

New Nitrogen-, Oxygen-, and Sulfur-Containing Heterocyclic Compounds as Anti-Colon Cancer Agents: Synthesis, Multitargeted Evaluations, Molecular Docking Simulations and ADMET Predictions

 , , ,
, , , 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
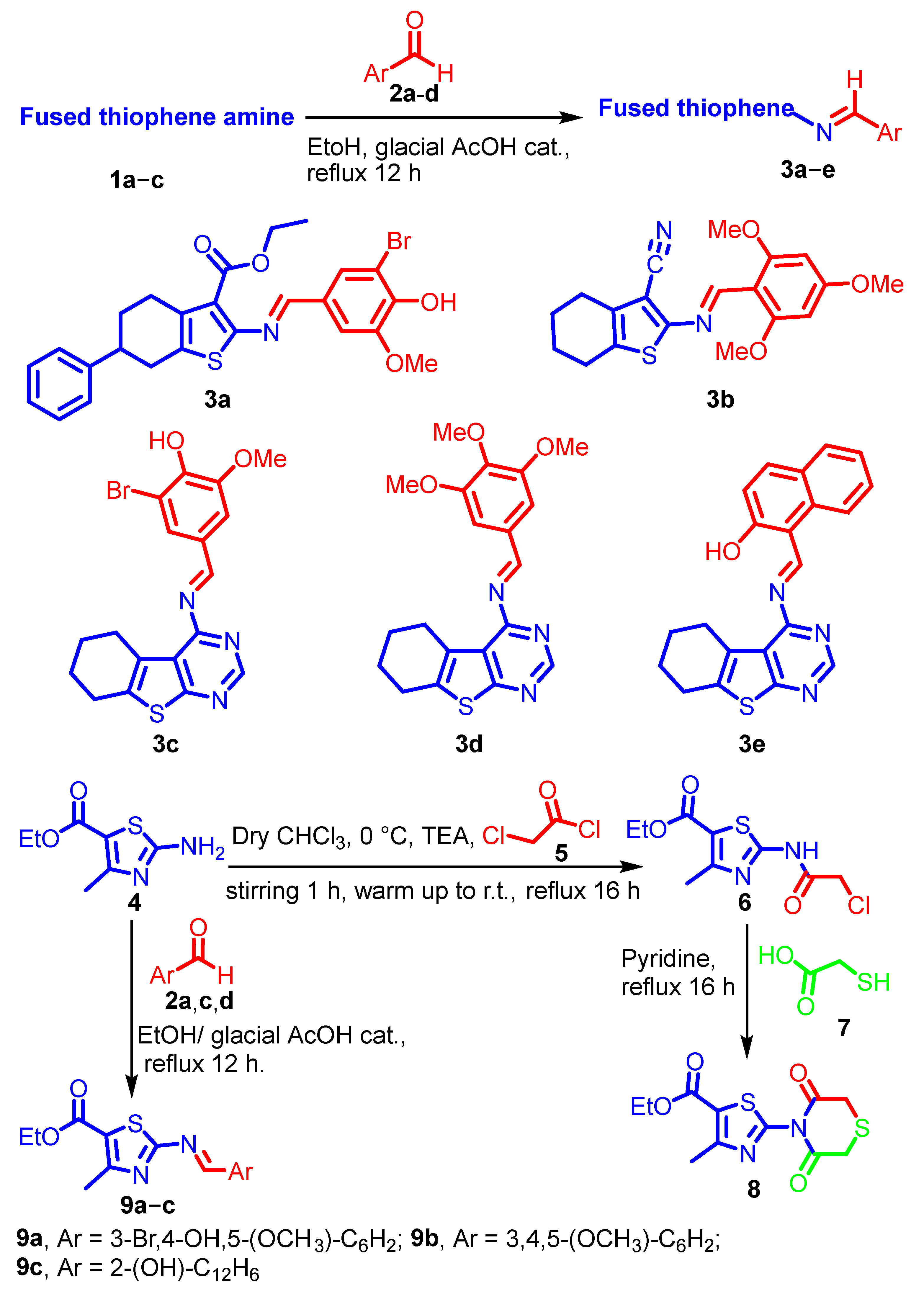
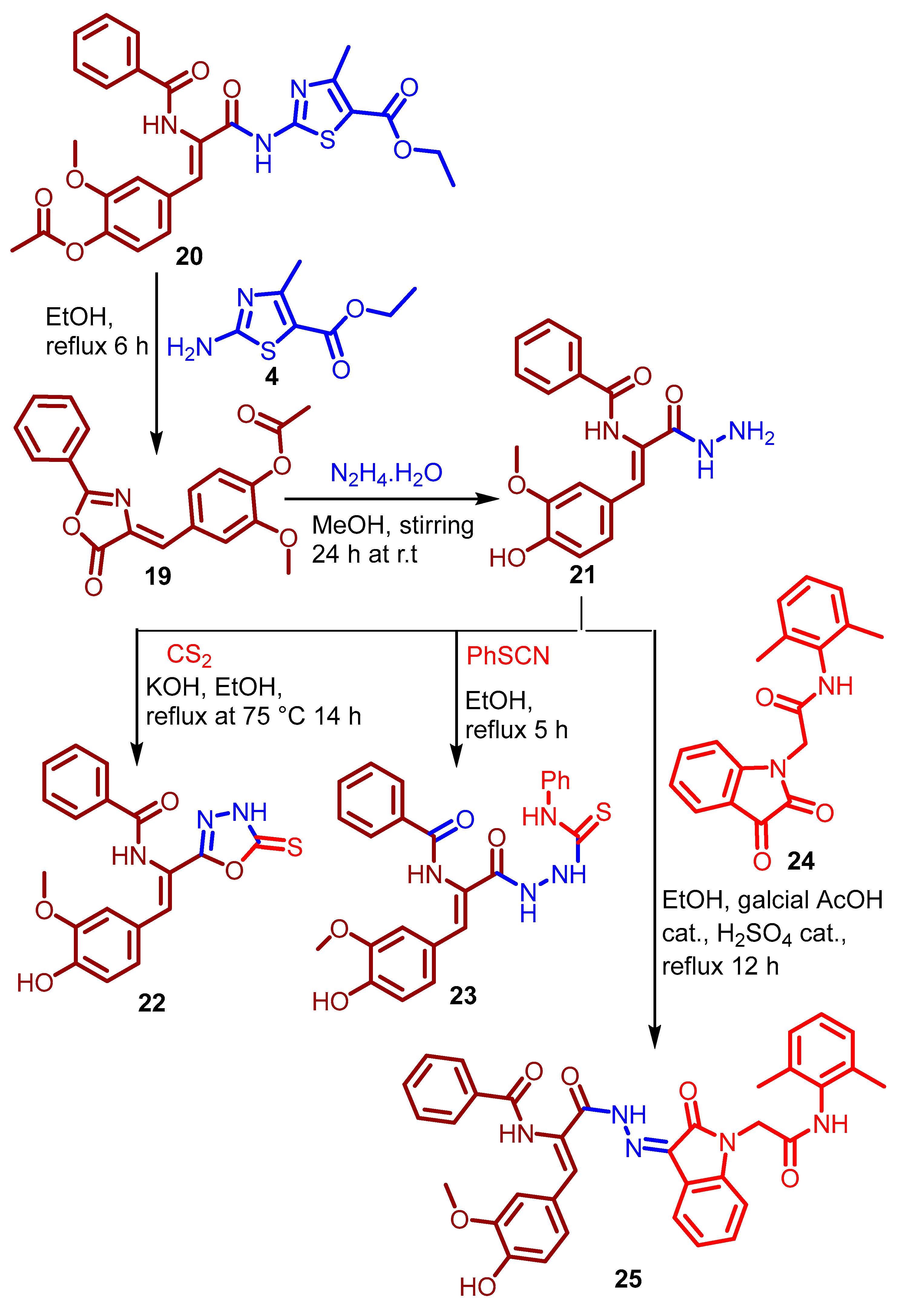
2.1. Chemistry
2.2. In Vitro Biological Evaluations
2.2.1. Antioxidant Activity
2.2.2. PDK-1 and LDHA Inhibitory Activities
2.2.3. Cytotoxic Activities
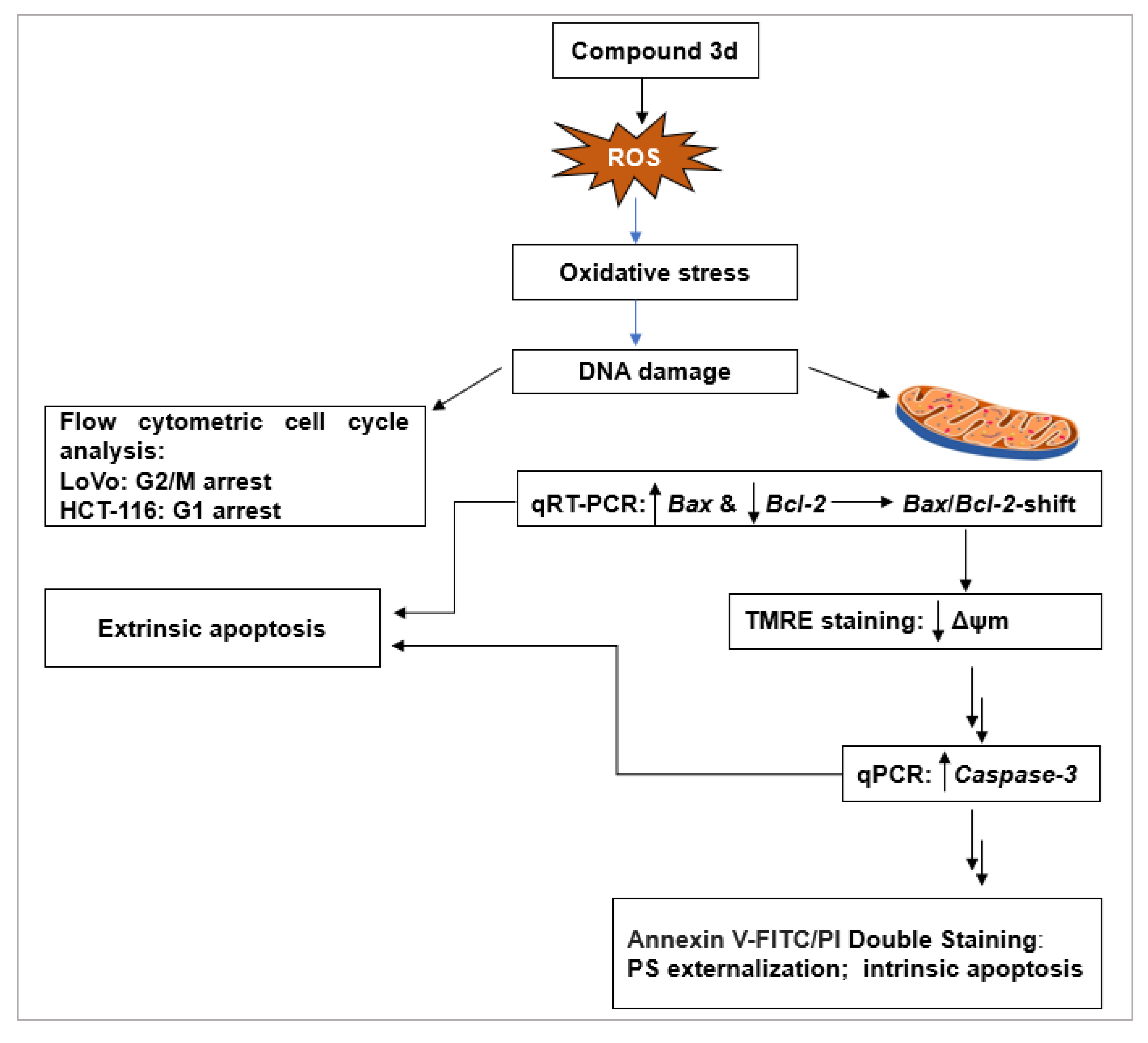
2.2.4. Investigation of Anticancer Mechanism of 3d
Effect of Compound 3d on Cell Cycle Progression
Induction of Apoptosis by Compound 3d
- ROS Generation as an Upstream Apoptotic Trigger
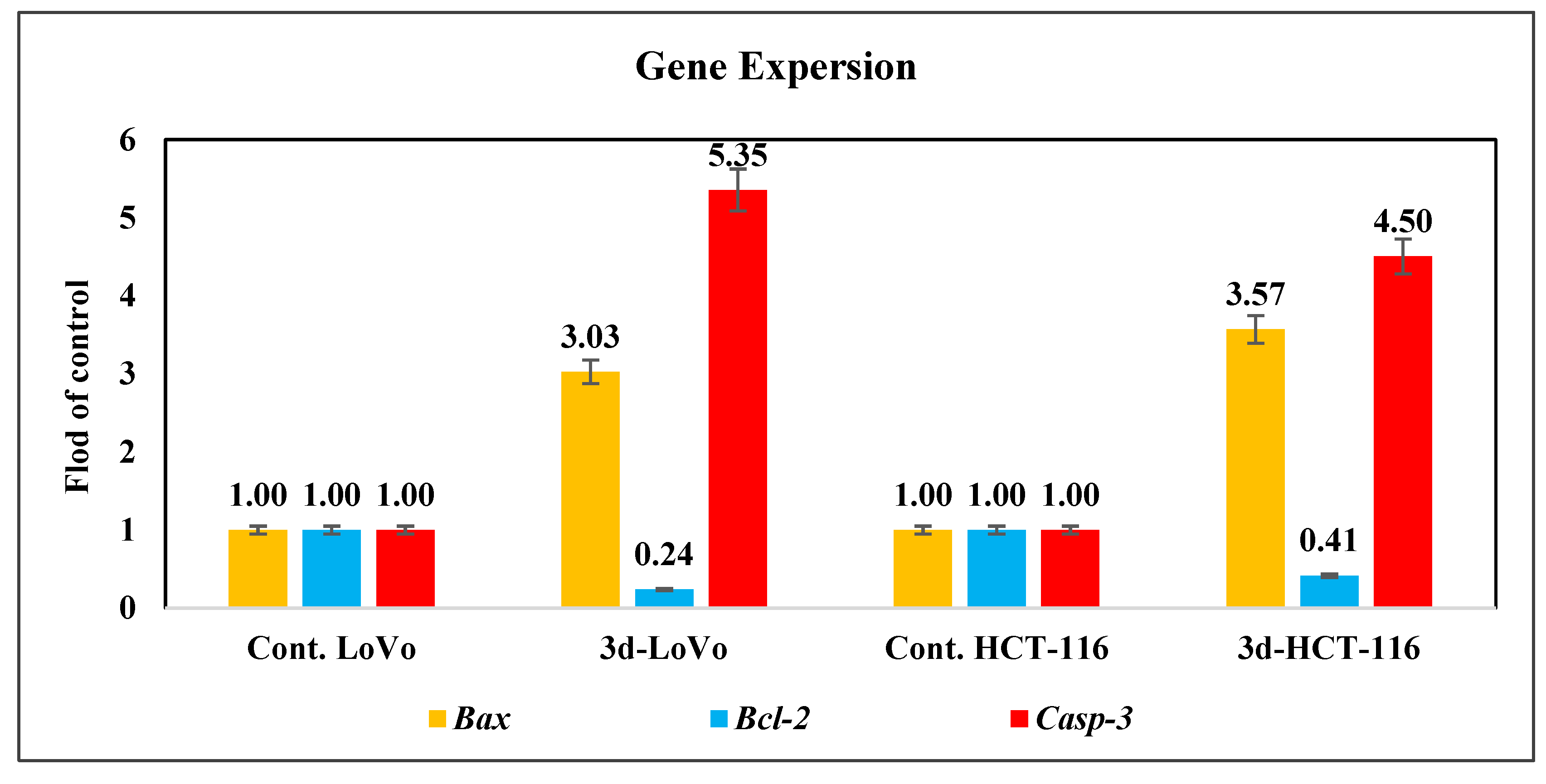
- Modulation of Apoptosis-Related Gene Expression
- Loss of Mitochondrial Membrane Potential (ΔΨm)
- Upregulation of Caspase-3 Expression
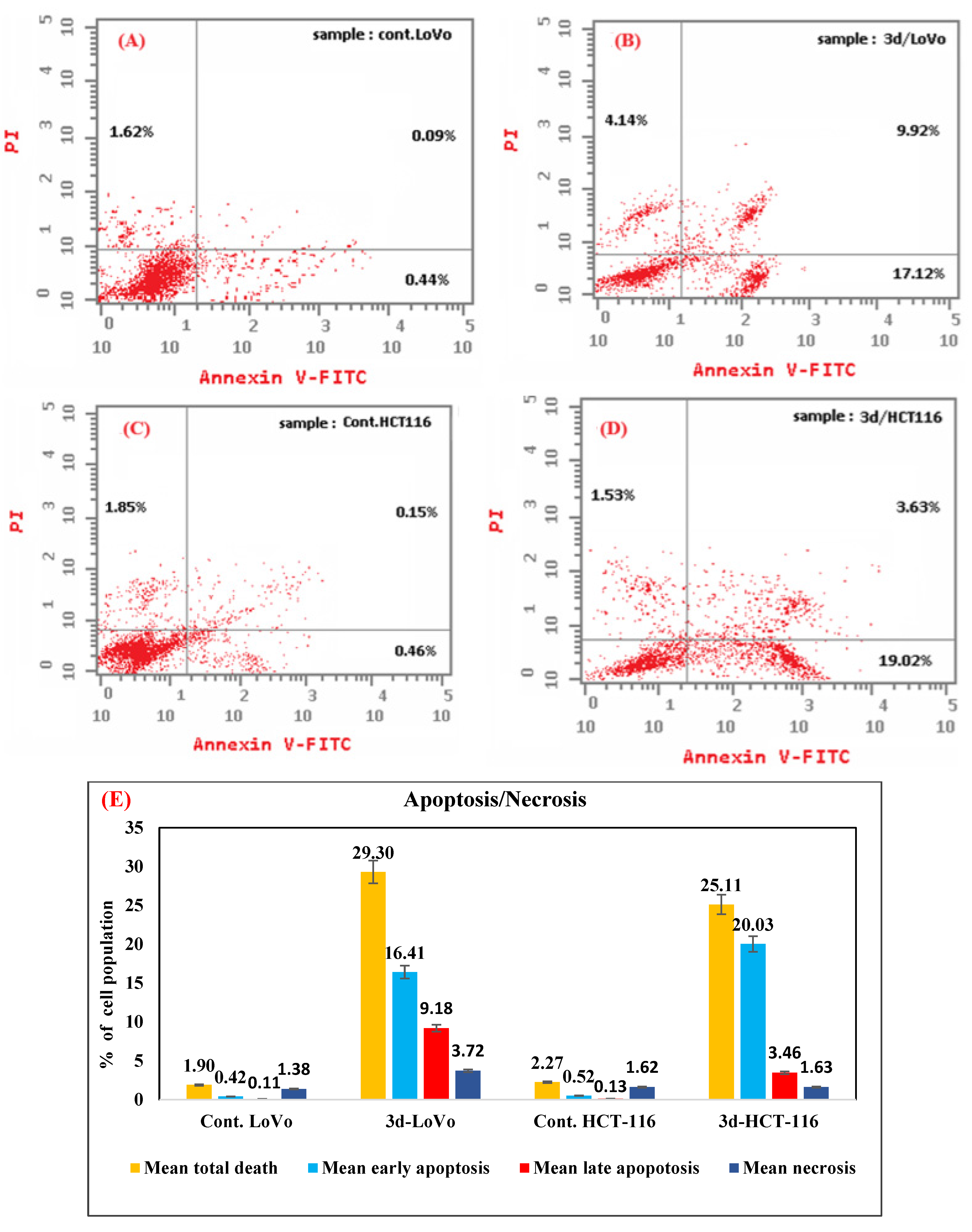
- Phosphatidylserine Externalization by Annexin V-FITC/PI Staining
2.3. In Silico Studies
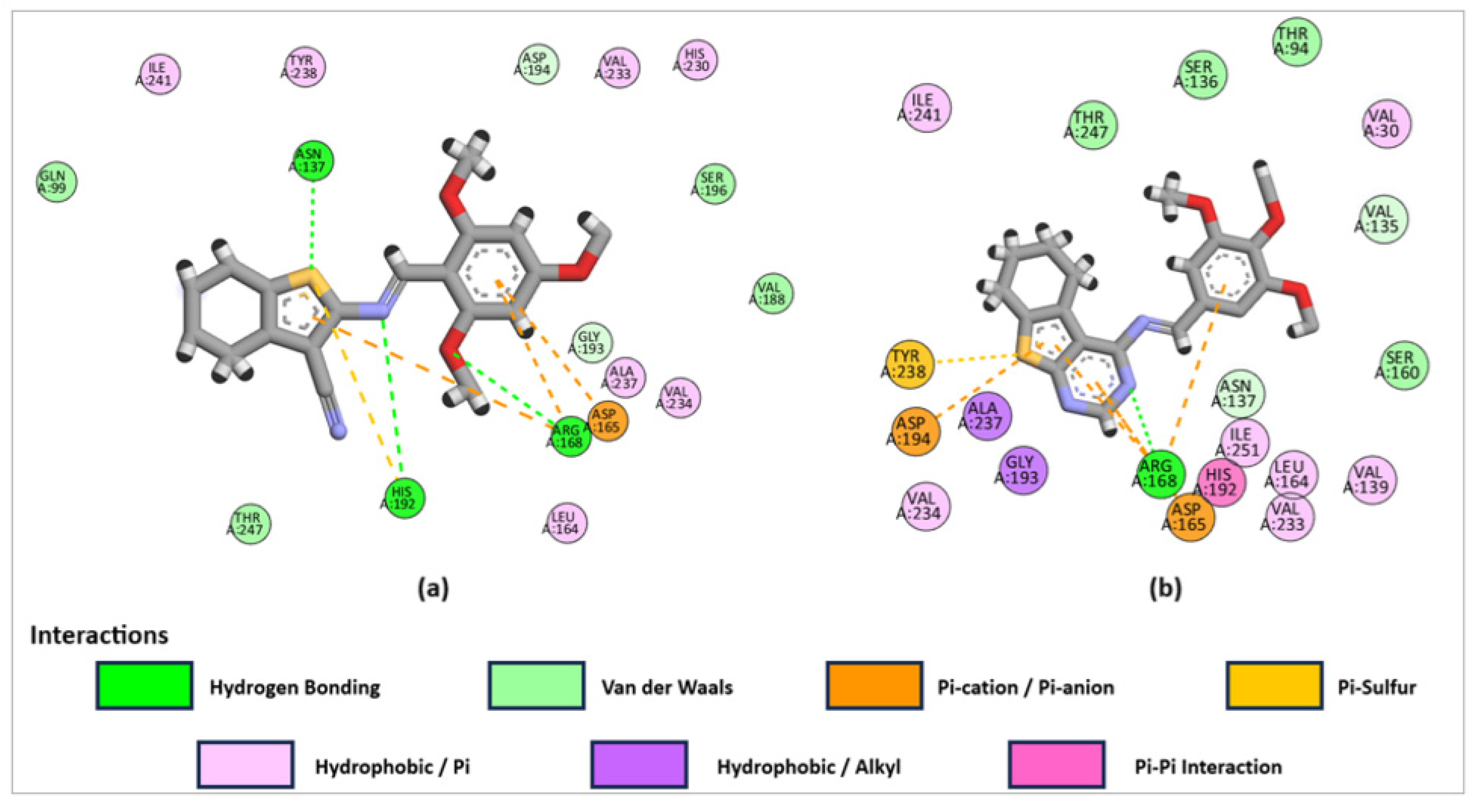
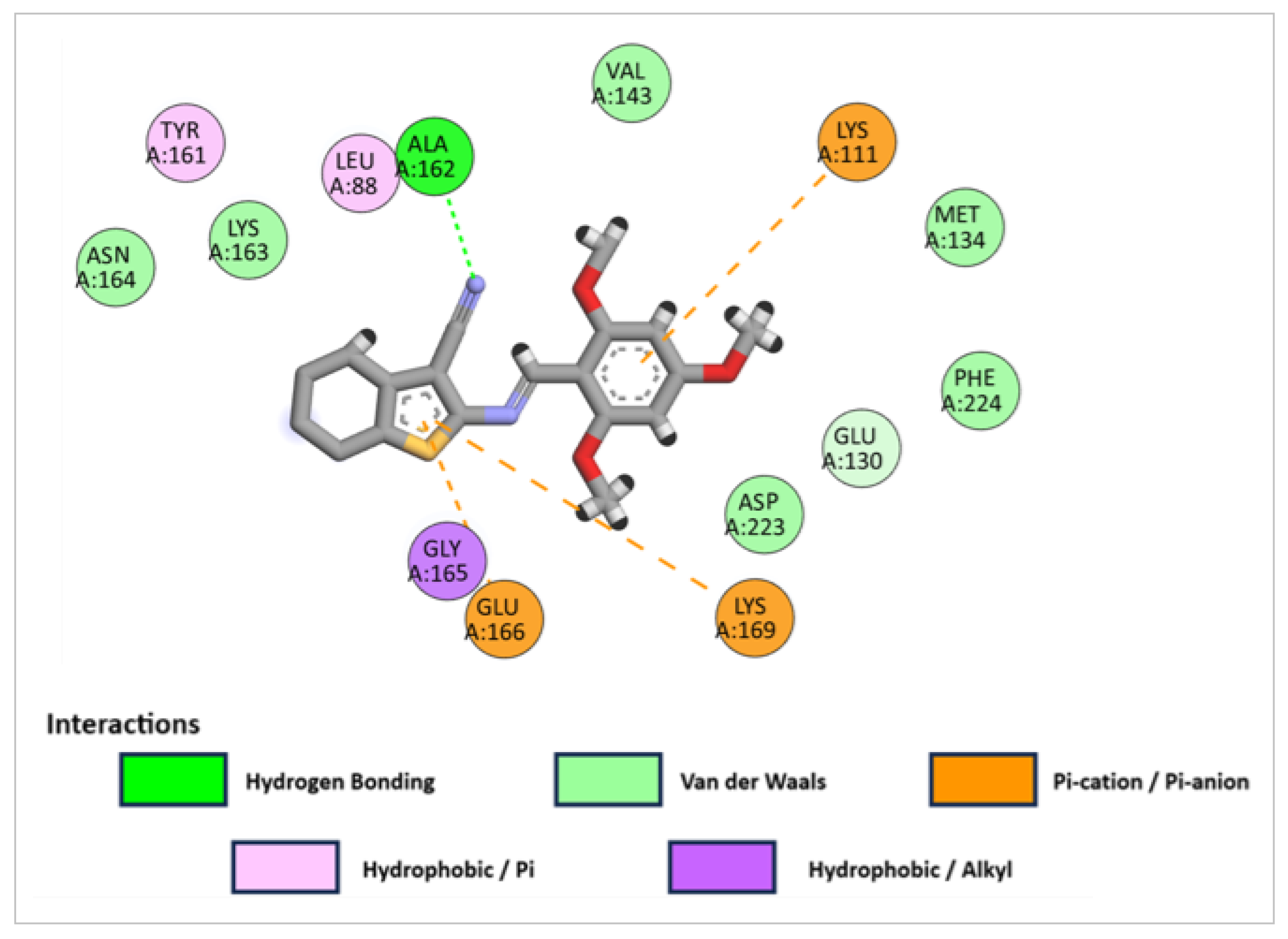
2.3.1. Molecular Docking Studies
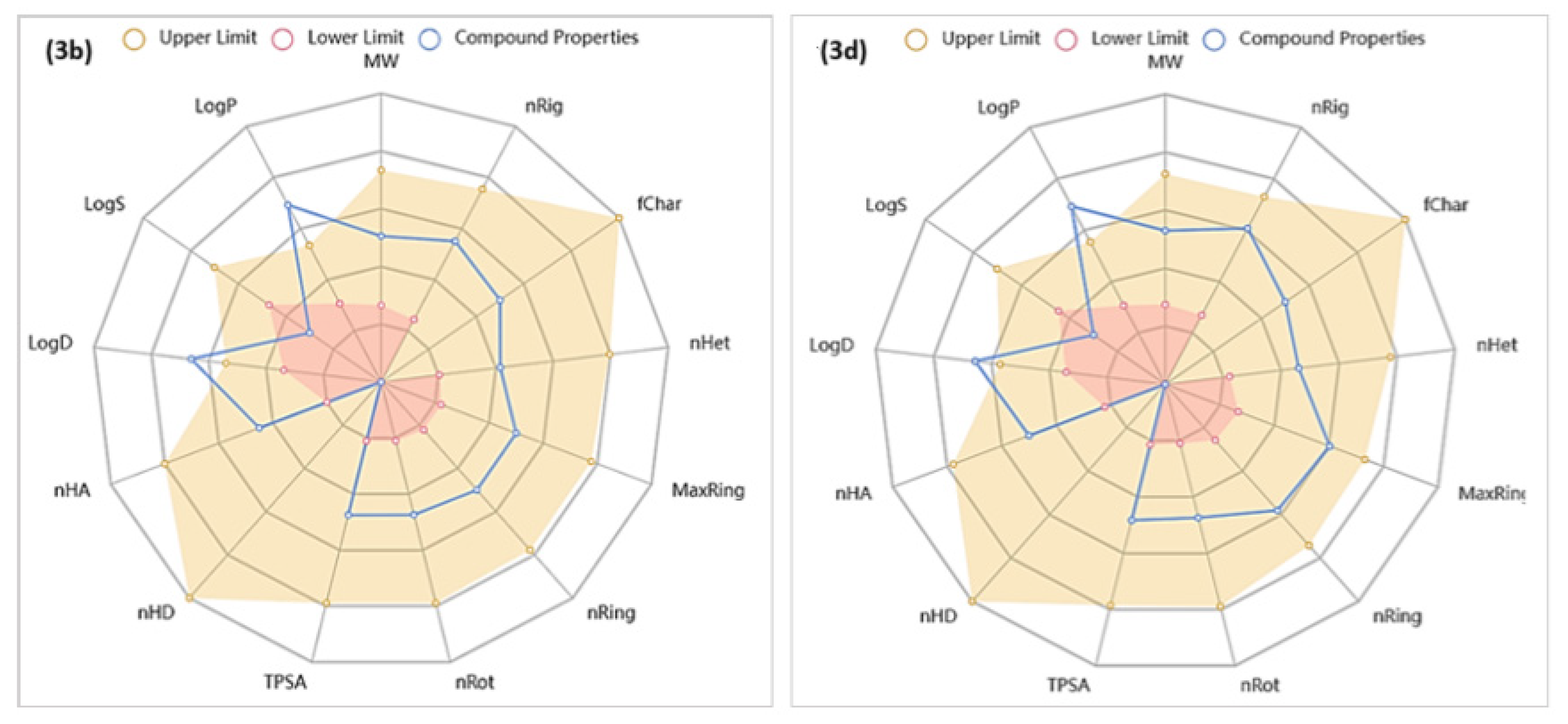
2.3.2. In Silico Predictions of the Physicochemical, Drug-Likeness, and ADMET Properties of Compounds 3b and 3d
Analyses of Physicochemical Characteristics
Analyses of Drug-Likeness Characteristics for Compounds 3b and 3d
Analyses of ADMET Characteristics for Compounds 3b and 3d
- Absorption
- Distribution
- Metabolism
- Excretion
- Toxicity
3. Materials and Methods
3.1. Chemistry
3.1.1. Instrumentation
General Procedure A for Synthesis of Various Schiff Bases
3.1.2. Synthesis of Thiophene Schiff Bases 3a–e
- (E)-Ethyl2-((3-bromo-4-hydroxy-5-methoxybenzylidene)amino)-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate 3a
- (E)-2-((2,4,6-Trimethoxybenzylidene)amino)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carbonitrile 3b
- (E)-2-Bromo-6-methoxy-4-(((5,6,7,8-tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-4-yl) imino)methyl)phenol 3c
- (E)-N-(5,6,7,8-Tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-4-yl)-1-(3,4,5-trimethoxyphenyl)methanimine 3d
- (E)-1-(((5,6,7,8-Tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-4-yl)imino)methyl) naphthalene-2-ol 3e
3.1.3. Synthesis of Ethyl 2-(3,5-Dioxothiomorpholino)-4-methylthiazole-5-carboxylate 8
3.1.4. Synthesis of Thiazolyl Schiff Bases 9a–c
- Ethyl (E)-2-((3-bromo-4-hydroxy-5-methoxybenzylidene)amino)-4-methyl thiazole-5-carboxylate 9a
- Ethyl(E)-4-methyl-2-((3,4,5-trimethoxybenzylidene)amino)thiazole-5-carboxylate 9b
- Ethyl(E)-2-(((2-hydroxynaphthalen-1-yl)methylene)amino)-4-methylthiazole-5-carboxylate 9c
3.1.5. General Procedure B for the Synthesis of 3-Substituted-6,7-dimethoxy-2-methyl quinazolin-4(3H)-one Derivatives 13 and 14
