

Medicinal Chemistry Strategies in Targeting TGF-βR1 Kinase Domain: Unveiling Insights into Inhibitor Structure–Activity Relationship (SAR)

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. TGF-β Receptor Isoforms
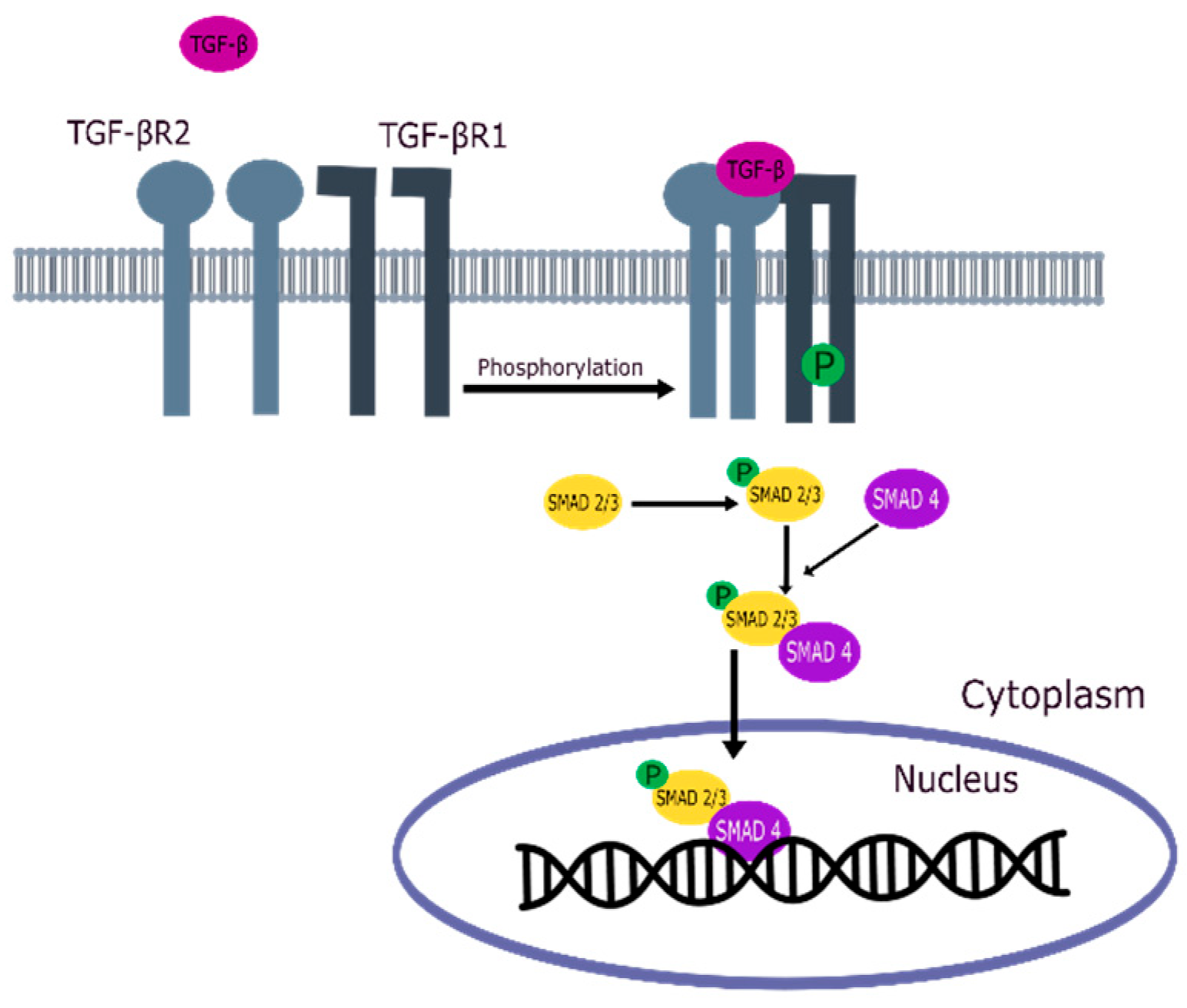
3. TGF-β Signaling Pathway
4. Role of the TGF-β Signaling Pathway in