
Game Changers: Blockbuster Small-Molecule Drugs Approved by the FDA in 2024


Abstract
1. Introduction
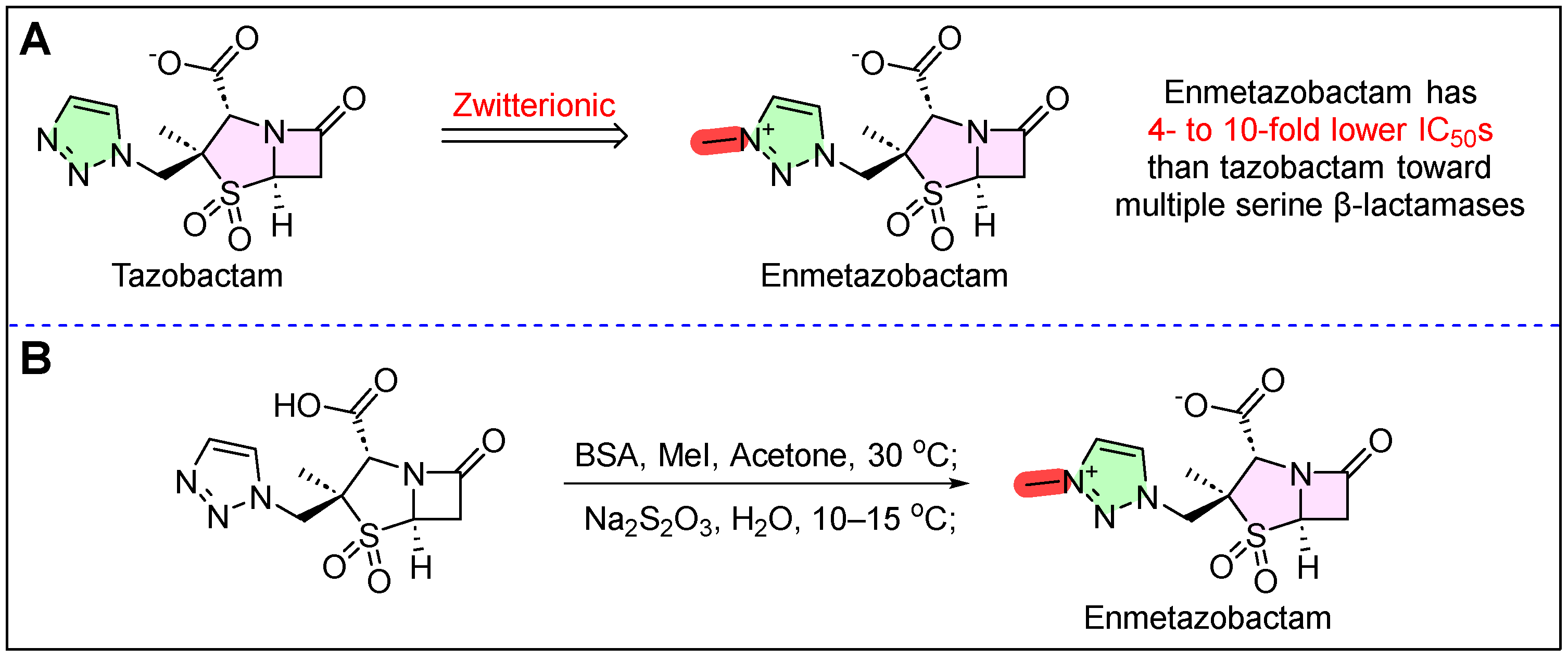
2. Exblifep (Cefepime/Enmetazobactam)
3. Rezdiffra (Resmetirom)
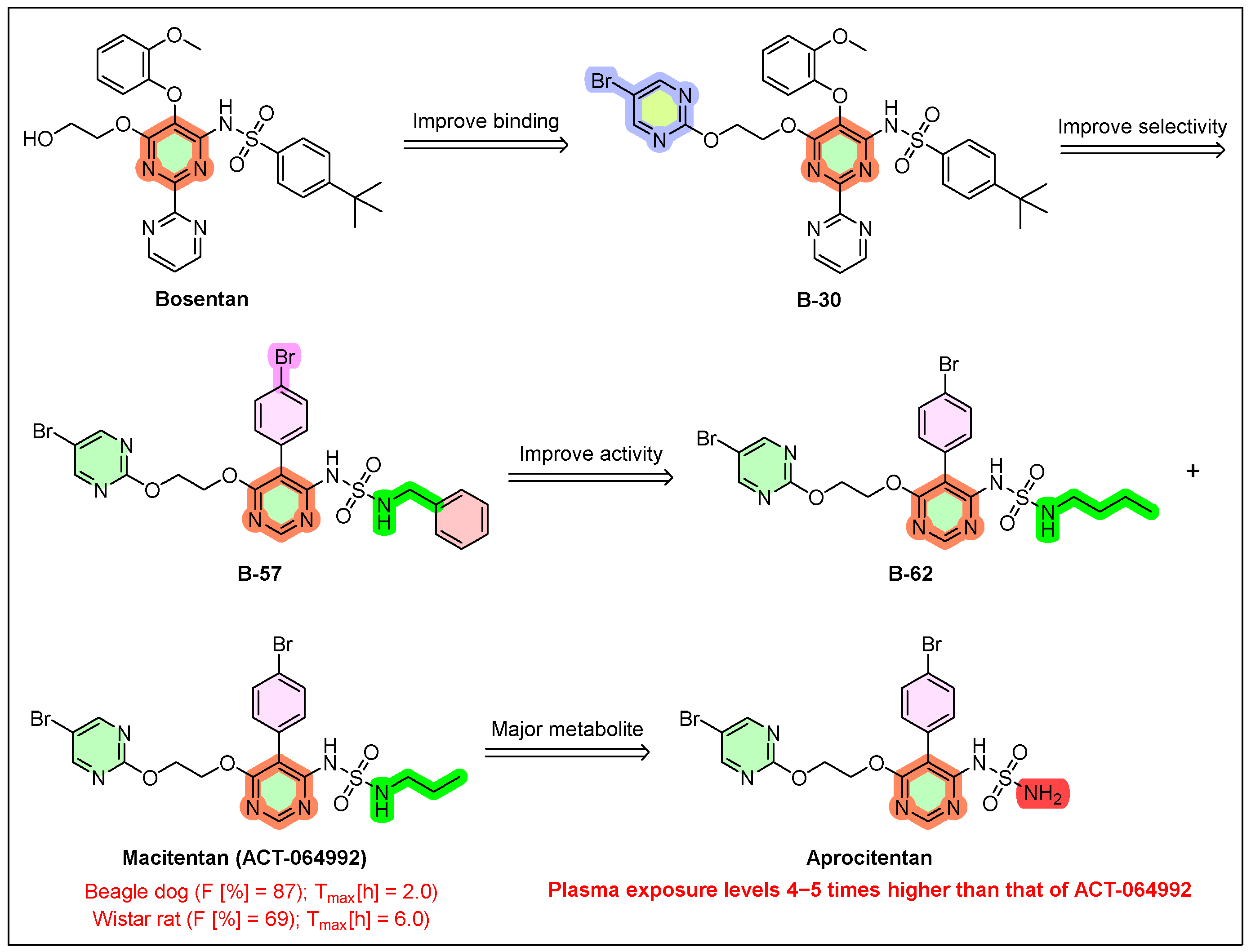
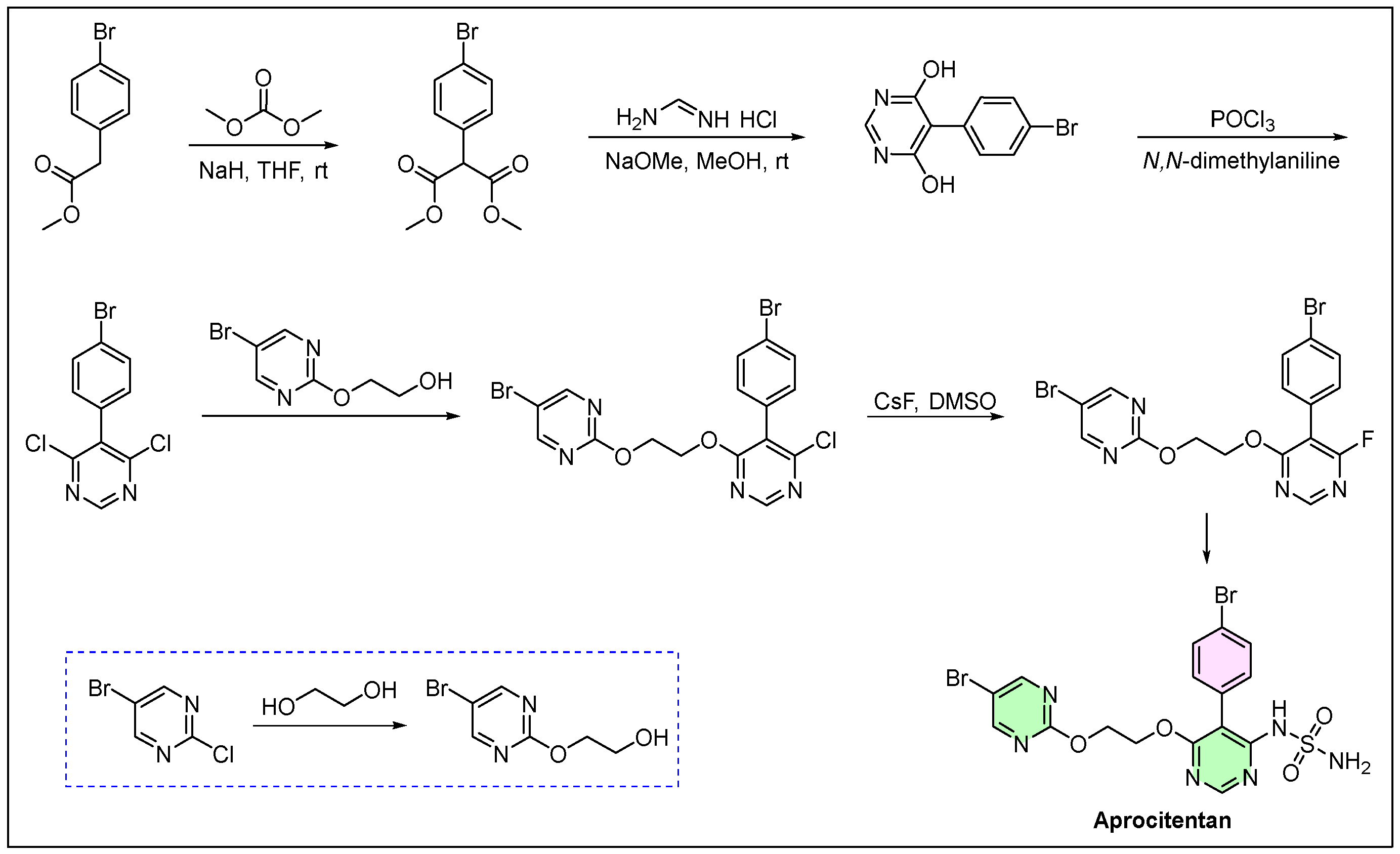
4. Tryvio (Aprocitentan)
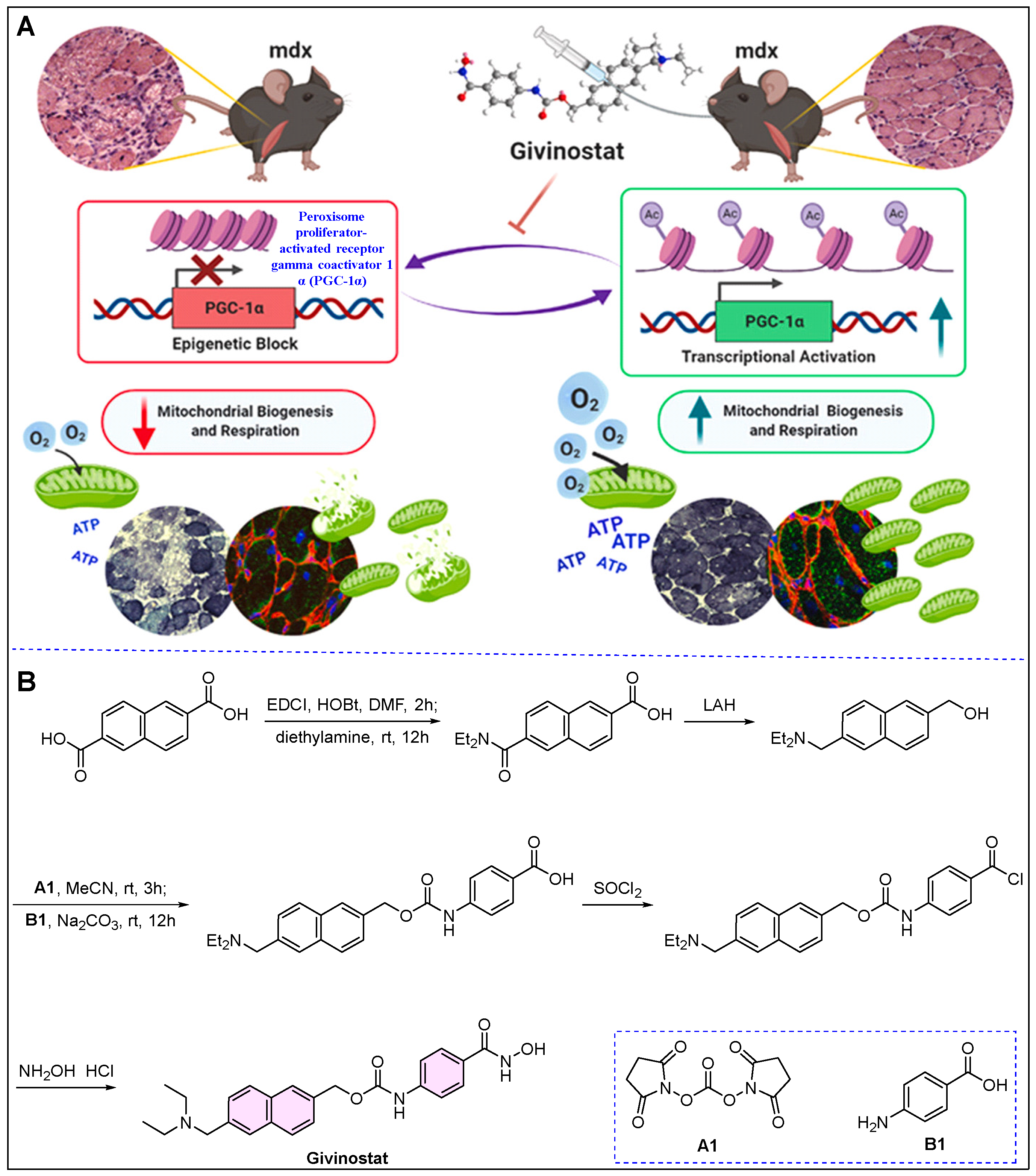
5. Duvyzat (Givinostat)
6. Vafseo (Vadadustat)
7. Voydeya (Danicopan)
8. Zevtera (Ceftobiprole)
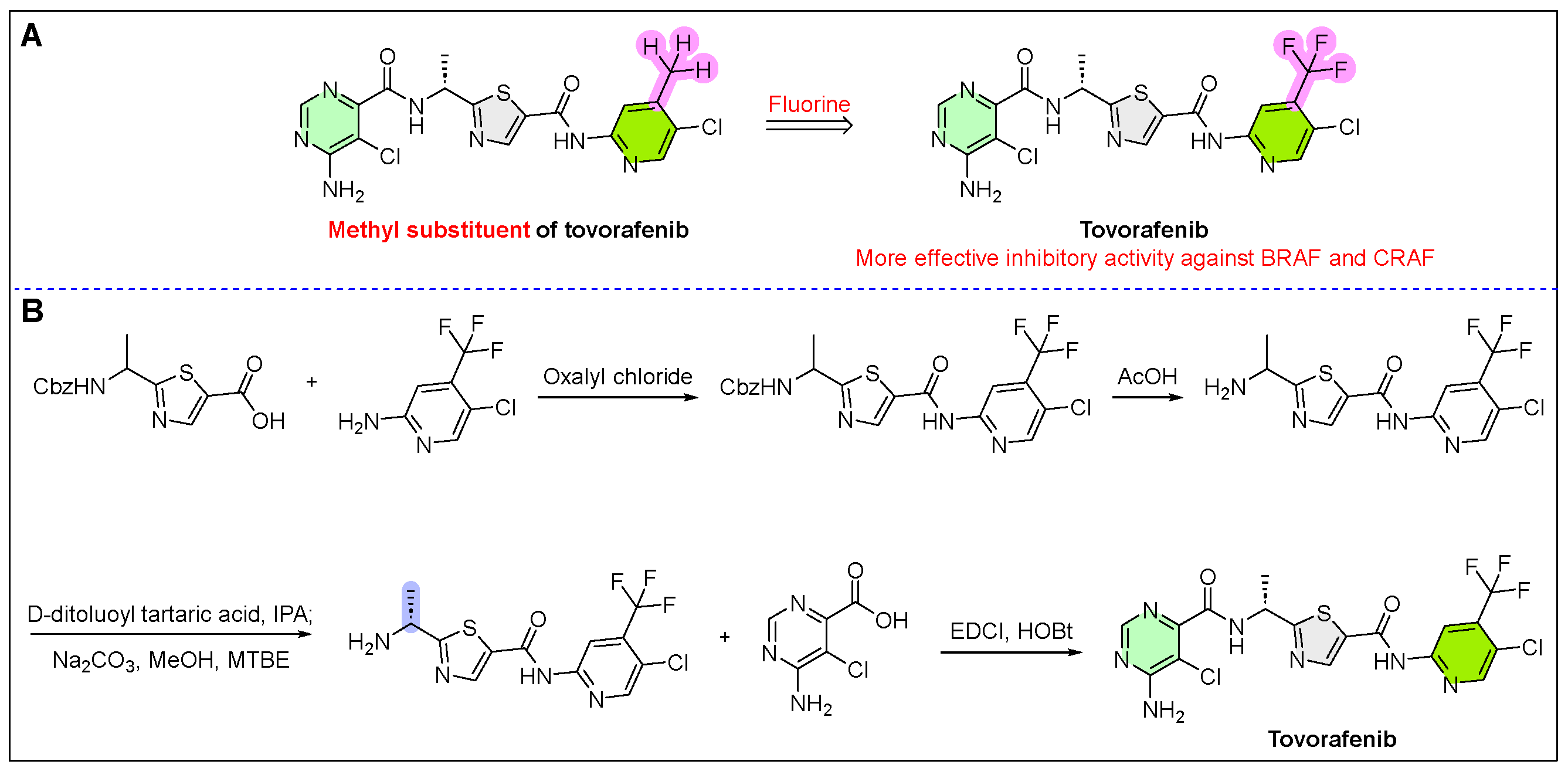
9. Ojemda (Tovorafenib)
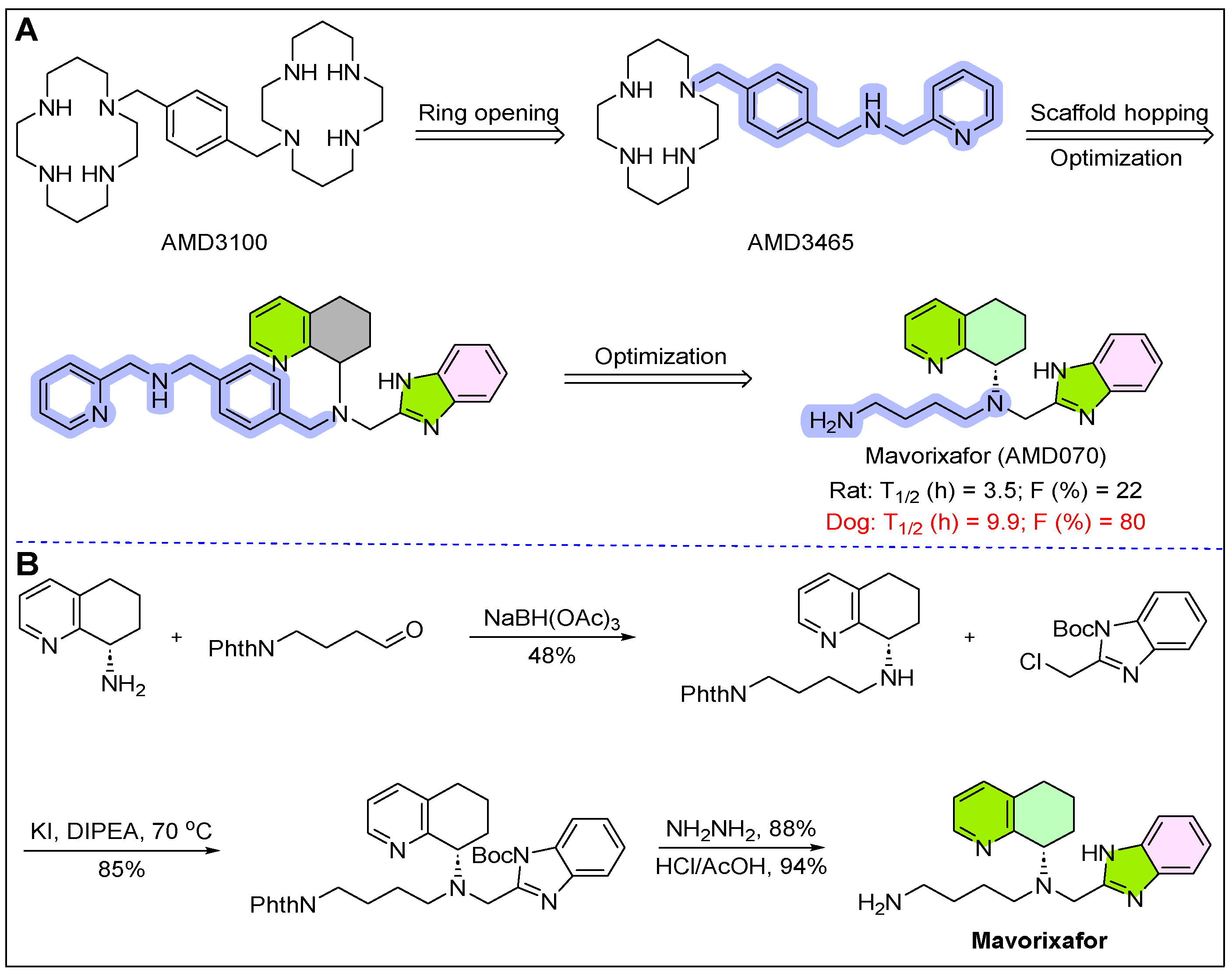
10. Xolremdi (Mavorixafor)
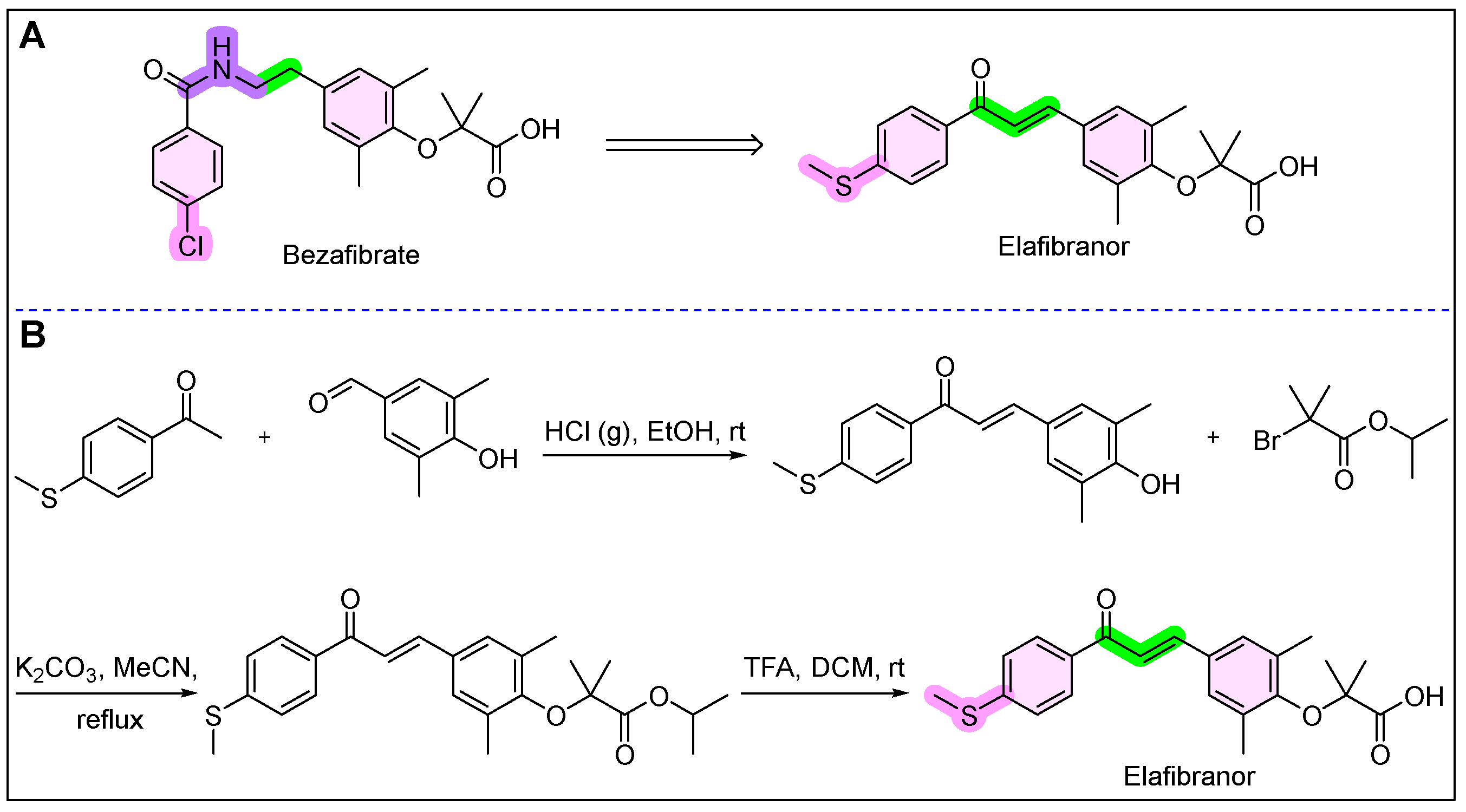
11. Iqirvo (Elafibranor)
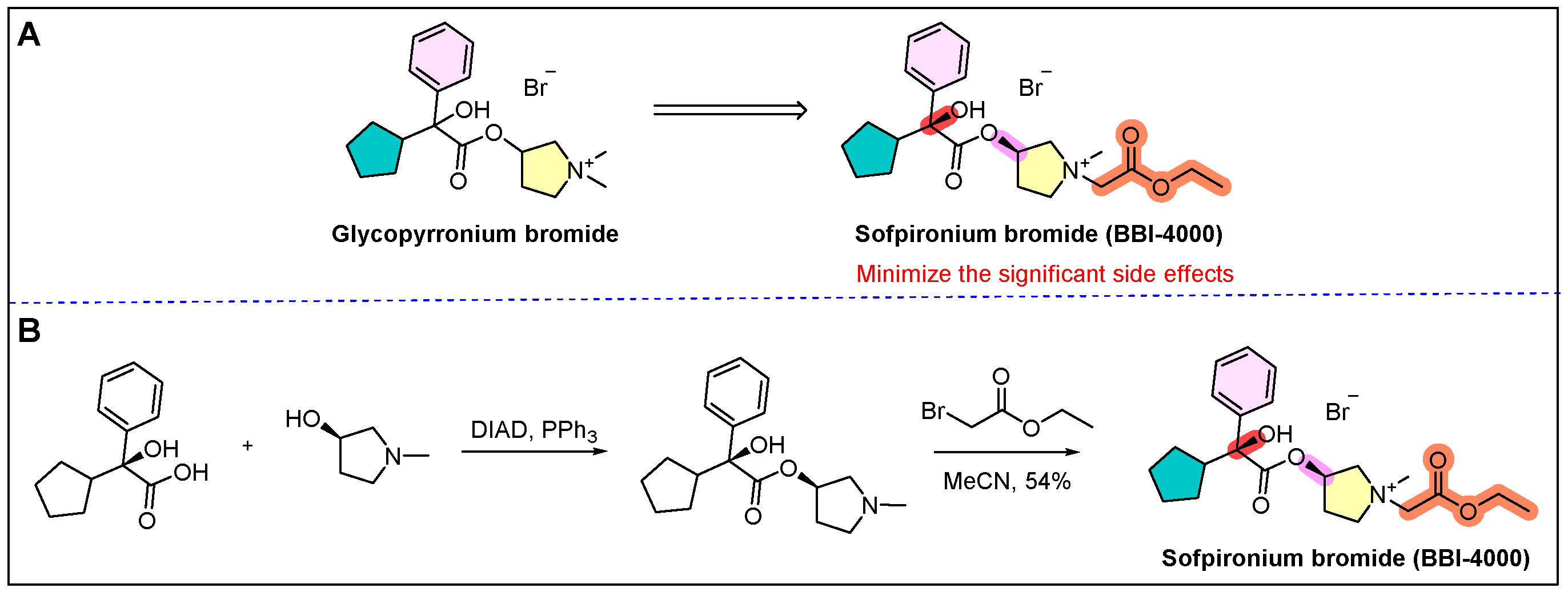
12. Sofdra (Sofpironium)
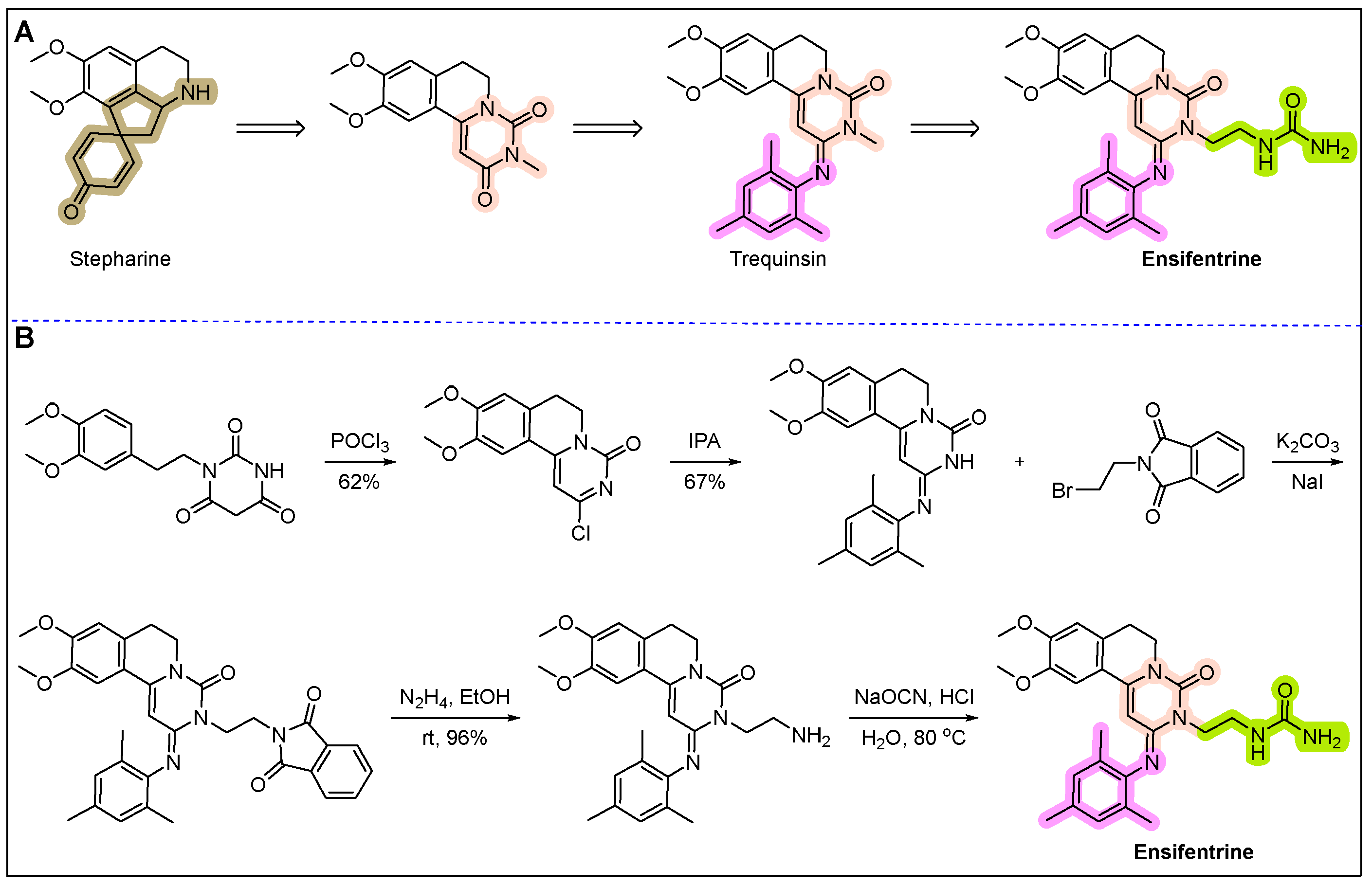
13. Ohtuvayre (Ensifentrine)
14. Leqselvi (Deuruxolitinib)
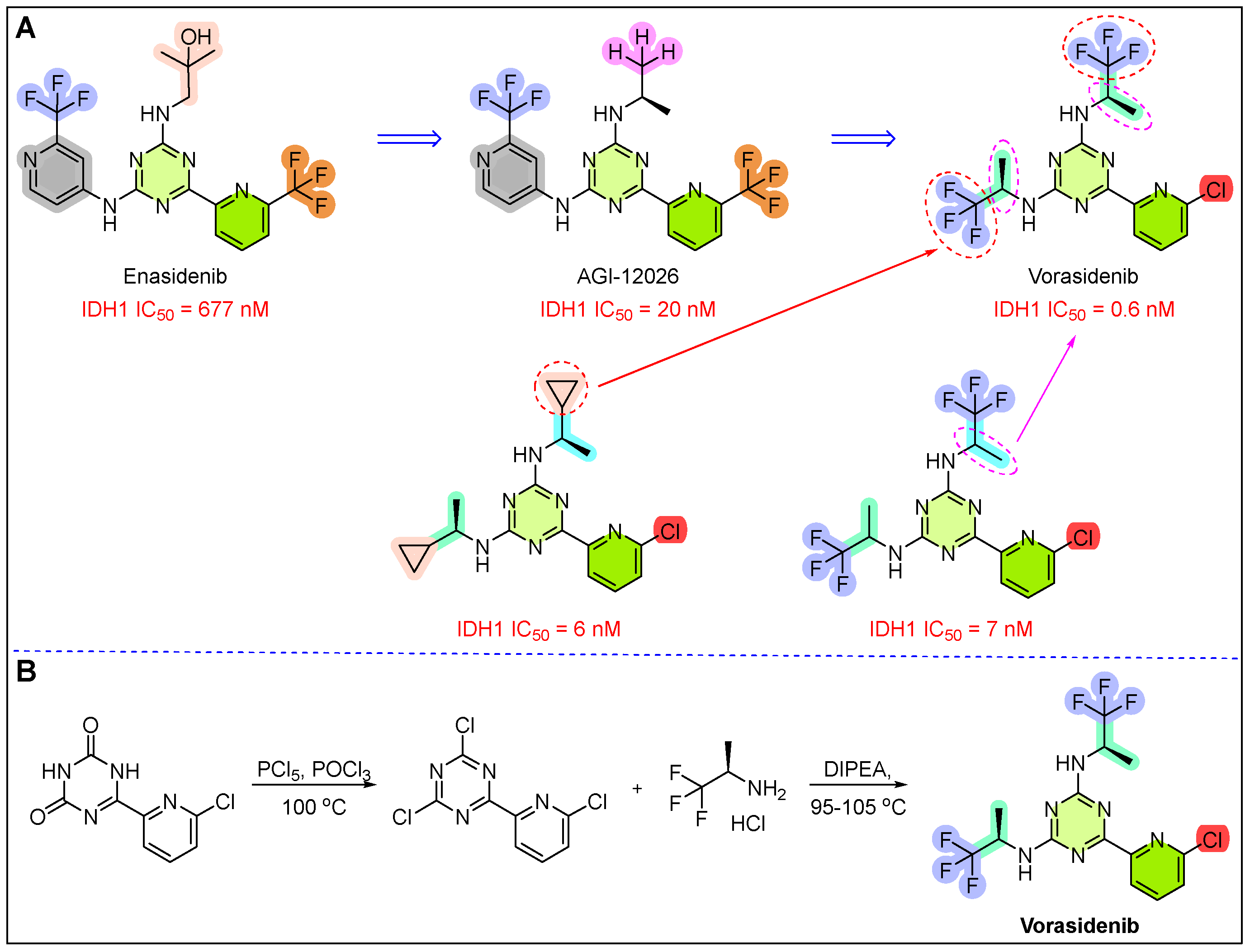
15. Voranigo (Vorasidenib)
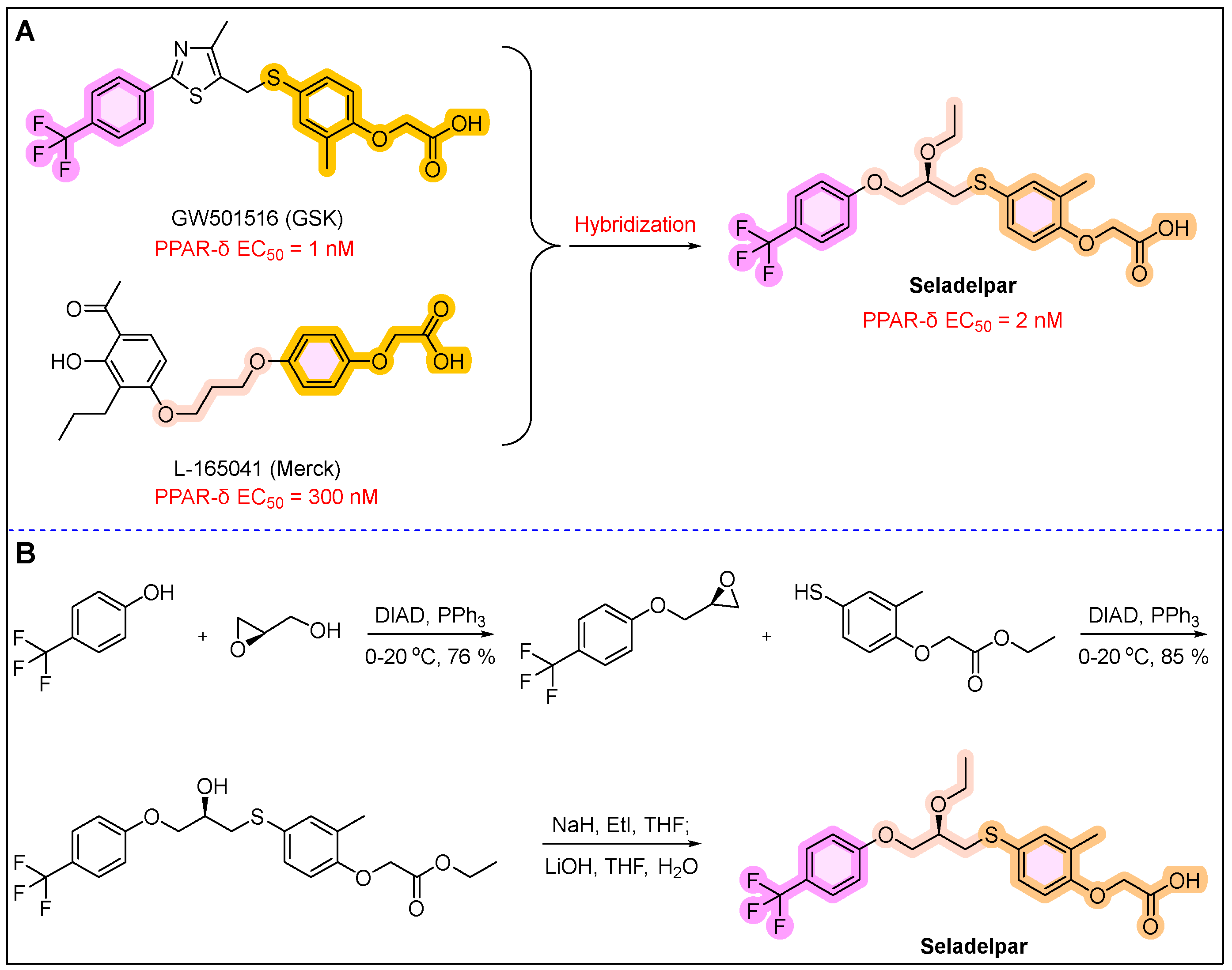
16. Livdelzi (Seladelpar)
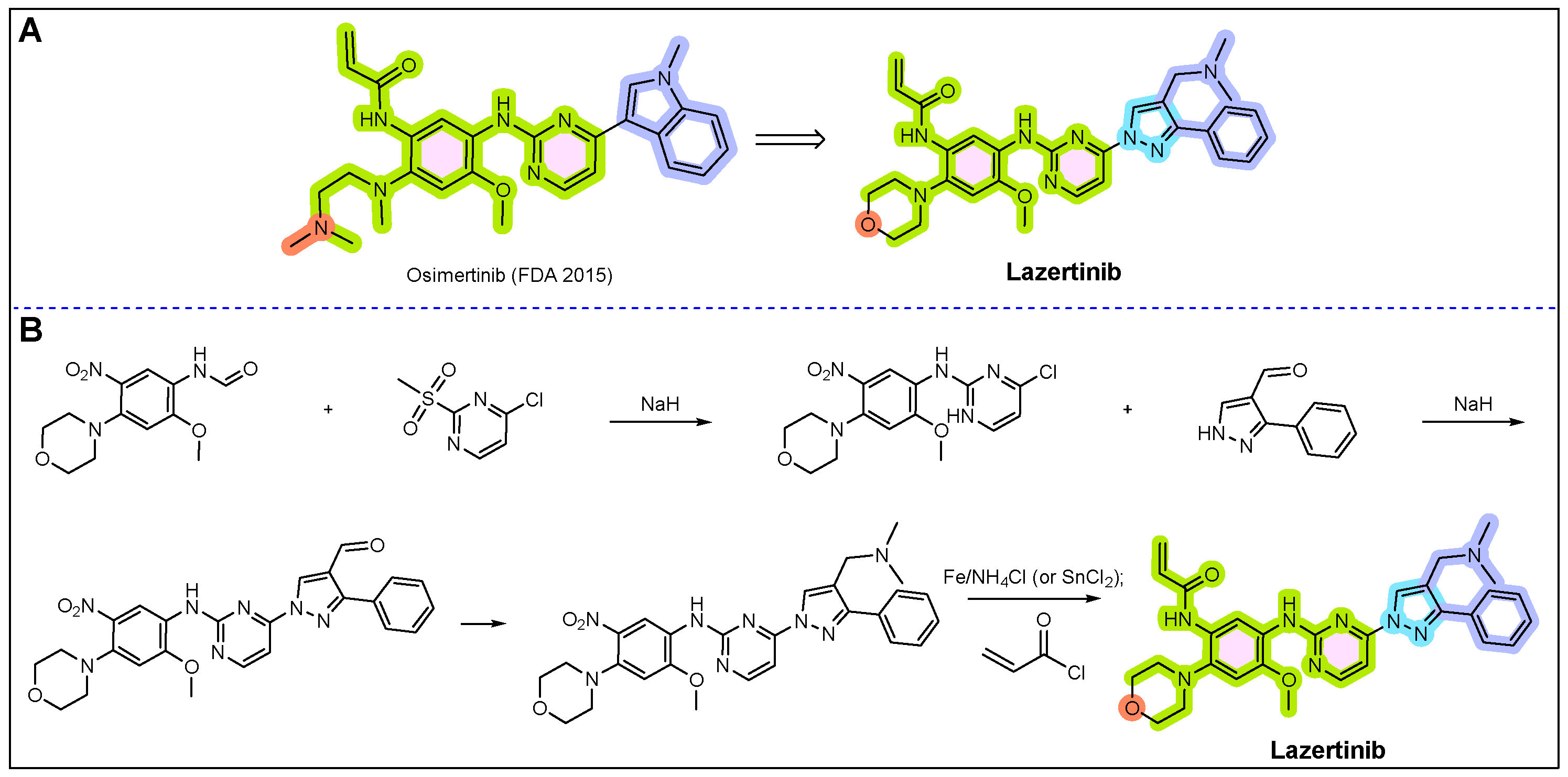
17. Rybrevant (Amivantamab)
18. Miplyffa (Arimoclomol)
19. Aqneursa (Levacetylleucine)
20. Cobenfy (Xanomeline/Trospium Chloride)
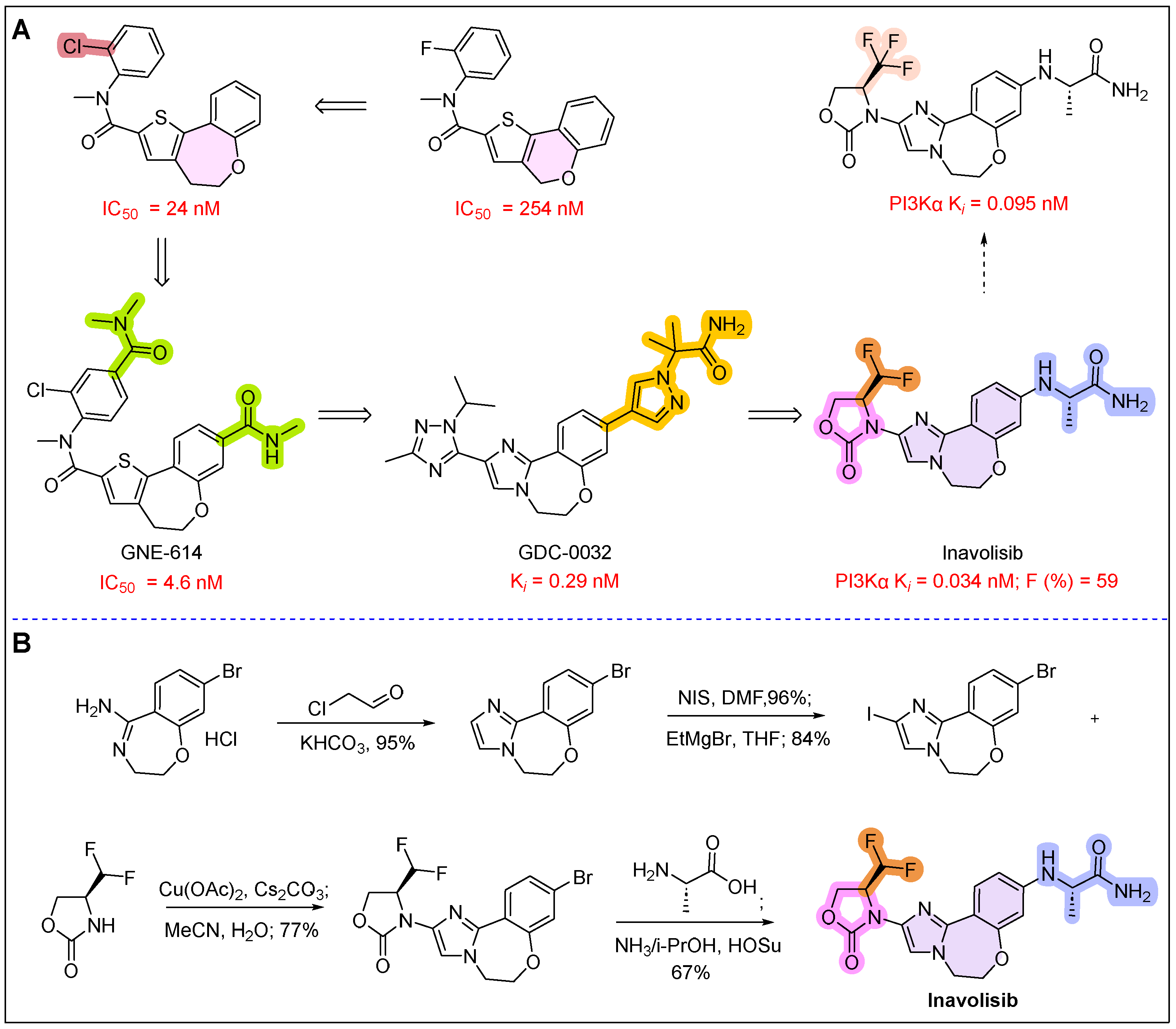
21. Itovebi (Inavolisib)
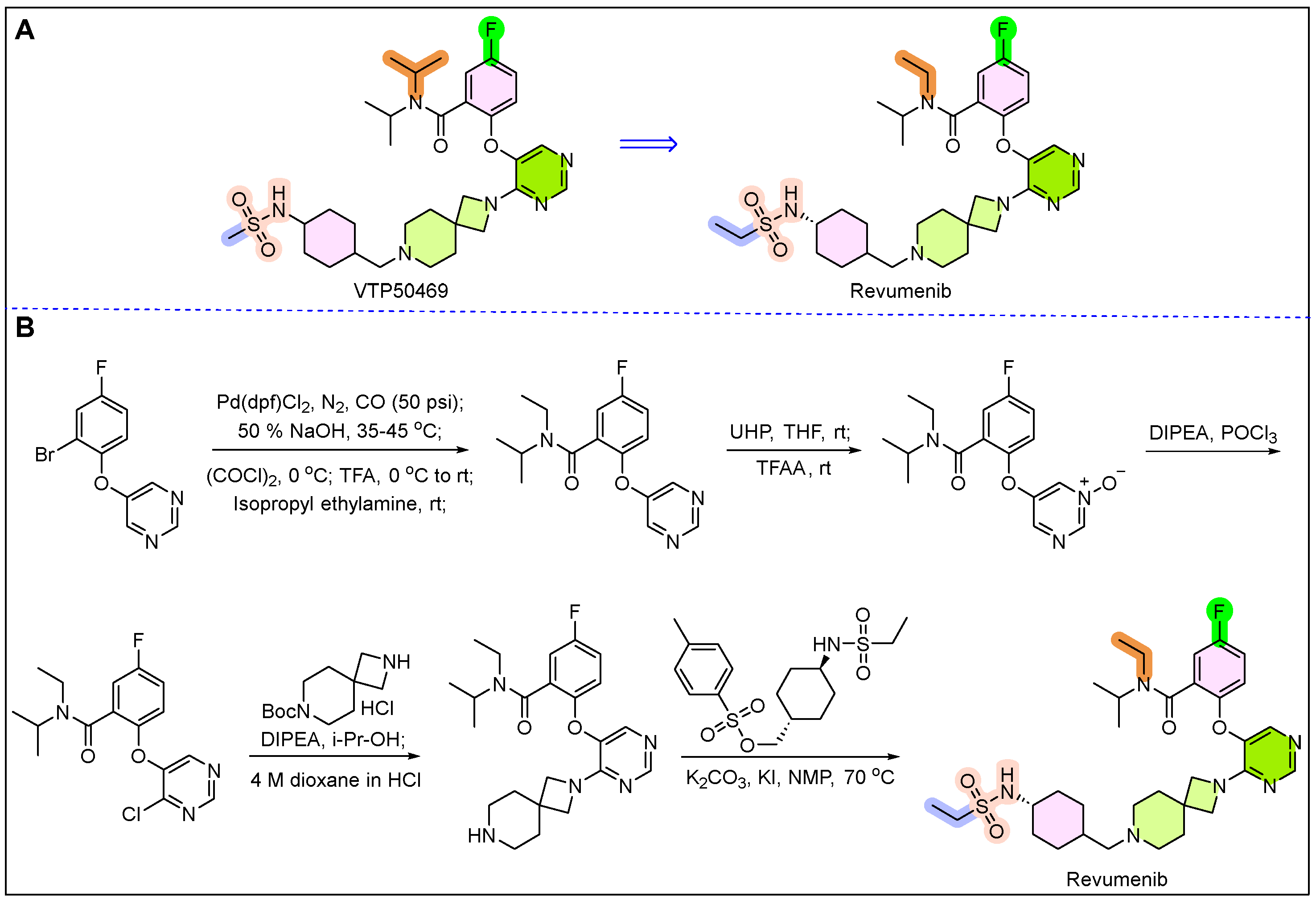
22. Orlynvah (Sulopenem Etzadroxil and Probenecid)
23. Revuforj (Revumenib)
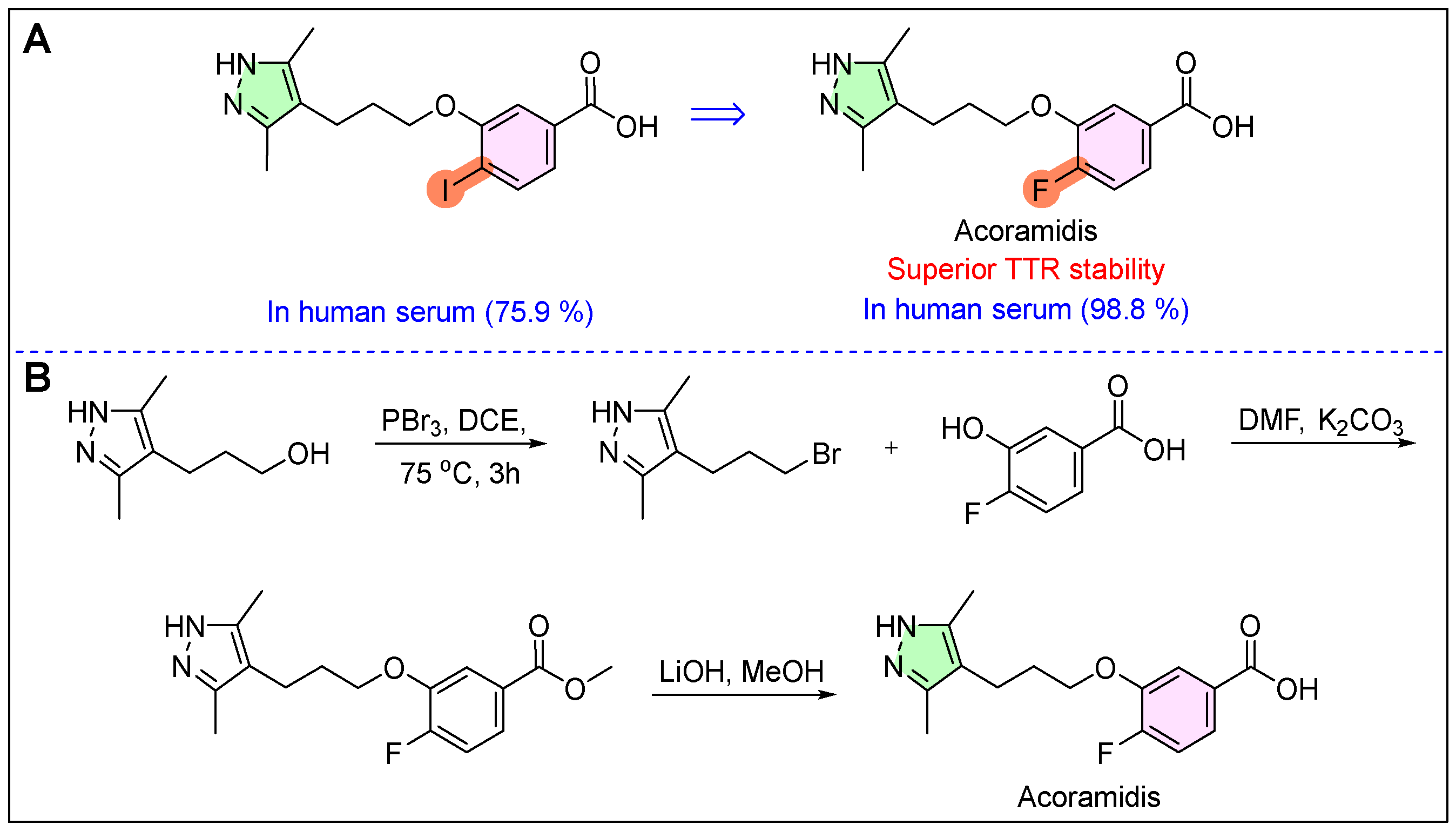
24. Rapiblyk (Landiolol)
25. Attruby (Acoramidis)
26. Crenessity (Crinecerfont)
27. Ensacove (Ensartinib)
28. Alyftrek (Vanzacaftor/Tezacaftor/Deutivacaftor)
29. Conclusions and Outlook
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Agrawal, S.; Park, E.; Kluetz, P.G. FDA approvals in 2024: New options for patients across cancer types and therapeutic classes. Nat. Rev. Clin. Oncol. 2025, in press. [Google Scholar] [CrossRef] [PubMed]
- Topouzis, S.; Papapetropoulos, A.; Alexander, S.P.; Cortese-Krott, M.; Kendall, D.A.; Martemyanov, K.; Mauro, C.; Nagercoil, N.; Panettieri, R.A., Jr.; Patel, H.H.; et al. Novel drugs approved by the EMA, the FDA and the MHRA in 2024: A year in review. Br. J. Pharmacol. 2025, 182, 1416–1445. [Google Scholar] [CrossRef]
- Zhai, D.; Zhang, Q.; Lu, X.; You, Q.; Wang, L. Global first-in-class drugs approved in 2023–2024: Breakthroughs and insights. Innovation 2025, 6, 100801. [Google Scholar] [CrossRef]
- Bai, Y.-R.; Duan, Q.-C.; Seng, D.-J.; Xu, Y.; Ren, H.-B.; Zhang, J.; Shen, D.-D.; Yang, L.; Liu, H.-M.; Yuan, S. A comprehensive review of small molecule drugs approved by the FDA in 2024: Advance and prospect. Chin. Chem. Lett. 2025, in press. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, F.; Wang, B.; Xie, L.; Chen, W. New FDA drug approvals for 2024: Synthesis and clinical application. Eur. J. Med. Chem. 2025, 285, 117241. [Google Scholar] [CrossRef]
- de la Torre, B.G.; Albericio, F. The Pharmaceutical Industry in 2024: An Analysis of the FDA Drug Approvals from the Perspective of Molecules. Molecules 2025, 30, 482. [Google Scholar] [CrossRef]
- Baedeker, M.; Ringel, M.S.; Möller, C.C. 2024 FDA approvals exceed average number but have lower sales projections. Nat. Rev. Drug Discov. 2025, 24, 85. [Google Scholar] [CrossRef]
- Mullard, A. 2024 FDA approvals. Nat. Rev. Drug Discov. 2025, 24, 75–82. [Google Scholar] [CrossRef]
- Keam, S.J. Cefepime/enmetazobactam: First approval. Drugs 2024, 84, 737–744. [Google Scholar] [CrossRef]
- Niu, Z.X.; Wang, Y.T.; Zhang, S.N.; Li, Y.; Chen, X.B.; Wang, S.Q.; Liu, H.M. Application and synthesis of thiazole ring in clinically approved drugs. Eur. J. Med. Chem. 2023, 250, 115172. [Google Scholar] [CrossRef]
- Shapiro, S. Methyls and Me. J. Med. Chem. 2025, 68, 6857–6859. [Google Scholar] [CrossRef] [PubMed]
- Udayampalayam Palanisamy, S.; Paul-Satyaseela, M.; Narayanan, S.; Balasubramanian, G.; Appu, A.; Manickam, S.; Periasamy, H. Compounds and Their Use. U.S. Patent 20140057888A1, 12 February 2019. [Google Scholar]
- Darlow, C.A.; Hope, W.; Dubey, V. Cefepime/Enmetazobactam: A microbiological, pharmacokinetic, pharmacodynamic, and clinical evaluation. Expert. Opin. Drug Metab. Toxicol. 2025, 21, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Tilahun, M.; Kassa, Y.; Gedefie, A.; Ashagire, M. Emerging carbapenem-resistant Enterobacteriaceae infection, its epidemiology and novel treatment options: A review. Infect. Drug Resist. 2021, 14, 4363–4374. [Google Scholar] [CrossRef]
- Zhang, S.; Liao, X.; Ding, T.; Ahn, J. Role of β-lactamase inhibitors as potentiators in antimicrobial chemotherapy targeting Gram-negative bacteria. Antibiotics 2024, 13, 260. [Google Scholar] [CrossRef]
- Sansone, P.; Giaccari, L.G.; Di Flumeri, G.; Pace, M.C.; Pota, V.; Coppolino, F.; Brunetti, S.; Aurilio, C. Imipenem/Cilastatin/Relebactam for Complicated Infections: A Real-World Evidence. Life 2024, 14, 614. [Google Scholar] [CrossRef]
- Kaye, K.S.; Belley, A.; Barth, P.; Lahlou, O.; Knechtle, P.; Motta, P.; Velicitat, P. Effect of cefepime/enmetazobactam vs piperacillin/tazobactam on clinical cure and microbiological eradication in patients with complicated urinary tract infection or acute pyelonephritis: A randomized clinical trial. JAMA 2022, 328, 1304–1314. [Google Scholar] [CrossRef]
- Ding, M.; Zhou, F.; Li, Y.; Liu, C.; Gu, Y.; Wu, J.; Fan, G.; Li, Y.; Li, X. Cassiae Semen improves non-alcoholic fatty liver disease through autophagy-related pathway. Chin. Herb. Med. 2023, 15, 421–429. [Google Scholar] [CrossRef]
- Fraile, J.M.; Palliyil, S.; Barelle, C.; Porter, A.J.; Kovaleva, M. Non-alcoholic steatohepatitis (NASH)–A review of a crowded clinical landscape, driven by a complex disease. Drug Des. Dev. Ther. 2021, 15, 3997–4009. [Google Scholar] [CrossRef]
- Keam, S.J. Resmetirom: First approval. Drugs 2024, 84, 729–735. [Google Scholar] [CrossRef]
- Brisnovali, N.F.; Haney, C.; Goedeke, L. Rezdiffra™(resmetirom): A THR-β agonist for non-alcoholic steatohepatitis. Trends Pharmacol. Sci. 2024, 45, 1081–1082. [Google Scholar] [CrossRef]
- Zhao, M.; Xie, H.; Shan, H.; Zheng, Z.; Li, G.; Li, M.; Hong, L. Development of Thyroid Hormones and Synthetic Thyromimetics in Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 1102. [Google Scholar] [CrossRef] [PubMed]
- Boulos, M.; Mousa, R.S.; Jeries, N.; Simaan, E.; Alam, K.; Bulus, B.; Assy, N. Hidden in the Fat: Unpacking the Metabolic Tango Between Metabolic Dysfunction-Associated Steatotic Liver Disease and Metabolic Syndrome. Int. J. Mol. Sci. 2025, 26, 3448. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Targher, G. Hepatic thyroid hormone receptor-β signalling: Mechanisms and recent advancements in the treatment of metabolic dysfunction-associated steatohepatitis. Diabetes Obes. Metab. 2025, 27, 1635–1647. [Google Scholar] [CrossRef]
- Dahiya, K.; Palkar, M.; Sharma, S. Unlocking the potential of THR-β agonist therapies: Resmetirom’s chemistry, biology, and patent insights. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2025, 1–18. [Google Scholar] [CrossRef]
