


Identification of Potential Inhibitors Targeting Non-Structural Proteins NS3 and NS5 of Dengue Virus Using Docking and Deep Learning Approaches

 ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Virtual Screening and Molecular Docking
2.2. Re-Screening Through ML
2.3. ADME/Toxicity Analysis
2.4. Molecular Dynamics Simulation
3. Materials and Methods
3.1. Retrieval and Preparation of Proteins
3.2. Preparation of Ligand
3.3. Structure-Based Virtual Screening
3.4. Re-Screening Through Machine Learning (ML)
3.5. ADME/Tox Analysis of Phytochemicals
3.6. Computational Dynamics Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Vasilakis, N.; Cardosa, J.; Hanley, K.A.; Holmes, E.C.; Weaver, S.C. Fever from the forest: Prospects for the continued emergence of sylvatic dengue virus and its impact on public health. Nat. Rev. Microbiol. 2011, 9, 532–541. [Google Scholar] [CrossRef]
- Gould, E.A.; Solomon, T. Pathogenic flaviviruses. Lancet 2008, 371, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Waterman, S.H.; Gubler, D.J. Dengue fever. Clin. Dermatol. 1989, 7, 117–122. [Google Scholar] [CrossRef]
- Ali, F.; Chorsiya, A.; Anjum, V.; Khasimbi, S.; Ali, A. A systematic review on phytochemicals for the treatment of dengue. Phytother. Res. 2021, 35, 1782–1816. [Google Scholar] [CrossRef] [PubMed]
- WHO. Dengue: Guidelines for Diagnosis, Treatment. Prevention and Control; World Health Organization: Geneva, Switzerland, 2009. [Google Scholar]
- Tahir ul Qamar, M.; Maryam, A.; Muneer, I.; Xing, F.; Ashfaq, U.A.; Khan, F.A.; Anwar, F.; Geesi, M.H.; Khalid, R.R.; Rauf, S.A. Computational screening of medicinal plant phytochemicals to discover potent pan-serotype inhibitors against dengue virus. Sci. Rep. 2019, 9, 1433. [Google Scholar] [CrossRef]
- Guzman, M.G.; Gubler, D.J.; Izquierdo, A.; Martinez, E.; Halstead, S.B. Dengue infection. Nat. Rev. Dis. Primers 2016, 2, 16055. [Google Scholar] [CrossRef] [PubMed]
- Norshidah, H.; Leow, C.H.; Ezleen, K.E.; Wahab, H.A.; Vignesh, R.; Rasul, A.; Lai, N.S. Assessing the potential of NS2B/NS3 protease inhibitors biomarker in curbing dengue virus infections: In silico vs. In vitro approach. Front. Cell. Infect. Microbiol. 2023, 13, 1061937. [Google Scholar] [CrossRef]
- Silva, E.M.; Conde, J.N.; Allonso, D.; Ventura, G.T.; Coelho, D.R.; Carneiro, P.H.; Silva, M.L.; Paes, M.V.; Rabelo, K.; Weissmuller, G. Dengue virus nonstructural 3 protein interacts directly with human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and reduces its glycolytic activity. Sci. Rep. 2019, 9, 2651. [Google Scholar] [CrossRef]
- Gan, S.J.; Leong, Y.Q.; bin Barhanuddin, M.F.H.; Wong, S.T.; Wong, S.F.; Mak, J.W.; Ahmad, R.B. Dengue fever and insecticide resistance in Aedes mosquitoes in Southeast Asia: A review. Parasites Vectors 2021, 14, 315. [Google Scholar] [CrossRef]
- Ullah, A.; Gong, P.; Khan, A.M.; Choudhary, M.I. Identification of new inhibitors of NS5 from dengue virus using saturation transfer difference (STD-NMR) and molecular docking studies. RSC Adv. 2023, 13, 355–369. [Google Scholar] [CrossRef]
- Darwish, N.T.; Sekaran, S.D.; Khor, S.M. Point-of-care tests: A review of advances in the emerging diagnostic tools for dengue virus infection. Sens. Actuators B Chem. 2018, 255, 3316–3331. [Google Scholar] [CrossRef]
- Henchal, E.A.; Putnak, J.R. The dengue viruses. Clin. Microbiol. Rev. 1990, 3, 376–396. [Google Scholar] [CrossRef]
- Kautner, I.; Robinson, M.J.; Kuhnle, U. Dengue virus infection: Epidemiology, pathogenesis, clinical presentation, diagnosis, and prevention. J. Pediatr. 1997, 131, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, S. NS3 protease from flavivirus as a target for designing antiviral inhibitors against dengue virus. Genet. Mol. Biol. 2010, 33, 214–219. [Google Scholar] [CrossRef]
- Pujar, G.V.; Sethu, A.K.; Bhagyalalitha, M.; Singh, M. Dengue structural proteins as antiviral drug targets: Current status in the drug discovery & development. Eur. J. Med. Chem. 2021, 221, 113527. [Google Scholar]
- Teramoto, T. Dengue virus serotypic replacement of NS3 protease or helicase domain causes chimeric viral attenuation but can be recovered by a compensated mutation at helicase domain or NS2B, respectively. J. Virol. 2023, 97, e0085423. [Google Scholar] [CrossRef]
- Mirza, S.B.; Lee, R.C.H.; Chu, J.J.H.; Salmas, R.E.; Mavromoustakos, T.; Durdagi, S. Discovery of selective dengue virus inhibitors using combination of molecular fingerprint-based virtual screening protocols, structure-based pharmacophore model development, molecular dynamics simulations and in vitro studies. J. Mol. Graph. Model. 2018, 79, 88–102. [Google Scholar] [CrossRef]
- Ul Qamar, M.T.; Mumtaz, A.; Ashfaq, U.A.; Adeel, M.M.; Fatima, T. Potential of plant alkaloids as dengue ns3 protease inhibitors: Molecular docking and simulation approach. Bangladesh J. Pharmacol. 2014, 9, 262–267. [Google Scholar] [CrossRef]
- Ul Qamar, T.; Mumtaz, A.; Ashfaq, U.A.; Azhar, S.; Fatima, T.; Hassan, M.; Hussain, S.S.; Akram, W.; Idrees, S. Computer aided screening of phytochemicals from Garcinia against the dengue NS2B/NS3 protease. Bioinformation 2014, 10, 115. [Google Scholar] [CrossRef]
- Erbel, P.; Schiering, N.; D’Arcy, A.; Renatus, M.; Kroemer, M.; Lim, S.P.; Yin, Z.; Keller, T.H.; Vasudevan, S.G.; Hommel, U. Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat. Struct. Mol. Biol. 2006, 13, 372–373. [Google Scholar] [CrossRef]
- Gebhard, L.G.; Filomatori, C.V.; Gamarnik, A.V. Functional RNA elements in the dengue virus genome. Viruses 2011, 3, 1739–1756. [Google Scholar] [CrossRef]
- Davidson, A.D. New insights into flavivirus nonstructural protein 5. Adv. Virus Res. 2009, 74, 41–101. [Google Scholar] [PubMed]
- Issur, M.; Geiss, B.J.; Bougie, I.; Picard-Jean, F.; Despins, S.; Mayette, J.; Hobdey, S.E.; Bisaillon, M. The flavivirus NS5 protein is a true RNA guanylyltransferase that catalyzes a two-step reaction to form the RNA cap structure. Rna 2009, 15, 2340–2350. [Google Scholar] [CrossRef] [PubMed]
- Henderson, B.R.; Saeedi, B.J.; Campagnola, G.; Geiss, B.J. Analysis of RNA binding by the dengue virus NS5 RNA capping enzyme. PLoS ONE 2011, 6, e25795. [Google Scholar] [CrossRef] [PubMed]
- Coutard, B.; Decroly, E.; Li, C.; Sharff, A.; Lescar, J.; Bricogne, G.; Barral, K. Assessment of Dengue virus helicase and methyltransferase as targets for fragment-based drug discovery. Antivir. Res. 2014, 106, 61–70. [Google Scholar] [CrossRef]
- Liu, L.; Dong, H.; Chen, H.; Zhang, J.; Ling, H.; Li, Z.; Shi, P.-Y.; Li, H. Flavivirus RNA cap methyltransferase: Structure, function, and inhibition. Front. Biol. 2010, 5, 286–303. [Google Scholar] [CrossRef]
- Zhou, Y.; Ray, D.; Zhao, Y.; Dong, H.; Ren, S.; Li, Z.; Guo, Y.; Bernard, K.A.; Shi, P.-Y.; Li, H. Structure and function of flavivirus NS5 methyltransferase. J. Virol. 2007, 81, 3891–3903. [Google Scholar] [CrossRef]
- Barral, K.; Sallamand, C.; Petzold, C.; Coutard, B.; Collet, A.; Thillier, Y.; Zimmermann, J.; Vasseur, J.J.; Canard, B.; Rohayem, J. Development of specific dengue virus 2′-O-and N7-methyltransferase assays for antiviral drug screening. Antivir. Res. 2013, 99, 292–300. [Google Scholar] [CrossRef]