Tumor Suppression
5. TGF-β Signaling Pathway in Tumor Promotion
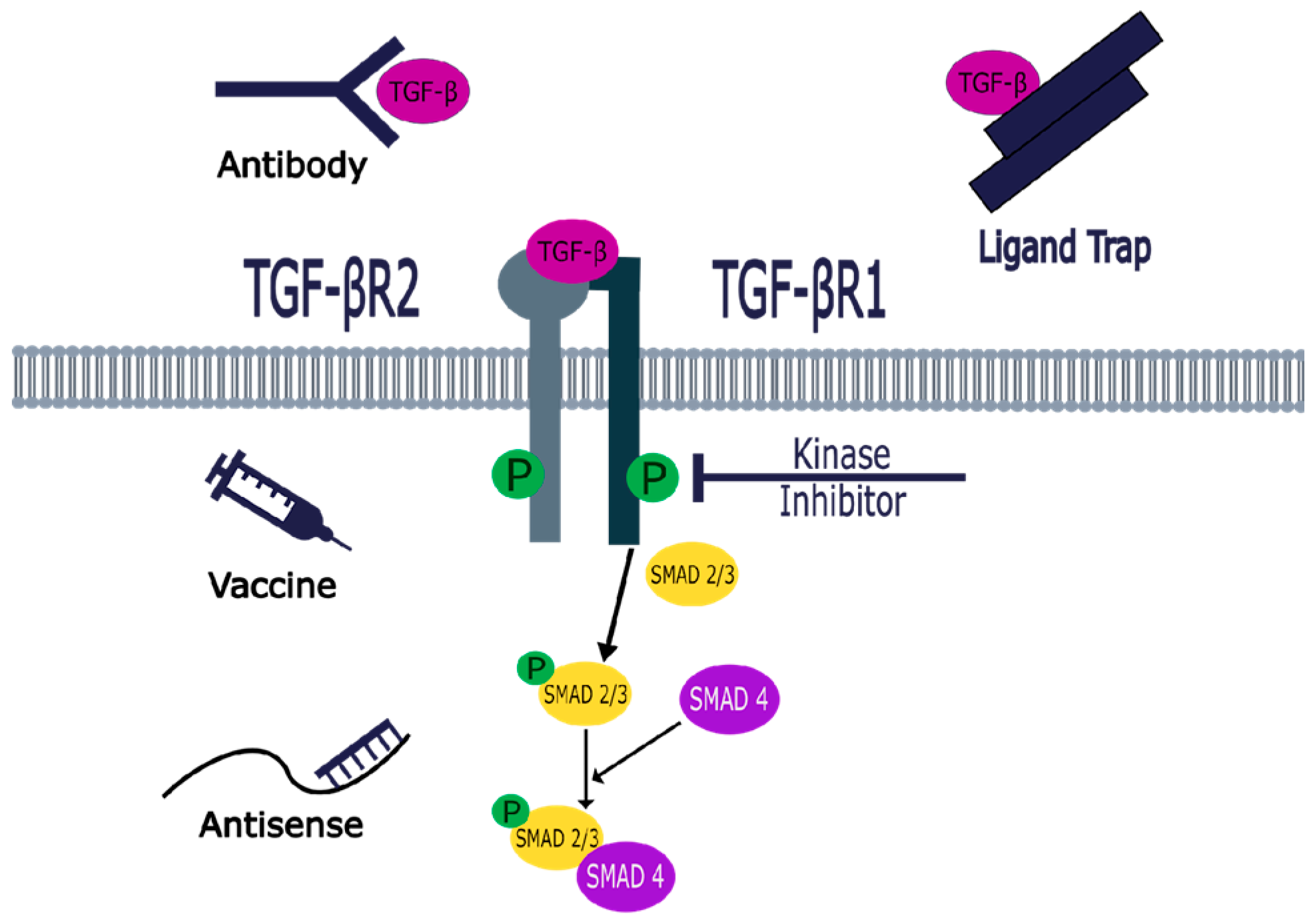