- 3-(4-(Benzo[d]thiazol-2-yl)phenyl)-6,7-dimethoxy-2-methylquinazolin-4(3H)-one 13
- 3-Cyclohexyl-6,7-dimethoxy-2-methylquinazolin-4(3H)-one 14
3.1.6. Synthesis of Ethyl (6,7-Dimethoxy-2-methyl-4-oxoquinazolin-3(4H)-yl)carbamate 17
3.1.7. Synthesis of Quinazolinone Based-Schiff Bases 18a–h
- (E)-6,7-Dimethoxy-2-methyl-3-((2,4,6-trimethoxybenzylidene)amino)quinazolin-4(3H)-one 18a
- (E)-3-(((2-Hydroxynaphthalen-1-yl)methylene)amino)-6,7-dimethoxy-2-methylquin- azolin-4(3H)-one 18b
- (E)-6,7-Dimethoxy-2-methyl-3-((4-(trifluoromethyl)benzylidene)amino)quinazolin-4(3H)-one 18c
- (E)-3-((4-Hydroxy-3-methoxybenzylidene)amino)-6,7-dimethoxy-2-methylquinazoli-N-4(3H)-one 18d
- (E)-6-Fluoro-2-methyl-3-((2,4,6-trimethoxybenzylidene)amino)quinazolin-4(3H)-one 18e
- (E)-6-Fluoro-3-(((2-hydroxynaphthalen-1-yl)methylene)amino)-2-methylquinazolin-4(3H)-one 18f
- (E)-6-Fluoro-3-((3-hydroxy-4-methoxybenzylidene)amino)-2-methylquinazolin-4(3H)-one 18g
- (E)-6-Fluoro-2-methyl-3-((thiophen-2-ylmethylene)amino)quinazolin-4(3H)-one 18h
3.1.8. Synthesis of Ethyl (Z)-2-(3-(4-Acetoxy-3-methoxyphenyl)-2-benzamido-acrylamido)- 4-methylthiazole-5-carboxylate 20
3.1.9. Synthesis of N-(2-(4-Hydroxy-3-methoxyphenyl)-1-(5-thioxo-4,5-dihydro-1,3,4-oxa-diazol-2-yl)vinyl)benzamide 22
3.1.10. Synthesis of N-(1-(4-Hydroxy-3-methoxyphenyl)-3-oxo-3-(2-(phenylcarbamothionyl)hydrazinyl)prop-1-en-2-yl)benzamide 23
3.1.11. Synthesis of N-(3-(2-(1-(2-((2,4-Dimethylphenyl)amino)-2-oxoethyl)-2-oxoindolin-3-ylidene)hydrazinyl)-1-(4-hydroxy-3-methoxyphenyl)-3-oxoprop-1-en-2-yl)benzamide 25
3.2. Evaluation of Biological Activities
3.2.1. In Vitro DPPH Radical Scavenging Assay
3.2.2. In Vitro PDK-1 Inhibition Assay
3.2.3. In Vitro LDHA Inhibitory Assay
3.2.4. Cell Culture and Viability Assay
3.2.5. Cell Cycle Assay
3.2.6. Annexin-V-FITC Assay
3.2.7. Analysis of Reactive Oxygen Species (ROS) Levels
3.2.8. Mitochondrial Transmembrane Potential (MMP) Measurement
3.2.9. Quantification of the Expression Levels of Bax, Bcl-2 and Caspase-3 Genes
3.3. In Silico Studies
3.3.1. Molecular Docking Simulations
3.3.2. Physicochemical, Drug-Likeness and ADMET Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xi, Y.; Xu, P. Global colorectal cancer burden in 2020 and projections to 2040. Trans. Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Nelson, V.K.; Nuli, M.V.; Mastanaiah, J.; Saleem, T.S.M.; Birudala, G.; Jamous, Y.F.; Alshargi, O.; Kotha, K.K.; Sudhan, H.H.; Mani, R.R.; et al. Reactive oxygen species mediated apoptotic death of colon cancer cells: Therapeutic potential of plant derived alkaloids. Front. Endocrinol. 2023, 14, 1201198. [Google Scholar] [CrossRef]