- Tay, M.Y.F.; Fraser, J.E.; Chan, W.K.K.; Moreland, N.J.; Rathore, A.P.; Wang, C.; Vasudevan, S.G.; Jans, D.A. Nuclear localization of dengue virus (DENV) 1–4 non-structural protein 5; protection against all 4 DENV serotypes by the inhibitor Ivermectin. Antivir. Res. 2013, 99, 301–306. [Google Scholar] [CrossRef]
- Lim, S.P.; Noble, C.G.; Shi, P.-Y. The dengue virus NS5 protein as a target for drug discovery. Antivir. Res. 2015, 119, 57–67. [Google Scholar] [CrossRef]
- Celegato, M.; Sturlese, M.; Vasconcelos Costa, V.; Trevisan, M.; Lallo Dias, A.S.; Souza Passos, I.B.; Queiroz-Junior, C.M.; Messa, L.; Favaro, A.; Moro, S.; et al. Small-Molecule Inhibitor of Flaviviral NS3-NS5 Interaction with Broad-Spectrum Activity and Efficacy In Vivo. mBio 2023, 14, e0309722. [Google Scholar] [CrossRef]
- Shimizu, H.; Saito, A.; Mikuni, J.; Nakayama, E.E.; Koyama, H.; Honma, T.; Shirouzu, M.; Sekine, S.-i.; Shioda, T. Discovery of a small molecule inhibitor targeting dengue virus NS5 RNA-dependent RNA polymerase. PLoS Neglected Trop. Dis. 2019, 13, e0007894. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Lee, L.K.; Lye, D.C.; Leo, Y.S. Current management of severe dengue infection. Expert Rev. Anti-Infect. Ther. 2017, 15, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Sivaraman, D.; Pradeep, P.S. Exploration of bioflavonoids targeting dengue virus NS5 RNA-dependent RNA polymerase: In silico molecular docking approach. J. Appl. Pharm. Sci. 2020, 10, 016–022. [Google Scholar]
- Lee, S.H.; Tang, Y.Q. Effects of Cocktail of four local Malaysian Medicinal Plants (Phyllanthus spp.) against Dengue Virus 2 [Cited 2015]. BMC Complement. Altern. Med. 2013, 13, 192. Available online: http://www.biomedcentral.com/content/pdf/1472-6882-13-192.pdf (accessed on 25 July 2024). [CrossRef]
- Hossain, A.; Rahman, M.E.; Rahman, M.S.; Nasirujjaman, K.; Matin, M.N.; Faruqe, M.O.; Rabbee, M.F. Identification of medicinal plant-based phytochemicals as a potential inhibitor for SARS-CoV-2 main protease (Mpro) using molecular docking and deep learning methods. Comput. Biol. Med. 2023, 157, 106785. [Google Scholar] [CrossRef] [PubMed]
- Earlia, N.; Muslem Suhendra, R.; Amin, M.; Prakoeswa, C.R.S.; Khairan; Idroes, R. GC/MS Analysis of Fatty Acids on Pliek U Oil and Its Pharmacological Study by Molecular Docking to Filaggrin as a Drug Candidate in Atopic Dermatitis Treatment. Sci. World J. 2019, 2019, 8605743. [Google Scholar] [CrossRef]
- Earlia, N.; Rahmad, R.; Amin, M.; Prakoeswa, C.R.S.; Khairan, K.; Idroes, R. The potential effect of fatty acids from Pliek U on epidermal fatty acid binding protein: Chromatography and bioinformatic studies. Sains Malays. 2019, 48, 1019–1024. [Google Scholar] [CrossRef]
- Nouroz, F.; Mehboob, M.; Mobin, T.; Khan, S. In silico exploitation of antiviral phytochemicals against dengue. Pak. J. Bot. 2021, 53, 309–319. [Google Scholar] [CrossRef]
- Xue, Q.; Liu, X.; Russell, P.; Li, J.; Pan, W.; Fu, J.; Zhang, A. Evaluation of the binding performance of flavonoids to estrogen receptor alpha by Autodock, Autodock Vina and Surflex-Dock. Ecotoxicol. Environ. Saf. 2022, 233, 113323. [Google Scholar] [CrossRef]
- Shannon, A.E.; Pedroso, M.M.; Chappell, K.J.; Watterson, D.; Liebscher, S.; Kok, W.M.; Fairlie, D.P.; Schenk, G.; Young, P.R. Product release is rate-limiting for catalytic processing by the Dengue virus protease. Sci. Rep. 2016, 6, 37539. [Google Scholar] [CrossRef]
- Bazan, J.F.; Fletterick, R.J. Detection of a trypsin-like serine protease domain in flaviviruses and pestviruses. Virology 1989, 171, 637–639. [Google Scholar] [CrossRef] [PubMed]
- Valle Rosaura, P.C.; Falgout, B. Mutagenesis of the NS3 Protease of Dengue Virus Type 2. J. Virol. 1998, 72, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Matusan Anita, E.; Pryor Melinda, J.; Davidson Andrew, D.; Wright Peter, J. Mutagenesis of the Dengue Virus Type 2 NS3 Protein within and outside Helicase Motifs: Effects on Enzyme Activity and Virus Replication. J. Virol. 2001, 75, 9633–9643. [Google Scholar] [CrossRef] [PubMed]
- Niyomrattanakit, P.; Winoyanuwattikun, P.; Chanprapaph, S.; Angsuthanasombat, C.; Panyim, S.; Katzenmeier, G. Identification of Residues in the Dengue Virus Type 2 NS2B Cofactor That Are Critical for NS3 Protease Activation. J. Virol. 2004, 78, 13708–13716. [Google Scholar] [CrossRef]
- Lescar, J.; Luo, D.; Xu, T.; Sampath, A.; Lim, S.P.; Canard, B.; Vasudevan, S.G. Towards the design of antiviral inhibitors against flaviviruses: The case for the multifunctional NS3 protein from Dengue virus as a target. Antivir. Res. 2008, 80, 94–101. [Google Scholar] [CrossRef]
- Salaemae, W.; Junaid, M.; Angsuthanasombat, C.; Katzenmeier, G. Structure-guided mutagenesis of active site residues in the dengue virus two-component protease NS2B-NS3. J. Biomed. Sci. 2010, 17, 68. [Google Scholar] [CrossRef]
- Walters, W.P.; Barzilay, R. Applications of Deep Learning in Molecule Generation and Molecular Property Prediction. Acc. Chem. Res. 2021, 54, 263–270. [Google Scholar] [CrossRef]
- Kim, J.; Park, S.; Min, D.; Kim, W. Comprehensive Survey of Recent Drug Discovery Using Deep Learning. Int. J. Mol. Sci. 2021, 22, 9983. [Google Scholar] [CrossRef]
- Néstor, M.; Tania, C.-R.; Ophir, M.; James, G.-P. A machine learning method for predicting the IC50 values of novel designed analogs of Non-Nucleoside Reverse-Transcriptase Inhibitors (NNRTIs) as potentially safer drugs. Inform. Med. Unlocked 2024, 48, 101512. [Google Scholar] [CrossRef]
- Ursu, O.; Rayan, A.; Goldblum, A.; Oprea, T.I. Understanding drug-likeness. WIREs Comput. Mol. Sci. 2011, 1, 760–781. [Google Scholar] [CrossRef]
- Wang, Y.; Xing, J.; Xu, Y.; Zhou, N.; Peng, J.; Xiong, Z.; Liu, X.; Luo, X.; Luo, C.; Chen, K.; et al. In silico ADME/T modelling for rational drug design. Q. Rev. Biophys. 2015, 48, 488–515. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Biswas, S.; Islam, K.J.; Paul, A.S.; Mahato, S.K.; Ali, M.A.; Halim, M.A. Antiviral phytochemicals as potent inhibitors against NS3 protease of dengue virus. Comput. Biol. Med. 2021, 134, 104492. [Google Scholar] [CrossRef]
- Shimu, M.S.; Mahmud, S.; Tallei, T.E.; Sami, S.A.; Adam, A.A.; Acharjee, U.K.; Paul, G.K.; Emran, T.B.; Zaman, S.; Uddin, M.S.; et al. Phytochemical Compound Screening to Identify Novel Small Molecules against Dengue Virus: A Docking and Dynamics Study. Molecules 2022, 27, 653. [Google Scholar] [CrossRef] [PubMed]
- Hossain, A.; Rahman, M.E.; Faruqe, M.O.; Saif, A.; Suhi, S.; Zaman, R.; Hirad, A.H.; Matin, M.N.; Rabbee, M.F.; Baek, K.-H. Characterization of Plant-Derived Natural Inhibitors of Dipeptidyl Peptidase-4 as Potential Antidiabetic Agents: A Computational Study. Pharmaceutics 2024, 16, 483. [Google Scholar] [CrossRef]
- Fouzia, I.; Anam, T.; Aqsa, S.; Raheem, U.; Kayode, R.; Muhammad, M.; Sadia, M.; Wasim, A.; Mazhar, I.; Moazur, R. Inhibition of NS2B-NS3 protease from all four serotypes of dengue virus by punicalagin, punicalin and ellagic acid identified from Punica granatum. J. Biomol. Struct. Dyn. 2024, 1–16. [Google Scholar] [CrossRef]