6. Therapeutic Targets of the TGF-β Signaling Pathway
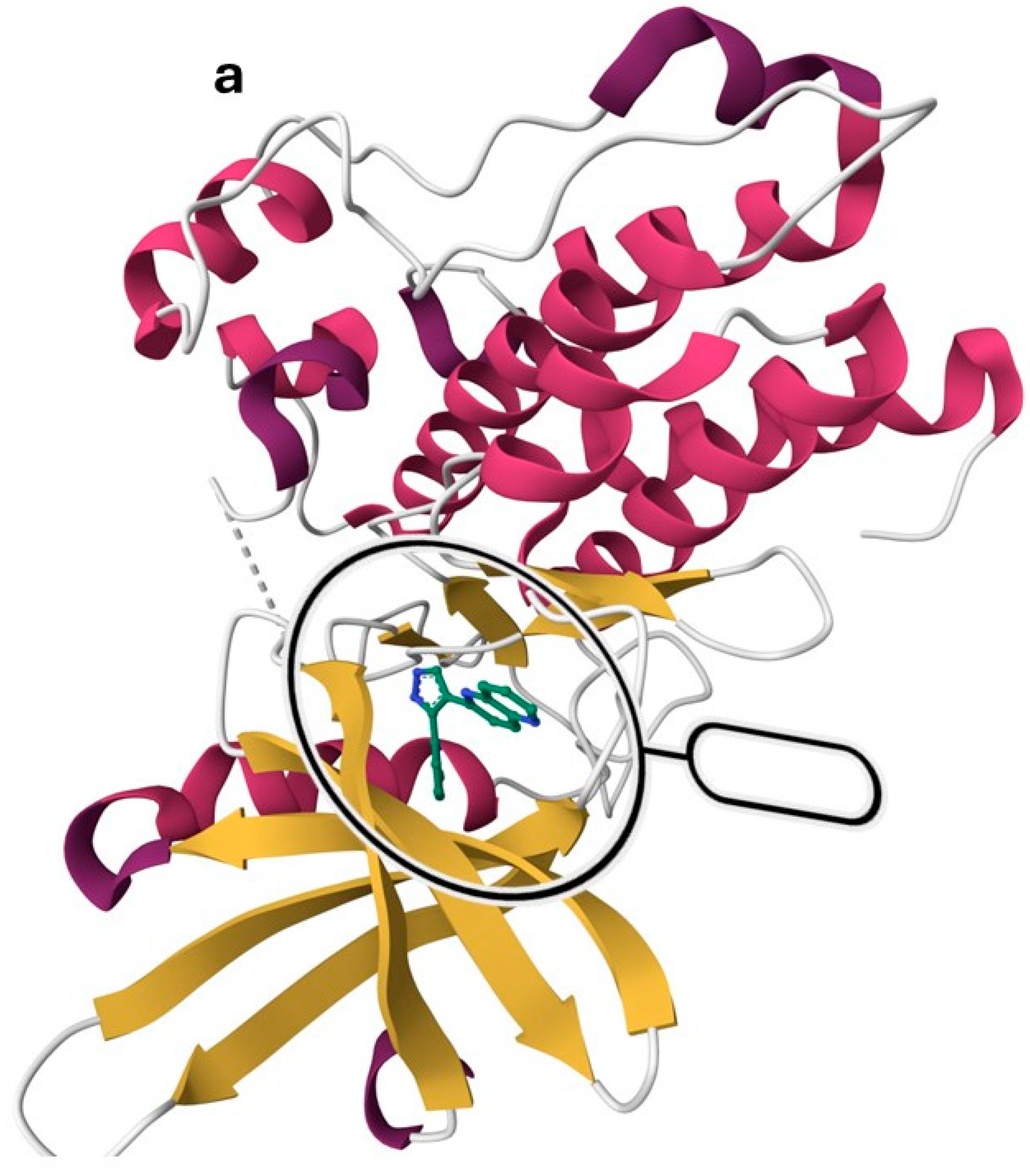
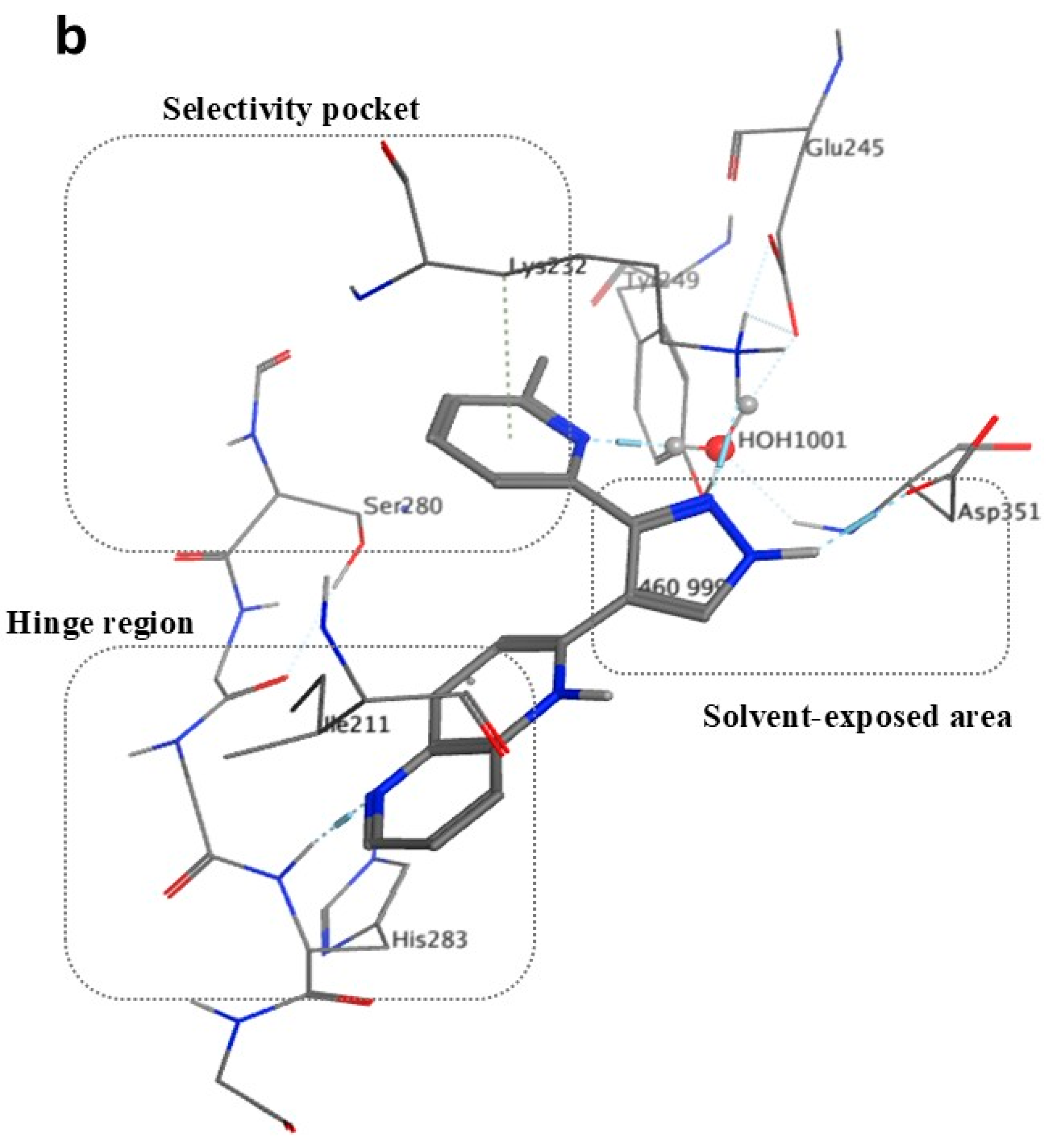