- Kelly, M.J.; Pietranico-Cole, S.; Larigan, J.D.; Haynes, N.-E.; Reynolds, C.H.; Scott, N.; Vermeulen, J.; Dvorozniak, M.; Conde-Knape, K.; Huang, K.-S.; et al. Discovery of 2-[3,5-Dichloro-4-(5-isopropyl-6-oxo-1,6-dihydropyridazin-3-yloxy)phenyl]-3,5-dioxo-2,3,4,5-tetrahydro [1,2,4]triazine-6-carbonitrile (MGL-3196), a highly selective thyroid hormone receptor β agonist in clinical trials for the treatment of dyslipidemia. J. Med. Chem. 2014, 57, 3912–3923. [Google Scholar]
- Younossi, Z.M.; Stepanova, M.; Racila, A.; Henry, L.; Labriola, D.; Taub, R.; Nader, F. Health-related quality of life (HRQL) assessments in a 52-week, double-blind, randomized, placebo-controlled phase III study of resmetirom (MGL-3196) in patients with metabolic dysfunction–associated steatohepatitis (MASH) and fibrosis. Hepatology 2025, 81, 1318–1327. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, L.; Cui, S.; Liang, Y.; Zhang, X. Synthesis and Anti-hypertensive Effects of the Twin Drug of Nicotinic Acid and Quercetin Tetramethyl Ether. Molecules 2014, 19, 4791–4801. [Google Scholar] [CrossRef]
- Song, Y.; Chen, C.; Li, W. Ginsenoside Rb1 in cardiovascular and cerebrovascular diseases: A review of therapeutic potentials and molecular mechanisms. Chin. Herb. Med. 2024, 16, 489–504. [Google Scholar] [CrossRef]
- Gudayneh, Y.A.; Shumye, A.F.; Gelaye, A.T.; Tegegn, M.T. Prevalence of hypertensive retinopathy and its associated factors among adult hypertensive patients attending at Comprehensive Specialized Hospitals in Northwest Ethiopia, 2024, a multicenter cross-sectional study. Int. J. Retin. Vitr. 2025, 11, 17. [Google Scholar] [CrossRef]
- Hanif, A.A.M.; Shamim, A.A.; Hossain, M.M.; Hasan, M.; Khan, M.S.A.; Hossaine, M.; Mridha, M.K. Gender-specific prevalence and associated factors of hypertension among elderly Bangladeshi people: Findings from a nationally representative cross-sectional survey. BMJ Open 2021, 11, e038326. [Google Scholar] [CrossRef]
- Alrashed, S.F.M.; Alruwaili, R.R.M.; Alrwaili, N.A.K.; Alruwaili, R.J.F.; Alhazmi, G.A.A.; Alruwaili, S.A.D. Hypertension and Heart Health. Gland. Surg. 2024, 9, 367–373. [Google Scholar]
- Redon, J.; Carmena, R. Present and future of drug therapy in hypertension: An overview. Blood Press. 2024, 33, 2320401. [Google Scholar] [CrossRef]
- Tamargo, J. After 30 years, the first endothelin-receptor antagonist (Aprocitentan) is approved for the treatment of arterial hypertension. Eur. Heart J. Cardiovasc. Pharmacother. 2024, 10, 371–373. [Google Scholar] [CrossRef]
- Dhillon, S. Aprocitentan: First approval. Drugs 2024, 84, 841–847. [Google Scholar] [CrossRef]
- Yao, Y.; Fan, B.; Yang, B.; Jia, Z.; Li, B. Aprocitentan: A new development of resistant hypertension. J. Clin. Hypertens. 2023, 25, 587–590. [Google Scholar] [CrossRef]
- McCoy, E.K.; Lisenby, K.M. Aprocitentan (a dual endothelin-receptor antagonist) for treatment-resistant hypertension. J. Cardiovasc. Pharmacol. 2021, 77, 699–706. [Google Scholar] [CrossRef]
- Mekuria, A.B.; Demelash Kifle, Z.; Abdelwuhab, M. Endothelin System and Therapeutic Application of Endothelin Receptor Antagonists. J. Clin. Exp. Pharmacol. 2021, 10, 272. [Google Scholar]
- Bolli, M.H.; Boss, C.; Binkert, C.; Buchmann, S.; Bur, D.; Hess, P.; Iglarz, M.; Meyer, S.; Rein, J.; Rey, M.; et al. The Discovery of N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N′-propylsulfamide (Macitentan), an Orally Active, Potent Dual Endothelin Receptor Antagonist. J. Med. Chem. 2012, 55, 7849–7861. [Google Scholar] [CrossRef]
- Naseralallah, L.; Koraysh, S. Aprocitentan: A new emerging prospect in the pharmacotherapy of hypertension. Blood Press. 2024, 33, 2424824. [Google Scholar] [CrossRef]
- Schlaich, M.P.; Bellet, M.; Weber, M.A.; Danaietash, P.; Bakris, G.L.; Flack, J.M.; Dreier, R.F.; Sassi-Sayadi, M.; Haskell, L.P.; Narkiewicz, K.; et al. Dual endothelin antagonist aprocitentan for resistant hypertension (PRECISION): A multicentre, blinded, randomised, parallel-group, phase 3 trial. Lancet 2022, 400, 1927–1937. [Google Scholar] [CrossRef]
- Tong, H.; Fan, S.; Hu, W.; Wang, H.; Guo, G.; Huang, X.; Yu, Q. Diarylpropionitrile-stimulated ERβ nuclear accumulation promotes MyoD-induced muscle regeneration in mdx mice by interacting with FOXO3A. Pharmacol. Res. 2024, 208, 107376. [Google Scholar] [CrossRef] [PubMed]
- Longmuir, K.; Park, J.; Fitzgerald, R.; Legge, M.; Shore, C. Hope: Valuing lives and persons with degenerative conditions—Duchenne muscular dystrophy. Sites A J. Soc. Anthropol. Cult. Stud. 2024, 20, 1–26. [Google Scholar] [CrossRef]
- Lamb, Y.N. Givinostat: First approval. Drugs 2024, 84, 849–856. [Google Scholar] [CrossRef]
- Syed, N.; Mughal, Z.U.N.; Haseeb, A.; Manhal, G.A.; Eissa, A.Y.; Ahmed, K.A.H.M. Duvyzat (givinostat): A new hope for Duchenne muscular dystrophy patients. Ann. Med. Surg. 2025, 87, 2529–2531. [Google Scholar] [CrossRef]
- Giovarelli, M.; Zecchini, S.; Catarinella, G.; Moscheni, C.; Sartori, P.; Barbieri, C.; De Palma, C. Givinostat as metabolic enhancer reverting mitochondrial biogenesis deficit in Duchenne Muscular Dystrophy. Pharmacol. Res. 2021, 170, 105751. [Google Scholar] [CrossRef]
- Chemi SpA; Italfarmaco SpA. Process for Preparing {6-[(Diethylamino)methyl]naphthalen-2-yl}methyl [4-(Hydroxycarbamoyl) phenyl]carbamate Having High Purity. Patent WO2020178776A1, 10 September 2020. [Google Scholar]
- Mercuri, E.; Vilchez, J.J.; Boespflug-Tanguy, O.; Zaidman, C.M.; Mah, J.K.; Goemans, N.; Willis, T. Safety and efficacy of givinostat in boys with Duchenne muscular dystrophy (EPIDYS): A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2024, 23, 393–403. [Google Scholar] [CrossRef]
- Badro, D.A. Chronic kidney disease management in developing countries. In Handbook of Medical and Health Sciences in Developing Countries: Education, Practice, and Research; Springer International Publishing: Cham, Switzerland, 2023; pp. 1–146. [Google Scholar]
- Jiang, C.; Wang, L.; Wang, Y.; Xu, R.; Yang, H.; Peng, J. Therapeutic effects of Chinese herbal medicines for treatment of urolithiasis: A review. Chin. Herb. Med. 2023, 15, 526–532. [Google Scholar] [CrossRef]
- Fishbane, S.; Spinowitz, B. Update on anemia in ESRD and earlier stages of CKD: Core curriculum 2018. Am. J. Kidney Dis. 2018, 71, 423–435. [Google Scholar] [CrossRef]
- Markham, A. Vadadustat: First approval. Drugs 2020, 80, 1365–1371. [Google Scholar] [CrossRef]
- Sayaf, A.M.; Kousar, K.; Suleman, M.; Albekairi, N.A.; Alshammari, A.; Mohammad, A.; Yeoh, K.K. Molecular exploration of natural and synthetic compounds databases for promising hypoxia inducible factor (HIF) Prolyl-4-hydroxylase domain (PHD) inhibitors using molecular simulation and free energy calculations. BMC Chem. 2024, 18, 236. [Google Scholar] [CrossRef]