- Bupesh, G.; Raja, R.S.; Saravanamurali, K.; Kumar, V.S.; Saran, N.; Kumar, M.; Vennila, S.; Sheriff, K.; Kaveri, K.; Gunasekaran, P. Antiviral activity of Ellagic Acid against envelope proteins from Dengue Virus through Insilico Docking. Int. J. Drug Dev. Res. 2014, 6, 205–210. [Google Scholar]
- Umar, A.K.; Zothantluanga, J.H.; Aswin, K.; Maulana, S.; Sulaiman Zubair, M.; Lalhlenmawia, H.; Rudrapal, M.; Chetia, D. Antiviral phytocompounds “ellagic acid” and “(+)-sesamin” of Bridelia retusa identified as potential inhibitors of SARS-CoV-2 3CL pro using extensive molecular docking, molecular dynamics simulation studies, binding free energy calculations, and bioactivity prediction. Struct. Chem. 2022, 33, 1445–1465. [Google Scholar]
- Watroly, M.N.; Sekar, M.; Fuloria, S.; Gan, S.H.; Jeyabalan, S.; Wu, Y.S.; Subramaniyan, V.; Sathasivam, K.V.; Ravi, S.; Mat Rani, N.N.I.; et al. Chemistry, Biosynthesis, Physicochemical and Biological Properties of Rubiadin: A Promising Natural Anthraquinone for New Drug Discovery and Development. Drug Des. Dev. Ther. 2021, 15, 4527–4549. [Google Scholar] [CrossRef]
- Che, S.; Zhou, N.; Liu, Y.; Xie, J.; Liu, E. Andrographolide exerts anti-respiratory syncytial virus activity by up-regulating heme oxygenase-1 independent of interferon responses in human airway epithelial cells. Mol. Biol. Rep. 2023, 50, 4261–4272. [Google Scholar] [CrossRef]
- Panraksa, P.; Ramphan, S.; Khongwichit, S.; Smith, D.R. Activity of andrographolide against dengue virus. Antivir. Res. 2017, 139, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, R.; Muthumanickam, S.; Anuradha, V.; Pandi, B.; Ramachandran, B. An Investigation of Fucoidan as a Potential Inhibitor Against DENV/NS3 Proteases Through Molecular Dynamics Simulations and DFT Studies; Research Square: Durham, NC, USA, 2022. [Google Scholar]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Murugesan, S.; Kottekad, S.; Crasta, I.; Sreevathsan, S.; Usharani, D.; Perumal, M.K.; Mudliar, S.N. Targeting COVID-19 (SARS-CoV-2) main protease through active phytocompounds of ayurvedic medicinal plants–Emblica officinalis (Amla), Phyllanthus niruri Linn. (Bhumi Amla) and Tinospora cordifolia (Giloy)–A molecular docking and simulation study. Comput. Biol. Med. 2021, 136, 104683. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Mehta, V.; Raj, U.; Varadwaj, P.K.; Udayabanu, M.; Yennamalli, R.M.; Singh, T.R. Computational and in-vitro validation of natural molecules as potential acetylcholinesterase inhibitors and neuroprotective agents. Curr. Alzheimer Res. 2019, 16, 116–127. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. In Chemical Biology: Methods and Protocols; Hempel, J.E., Williams, C.H., Hong, C.C., Eds.; Springer: New York, NY, USA, 2015; pp. 243–250. [Google Scholar]
- Mustafa, N.F.; Cheng, K.K.; Razali, S.A.; Nadri, M.H. Molecular docking and pharmacokinetics analysis of phytochemicals from Piper caninum as dengue NS2B-NS3 protease inhibitors: Evaluation of phytochemicals as inhibitor for NS2B-NS3. J. Trop. Life Sci. 2024, 14, 131–142. [Google Scholar] [CrossRef]
- Purohit, P.; Sahoo, S.; Panda, M.; Sahoo, P.S.; Meher, B.R. Targeting the DENV NS2B-NS3 protease with active antiviral phytocompounds: Structure-based virtual screening, molecular docking and molecular dynamics simulation studies. J. Mol. Model. 2022, 28, 365. [Google Scholar] [CrossRef]
- Mukhtar, M.; Khan, H.A. Exploring the inhibitory potential of Nigella sativa against dengue virus NS2B/NS3 protease and NS5 polymerase using computational approaches. RSC Adv. 2023, 13, 18306–18322. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Du, J.; Fang, J.; Yin, Y.; Xu, G.; Xie, L. DeepScreening: A deep learning-based screening web server for accelerating drug discovery. Database 2019, 2019, baz104. [Google Scholar] [CrossRef]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Fu, T.-T.; Tu, G.; Ping, M.; Zheng, G.-X.; Yang, F.-Y.; Yang, J.-Y.; Zhang, Y.; Yao, X.-J.; Xue, W.-W.; Zhu, F. Subtype-selective mechanisms of negative allosteric modulators binding to group I metabotropic glutamate receptors. Acta Pharmacol. Sin. 2021, 42, 1354–1367. [Google Scholar] [CrossRef]
- Xue, W.; Wang, P.; Tu, G.; Yang, F.; Zheng, G.; Li, X.; Li, X.; Chen, Y.; Yao, X.; Zhu, F. Computational identification of the binding mechanism of a triple reuptake inhibitor amitifadine for the treatment of major depressive disorder. Phys. Chem. Chem. Phys. 2018, 20, 6606–6616. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham Iii, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Dutta, M.; Tareq, A.M.; Rakib, A.; Mahmud, S.; Sami, S.A.; Mallick, J.; Islam, M.N.; Majumder, M.; Uddin, M.Z.; Alsubaie, A.; et al. Phytochemicals from Leucas zeylanica Targeting Main Protease of SARS-CoV-2: Chemical Profiles, Molecular Docking, and Molecular Dynamics Simulations. Biology 2021, 10, 789. [Google Scholar] [CrossRef]
- Mahmud, S.; Parves, M.R.; Riza, Y.M.; Sujon, K.M.; Ray, S.; Tithi, F.A.; Zaoti, Z.F.; Alam, S.; Absar, N. Exploring the potent inhibitors and binding modes of phospholipase A2 through in silico investigation. J. Biomol. Struct. Dyn. 2020, 38, 4221–4231. [Google Scholar] [CrossRef]
- Khan, M.A.; Mahmud, S.; Alam, A.R.U.; Rahman, M.E.; Ahmed, F.; Rahmatullah, M. Comparative molecular investigation of the potential inhibitors against SARS-CoV-2 main protease: A molecular docking study. J. Biomol. Struct. Dyn. 2021, 39, 6317–6323. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.J.; Parves, M.R.; Mahmud, S.; Tithi, F.A.; Reza, M.A. Assessment of structurally and functionally high-risk nsSNPs impacts on human bone morphogenetic protein receptor type IA (BMPR1A) by computational approach. Comput. Biol. Chem. 2019, 80, 31–45. [Google Scholar] [CrossRef]
- Margret, I.U.J.; Nnaemeka, U.S. Identification, Quantification of Phytochemicals and Elemental Analysis from Ethanolic Leaf Extract of Andrographis paniculate. S. Asian Res. J. Nat. Prod. 2023, 6, 144–156. [Google Scholar]
- Kalaivani, C.S.; Sathish, S.S.; Janakiraman, N.; Johnson, M. GC-MS studies on Andrographis paniculata (Burm.f.) Wall. ex Nees—A medicinally important plant. Int. J. Med. Arom. Plants 2012, 2, 69–74. [Google Scholar]
- Singh, P.; Bhat, S.S.; Punnapuzha, A.; Bhagavatula, A.; Venkanna, B.U.; Mohamed, R.; Rao, R.P. Effect of Key Phytochemicals from Andrographis paniculata, Tinospora cordifolia, and Ocimum sanctum on PLpro-ISG15 De-Conjugation Machinery—A Computational Approach. Computation 2022, 10, 109. [Google Scholar] [CrossRef]
- Owoade, A.O. Phytochemical Characterization and Antioxidant Bioactivity of Andrographis paniculata (Nees). Pan Afr. J. Life Sci. 2021, 5, 246–256. [Google Scholar] [CrossRef]
- Dulara; Kumar, B.; Godara, P.; Barwer, N. In-Vivo and In-Vitro Phytochemical GC-MS Analysis of Volatile Constituents of Andrographis paniculata (Burm. f.) Nees. The Pharma Innovation. 18 April 2019. Available online: https://www.thepharmajournal.com/archives/2019/vol8issue5/PartD/8-10-11-667.pdf (accessed on 1 February 2025).