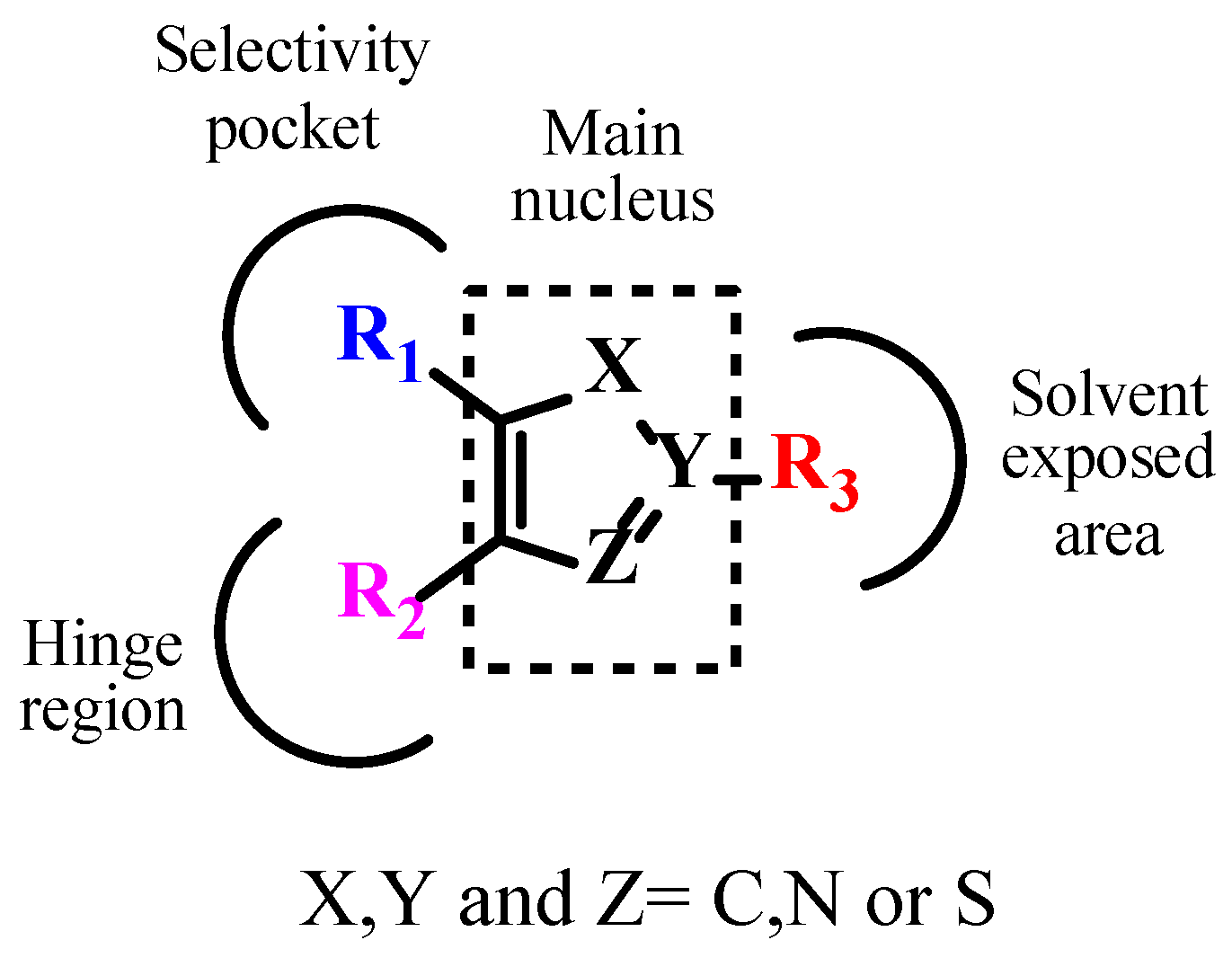
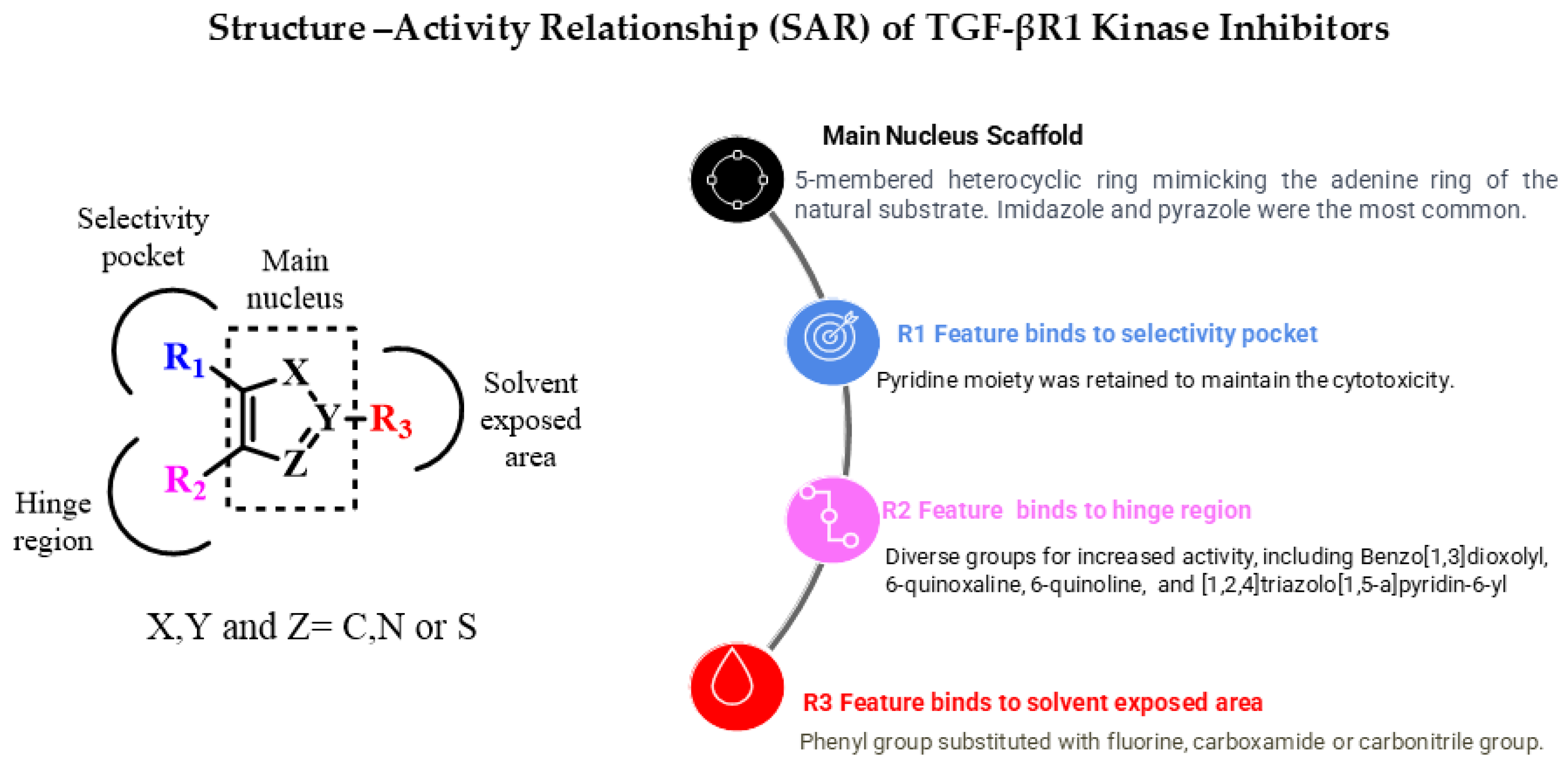