- Yeh, T.L.; Leissing, T.M.; Abboud, M.I.; Thinnes, C.C.; Atasoylu, O.; Holt-Martyn, J.P.; Schofield, C.J. Molecular and cellular mechanisms of HIF prolyl hydroxylase inhibitors in clinical trials. Chem. Sci. 2017, 8, 7651–7668. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Kou, J.; Wu, S.; Cai, X.; Xiao, Q.; Li, Y.; Hu, J.; Li, J.; Wang, Z. Development of a Robust and Scalable Process for the Large-Scale Preparation of Vadadustat. Org. Process Res. Dev. 2021, 25, 960–968. [Google Scholar] [CrossRef]
- Zuk, A.; Si, Z.; Loi, S.; Bommegowda, S.; Hoivik, D.; Danthi, S.; Rabinowitz, M. Preclinical characterization of vadadustat (AKB-6548), an oral small molecule hypoxia-inducible factor prolyl-4-hydroxylase inhibitor, for the potential treatment of renal anemia. J. Pharmacol. Exp. Ther. 2022, 383, 11–24. [Google Scholar] [CrossRef]
- Winkelmayer, W.C.; Burke, S.K.; Chertow, G.M.; Eckardt, K.U.; Luo, W.; Minga, T.; Roy-Chaudhury, P. Vascular Access Thrombosis Events in Patients with Dialysis-Dependent Chronic Kidney Disease Treated with Vadadustat or Darbepoetin Alfa: The INNO2VATE Trial Program. Kidney Med. 2025, 7, 100997. [Google Scholar] [CrossRef]
- Vallejo, C.; Boix, S.B.; Carnicero, F.; Gaya, A.; Gonzalez, A.P.; Fernandez, F.A.G.; Vara, M. Patients with Paroxysmal Nocturnal Hemoglobinuria (PNH) Treated with Pegcetacoplan: Real Life Experience. Blood 2023, 142, 5653. [Google Scholar] [CrossRef]
- Oliver, M.; Patriquin, C.J. Paroxysmal nocturnal hemoglobinuria: Current management, unmet needs, and recommendations. J. Blood Med. 2023, 14, 613–628. [Google Scholar] [CrossRef]
- Kang, C. Danicopan: First approval. Drugs 2024, 84, 613–618. [Google Scholar] [CrossRef]
- Xu, B.; Zhou, J. Oral complement factor D inhibitor danicopan for paroxysmal nocturnal hemoglobinuria. Expert. Rev. Clin. Pharmacol. 2024, 17, 857–864. [Google Scholar] [CrossRef]
- Zhang, W.; Wu, M.; Vadlakonda, S.; Juarez, L.; Cheng, X.; Muppa, S.; Kotian, P. Scaffold hopping via ring opening enables identification of acyclic compounds as new complement Factor D inhibitors. Bioorganic Med. Chem. 2022, 74, 117034. [Google Scholar] [CrossRef]
- Liu, Y.C.; Tipparaju, S.K.; Tracey, M.R.; Carr, R.; Damarancha, A.; Hashimoto, A.; Hubbs, S.; Keyes, C.; Lucas, J.; Nandialath, V.; et al. Technology Transfer and Process Development of Danicopan to Enable Process Validation. Org. Process. Res. Dev. 2024, 28, 2394–2405. [Google Scholar] [CrossRef]
- Du, Y.; Bian, Y.; Baecker, D.; Dhawan, G.; Semghouli, A.; Kiss, L.; Zhang, W.; Sorochinsky, A.E.; Soloshonok, V.A.; Han, J. Fluorine in the Pharmaceutical Industry: FDA-Approved Fluorine-Containing Drugs in 2024. Chem. Eur. J. 2025, 31, e202500662. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, J.; Giuliano, C.; Kale-Pradhan, P.B. Ceftobiprole Medocaril: A New Fifth-Generation Cephalosporin. Ann. Pharmacother. 2024. [Google Scholar] [CrossRef] [PubMed]
- Holland, T.L.; Cosgrove, S.E.; Doernberg, S.B.; Jenkins, T.C.; Turner, N.A.; Boucher, H.W.; Fowler, V.G., Jr. Ceftobiprole for treatment of complicated Staphylococcus aureus bacteremia. New Engl. J. Med. 2023, 389, 1390–1401. [Google Scholar] [CrossRef]
- Bassetti, M.; Peghin, M.; Castaldo, N.; Giacobbe, D.R. The safety of treatment options for acute bacterial skin and skin structure infections. Expert. Opin. Drug Saf. 2019, 18, 635–650. [Google Scholar] [CrossRef]
- Scheeren, T.W.; Welte, T.; Saulay, M.; Engelhardt, M.; Santerre-Henriksen, A.; Hamed, K. Early improvement in severely ill patients with pneumonia treated with ceftobiprole: A retrospective analysis of two major trials. BMC Infect. Dis. 2019, 19, 195. [Google Scholar] [CrossRef]
- Hebeisen, P.; Heinze-Krauss, I.; Angehrn, P.; Hohl, P.; Page, M.G.; Then, R.L. In vitro and in vivo properties of Ro 63-9141, a novel broad-spectrum cephalosporin with activity against methicillin-resistant staphylococci. Antimicrob. Agents Chemother. 2001, 45, 825–836. [Google Scholar] [CrossRef]
- Kremminger, P.; Ludescher, J.; Sturm, H. Method for the Production of Ceftobiprole Medocaril. Patent WO2010136422A1, 25 May 2010. [Google Scholar]
- Singh, S.; Bradford, D.; Chatterjee, S.; Li, X.; Aungst, S.L.; Skinner, A.M.; Drezner, N. FDA Approval Summary: Tovorafenib for Relapsed or Refractory BRAF-Altered Pediatric Low-Grade Glioma. Clin. Cancer Res. 2025, 31, 1383–1389. [Google Scholar] [CrossRef]
- Dhillon, S. Tovorafenib: First approval. Drugs 2024, 84, 985–993. [Google Scholar] [CrossRef]
- Chen, W.; Cossrow, J.; Franklin, L.; Guan, B.; John, H.J.; Kumaravel, G.; Lane, B.; Littke, A.; Lugovskoy, A.; Peng, H.; et al. Pyrimidine Derivatives Useful as RAF Kinase Inhibitors. Patent CN 101784545 A, 20 April 2016. [Google Scholar]
- Chen, W.; Cossrow, J.; Franklin, L.; Guan, B.; John, H.J.; Kumaravel, G.; Lane, B.; Littke, A.; Lugovskoy, A.; Peng, H.; et al. As the Pyrimidine Derivatives of RAF Kinase Inhibitor. Patent CN 101784545 B, 20 April 2016. [Google Scholar]
- Hirano, S.; Takeda, Y.; Nakamoto, K.; Ikeuchi, M.; Kitayama, M.; Yamada, M.; Kawakami, J.I. Method for Producing Optically Active Compound. U.S. Patent 20200317659 A1, 27 April 2021. [Google Scholar]
- Kilburn, L.B.; Khuong-Quang, D.A.; Hansford, J.R.; Landi, D.; van Der Lugt, J.; Leary, S.E.; Nysom, K. The type II RAF inhibitor tovorafenib in relapsed/refractory pediatric low-grade glioma: The phase 2 FIREFLY-1 trial. Nat. Med. 2024, 30, 207–217. [Google Scholar] [CrossRef]
- Saotome, K.; McGoldrick, L.L.; Ho, J.H.; Ramlall, T.F.; Shah, S.; Moore, M.J.; Franklin, M.C. Structural insights into CXCR4 modulation and oligomerization. Nat. Struct. Mol. Biol. 2025, 32, 315–325. [Google Scholar] [CrossRef]
- Roland, L.; Nguyen, C.H.; Zmajkovicova, K.; Khamyath, M.; Kalogeraki, M.; Schell, B.; Balabanian, K. CXCR4 antagonism ameliorates leukocyte abnormalities in a preclinical model of WHIM syndrome. Front. Immunol. 2024, 15, 1468823. [Google Scholar] [CrossRef] [PubMed]
- Bridger, G.J.; Skerlj, R.T.; Hernandez-Abad, P.E.; Bogucki, D.E.; Wang, Z.; Zhou, Y.; Schols, D. Synthesis and structure−activity relationships of azamacrocyclic CXC chemokine receptor 4 antagonists: Analogues containing a single azamacrocyclic ring are potent inhibitors of T-cell tropic (X4) HIV-1 replication. J. Med. Chem. 2010, 53, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Skerlj, R.T.; Bridger, G.J.; Kaller, A.; McEachern, E.J.; Crawford, J.B.; Zhou, Y.; Schols, D. Discovery of novel small molecule orally bioavailable C− X− C chemokine receptor 4 antagonists that are potent inhibitors of T-tropic (X4) HIV-1 replication. J. Med. Chem. 2010, 53, 3376–3388. [Google Scholar] [CrossRef]
- Crawford, J.B.; Chen, G.; Gauthier, D.; Wilson, T.; Carpenter, B.; Baird, I.R.; Bridger, G.J. AMD070, a CXCR4 chemokine receptor antagonist: Practical large-scale laboratory synthesis. Org. Process Res. Dev. 2008, 12, 823–830. [Google Scholar] [CrossRef]
- Badolato, R.; Alsina, L.; Azar, A.; Bertrand, Y.; Bolyard, A.A.; Dale, D.; Donadieu, J. A phase 3 randomized trial of mavorixafor, a CXCR4 antagonist, for WHIM syndrome. Blood 2024, 144, 35–45. [Google Scholar] [CrossRef]