- Ghosh, B.K.; Datta, A.K.; Mandal, A.; Dubey, P.K.; Halder, S. AN OVERVIEW ON ANDROGRAPHIS PANICULATA (BURM. F.) NEES. Int. J. Res. Ayurveda Pharm. 2012, 3, 752–760. [Google Scholar] [CrossRef]
- Al-Khayri, J.M.; Dubey, S.; Thirumoorthy, G.; Nagella, P.; Rezk, A.A.-S.; Shehata, W.F. In Silico Identification of 1-DTP Inhibitors of Corynebacterium diphtheriae Using Phytochemicals from Andrographis paniculata. Molecules 2023, 28, 909. [Google Scholar] [CrossRef]
- Ali, S.K.; Makeen, H.A.; Khuwaja, G.; Alhazmi, H.A.; Sharma, M.; Koty, A.; Mazahirul, I.; Parveen, H.; Mohammed, A.; Mukhtar, S.; et al. Assessment of the Phytochemical Profile, Antioxidant Capacity, and Hepatoprotective Effect of Andrographis paniculata against CCl4-Induced Liver Dysfunction in Wistar Albino Rats. Medicina 2023, 59, 1260. [Google Scholar] [CrossRef] [PubMed]
- Akbar, M.; Amin, A.; Khalil, T.; Iqbal, M.S.; Nazir, A.; Taswar, A. Antibacterial activity of Alternanthera philoxeroides (Mart.) Griseb. against bacterial phytopathogens: Erwinia carotovora, Ralstonia solanacearum and Xanthomonas axonopodis. Allelopath. J. 2021, 53, 83–92. [Google Scholar] [CrossRef]
- Nahar, L.; Nath, S.; Sarker, S.D. “Malancha” [Alternanthera philoxeroides (Mart.) Griseb.]: A Potential Therapeutic Option against Viral Diseases. Biomolecules 2022, 12, 582. [Google Scholar] [CrossRef]
- Kong, Y.R.; Jong, Y.X.; Balakrishnan, M.; Bok, Z.K.; Weng, J.K.K.; Tay, K.C.; Goh, B.H.; Ong, Y.S.; Chan, K.G.; Lee, L.H.; et al. Beneficial Role of Carica papaya Extracts and Phytochemicals on Oxidative Stress and Related Diseases: A Mini Review. Biology 2021, 10, 287. [Google Scholar] [CrossRef]
- Akhila, S.; Vijayalakshmi, N.G. Phytochemical studies on carica papaya leaf juice. Int. J. Pharm. Sci. Res. 2015, 6, 880. [Google Scholar] [CrossRef]
- Sharma, A.; Bachheti, A.; Sharma, P.; Bachheti, R.K.; Husen, A. Phytochemistry, pharmacological activities, nanoparticle fabrication, commercial products and waste utilization of Carica papaya L.: A comprehensive review. Curr. Res. Biotechnol. 2020, 2, 145–160. [Google Scholar] [CrossRef]
- Oyelere, S.F.; Ajayi, O.H.; Ayoade, T.E.; Pereira, G.B.S.; Owoyemi, B.C.D.; Ilesanmi, A.O.; Akinyemi, O.A. A detailed review on the phytochemical profiles and anti-diabetic mechanisms of Momordica charantia. Heliyon 2022, 8, e09253. [Google Scholar] [CrossRef]
- Mozaniel, S.d.O.; Wanessa, A.d.C.; Fernanda, W.F.B.; Marilena, E.A.; Gracialda, C.F.; Raul, N.d.C.J. Phytochemical profile and biological activities of Momordica charantia L. (Cucurbitaceae): A review. Afr. J. Biotechnol. 2018, 17, 829–846. [Google Scholar] [CrossRef]
- Mukherjee, S.; Karati, D. Exploring the phytochemistry, pharmacognostic properties, and pharmacological activities of medically important plant Momordica charantia. Pharmacol. Res. Mod. Chin. Med. 2023, 6, 100226. [Google Scholar] [CrossRef]
- Criste, A.; Urcan, A.C.; Bunea, A.; Furtuna, F.R.P.; Olah, N.K.; Madden, R.H.; Corcionivoschi, N. Phytochemical Composition and Biological Activity of Berries and Leaves from Four Romanian Sea Buckthorn (Hippophae rhamnoides L.) Varieties. Molecules 2020, 25, 1170. [Google Scholar] [CrossRef] [PubMed]
- He, N.; Wang, Q.; Huang, H.; Chen, J.; Wu, G.; Zhu, M.; Shao, F.; Yan, Z.; Sang, Z.; Cao, L.; et al. A Comprehensive Review on Extraction, Structure, Detection, Bioactivity, and Metabolism of Flavonoids from Sea Buckthorn (Hippophae rhamnoides L.). J. Food Biochem. 2023, 2023, 4839124. [Google Scholar] [CrossRef]