7. Exploration of TGF-βR1 Binding Site
8. Reported TGF-βR1 Inhibitors
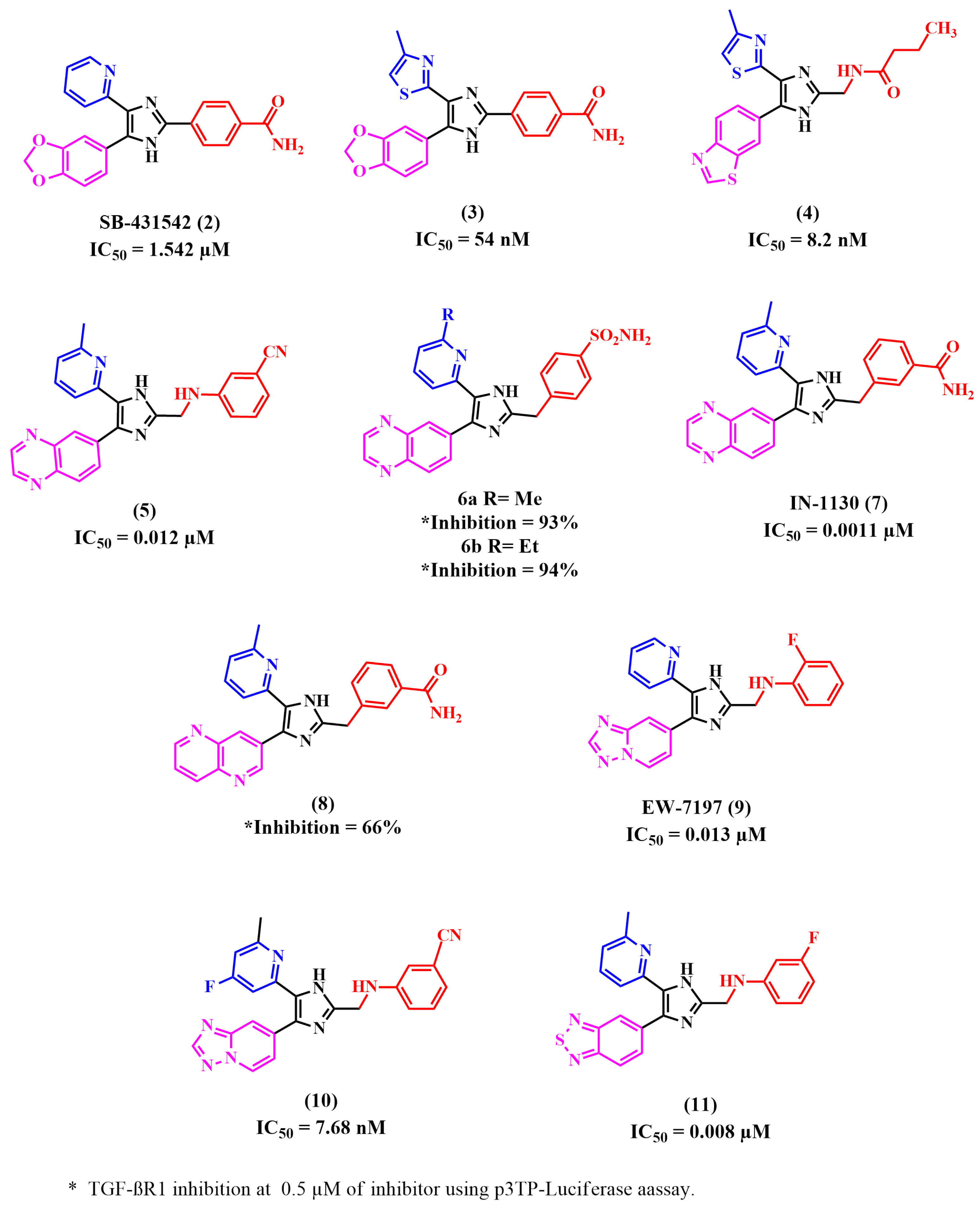