- Wan, M.L.; Wang, Y.; Zeng, Z.; Deng, B.; Zhu, B.S.; Cao, T.; Li, Y.K.; Xiao, J.; Han, Q.; Wu, Q. Colorectal cancer (CRC) as a multifactorial disease and its causal correlations with multiple signaling pathways. Biosci. Rep. 2020, 40, BSR20200265. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ma, X.; Chakravarti, D.; Shalapour, S.; DePinho, R.A. Genetic and biological hallmarks of colorectal cancer. Genes Dev. 2021, 35, 787–820. [Google Scholar] [CrossRef]
- Bardelčíková, A.; Šoltys, J.; Mojžiš, J. Oxidative Stress, Inflammation and Colorectal Cancer: An Overview. Antioxidants 2023, 12, 901. [Google Scholar] [CrossRef]
- Catalano, T.; Selvaggi, F.; Cotellese, R.; Aceto, G.M. The Role of Reactive Oxygen Species in Colorectal Cancer Initiation and Progression: Perspectives on Theranostic Approaches. Cancers 2025, 17, 752. [Google Scholar] [CrossRef]
- Guzmán-Carrasco, A.; Mesas, C.; Doello, K.; Porres, J.M.; García-Beltrán, A.; Martínez, R.; Bermúdez, F.; Peña, M.; Melguizo, C.; Prados, J. The Antioxidant and Chemopreventive Activity of a Nutraceutical Derived from Brassicaceae Seed Extracts for Colorectal Cancer. Nutrients 2025, 17, 1358. [Google Scholar] [CrossRef]
- Fang, S.; Fang, X. Advances in glucose metabolism research in colorectal cancer. Biomed. Rep. 2016, 5, 289–295. [Google Scholar] [CrossRef]
- Pérez-Tomás, R.; Pérez-Guillén, I. Lactate in the Tumor Microenvironment: An Essential Molecule in Cancer Progression and Treatment. Cancers 2020, 12, 3244. [Google Scholar] [CrossRef]
- Angulo-Elizari, E.; Gaviria-Soteras, L.; Zubiri, I.; Ramos-Inza, S.; Sanmartin, C.; Plano, D. Unmasking the Warburg Effect: Unleashing the Power of Enzyme Inhibitors for Cancer Therapy. Drugs Drug Candidates 2023, 2, 728–769. [Google Scholar] [CrossRef]
- Alvarado-Ortiz, E.; Ortiz-Sánchez, E.; Sarabia-Sánchez, M.A.; de la Cruz-López, K.G.; García-Carrancá, A.; Robles-Flores, M. Mutant p53 gain-of-function stimulates canonical Wnt signaling via PI3K/AKT pathway in colon cancer. J. Cell Commun. Signal. 2023, 17, 1389–1403. [Google Scholar] [CrossRef] [PubMed]
- Morin, P.J. Colorectal cancer: The APC-lncRNA link. J. Clin. Investig. 2019, 129, 503–505. [Google Scholar] [CrossRef]
- Yang, S.Y.; Sales, K.M.; Fuller, B.; Seifalian, A.M.; Winslet, M.C. Apoptosis and colorectal cancer: Implications for therapy. Trends Mol. Med. 2009, 15, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Zhang, X.; He, L.; Li, Y.; Qiu, Y.; Hu, W.; Lu, W.; Du, H.; Yang, D. Compound 225# inhibits the proliferation of human colorectal cancer cells by promoting cell cycle arrest and apoptosis induction. Oncol. Rep. 2024, 51, 70. [Google Scholar] [CrossRef]