- Dienaitė, L.; Pukalskas, A.; Pukalskienė, M.; Pereira, C.V.; Matias, A.A.; Venskutonis, P.R. Phytochemical Composition, Antioxidant and Antiproliferative Activities of Defatted Sea Buckthorn (Hippophaë rhamnoides L.) Berry Pomace Fractions Consecutively Recovered by Pressurized Ethanol and Water. Antioxidants 2020, 9, 274. [Google Scholar] [CrossRef]
- Cayona, R.; Creencia, E. Phytochemicals of Euphorbia hirta L. and Their Inhibitory Potential Against SARS-CoV-2 Main Protease. Front. Mol. Biosci. 2022, 8, 801401. [Google Scholar] [CrossRef]
- Mardanarian, R.A.; Melyani, P.I.; Ronny, L.; Jutti, L. Bioactive compounds of Boesenbergia sp. and their anti-inflammatory mechanism: A review. J. Appl. Pharm. Sci. 2020, 10, 116–126. [Google Scholar] [CrossRef]
- Gurung, A.B.; Ali, M.A.; Al-Hemaid, F.; El-Zaidy, M.; Lee, J. In silico analyses of major active constituents of fingerroot (Boesenbergia rotunda) unveils inhibitory activities against SARS-CoV-2 main protease enzyme. Saudi J. Biol. Sci. 2021, 29, 65–74. [Google Scholar] [CrossRef]
- Jing, L.J.; Mohamed, M.; Rahmat, A.; Bakar, M.F.A. Phytochemicals, antioxidant properties and anticancer investigations of the different parts of several gingers species (Boesenbergia rotunda, Boesenbergia pulchella var attenuata and Boesenbergia armeniaca). J. Med. Plants Res. 2010, 4, 27–32. [Google Scholar]
- Oladeji, O.S.; Adelowo, F.E.; Ayodele, D.T.; Odelade, K.A. Phytochemistry and pharmacological activities of Cymbopogon citratus: A review. Sci. Afr. 2019, 6, e00137. [Google Scholar] [CrossRef]
- Soares, M.O.; Alves, R.C.; Pires, P.C.; Oliveira, M.B.P.; Vinha, A.F. Angolan Cymbopogon citratus used for therapeutic benefits: Nutritional composition and influence of solvents in phytochemicals content and antioxidant activity of leaf extracts. Food Chem. Toxicol. 2013, 60, 413–418. [Google Scholar] [CrossRef]
- Halabi, M.F.; Sheikh, B.Y. Anti-Proliferative Effect and Phytochemical Analysis of Cymbopogon citratus Extract. BioMed Res. Int. 2014, 2014, 906239. [Google Scholar] [CrossRef]
- Boeira, C.P.; Piovesan, N.; Flores, D.C.B.; Soquetta, M.B.; Lucas, B.N.; Heck, R.T.; Alves, J.d.S.; Campagnol, P.C.B.; dos Santos, D.; Flores, E.M.M.; et al. Phytochemical characterization and antimicrobial activity of Cymbopogon citratus extract for application as natural antioxidant in fresh sausage. Food Chem. 2020, 319, 126553. [Google Scholar] [CrossRef] [PubMed]
- Bolade, O.P.; Akinsiku, A.A.; Adeyemi, A.O.; Williams, A.B.; Benson, N.U. Dataset on phytochemical screening, FTIR and GC–MS characterisation of Azadirachta indica and Cymbopogon citratus as reducing and stabilising agents for nanoparticles synthesis. Data Brief 2018, 20, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Batiha, G.E.-S.; Beshbishy, A.M.; Wasef, L.; Elewa, Y.H.A.; El-Hack, M.E.A.; Taha, A.E.; Al-Sagheer, A.A.; Devkota, H.P.; Tufarelli, V. Uncaria tomentosa (Willd. ex Schult.) DC.: A Review on Chemical Constituents and Biological Activities. Appl. Sci. 2020, 10, 2668. [Google Scholar] [CrossRef]
- Inthi, P.; Pandith, H.; Kongtawelert, P.; Banjerdpongchai, R. Anti-cancer Effect and Active Phytochemicals of Houttuynia cordata Thunb. Against Human Breast Cancer Cells. Asian Pac. J. Cancer Prev. 2023, 24, 1265–1274. [Google Scholar] [CrossRef]
- Fu, J.; Dai, L.; Lin, Z.; Lu, H. Houttuynia cordata Thunb: A Review of Phytochemistry and Pharmacology and Quality Control. Chin. Med. 2013, 04, 101–123. [Google Scholar] [CrossRef]