- Bidiuk, J.; Szmigielski, C.; Lewandowski, J.; Sinski, M. Cardiac morphology and function in patients with primary biliary cholangitis (PBC) without cirrhosis. Eur. Heart J. 2024, 45, ehae666.3039. [Google Scholar] [CrossRef]
- Graf, M.; Lange, C.M.; Langer, M.M.; Schattenberg, J.M.; Seessle, J.; Dietz, J.; Graf, C. Primary biliary cholangitis (PBC)-autoimmune hepatitis (AIH) variant syndrome: Clinical features, response to therapy and long-term outcome. J. Clin. Med. 2023, 12, 7047. [Google Scholar] [CrossRef]
- Blair, H.A. Elafibranor: First Approval. Drugs 2024, 84, 1143–1148. [Google Scholar] [CrossRef]
- Ali, J.S.; Ali, T.S.; Al Hasibuzzaman, M. Latest FDA approved drug Elafibranor (Iqirvo): A novel prospect for treatment of primary biliary cholangitis. Ann. Med. Surg. 2025, 87, 30–32. [Google Scholar]
- Kamata, S.; Honda, A.; Ishikawa, R.; Akahane, M.; Fujita, A.; Kaneko, C.; Ishii, I. Functional and structural insights into the human PPARα/δ/γ targeting preferences of anti-NASH investigational drugs, lanifibranor, seladelpar, and elafibranor. Antioxidants 2023, 12, 1523. [Google Scholar] [CrossRef]
- Kamata, S.; Oyama, T.; Saito, K.; Honda, A.; Yamamoto, Y.; Suda, K.; Ishii, I. PPARα ligand-binding domain structures with endogenous fatty acids and fibrates. iScience 2020, 23, 101727. [Google Scholar] [CrossRef] [PubMed]
- Caumont-Bertrand, K.; Delhomel, J.-F. Preparation of 1,3-Diphenyl Prop-2-En-1-One Derivatives. U.S. Patent 7385082B2, 10 June 2008. [Google Scholar]
- Schattenberg, J.M.; Pares, A.; Kowdley, K.V.; Heneghan, M.A.; Caldwell, S.; Pratt, D.; Luketic, V. A randomized placebo-controlled trial of elafibranor in patients with primary biliary cholangitis and incomplete response to UDCA. J. Hepatol. 2021, 74, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Davis, H.; Wilson, P. Axillary hyperhidrosis: A review of the extent of the problem and treatment modalities. Surgeon 2015, 13, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Paik, J. Sofpironium bromide: First approval. Drugs 2020, 80, 1981–1986. [Google Scholar] [CrossRef]
- Odat, R.M.; Yousef Aldalati, A.; Hammadeh, B.M.; Mohammad Hussein, A.; Idrees, M.; Marzouk, H.; Hanifa, H. Efficacy and safety of sofpironium in treatment of primary hyperhidrosis: A systematic review. J. Dermatol. Treat. 2025, 36, 2441258. [Google Scholar] [CrossRef]
- Kruse, A.C.; Hu, J.; Pan, A.C.; Arlow, D.H.; Rosenbaum, D.M.; Rosemond, E.; Kobilka, B.K. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 2012, 482, 552–556. [Google Scholar] [CrossRef]
- Bodor, N.S.; Esters, S.A. Soft Anticholinergic Esters. U.S. Patent 20120177590A1, 14 January 2014. [Google Scholar]
- Yokozeki, H.; Fujimoto, T.; Abe, Y.; Igarashi, M.; Ishikoh, A.; Omi, T.; Takayama, S. A phase 3, multicenter, randomized, double-blind, vehicle-controlled, parallel-group study of 5% sofpironium bromide (BBI-4000) gel in Japanese patients with primary axillary hyperhidrosis. J. Dermatol. 2021, 48, 279–288. [Google Scholar] [CrossRef]
- Fujimoto, T.; Abe, Y.; Igarashi, M.; Ishikoh, A.; Omi, T.; Kanda, H.; Yokozeki, H. A phase III, 52-week, open-label study to evaluate the safety and efficacy of 5% sofpironium bromide (BBI-4000) gel in Japanese patients with primary axillary hyperhidrosis. J. Dermatol. 2021, 48, 1149–1161. [Google Scholar] [CrossRef]
- Ferrera, M.C.; Labaki, W.W.; Han, M.K. Advances in chronic obstructive pulmonary disease. Annu. Rev. Med. 2021, 72, 119–134. [Google Scholar] [CrossRef]
- Hubert, S.; Kök-Carrière, A.; De Ceuninck, F. Ensifentrine (Ohtuvayre™) for chronic obstructive pulmonary disease. Trends Pharmacol. Sci. 2024, 45, 941–942. [Google Scholar] [CrossRef]
- Dransfield, M.; Marchetti, N.; Kalhan, R.; Reyner, D.; Dixon, A.L.; Rheault, T.; Rickard, K.A.; Anzueto, A. Ensifentrine in COPD patients taking long-acting bronchodilators: A pooled post-hoc analysis of the ENHANCE-1/2 studies. Chron. Resp. Dis. 2025, 22, 14799731251314874. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Wang, C.; Zhang, J.; Wang, X.; Sun, B.; Xiao, X.; Zhang, H.; Song, Y.; Jia, Y. Chinese herbal medicines for prostate cancer therapy: From experimental research to clinical practice, Chin. Herb. Med. 2023, 15, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Ahmed, W.; Ahmed, T.; Chen, L. Nanoparticles derived from herbal preparations may represent a novel nucleic acid therapy. Interdiscip. Med. 2024, 2, e20230029. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, L. Chinese herbal medicine: Fighting SARS-CoV-2 infection on all fronts. J. Ethnopharmacol. 2021, 270, 113869. [Google Scholar] [CrossRef]
- Miao, X.; Jin, C.; Liu, J.; Wang, J.; Chen, Y. Honokiol attenuates acetaminophen-induced acute liver injury by inhibiting hepatic CYP1A2 activity and improving liver mitochondrial dysfunction. Chin. Herb. Med. 2023, 15, 231–239. [Google Scholar] [CrossRef]
- Wang, J.; Liao, Z.X. Research progress of microrobots in tumor drug delivery. Food Med. Homol. 2024, 1, 9420025. [Google Scholar] [CrossRef]
- Li, D.; Guo, H.; Niu, L.; Yin, Q.; Zhang, Y.; Zhuang, P. Clinical value-oriented research paradigm about inheritance and innovation development of TCM dominant diseases. Chin. Herb. Med. 2023, 15, 476–484. [Google Scholar] [CrossRef]
- Wang, Z.; Song, X.-Q.; Xu, W.; Lei, S.; Zhang, H.; Yang, L. Stand Up to Stand Out: Natural Dietary Polyphenols Curcumin, Resveratrol, and Gossypol as Potential Therapeutic Candidates against Severe Acute Respiratory Syndrome Coronavirus 2 Infection. Nutrients 2023, 15, 3885. [Google Scholar] [CrossRef]
- Zhan, Y.; Xu, X.; Luo, X.; Liu, R.; Lin, Y.; Zhao, P.; Shi, J. Preparation of tanshinone IIA self-soluble microneedles and its inhibition on proliferation of human skin fibroblasts. Chin. Herb. Med. 2023, 15, 251–262. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, L. GS-5734: A potentially approved drug by FDA against SARS-CoV-2. New J. Chem. 2020, 44, 12417–12429. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, S.; Chen, K.; Ji, L.; Cui, S. Magnolol and 5fluorouracil synergy inhibition of metastasis of cervical cancer cells by targeting PI3K/AKT/mTOR and EMT pathways. Chin. Herb. Med. 2024, 16, 94–105. [Google Scholar] [PubMed]
- Wang, Z.; Wang, N.; Yang, L.; Song, X.Q. Bioactive natural products in COVID-19 therapy. Front. Pharmacol. 2022, 13, 926507. [Google Scholar] [CrossRef]
- Song, X.; Wang, J.; Zhang, Y.; Du, X.; Qian, Q. Protective effect of hydroxysafflor yellow A on MSCs against senescence induced by D-galactose. Chin. Herb. Med. 2023, 15, 86–93. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, L. The therapeutic potential of natural dietary flavonoids against SARS-CoV-2 infection. Nutrients 2023, 15, 3443. [Google Scholar] [CrossRef]