- Doostmohammadi, A.; Jooya, H.; Ghorbanian, K.; Gohari, S.; Dadashpour, M. Potentials and future perspectives of multi-target drugs in cancer treatment: The next generation anti-cancer agents. Cell. Commun. Signal. 2024, 22, 228. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, A.K.; Singh, H.; Vijayan, V.; Kumar, D.; Naik, J.; Thareja, S.; Yadav, J.P.; Pathak, P.; Grishina, M.; et al. Nitrogen Containing Heterocycles as Anticancer Agents: A Medicinal Chemistry Perspective. Pharmaceuticals 2023, 16, 299. [Google Scholar] [CrossRef]
- Bansal, R.; Malhotra, A. Therapeutic progression of quinazolines as targeted chemotherapeutic agents. Eur. J. Med. Chem. 2021, 211, 113016. [Google Scholar] [CrossRef]
- Abdel-Wahab, S.M.; Yassen, A.S.; Elrayessb, R.A.; Attiaa, K.M.; Elshihawy, H.A. 4,5,6,7-Tetrahydro benzo[b]thiophene-Based Derivatives as Anticancer Agents: An overview. Rec. Pharm. Biomed. Sci. 2023, 7, 173–180. [Google Scholar] [CrossRef]
- Sayed, M.T.M.; Hassan, R.A.; Halim, P.A.; El-Ansary, A.K. Recent updates on thienopyrimidine derivatives as anticancer agents. Med. Chem. Res. 2023, 32, 659–681. [Google Scholar] [CrossRef]
- Stecoza, C.E.; Nitulescu, G.M.; Draghici, C.; Caproiu, M.T.; Olaru, O.T.; Bostan, M.; Mihaila, M. Synthesis and Anticancer Evaluation of New 1,3,4-Oxadiazole Derivatives. Pharmaceuticals 2021, 14, 438. [Google Scholar] [CrossRef]
- Carradori, S.; Guglielmi, P.; Luisi, G.; Secci, D. Nitrogen- and Sulfur-Containing Heterocycles as Dual Anti-oxidant and Anti-cancer Agents. In Handbook of Oxidative Stress in Cancer: Mechanistic Aspects; Chakraborti, S., Ray, B.K., Roychowdhury, S., Eds.; Springer: Singapore, 2021; pp. 1–18. [Google Scholar] [CrossRef]
- El-Sayed, N.N.E.; Al-Otaibi, T.M.; Alonazi, M.; Masand, V.H.; Barakat, A.; Almarhoon, Z.M.; Ben Bacha, A. Synthesis and Characterization of Some New Quinoxalin-2(1H)one and 2-Methyl-3H-quinazolin-4-one Derivatives Targeting the Onset and Progression of CRC with SAR, Molecular Docking, and ADMET Analyses. Molecules 2021, 26, 3121. [Google Scholar] [CrossRef]
- Briel, D.; Rybak, A.; Kronbach, C.; Unverferth, K. Substituted 2-aminothiopen-derivatives: A potential new class of GluR6-antagonists. Eur. J. Med. Chem. 2010, 45, 69–77. [Google Scholar] [CrossRef]
- Fondjo, E.S.; Döpp, D.; Henkel, G. Reactions of some anellated 2-aminothiophenes with electron poor acetylenes. Tetrahedron 2006, 62, 7121–7131. [Google Scholar] [CrossRef]
- Pfeiffer, W.D.; Dollinger, H.; Langer, P. Synthesis of 5-Thioxo-hexahydrobenzo[b]thiopheno[2,3-d]-1,2,4-triazolo[1,5-c]pyrimidines and Related Compounds Based on Cyclocondensations of 2-Isothiocyanato-3-cyano-4,5,6,7-tetrahydrobenzo[b]thiophene. Phosphorus Sulfur Silicon Relat. Elem. 2009, 184, 626–637. [Google Scholar] [CrossRef]
- King, L.C.; Hlavacek, R.J. The reaction of ketones with iodine and thiourea. J. Am. Chem. Soc. 1950, 72, 3722–3725. [Google Scholar] [CrossRef]
- Lin, G.W.; Wang, Y.; Jin, Q.M.; Yang, T.T.; Song, J.M.; Lu, Y.; Huang, Q.J.; Song, K.; Zhou, J.; Lu, T. Synthesis, structures and the biological activity study on the metal complexes of 2-(4-aminophenyl) benzothiazole derivative. Inorg. Chim. Acta 2012, 382, 35–42. [Google Scholar] [CrossRef]