- Neto, A.C.; Netto, J.C.; Pereira, P.S.; Pereira, A.M.; Taleb-Contini, S.H.; França, S.C.; Marques, M.O.; Beleboni, R.O. The role of polar phytocomplexes on anticonvulsant effects of leaf extracts of Lippia alba (Mill.) N.E. Brown chemotypes. J. Pharm. Pharmacol. 2010, 61, 933–939. [Google Scholar] [CrossRef]
- Filho, J.G.S.; Duringer, J.M.; Souza, I.A.; da Cunha, E.V.; Craig, A.M.; Silva, M.S.; Barbosa-Filho, J.M.; Xavier, H.S. Phytochemistry and acute toxicity from the roots of Lippia alba. Pharm. Biol. 2009, 47, 142–145. [Google Scholar] [CrossRef]
- dos Santos, N.O.; Pascon, R.C.; Vallim, M.A.; Figueiredo, C.R.; Soares, M.G.; Lago, J.H.G.; Sartorelli, P. Cytotoxic and Antimicrobial Constituents from the Essential Oil of Lippia alba (Verbenaceae). Medicines 2016, 3, 22. [Google Scholar] [CrossRef]
- Sourki, A.H.; Ghani, A.; Kiani, F.; Alipour, A. Phytochemical profiles of lemon verbena (Lippia citriodora H.B.K.) and its potential application to cookie enrichment. Food Sci. Nutr. 2021, 9, 3100–3113. [Google Scholar] [CrossRef]
- Abdoul-Latif, F.M.; Ainane, A.; Abdoul-Latif, T.M.; Ainane, T. Chemical study and evaluation of insectical properties of african lippia citriodora essential oil. J. Biopestic. 2020, 13, 119–126. [Google Scholar] [CrossRef]
- Zayed, M.Z.; Samling, B. Phytochemical constituents of the leaves of leucaena leucocephala from malaysia. Int. J. Pharm. Pharm. Sci. 2016, 8, 174. [Google Scholar] [CrossRef]
- Bassey, R.A.; Ndarake, E.I.I.; Ngele, B.A.; Iniobong, E.A. GC-MS Analysis and Phytochemical Constituents in Anthocleista vogelii and Leuceana leucocephala obtained from Calabar, Cross River State, Southern Nigeria. Res. J. Sci. Technol. 2023, 3, 68–81. Available online: https://rejost.com.ng (accessed on 1 February 2025).
- Burlacu, E.; Nisca, A.; Tanase, C. A Comprehensive Review of Phytochemistry and Biological Activities of Quercus Species. Forests 2020, 11, 904. [Google Scholar] [CrossRef]
- Ameera, O.H.; Ghaidaa, J.M.; Mohammed, Y.H.; Hameed, I.H. Phytochemical screening of methanolic dried galls extract of Quercus infectoria using gas chromatography-mass spectrometry (GC-MS) and Fourier transform-infrared (FT-IR). J. Pharmacogn. Phytother. 2016, 8, 49–59. [Google Scholar] [CrossRef]
- Elham, A.; Arken, M.; Kalimanjan, G.; Arkin, A.; Iminjan, M. A review of the phytochemical, pharmacological, pharmacokinetic, and toxicological evaluation of Quercus infectoria galls. J. Ethnopharmacol. 2021, 273, 113592. [Google Scholar] [CrossRef] [PubMed]
- Satti, A.A.; Abdelgadir, A.A.; Hago, S.A.; Ahmed, E.M.; Elimam, Y.M. GC-MS Analysis, Antioxidant and Antibacterial activity GC-MS analysis, antioxidant and antibacterial activity of acetone fractions obtained from Guiera Senegalensis leaves and Quercus infectoria Nutgalls extracts. Arab. J. Med. Aromat. Plants 2021, 7, 282–294. [Google Scholar]
- Nursanty, R.; Padzil, K.N.B.M.; Ramli, N.I.B.; Mahyudin, N.A.; BIN Jaafar, A.H.; Rukayadi, Y. Phytochemical analysis of ethanolic Psidium guajava leaves extract using GC-MS and LC-MS. Biodiversitas J. Biol. Divers. 2023, 24. [Google Scholar] [CrossRef]
- Vandayar, M.S.A.V. Phytochemicals analysis and GC–MS analysis of identification and characterization of bioactive compounds present in methanolic leaf extract Azadirachta indica. Int. J. Pharm. Sci. Drug Anal. 2021, 1, 39–50. [Google Scholar]