- Shi, L.; Cui, T.; Wang, X.; Wu, R.; Wu, J.; Wang, Y.; Wang, W. Biotransformation and pharmacological activities of platycosides from Platycodon grandiflorum roots. Chin. Herb. Med. 2024, 16, 392–400. [Google Scholar] [CrossRef]
- Yang, L.; Wang, Z. Bench-to-bedside: Innovation of small molecule anti-SARS-CoV-2 drugs in China. Eur. J. Med. Chem. 2023, 257, 115503. [Google Scholar] [CrossRef]
- Lal, B.; Dohadwalla, A.N.; Dadkar, N.K.; D’Sa, A.; De Souza, N.J. Trequinsin, a Potent New Antihypertensive Vasodilator in the Series of 2-(Arylimino)-3-alkyl-9, 10-dimethoxy-3, 4, 6, 7-tetrahydro-2H-pyrimido [6, 1-a] isoquinolin-4-ones. J. Med. Chem. 1984, 27, 1470–1480. [Google Scholar] [CrossRef]
- Cipla Limited. Solid State Forms of Ensifentrine. Patent WO2024127413 A1, 20 June 2024. [Google Scholar]
- Anzueto, A.; Barjaktarevic, I.Z.; Siler, T.M.; Rheault, T.; Bengtsson, T.; Rickard, K.; Sciurba, F. Ensifentrine, a novel phosphodiesterase 3 and 4 inhibitor for the treatment of chronic obstructive pulmonary disease: Randomized, double-blind, placebo-controlled, multicenter phase III trials (the ENHANCE trials). Am. J. Respir. Crit. Care Med. 2023, 208, 406–416. [Google Scholar] [CrossRef]
- Pratt, C.H.; King, L.E.; Messenger, A.G.; Christiano, A.M.; Sundberg, J.P. Alopecia areata. Nat. Rev. Dis. Primers 2017, 3, 17011. [Google Scholar] [CrossRef]
- Gupta, A.K.; Bamimore, M.A.; Mirmirani, P.; Piguet, V.; Talukder, M. The Relative Efficacy and Safety of Monotherapies for Alopecia Areata: A Network Meta-Analysis Study. J. Cosmet. Dermatol. 2025, 24, e70185. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, L.; Song, X.Q. Oral GS-441524 derivatives: Next-generation inhibitors of SARS-CoV-2 RNA-dependent RNA polymerase. Front. Immunol. 2022, 13, 1015355. [Google Scholar] [CrossRef]
- Di Martino, R.M.C.; Maxwell, B.D.; Pirali, T. Deuterium in drug discovery: Progress, opportunities and challenges. Nat. Rev. Drug Discov. 2023, 22, 562–584. [Google Scholar] [CrossRef]
- Davis, R.R.; Li, B.; Yun, S.Y.; Chan, A.; Nareddy, P.; Gunawan, S.; Schonbrunn, E. Structural insights into JAK2 inhibition by ruxolitinib, fedratinib, and derivatives thereof. J. Med. Chem. 2021, 64, 2228–2241. [Google Scholar] [CrossRef]
- Concert Pharmaceuticals, Inc. Process for Preparing Enantiomerically Enriched JAK Inhibitors. Patent WO2022036030 A1, 17 February 2022. [Google Scholar]
- King, B.; Senna, M.M.; Mesinkovska, N.A.; Lynde, C.; Zirwas, M.; Maari, C.; Cassella, J. Efficacy and safety of deuruxolitinib, an oral selective Janus kinase inhibitor, in adults with alopecia areata: Results from the Phase 3 randomized, controlled trial (THRIVE-AA1). J. Am. Acad. Dermatol. 2024, 91, 880–888. [Google Scholar] [CrossRef]
- Yamaguchi, H.L.; Yamaguchi, Y.; Peeva, E. Hair regrowth in alopecia areata and re-pigmentation in vitiligo in response to treatment: Commonalities and differences. J. Eur. Acad. Dermatol. Venereol. 2025, 39, 498–511. [Google Scholar] [CrossRef]
- Weller, M.; Wen, P.Y.; Chang, S.M.; Dirven, L.; Lim, M.; Monje, M.; Reifenberger, G. Glioma. Nat. Rev. Dis. Primers 2024, 10, 33. [Google Scholar] [CrossRef]
- Eisenstein, M. FDA approves first IDH-targeted glioma drug. Nat. Biotechnol. 2024, 42, 1325. [Google Scholar]
- Lamb, Y.N. Vorasidenib: First Approval. Drugs 2024, 84, 1325–1331. [Google Scholar] [CrossRef]
- Konteatis, Z.; Artin, E.; Nicolay, B.; Straley, K.; Padyana, A.K.; Jin, L.; Chen, Y.; Narayaraswamy, R.; Tong, S.; Wang, F.; et al. Vorasidenib (AG-881): A first-in-class, brain-penetrant dual inhibitor of mutant IDH1 and 2 for treatment of glioma. ACS Med. Chem. Lett. 2020, 11, 101–107. [Google Scholar] [CrossRef]
- Wen, P.; Mellinghoff, I.; van den Bent, M.; Blumenthal, D.; Touat, M.; Peters, K.; Ellingson, B. LTBK-06. Impact of vorasidenib treatment on mutant IDH1 OR IDH2 diffuse glioma tumor growth rate: Results from the randomized, double-blind, phase 3 indigo study. Neuro-Oncol. 2023, 25, 310–311. [Google Scholar] [CrossRef]
- Nevzorova, Y.A.; Cubero, F.J. Seladelpar: New hope for patients with primary biliary cholangitis. Medicine 2024, 5, 377–379. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Seladelpar: First approval. Drugs 2024, 84, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Kotsiliti, E. Seladelpar in primary biliary cholangitis. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 300. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, A.; DeAngelis, A.; Pelton, P.; Xu, J.; Zhu, P.; Kuo, G.H. Discovery of para-alkylthiophenoxyacetic acids as a novel series of potent and selective PPARδ agonists. Bioorganic Med. Chem. Lett. 2007, 17, 3855–3859. [Google Scholar] [CrossRef] [PubMed]
- Kuo, G.-H.; Zhang, R.; Wang, A.; Deangelis, A.R. Preparation of 4-((Phenoxyalkyl) thio)phenoxyacetic Acids and Analogs as PPARδ Agonists for Treating Conditions like Dyslipidemia. U.S. Patent 20160199342A1, 8 August 2017. [Google Scholar]
- Hirschfield, G.M.; Bowlus, C.L.; Mayo, M.J.; Kremer, A.E.; Vierling, J.M.; Kowdley, K.V.; McWherter, C.A. A phase 3 trial of seladelpar in primary biliary cholangitis. New Engl. J. Med. 2024, 390, 783–794. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, L. Natural-product-based, carrier-free, noncovalent nanoparticles for tumor chemo-photodynamic combination therapy. Pharmacol. Res. 2024, 203, 107150. [Google Scholar] [CrossRef]
- Kong, F.; Wang, C.; Zhao, L.; Liao, D.; Wang, X.; Sun, B.; Yang, P.; Jia, Y. Traditional Chinese medicines for non-small cell lung cancer: Therapies and mechanisms. Chin. Herb. Med. 2023, 15, 509–515. [Google Scholar] [CrossRef]
- Zhang, Y.; You, P.; Liu, R.; Lu, Y.; Li, J.; Lei, Y.; Wu, S.; Zhou, H. Artificial Intelligence in Clinical Trials of Lung Cancer: Current and Future Prospects. Intell. Oncol. 2025, 1, 34–51. [Google Scholar] [CrossRef]
- Zhou, D.; Chang, W.; Qi, J.; Chen, G.; Li, N. Lung protective effects of dietary malate esters derivatives from Bletilla striata against SiO2 nanoparticles through activation of Nrf2 pathway. Chin. Herb. Med. 2023, 15, 76–85. [Google Scholar] [CrossRef]
- Yang, L.; Wang, Z. Natural products, alone or in combination with FDA-approved drugs, to treat COVID-19 and lung cancer. Biomedicines 2021, 9, 689. [Google Scholar] [CrossRef]
- Liu, Y.C.; Yang, J.Y.; Zhou, X.C.; Qian, X.; Chen, Z.; Sun, M.Y.; Tong, Y.B.; Zhang, A.Q. Exploring the mechanism of action of bei shashen-maidong in the treatment of non-small cell lung cancer based on network pharmacology and experimental validation. TMR Mod. Herb. Med. 2023, 6, 20. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, L.; Li, Y.; Song, S.; Qu, J.; He, R.; Ren, S.; Gong, P. An activatable, carrier-free, triple-combination nanomedicine for ALK/EGFR-mutant non-small cell lung cancer highly permeable targeted chemotherapy. New J. Chem. 2022, 46, 17673–17677. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y.; Lai, Y.; Xu, W.; Lei, S.; Wang, Z. A computer-aided, heterodimer-based “triadic” carrier-free drug delivery platform to mitigate multidrug resistance in lung cancer and enhance efficiency. J. Colloid. Interf. Sci. 2025, 677, 523–540. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, W.; Lei, S.; Lai, Y.; Zhang, Y.; Wang, Y.; Xiang, Z.; Fu, X.; Yang, L. A computer-aided, carrier-free drug delivery system with enhanced cytotoxicity and biocompatibility: A universal model for multifunctional lung cancer therapy. Colloid. Surf. B 2025, 250, 114557. [Google Scholar] [CrossRef]
