −3.89 |
| Lys232 | 1.20 | 2.05 | 1.50 | 2.34 | 1.85 | 1.75 |
| Ser245 | −2.05 | −0.20 | −1.78 | −0.50 | −2.10 | −0.85 |
| Pro248 | 3.75 | 2.50 | 1.98 | 3.75 | 2.25 | 4.00 |
| Tyr264 | 1.10 | 2.85 | 1.45 | 3.15 | 1.85 | 3.60 |
| Tyr268 | −2.40 | −0.10 | −0.75 | −0.50 | −2.00 | −0.80 |
| Tyr269 | −2.04 | −0.02 | −1.88 | −0.24 | −2.07 | −0.14 |
| Gln270 | −1.98 | −0.06 | −1.62 | −0.19 | −2.17 | −0.08 |
| Tyr273 | −2.05 | −0.01 | −2.03 | −0.20 | −1.89 | −0.05 |
| Asp302 | 2.10 | 2.05 | 2.50 | 2.34 | 2.85 | 2.70 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alici, H. Structure-Based Design and In-Silico Evaluation of Computationally Proposed Curcumin Derivatives as Potential Inhibitors of the Coronaviral PLpro Enzymes. Pharmaceuticals 2025, 18, 798. https://doi.org/10.3390/ph18060798
Alici H. Structure-Based Design and In-Silico Evaluation of Computationally Proposed Curcumin Derivatives as Potential Inhibitors of the Coronaviral PLpro Enzymes. Pharmaceuticals. 2025; 18(6):798. https://doi.org/10.3390/ph18060798
Chicago/Turabian StyleAlici, Hakan. 2025. "Structure-Based Design and In-Silico Evaluation of Computationally Proposed Curcumin Derivatives as Potential Inhibitors of the Coronaviral PLpro Enzymes" Pharmaceuticals 18, no. 6: 798. https://doi.org/10.3390/ph18060798
APA StyleAlici, H. (2025). Structure-Based Design and In-Silico Evaluation of Computationally Proposed Curcumin Derivatives as Potential Inhibitors of the Coronaviral PLpro Enzymes. Pharmaceuticals, 18(6), 798. https://doi.org/10.3390/ph18060798