- Ahammad, F.; Alam, R.; Mahmud, R.; Akhter, S.; Talukder, E.K.; Tonmoy, A.M.; Fahim, S.; Al-Ghamdi, K.; Samad, A.; Qadri, I. Pharmacoinformatics and molecular dynamics simulation-based phytochemical screening of neem plant (Azadiractha indica) against human cancer by targeting MCM7 protein. Brief. Bioinform. 2021, 22, bbab098. [Google Scholar] [CrossRef]
- Rahman, S.; Islam, R.; Kamruzzaman, M.; Alam, K.; Jamal, A.H.M. Ocimum sanctum L.: A Review of Phytochemical and Pharmacological Profile. Am. J. Drug Discov. Dev. 2011, 1, 1–15. [Google Scholar]
- Cohen, M.C. Ocimum sanctum (Holy Basil)—A Herb for all Reasons. Brief. Bioinform. 2014, 5, 291–299. [Google Scholar]


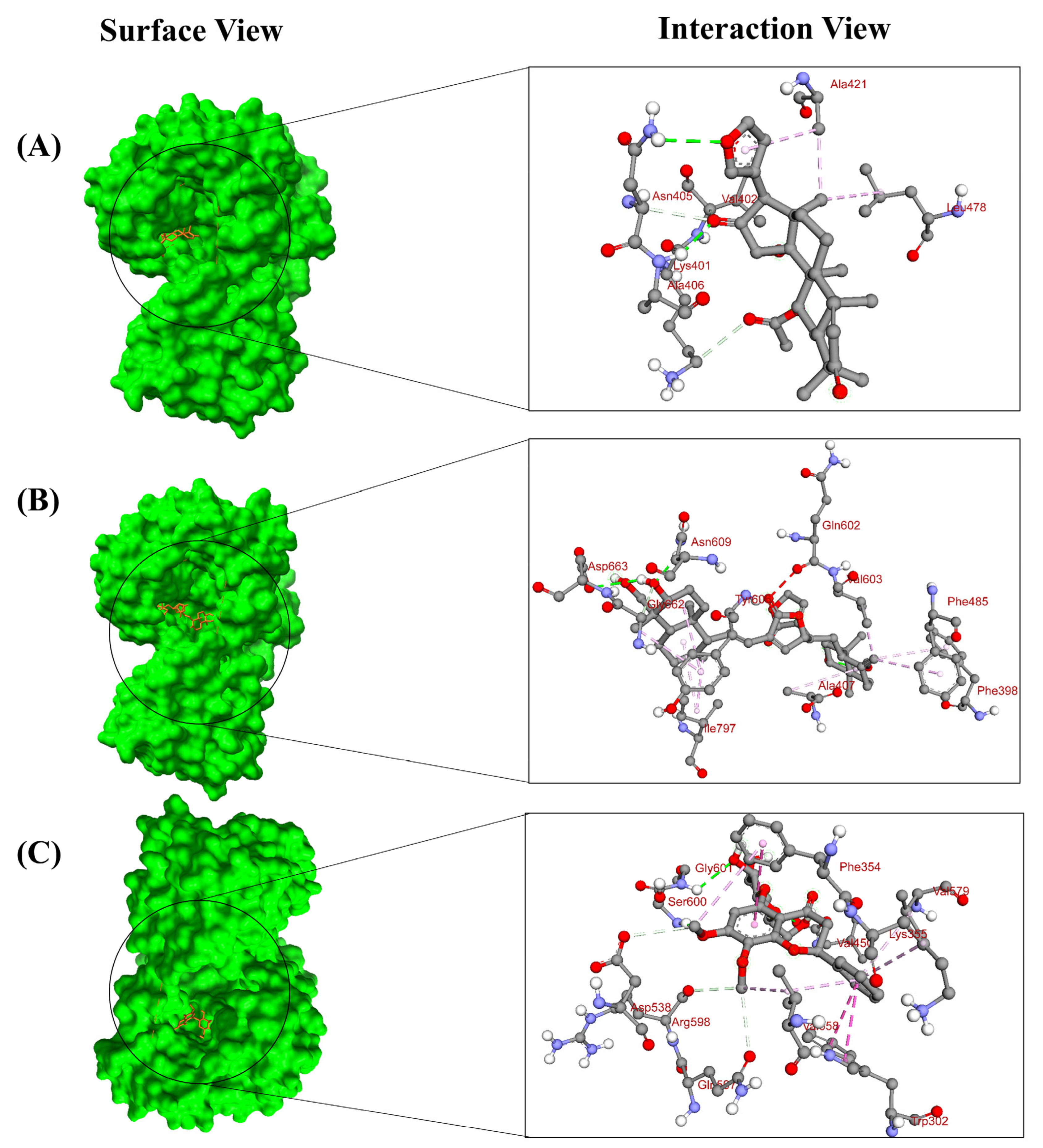
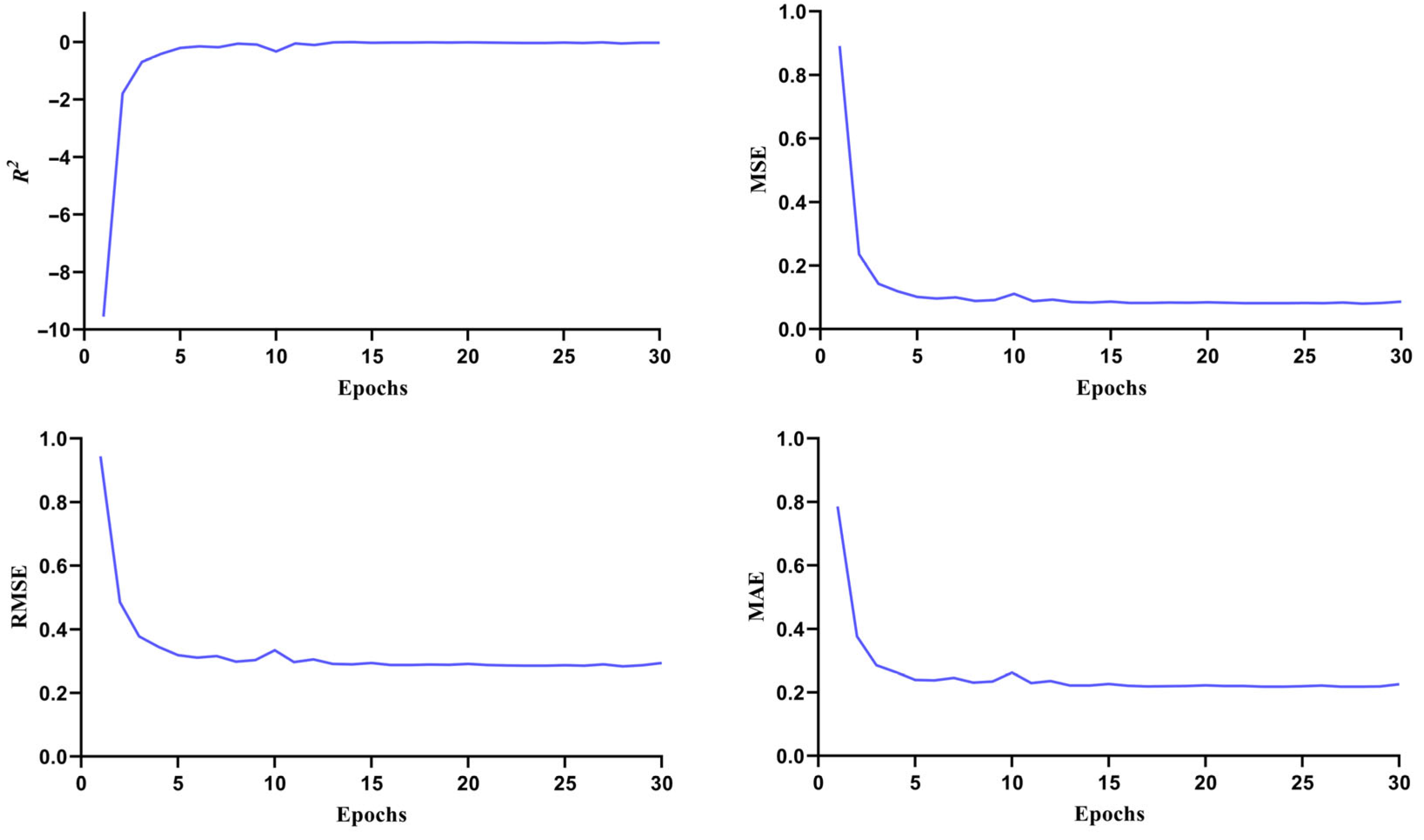
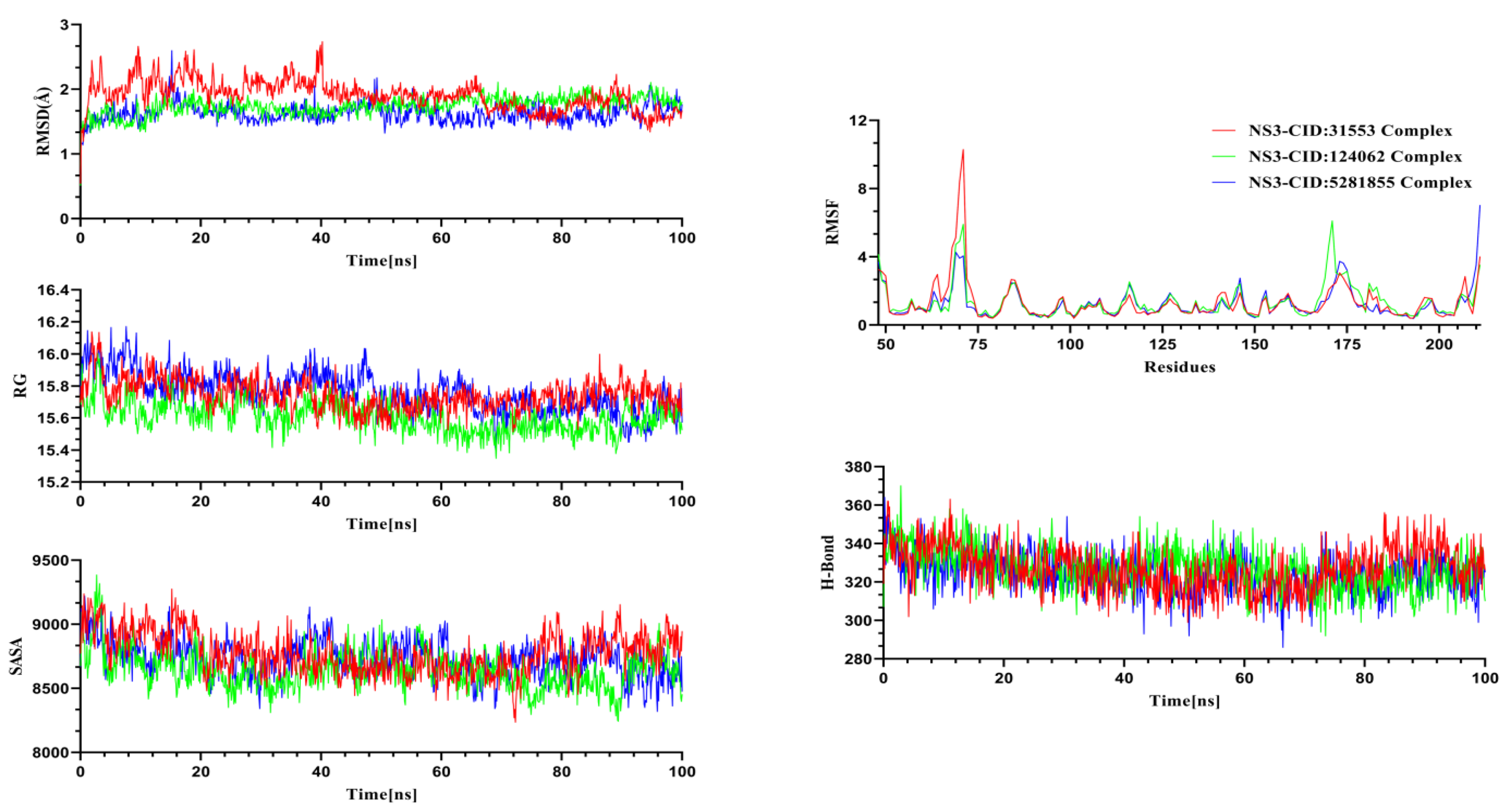
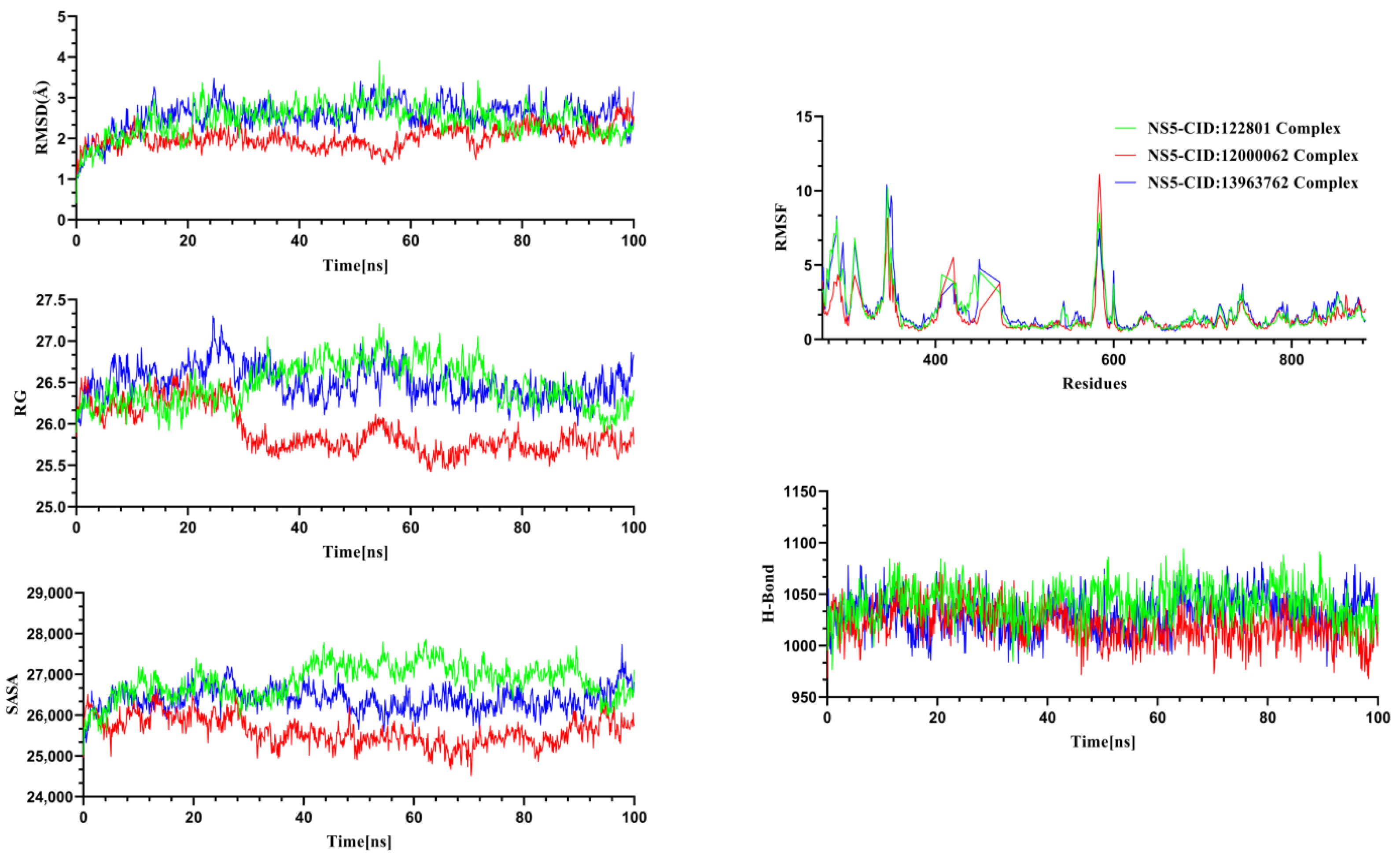

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name and CID | Binding Affinity (kcal/mol) | Predicted pIC50 Score | Residue in Contact | Interaction Type | Bond Distance in Å |
|---|---|---|---|---|---|
| Silibinin (31553) | −8.5 | 4.76 | TRP1069 | H Bond | 2.72728 |
| LYS1074 | H Bond | 2.15489 | |||
| LEU1149 | H Bond | 2.91918 | |||
| LEU1085 | H Bond | 2.95107 | |||
| TRP1083 | hydrophobic | 4.31848 | |||
| VAL1146 | hydrophobic | 5.21579 | |||
| LEU1076 | hydrophobic | 4.46035 | |||
| Rubiadin (124062) | −8.3 | 4.7 | LEU1149 | H Bond | 2.51707 |
| ASN1167 | H Bond | 2.13321 | |||
| LYS1074 | H Bond | 2.54066 | |||
| LEU1076 | hydrophobic | 2.86341 | |||
| TRP1083 | hydrophobic | 5.94837 | |||
| VAL1147 | hydrophobic | 3.92637 | |||
| ILE1165 | hydrophobic | 5.16293 | |||
| LEU1085 | hydrophobic | 4.94692 | |||
| Ellagic acid (5281855) | −8.2 | 4.35 | LEU1149 | H Bond | 2.23803 |
| ASN1167 | H Bond | 2.53243 | |||
| LEU1085 | H Bond | 2.04294 | |||
| LEU1076 | hydrophobic | 2.66139 | |||
| TRP1083 | hydrophobic | 5.50333 | |||
| VAL1147 | hydrophobic | 4.24805 | |||
| LYS1074 | hydrophobic | 4.87666 | |||
| ILE1165 | hydrophobic | 5.22632 | |||
| Balapiravir (11691726) | −6.6 | 4.11 | GLY1087 | H Bond | 2.03115 |
| VAL1146 | H Bond | 2.21827 | |||
| LYS1074 | H Bond | 2.81251 | |||
| LEU1149 | H Bond | 2.76025 | |||
| ASN1167 | hydrophobic | 2.07438 | |||
| GLY1148 | hydrophobic | 4.41981 | |||
| LEU1176 | hydrophobic | 4.12744 | |||
| ILE1165 | hydrophobic | 4.10504 | |||
| Bisandrographolide A (12000062) | −10.1 | 4.43 | ASN609 | H Bond | 2.23313 |
| ASP663 | H Bond | 3.03945 | |||
| VAL603 | H Bond | 2.51134 | |||
| ALA407 | hydrophobic | 5.0847 | |||
| ILE797 | hydrophobic | 4.3585 | |||
| VAL603 | hydrophobic | 4.12083 | |||
| ILE797 | hydrophobic | 4.55821 | |||
| PHE398 | hydrophobic | 5.22525 | |||
| PHE485 | hydrophobic | 4.97929 | |||
| TYR606 | hydrophobic | 5.23019 | |||
| TYR606 | hydrophobic | 5.2332 | |||
| NSC 640467 (122801) | −9.3 | 4.71 | ASN405 | H Bond | 2.89725 |
| ALA406 | H Bond | 2.31095 | |||
| LYS401 | H Bond | 2.64717 | |||
| VAL402 | H Bond | 2.27594 | |||
| ASN405 | H Bond | 2.26522 | |||
| ALA407 | H Bond | 3.05968 | |||
| ALA421 | hydrophobic | 3.65796 | |||
| LEU478 | hydrophobic | 4.98358 | |||
| ALA421 | hydrophobic | 4.94432 | |||
| Andrographidin A (13963762) | −8.9 | 4.84 | SER600 | H Bond | 2.3857 |
| GLY601 | H Bond | 2.66425 | |||
| VAL450 | H Bond | 2.78778 | |||
| ASP538 | H Bond | 2.61398 | |||
| GLN597 | H Bond | 2.73499 | |||
| ARG598 | H Bond | 2.43382 | |||
| TRP302 | hydrophobic | 4.19854 | |||
| PHE354 | hydrophobic | 5.25107 | |||
| VAL358 | hydrophobic | 4.62125 | |||
| LYS355 | hydrophobic | 5.30999 | |||
| VAL579 | hydrophobic | 4.86855 | |||
| Balapiravir (11691726) | −6.9 | 4.11 | GLN597 | H Bond | 2.87859 |