- Passaro, A.; Mok, T.; Peters, S.; Popat, S.; Ahn, M.J.; De Marinis, F. Recent advances on the role of EGFR tyrosine kinase inhibitors in the management of NSCLC with uncommon, non exon 20 insertions, EGFR mutations. J. Thorac. Oncol. 2021, 16, 764–773. [Google Scholar] [CrossRef]
- Heppner, D.E. Ascertaining a Structural Basis in Drug Discovery and Development. J. Med. Chem. 2025, 68, 4991–4995. [Google Scholar] [CrossRef]
- Beyett, T.S.; To, C.; Heppner, D.E.; Rana, J.K.; Schmoker, A.M.; Jang, J.; Eck, M.J. Molecular basis for cooperative binding and synergy of ATP-site and allosteric EGFR inhibitors. Nat. Commun. 2022, 13, 2530. [Google Scholar] [CrossRef]
- Oh, S.-H.; Khoo, J.-H.; Lim, J.-C.; Lee, S.-R.; Ju, H.; Shin, W.-S.; Park, D.-G.; Park, S.-M.; Hwang, Y.-A. Improved Process for Preparing Lazertinib and Intermediates Thereof. Patent WO2019022485, 31 January 2019. [Google Scholar]
- Cho, B.C.; Lu, S.; Felip, E.; Spira, A.I.; Girard, N.; Lee, J.S.; Hayashi, H. Amivantamab plus lazertinib in previously untreated EGFR-mutated advanced NSCLC. N. Engl. J. Med. 2024, 391, 1486–1498. [Google Scholar] [CrossRef]
- Pallottini, V.; Pfrieger, F.W. Understanding and treating niemann–pick type c disease: Models matter. Int. J. Mol. Sci. 2020, 21, 8979. [Google Scholar] [CrossRef]
- Keam, S.J. Arimoclomol: First Approval. Drugs 2025, 85, 111–116. [Google Scholar] [CrossRef]
- Keppel Hesselink, J.M. Bimoclomol and arimoclomol: HSP-co-inducers for the treatment of protein misfolding disorders, neuropathy and neuropathic pain. J. Pain. Relief 2016, 6, 279. [Google Scholar]
- Atkinson, B.N.; Woodward, H.L.; Sipthorp, J.; Fish, P.V. Regioselective and enantiospecific synthesis of the HSP co-inducer arimoclomol from chiral glycidyl derivatives. Org. Biomol. Chem. 2017, 15, 9794–9799. [Google Scholar] [CrossRef] [PubMed]
- Mengel, E.; Patterson, M.C.; Da Riol, R.M.; Del Toro, M.; Deodato, F.; Gautschi, M.; í Dali, C. Efficacy and safety of arimoclomol in Niemann-Pick disease type C: Results from a double-blind, randomised, placebo-controlled, multinational phase 2/3 trial of a novel treatment. J. Inherit. Metab. Dis. 2021, 44, 1463–1480. [Google Scholar] [CrossRef]
- Beninger, P. Aqneursa (levacetylleucine). Clin. Ther. 2024, 46, 1091–1092. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, L. Broad-spectrum prodrugs with anti-SARS-CoV-2 activities: Strategies, benefits, and challenges. J. Med. Virol. 2022, 94, 1373–1390. [Google Scholar] [CrossRef]
- Zhang, R.; Yao, X.; Li, Q.; Li, X.; Ma, Q.; Huang, W.; Shi, X.; Yang, Y.; Liu, H. Self-assembled nanoparticles of rapamycin prodrugs for the treatment of multiple sclerosis. J. Colloid. Interf. Sci. 2025, 683, 448–459. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, L. Post-acute sequelae of SARS-CoV-2 infection: A neglected public health issue. Front. Public Health 2022, 10, 908757. [Google Scholar] [CrossRef]
- Al Musaimi, O.; AlShaer, D.; de la Torre, B.G.; Albericio, F. 2024 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2025, 18, 291. [Google Scholar] [CrossRef]
- Fetalvero, K.M.; Narayan, S.; O’Neill, D.J.; Saiah, E.; Sengupta, S. Modulators of Sestrin-Gator2 Interaction and Uses Thereof. U.S. Patent 20170114080A1, 16 October 2018. [Google Scholar]
- Bremova-Ertl, T.; Ramaswami, U.; Brands, M.; Foltan, T.; Gautschi, M.; Gissen, P.; Martakis, K. Trial of N-Acetyl-l-Leucine in Niemann–Pick disease type C. N. Engl. J. Med. 2024, 390, 421–431. [Google Scholar] [CrossRef]
- Suchonova, D.; Kéri, P. Rethinking Schizophrenia: Beyond the Voices of Schizophrenia. Eur. Psychiatry 2024, 67, S761–S762. [Google Scholar] [CrossRef]
- Syed, Y.Y. Xanomeline/Trospium Chloride: First Approval. Drugs 2025, 85, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Kaul, I.; Sawchak, S.; Correll, C.U.; Kakar, R.; Breier, A.; Zhu, H.; Brannan, S.K. Efficacy and safety of the muscarinic receptor agonist KarXT (xanomeline–trospium) in schizophrenia (EMERGENT-2) in the USA: Results from a randomised, double-blind, placebo-controlled, flexible-dose phase 3 trial. Lancet 2024, 403, 160–170. [Google Scholar] [CrossRef]
- Blair, H.A. Inavolisib: First Approval. Drugs 2025, 85, 271–278. [Google Scholar] [CrossRef]
- Hanan, E.J.; Braun, M.G.; Heald, R.A.; MacLeod, C.; Chan, C.; Clausen, S.; Edgar, K.A.; Eigenbrot, C.; Elliott, R.; Endres, N.; et al. Discovery of GDC-0077 (inavolisib), a highly selective inhibitor and degrader of mutant PI3Kα. J. Med. Chem. 2022, 65, 16589–16621. [Google Scholar] [CrossRef]
- Kargbo, R.B. Effective Combination Therapies for the Treatment of HER2 Cancer. ACS Med. Chem. Lett. 2023, 14, 231–232. [Google Scholar] [CrossRef]
- Bedard, P.L.; Accordino, M.K.; Cervantes, A.; Gambardella, V.; Hamilton, E.P.; Italiano, A.; Jhaveri, K.L. Long-term safety of inavolisib (GDC-0077) in an ongoing phase 1/1b study evaluating monotherapy and in combination (combo) with palbociclib and/or endocrine therapy in patients (pts) with PIK3CA-mutated, hormone receptor-positive/HER2-negative (HR+/HER2-) metastatic breast cancer (BC). J. Clin. Oncol. 2022, 40, 1052. [Google Scholar]
- Martínez-Gamboa, D.A.; Kaner, J. Revumenib: A new era in acute leukemia treatment. Trends Cancer 2025, 11, 81–83. [Google Scholar] [CrossRef]
- Syed, Y.Y. Revumenib: First Approval. Drugs 2025, 85, 577–583. [Google Scholar] [CrossRef]
- Perner, F.; Stein, E.M.; Wenge, D.V.; Singh, S.; Kim, J.; Apazidis, A.; Cai, S.F. MEN1 mutations mediate clinical resistance to menin inhibition. Nature 2023, 615, 913–919. [Google Scholar] [CrossRef]
- Cacatian, S.; Claremon, D.A.; Dillard, L.W.; Dong, C.; Fan, Y.; Jia, L.; Lotesta, S.D.; Marcus, A.; Morales-Ramos, A.; Singh, S.B.; et al. Preparation of Inhibitors of the Menin-MLL Interaction. Patent WO2017214367Al, 14 December 2017. [Google Scholar]
- Zucenka, A.; Issa, G.C.; Arellano, M.; Khazal, S.; Khera, N.; Stock, W.; Stein, E.M. Revumenib maintenance therapy following revumenib-induced remission and transplant. Blood 2023, 142, 4950. [Google Scholar] [CrossRef]
- Ghallab, M.; Ahmed, M.S.; Ostrow, T.H.; Rasool, M.H.; Alagha, Z.; Miller, D.; Frenkel, D. Landiolol for Treating Arrhythmias: A State-of-The-Art Review. Cardiol. Rev. 2024. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Ståhlberg, J.; Henriksson, H.; Divne, C.; Isaksson, R.; Pettersson, G.; Johansson, G.; Jones, T.A. Structural basis for enantiomer binding and separation of a common β-blocker: Crystal structure of cellobiohydrolase Cel7A with bound (S)-propranolol at 1.9 Å resolution. J. Mol. Biol. 2001, 305, 79–93. [Google Scholar] [CrossRef]