- Osarodion, O.P.; Orile, E.M.; Odianosen, U.C. Electron impact ionization mass spectra of 3-amino 5,6-dimethoxyl-2-methylquinazolin-4-(3H)-one derivative. Am. J. Mater. Synth. Process. 2019, 4, 62–67. [Google Scholar] [CrossRef]
- Al-Maneai, N.M. Synthesis and Pharmacological Evaluation of Some Novel 4-(3H)-Quinazolinone Derivatives. Master’s Thesis, King Saud University, Riyadh, Saudi Arabia, 2017. [Google Scholar]
- Soujanya, M.; Rajitha, G.; Umamaheswari, A.; Kumar, K.S. Synthesis, Biological Evaluation and Docking Studies of N-(2-benzamido feruloyl) Aryl Hydrazone Analogues. Lett. Drug Des. Discov. 2018, 15, 875–886. [Google Scholar] [CrossRef]
- Mansour, M.A.; AboulMagd, A.M.; Abdel-Rahman, H.M. Quinazoline-Schiff base conjugates: In silico study and ADMET predictions as multi-target inhibitors of coronavirus (SARS-CoV-2) proteins. RSC Adv. 2020, 10, 34033–34045. [Google Scholar] [CrossRef] [PubMed]
- Prokof’ev, E.P.; Karpeiskaya, E.I. The proton coupled 13C NMR direct determination of Z-, E-configuration of 4-benzyliden-2-phenyl(methyl)-Δ2-oxazolin-5-ones and products of their solvolysis. Tetrahedron Lett. 1979, 20, 737–740. [Google Scholar] [CrossRef]
- Belskaya, N.P.; Dehaen, W.; Bakulev, V.A. Synthesis and properties of hydrazones bearing amide, thioamide and amidine functions. ARKIVOC 2010, 2010, 275–322. [Google Scholar] [CrossRef]
- Bersuder, P.; Hole, M.; Smith, G. Antioxidants from a heated histidine-glucose model system. I: Investigation of the antioxidant role of histidine and isolation of antioxidants by high-performance liquid chromatography. J. Am. Oil Chem. Soc. 1998, 75, 181–187. [Google Scholar] [CrossRef]
- Xu, B.; Yu, Z.; Xiang, S.; Li, Y.; Zhang, S.L.; He, Y. Rational design of mitochondria-targeted pyruvate dehydrogenase kinase 1 inhibitors with improved selectivity and antiproliferative activity. Eur. J. Med. Chem. 2018, 155, 275–284. [Google Scholar] [CrossRef]
- Zhang, S.L.; Yang, Z.; Hu, X.; Tam, K.Y. Dichloroacetophenones targeting at pyruvate dehydrogenase kinase 1 with improved selectivity and antiproliferative activity: Synthesis and structure-activity relationships. Bioorg. Med. Chem. Lett. 2018, 28, 3441–3445. [Google Scholar] [CrossRef]
- Al-Salam, S.; Kandhan, K.; Sudhadevi, M. Down regulation of lactate dehydrogenase initiates apoptosis in HeLa and MCF-7 cancer cells through increased voltage-dependent anion channel protein and inhibition of BCL2. Oncotarget 2021, 12, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, C.; Hu, X.; Lian, Y.; Ding, C.; Ming, L. Inhibition of LDHA suppresses cell proliferation and increases mitochondrial apoptosis via the JNK signaling pathway in cervical cancer cells. Oncol. Rep. 2022, 47, 77. [Google Scholar] [CrossRef]