| ARG598 | H Bond | 2.99243 | |||
| SER600 | H Bond | 3.02471 | |||
| GLN602 | H Bond | 2.32165 | |||
| GLY599 | H Bond | 2.86808 | |||
| LYS575 | hydrophobic | 4.67156 | |||
| VAL577 | hydrophobic | 4.42977 | |||
| VAL579 | hydrophobic | 3.64284 |
| Properties | Silibinin | Rubiadin | Ellagic Acid | NSC 640467 | Bisandrographolide A | Andrographidin A | |
|---|---|---|---|---|---|---|---|
| Physicochemical Properties | MW (g/mol) | 482.441 | 254.24 | 302.19 | 466.57 | 664.88 | 462.45 |
| Heavy Atoms | 35 | 19 | 22 | 34 | 48 | 33 | |
| Aromatic Atoms | 18 | 12 | 16 | 5 | 0 | 12 | |
| Rotatable bonds | 4 | 0 | 0 | 2 | 8 | 6 | |
| H-Bond acceptors | 10 | 4 | 8 | 6 | 8 | 10 | |
| H-Bond donors | 5 | 2 | 4 | 0 | 4 | 4 | |
| TPSA (Å2) | 155.14 Å2 | 74.60 Å2 | 141.34 Å2 | 86.11Å2 | 133.52 Å2 | 144.14 Å2 | |
| Lipophilicity | Log Po/w (Cons) | 2.36 | 2.18 | 1.31 | 4.62 | 5.36 | 0.58 |
| Water Solubility | Log S (ESOL) | −4.14 | −3.82 | −2.94 | −5.31 | −7.26 | −3.01 |
| Pharmacokinetics | GI Absorption | low | high | high | high | low | low |
| BBB permeant | no | yes | no | no | no | no | |
| P-GP Substrate | no | no | no | yes | yes | yes | |
| Drug likeness | Lipinski | yes | yes | yes | yes | yes | yes |
| Medi. Chemistry | Synth. accessibility | 4.92 | 2.52 | 3.17 | 6.34 | 8.00 | 5.33 |
| Target | Silibinin | Rubiadin | Ellagic Acid | NSC 640467 | Bisandrographolide A | Andrographidine A |
|---|---|---|---|---|---|---|
| Hepatotoxicity | inactive | inactive | inactive | inactive | inactive | inactive |
| Carcinogenicity | inactive | inactive | active | inactive | inactive | inactive |
| Mutagenicity | inactive | active | inactive | inactive | inactive | inactive |
| Cytotoxicity | inactive | inactive | inactive | inactive | inactive | active |
| LD50 (mg kg−1) | 2000 | 7000 | 2991 | 555 | 452 | 3000 |
| Immunotoxicity | active | active | inactive | active | active | active |
| Toxicity Class | 4 | 5 | 4 | 4 | 4 | 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hossain, A.; Joti, F.T.; Hossain, M.S.; Al-Noman, A.; Thowing, C.; Mursona, M.; Islam, M.R.; Rahman, M.E.; Matin, M.N.; Haque, M.A. Identification of Potential Inhibitors Targeting Non-Structural Proteins NS3 and NS5 of Dengue Virus Using Docking and Deep Learning Approaches. Pharmaceuticals 2025, 18, 566. https://doi.org/10.3390/ph18040566
Hossain A, Joti FT, Hossain MS, Al-Noman A, Thowing C, Mursona M, Islam MR, Rahman ME, Matin MN, Haque MA. Identification of Potential Inhibitors Targeting Non-Structural Proteins NS3 and NS5 of Dengue Virus Using Docking and Deep Learning Approaches. Pharmaceuticals. 2025; 18(4):566. https://doi.org/10.3390/ph18040566
Chicago/Turabian StyleHossain, Alomgir, Faria Tasnin Joti, Md. Shohag Hossain, Abdullah Al-Noman, Chomong Thowing, Mehjabin Mursona, Md. Robiul Islam, Md. Ekhtiar Rahman, Mohammad Nurul Matin, and Md Azizul Haque. 2025. "Identification of Potential Inhibitors Targeting Non-Structural Proteins NS3 and NS5 of Dengue Virus Using Docking and Deep Learning Approaches" Pharmaceuticals 18, no. 4: 566. https://doi.org/10.3390/ph18040566
APA StyleHossain, A., Joti, F. T., Hossain, M. S., Al-Noman, A., Thowing, C., Mursona, M., Islam, M. R., Rahman, M. E., Matin, M. N., & Haque, M. A. (2025). Identification of Potential Inhibitors Targeting Non-Structural Proteins NS3 and NS5 of Dengue Virus Using Docking and Deep Learning Approaches. Pharmaceuticals, 18(4), 566. https://doi.org/10.3390/ph18040566