- Iguchi, S.; Kawamura, M.; Miyamoto, T. Novel Esters of Phenylalkanoic Acid. Patent EP0397031A1, 20 July 1994. [Google Scholar]
- Lamb, Y.N.; Deeks, E.D. Tafamidis: A review in transthyretin amyloidosis with polyneuropathy. Drugs 2019, 79, 863–874. [Google Scholar] [CrossRef]
- Sinha, U.; Rao, S. Methods of Treating Ttr Amyloidosis Using Ag10. Patent WO 2019183463 A1, 26 September 2019. [Google Scholar]
- Graef, I.A.; Alhamadsheh, M.M. Compounds and Compositions that Bind and Stabilize Transthyretin and Their Use for Inhibiting Transthyretin Amyloidosis and Protein-Protein Interactions. U.S. Patent 20140179751A1, 27 October 2015. [Google Scholar]
- Lee, A. Acoramidis: First Approval. Drugs 2025. [Google Scholar] [CrossRef]
- Jaschke, N.P. Crinecerfont in Adult Congenital Adrenal Hyperplasia. N. Engl. J. Med. 2024, 391, 1557. [Google Scholar]
- Lee, A. Crinecerfont: First Approval. Drugs 2025. [Google Scholar] [CrossRef]
- Palmer, A.; Stirn, S.; Radisson, J. Synthetic Method for Preparation of 4-(2-chloro-4-methoxy-5-methylphenyl)-n-[(1s)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-n-prop-2-ynyl-1,3-thiazol-2-amine. Patent WO 2021111179 A1, 10 June 2021. [Google Scholar]
- Auchus, R.J.; Hamidi, O.; Pivonello, R.; Bancos, I.; Russo, G.; Witchel, S.F.; Farber, R.H. Phase 3 trial of crinecerfont in adult congenital adrenal hyperplasia. N. Engl. J. Med. 2024, 391, 504–514. [Google Scholar] [CrossRef]
- Besse, B.; Pons-Tostivint, E.; Park, K.; Hartl, S.; Forde, M.; Hochmair, M.J.; Awad, M.M.; Thomas, M.; Goss, G.; Wheatley-Price, P.; et al. Biomarker-directed targeted therapy plus durvalumab in advanced non-small-cell lung cancer: A phase 2 umbrella trial. Nat. Med. 2024, 30, 716–729. [Google Scholar] [CrossRef]
- Tian, Y.; Ma, B.; Yu, S.; Li, Y.; Pei, H.; Tian, S.; Zhao, X.; Liu, C.; Zuo, Z.; Wang, Z. Clinical antitumor application and pharmacological mechanisms of Dahuang Zhechong Pill. Chin. Herb. Med. 2023, 15, 169–180. [Google Scholar] [CrossRef]
- Yang, L.Y.; Lei, S.Z.; Xu, W.J.; Lai, Y.; Zhang, Y.; Wang, Y.; Wang, Z.L. Rising above: Exploring the therapeutic potential of natural product-based compounds in human cancer treatment. Tradit. Med. Res. 2025, 10, 18. [Google Scholar] [CrossRef]
- Gambacorti-Passerini, C.; Orlov, S.; Zhang, L.; Braiteh, F.; Huang, H.; Esaki, T.; Kim, T.M. Long-term effects of crizotinib in ALK-positive tumors (excluding NSCLC): A phase 1b open-label study. Am. J. Hematol. 2018, 93, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Musa, S.; Amara, N.; Selawi, A.; Wang, J.; Marchini, C.; Agbarya, A.; Mahajna, J. Overcoming Chemoresistance in Cancer: The Promise of Crizotinib. Cancers 2024, 16, 2479. [Google Scholar] [CrossRef]
- Yu, Z.Q.; Lv, Y.W.; Yu, C.M.; Su, W.K. Continuous flow reactor for Balz–Schiemann reaction: A new procedure for the preparation of aromatic fluorides. Tetrahedron Lett. 2013, 54, 1261–1263. [Google Scholar] [CrossRef]
- Horn, L.; Wang, Z.; Wu, G.; Poddubskaya, E.; Mok, T.; Reck, M.; Wu, Y.L. Ensartinib vs crizotinib for patients with anaplastic lymphoma kinase− positive non–small cell lung cancer: A randomized clinical trial. JAMA Oncol. 2021, 7, 1617–1625. [Google Scholar] [CrossRef]
- Gryspeert, K.; Dipalo, L.L.; Cunha, A.L.D.S.; Bulcaen, M.; Ensinck, M.M.; Carlon, M.S. Cystic fibrosis at a glance: From disease mechanism to therapy. Trends Mol. Med. 2025, 31, 399–400. [Google Scholar] [CrossRef]
- DeMattei, J.; Looker, A.R.; Neubert-Langille, B.; Trudeau, M.; Roeper, S.; Ryan, M.P.; Yap, D.M.L.; Krueger, B.R.; Grootenhuis, P.D.J.; Van Goor, F.F. Process for Making Modulators of Cystic Fibrosis Transmembrane Conductance Regulator. Patent WO 2010108162A1, 23 September 2010. [Google Scholar]
- Looker, A.R.; Wilde, N.; Ryan, M.P.; Roeper, S.; Ye, Z.; Lewandowski, B.L. Utilizing O-quinone methide chemistry: Synthesis of d 9-ivacaftor. J. Org. Chem. 2019, 85, 501–507. [Google Scholar] [CrossRef]
- Jordan, K.D.; Zemanick, E.T.; Taylor-Cousar, J.L.; Hoppe, J.E. Managing cystic fibrosis in children aged 6-11yrs: A critical review of elexacaftor/tezacaftor/ivacaftor combination therapy. Expert Rev. Respir. Med. 2023, 17, 97–108. [Google Scholar] [CrossRef]
- Xie, H.; Jia, Y.; Liu, S. Integration of artificial intelligence in clinical laboratory medicine: Advancements and challenges. Interdiscip. Med. 2024, 2, e20230056. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, X.; Li, X.; Wang, S.; Zhao, C.; Chen, Z.; You, L.; Jiang, Y.; Lin, H.; Wei, X.; et al. Roadmap for modern development of chinese medicine: Traditional, evidence-based, and digital-intelligent paths. Sci. Tradit. Chin. Med. 2024, 2, 14–19. [Google Scholar] [CrossRef]
- Yang, Y.; Guan, S.; Ou, Z.; Li, W.; Yan, L.; Situ, B. Advances in AI-based cancer cytopathology. Interdiscip. Med. 2023, 1, e20230013. [Google Scholar] [CrossRef]
- Yan, X.; Yue, T.; Winkler, D.A.; Yin, Y.; Zhu, H.; Jiang, G.; Yan, B. Converting nanotoxicity data to information using artificial intelligence and simulation. Chem. Rev. 2023, 123, 8575–8637. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, X.; Chen, G.; Zhang, Y.; Wei, M.; Chen, X.; Li, B.; Wu, Y.; Wu, L. Supramolecular framework membrane for precise sieving of small molecules, nanoparticles and proteins. Nat. Commun. 2023, 14, 975. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xu, X.; Wang, K.; Xue, M.; Wang, Q.; Zhong, W.; Wan, Y.; Liu, X.; Zheng, J.; Gao, G.; et al. Hypoxia-activated liposomes enable synergistic photodynamic therapy for oral cancer. Adv. Heal. Mater. 2025, 14, e2404395. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Approval Date | Brand Name (Company) | Active Ingredient (Chemical Structure) | Clinical Trials (Administration) | Indication (Mechanism) | Possible Side Effects |
|---|---|---|---|---|---|---|
| 1 | 2/22/2024 | Exblifep (Allecra Therapeutics) | 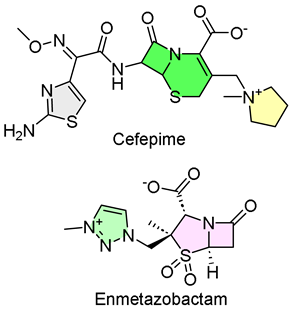 | NCT03687255, composite response was achieved in 273 of 345 patients (intravenous, IV) | Complicated urinary tract infections (cephalosporin and β-lactamase inhibitor) | Allergic reactions, neurological issues, and diarrhea |
| 2 | 3/14/2024 | Rezdiffra (Madrigal Pharmaceuticals) | 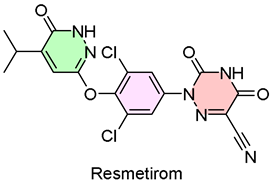 | NCT03900429, resolving NASH and reducing liver scarring (oral administration) | Nonalcoholic steatohepatitis (thyroid hormone receptor β agonist) | Liver toxicity and inflammation of the gallbladder or pancreas |
| 3 | 3/19/2024 | Tryvio (Idorsia Pharmaceuticals) |  | NCT03541174, lower sitting blood pressure (oral administration) | Hypertension (endothelin A and endothelin B receptors; antagonist) | Liver issues, harm to an unborn baby, and reduced sperm counts in males |