- Kozal, K.; Jóźwiak, P.; Krześlak, A. Contemporary Perspectives on the Warburg Effect Inhibition in Cancer Therapy. Cancer Control 2021, 28, 10732748211041243. [Google Scholar] [CrossRef]
- Kim, E.Y.; Choi, H.J.; Park, M.J.; Jung, Y.S.; Lee, S.O.; Kim, K.J.; Choi, J.H.; Chung, T.W.; Ha, K.T. Myristica fragrans Suppresses Tumor Growth and Metabolism by Inhibiting Lactate Dehydrogenase A. Am. J. Chin. Med. 2016, 44, 1063–1079. [Google Scholar] [CrossRef]
- Basu, A.; Haldar, S. The relationship between BcI2, Bax and p53: Consequences for cell cycle progression and cell death. Mol. Hum. Reprod. 1998, 4, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, M.; Ahmad, R.; Tantry, I.Q.; Ahmad, W.; Siddiqui, S.; Alam, M.; Abbas, K.; Hassan, M.I.; Habib, S.; Islam, S. Apoptosis: A Comprehensive Overview of Signaling Pathways, Morphological Changes, and Physiological Significance and Therapeutic Implications. Cells 2024, 13, 1838. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, E.; Eaves, C.J. Paradoxical roles of caspase-3 in regulating cell survival, proliferation, and tumorigenesis. J. Cell Biol. 2022, 221, e202201159. [Google Scholar] [CrossRef]
- Hussar, P. Apoptosis Regulators Bcl-2 and Caspase-3. Encyclopedia 2022, 2, 1624–1636. [Google Scholar] [CrossRef]
- Zhra, M.; Qasem, R.J.; Aldossari, F.; Saleem, R.; Aljada, A. A Comprehensive Exploration of Caspase Detection Methods: From Classical Approaches to Cutting-Edge Innovations. Int. J. Mol. Sci. 2024, 25, 5460. [Google Scholar] [CrossRef]
- Idelchik, M.D.P.S.; Begley, U.; Begley, T.J.; Melendez, J.A. Mitochondrial ROS control of cancer. Semin. Cancer Biol. 2017, 47, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Lázaro, D.; Sanz, B.; Seco-Calvo, J. Mechanisms of programmed cell death: Structural and functional pathways. A narrative review. Investig. Clín. 2024, 65, 230–252. [Google Scholar] [CrossRef]
- Tsuzuki, T.; Kambe, T.; Shibata, A.; Kawakami, Y.; Nakagawa, K.; Miyazawa, T. Conjugated EPA Activates Mutant p53 via Lipid Peroxidation and Induces p53-Dependent Apoptosis in DLD-1 Colorectal Adenocarcinoma Human Cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2007, 1771, 20–30. [Google Scholar] [CrossRef]
- Sheikh, B.Y.; Sarker, M.M.R.; Kamarudin, M.N.A.; Mohan, G. Antiproliferative and apoptosis inducing effects of citral via p53 and ROS-induced mitochondrial-mediated apoptosis in human colorectal HCT116 and HT29 cell lines. Biomed. Pharmacother. 2017, 96, 834–846. [Google Scholar] [CrossRef]
- Meng, X.; Zhang, J.; Wu, H.; Yu, D.; Fang, X. Akkermansia muciniphila Aspartic Protease Amuc_1434* Inhibits Human Colorectal Cancer LS174T Cell Viability via TRAIL-Mediated Apoptosis Pathway. Int. J. Mol. Sci. 2020, 21, 3385. [Google Scholar] [CrossRef]
- van Engeland, M.; Nieland, L.J.; Ramaekers, F.C.; Schutte, B.; Reutelingsperger, C.P. Annexin V-affinity assay: A review on an apoptosis detection system based on phosphatidylserine exposure. Cytometry 1998, 31, 1–9. [Google Scholar] [CrossRef]
- Zhu, S.; Li, T.; Tan, J.; Yan, X.; Zhang, D.; Zheng, C.; Chen, Y.; Xiang, Z.; Cui, H. Bax is essential for death receptor-mediated apoptosis in human colon cancer cells. Cancer Biother. Radiopharm. 2012, 27, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, A.; Purkey, H.E.; Hitz, A.; Robarge, K.; Peterson, D.; Labadie, S.; Kwong, M.; Hong, R.; Gao, M.; Del Nagro, C.; et al. Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat. Chem. Biol. 2016, 12, 779–786. [Google Scholar] [CrossRef]
- Kato, M.; Li, J.; Chuang, J.L.; Chuang, D.T. Distinct structural mechanisms for inhibition of pyruvate dehydrogenase kinase isoforms by AZD7545, dichloroacetate, and radicicol. Structure 2007, 15, 992–1004. [Google Scholar] [CrossRef]
- Rimac, H.; Grishina, M.; Potemkin, V. Use of the Complementarity Principle in Docking Procedures: A New Approach for Evaluating the Correctness of Binding Poses. J. Chem. Inf. Model. 2021, 61, 1801–1813. [Google Scholar] [CrossRef]
- Sharma, H.; Sharma, P.; Urquiza, U.; Chastain, L.R.; Ihnat, M.A. Exploration of a Large Virtual Chemical Space: Identification of Potent Inhibitors of Lactate Dehydrogenase-A against Pancreatic Cancer. J. Chem. Inf. Model. 2023, 63, 1028–1043. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Hernandez, R.D.; Genio, F.A.F.; Casanova, J.R.; Conato, M.T.; Paderes, M.C. Antiproliferative activities and SwissADME predictions of physicochemical properties of carbonyl group-modified rotenone analogues. ChemistryOpen 2024, 13, e202300087. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Skoraczyński, G.; Kitlas, M.; Miasojedow, B.; Gambin, A. Critical assessment of synthetic accessibility scores in computer-assisted synthesis planning. J. Cheminform. 2023, 15, 6. [Google Scholar] [CrossRef]
- Ivanenkov, Y.A.; Zagribelnyy, B.A.; Aladinskiy, V.A. Are We Opening the Door to a New Era of Medicinal Chemistry or Being Collapsed to a Chemical Singularity? J. Med. Chem. 2019, 62, 10026–10043. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Nissink, J.W.M. Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017-Utility and Limitations. ACS Chem. Biol. 2018, 13, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.B.; Zgair, A.; Taha, D.A.; Zang, X.; Kagan, L.; Kim, T.H.; Kim, M.G.; Yun, H.Y.; Fischer, P.M.; Gershkovich, P. Quantitative analysis of lab-to-lab variability in Caco-2 permeability assays. Eur. J. Pharm. Biopharm. 2017, 114, 38–42. [Google Scholar] [CrossRef]
- Kim, Y.; Chen, J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. [Google Scholar] [CrossRef]