| 4 | 3/21/2024 | Duvyzat (Italfarmaco SpA) | 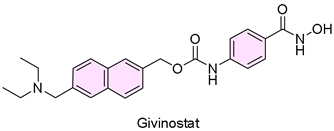 | NCT02851797, experienced less decline in muscle function tests (oral administration) | Duchenne muscular dystrophy (histone deacetylase inhibitor) | Diarrhea, abdominal pain, low platelets, or high triglycerides |
| 5 | 3/27/2024 | Vafseo (Akebia Therapeutics) | 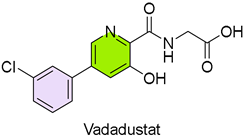 | NCT02865850, NCT02892149, maintaining correct hemoglobin levels (oral administration) | Anemia due to chronic kidney disease (hypoxia-inducible factor prolyl hydroxylase [HIF-PH] inhibitor) | Thrombotic vascular events |
| 6 | 3/29/2024 | Voydeya (Alexion Pharmaceuticals) | 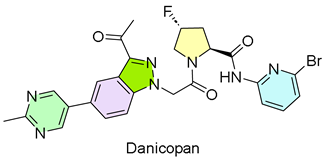 | ALXN2040-PNH-301, Voydeya’s superiority when combined with ravulizumab or eculizumab | Extravascular hemolysis (factor D inhibitor) | Hepatic enzyme increases and hyperlipidemia |
| 7 | 4/3/2024 | Zevtera (Basilea Pharmaceutica) | 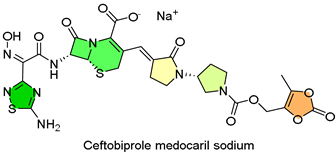 | BPR-CS-009, BPR-CS-008, CAP-3001, BPR-PIP-002, overall success (IV) | Staphylococcus aureus bloodstream infections, ABSSSI and CABP; (cephalosporin antibiotic) | Nausea, headache, diarrhea, and fever |
| 8 | 4/23/2024 | Ojemda (Day One Biopharmaceuticals) |  | NCT04775485/FIREFLY-1, 51% of patients had complete, partial, or minor tumor shrinkage | Pediatric low-grade glioma (Pan-RAF kinase inhibitor) | Bleeding issues, skin reactions, liver issues, or stunted growth |
| 9 | 4/26/2024 | Xolremdi (X4 Pharmaceuticals) | 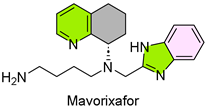 | NCT03995108, more white blood cells and fewer infections | WHIM syndrome (CXCR4 antagonist) | Fetal harm, QTc interval prolongation |
| 10 | 6/10/2024 | Iqirvo (Ipsen biopharm) | 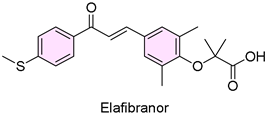 | NCT04526665, more patients achieved biochemical response | Primary biliary cholangitis (PPARα/δ agonist) | Weight gain, diarrhea, and rash |
| 11 | 6/18/2024 | Sofdra (Botanix SB) | 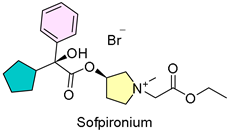 | NCT03836287, NCT03948646, greater reduction in GSP (topical gel) | Primary axillary hyperhidrosis (anti-cholinergic agent) | Dry mouth, blurry vision, mydriasis |
| 12 | 6/26/2024 | Ohtuvayre (Verona Pharma) |  | ENHANCE-1, ENHANCE-2, improved pulmonary function | Chronic obstructive pulmonary disease (phosphodiesterase 3/4 inhibitor) | Back pain, diarrhea, and mental health problems |
| 13 | 7/25/2024 | Leqselvi (Sun Pharma) | 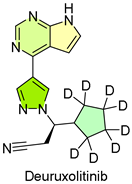 | About 30% of subjects have 80% or more scalp hair | Severe alopecia areata (JAK1 and JAK2 inhibitor) | Serious infections, cardiovascular events, and immune system problems |
| 14 | 8/6/2024 | Voranigo (Servier Pharmaceuticals) | 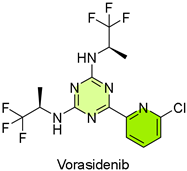 | NCT04164901, improved overall PFS | IDH-mutant grade 2 glioma (IDH1 and IDH2 inhibitor) | Hepatoxicity and embryo-fetal toxicity |
| 15 | 8/14/2024 | Livdelzi (CymaBay Therapeutics) | 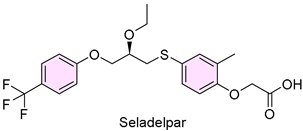 | NCT04620733, more patients achieved biochemical response | Primary biliary cholangitis (peroxisome proliferator-activated receptor-δ (PPARδ) agonist) | Headaches, abdominal pain, and dizziness |
| 16 | 8/19/2024 | Lazcluze (Johnson & Johnson) |  | NCT04487080, median PFS was 23.7 months | Non-small-cell lung cancer (EGFR kinase inhibitor) | Lung, skin, and eye problems; blood clots |
| 17 | 9/20/2024 | Miplyffa (Zevra Therapeutics) |  | Less worsening of symptoms | Niemann–Pick disease type C | Hypersensitivity reactions, diarrhea |
| 18 | 9/24/2024 | Aqneursa (IntraBio) |  | Less worsening of symptoms | Niemann–Pick disease type C | Abdominal pain, dysphagia, and vomiting |
| 19 | 9/26/2024 | Cobenfy (Bristol Myers Squibb) | 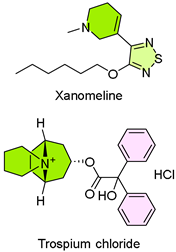 | Meaningful reduction in symptoms | Schizophrenia (M1/M4 mAChR modulator) | Urinary retention, increased heart rate, decreased gastric motility, or facial and lip angioedema |
| 20 | 10/10/2024 | Itovebi (Genentech) |  | NCT04191499, the median progression-free survival (PFS) was 15.0 months | Advanced or metastatic breast cancer (mutant PI3Kα inhibitor and degrader) | Neutropenia, decreased hemoglobin, thrombocytopenia, and lymphocytopenia |
| 21 | 10/25/2024 | Orlynvah (IterumTherapeutics) | 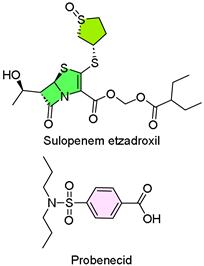 | Effective treatment for Urinary tract infection (UTI) | Uncomplicated urinary tract infection (penicillin antibiotics, tubular transport inhibitor) | Diarrhea, nausea, vulvovaginal candidiasis, headache, and vomiting |
| 22 | 11/15/2024 | Revuforj (Syndax Pharmaceuticals) | 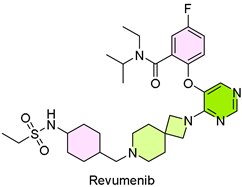 | The complete response (CR) rate combined with partial hematological recovery yielded a CRh rate of 21.2% | Relapsed or refractory acute leukemia (menin inhibitor) | Bleeding, nausea, musculoskeletal pain, infection |
| 23 | 11/22/2024 | Rapiblyk (AOP Health) |  | The patient’s heart rate decreases by 40% to 90% | Supraventricular tachycardia (β1-selective blocker) | Hypertension |
| 24 | 11/22/2024 | Attruby (Pfizer) | 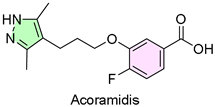 | NCT03860935, NCT04622046, NCT06563895, NCT04988386, reduce cardiovascular deaths and related hospitalizations | Transthyretin stabilizer | Cardiomyopathy |
| 25 | 12/13/2024 | Crenessity (Neurocrine Biosciences) |  | Serum androstenedione was statistically significantly lower | Congenital adrenal hyperplasia (CRF1 Antagonist) | Fatigue, headache, dizziness, joint pain, back pain, and myalgia |
| 26 | 12/18/2024 | Ensacove (Xcovery Holdings) | 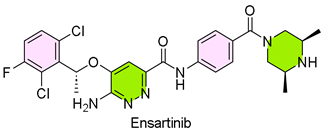 | NCT02767804, the objective response rate (ORR) was 75%; 59% of patients sustained remission for 36 months or longer | Non-small cell lung cancer (ALK/MET inhibitor) | Rash, constipation, cough, pruritus, nausea, edema, fever, and fatigue |
| 27 | 12/20/2024 | Alyftrek (Vertex Pharmaceuticals) |  | NCT05033080, NCT05076149, NCT05422222, better than Trikafta | Cystic fibrosis (CFTR, proofreader, and potentiator) | Cough, nasopharyngitis, upper respiratory tract infection, headache, oropharyngeal pain, influenza, fatigue, and rash |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Sun, X.; Sun, M.; Wang, C.; Yang, L. Game Changers: Blockbuster Small-Molecule Drugs Approved by the FDA in 2024. Pharmaceuticals 2025, 18, 729. https://doi.org/10.3390/ph18050729
Wang Z, Sun X, Sun M, Wang C, Yang L. Game Changers: Blockbuster Small-Molecule Drugs Approved by the FDA in 2024. Pharmaceuticals. 2025; 18(5):729. https://doi.org/10.3390/ph18050729
Chicago/Turabian StyleWang, Zhonglei, Xin Sun, Mingyu Sun, Chao Wang, and Liyan Yang. 2025. "Game Changers: Blockbuster Small-Molecule Drugs Approved by the FDA in 2024" Pharmaceuticals 18, no. 5: 729. https://doi.org/10.3390/ph18050729
APA StyleWang, Z., Sun, X., Sun, M., Wang, C., & Yang, L. (2025). Game Changers: Blockbuster Small-Molecule Drugs Approved by the FDA in 2024. Pharmaceuticals, 18(5), 729. https://doi.org/10.3390/ph18050729