- Kratochwil, N.A.; Huber, W.; Müller, F.; Kansy, M.; Gerber, P.R. Predicting plasma protein binding of drugs: A new approach. Biochem. Pharmacol. 2002, 64, 1355–1374. [Google Scholar] [CrossRef]
- Crettol, S.; Petrovic, N.; Murray, M. Pharmacogenetics of phase I and phase II drug metabolism. Curr. Pharm. Des. 2010, 16, 204–219. [Google Scholar] [CrossRef]
- Smith, D.A.; Beaumont, K.; Maurer, T.S.; Di, L. Clearance in Drug Design. J. Med. Chem. 2019, 62, 2245–2255. [Google Scholar] [CrossRef] [PubMed]
- Garrido, A.; Lepailleur, A.; Mignani, S.M.; Dallemagne, P.; Rochais, C. hERG toxicity assessment: Useful guidelines for drug design. Eur. J. Med. Chem. 2020, 195, 112290. [Google Scholar] [CrossRef]
- Gozalbes, R.; Jacewicz, M.; Annand, R.; Tsaioun, K.; Pineda-Lucena, A. QSAR-based permeability model for drug-like compounds. Bioorg. Med. Chem. 2011, 19, 2615–2624. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA; Dassault Systèmes. Discovery Studio Visualizer, v21.1.0.20298; Dassault Systemes: San Diego, CA, USA, 2005.



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Sayed, N.N.E.; Krayem, N.; Derbala, H.A.; Kamal, S.; Bukhari, S.N.A.; El-Ashrey, M.K.; Almarhoon, Z.M.; Soliman Alterary, S.; Ben Bacha, A. New Nitrogen-, Oxygen-, and Sulfur-Containing Heterocyclic Compounds as Anti-Colon Cancer Agents: Synthesis, Multitargeted Evaluations, Molecular Docking Simulations and ADMET Predictions. Pharmaceuticals 2025, 18, 801. https://doi.org/10.3390/ph18060801
El-Sayed NNE, Krayem N, Derbala HA, Kamal S, Bukhari SNA, El-Ashrey MK, Almarhoon ZM, Soliman Alterary S, Ben Bacha A. New Nitrogen-, Oxygen-, and Sulfur-Containing Heterocyclic Compounds as Anti-Colon Cancer Agents: Synthesis, Multitargeted Evaluations, Molecular Docking Simulations and ADMET Predictions. Pharmaceuticals. 2025; 18(6):801. https://doi.org/10.3390/ph18060801
Chicago/Turabian StyleEl-Sayed, Nahed Nasser Eid, Najeh Krayem, Hamed Ahmed Derbala, Shimaa Kamal, Syde Nasir Abbas Bukhari, Mohamed K. El-Ashrey, Zainab M. Almarhoon, Seham Soliman Alterary, and Abir Ben Bacha. 2025. "New Nitrogen-, Oxygen-, and Sulfur-Containing Heterocyclic Compounds as Anti-Colon Cancer Agents: Synthesis, Multitargeted Evaluations, Molecular Docking Simulations and ADMET Predictions" Pharmaceuticals 18, no. 6: 801. https://doi.org/10.3390/ph18060801
APA StyleEl-Sayed, N. N. E., Krayem, N., Derbala, H. A., Kamal, S., Bukhari, S. N. A., El-Ashrey, M. K., Almarhoon, Z. M., Soliman Alterary, S., & Ben Bacha, A. (2025). New Nitrogen-, Oxygen-, and Sulfur-Containing Heterocyclic Compounds as Anti-Colon Cancer Agents: Synthesis, Multitargeted Evaluations, Molecular Docking Simulations and ADMET Predictions. Pharmaceuticals, 18(6), 801. https://doi.org/10.3390/ph18060801