8.1. Imidazole Derivatives
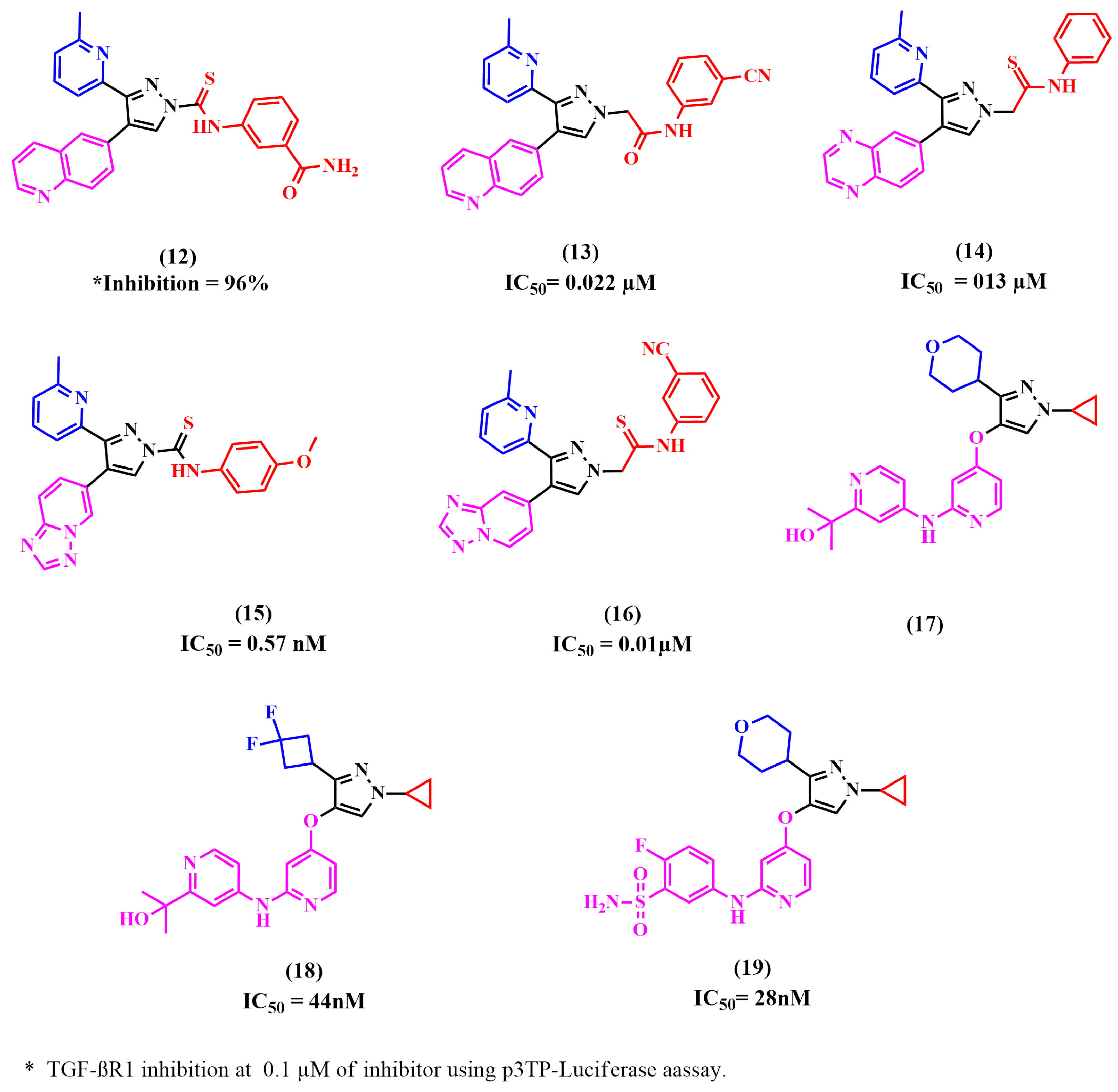
8.2. Pyrazole Derivatives
8.3. Thiazole Derivatives
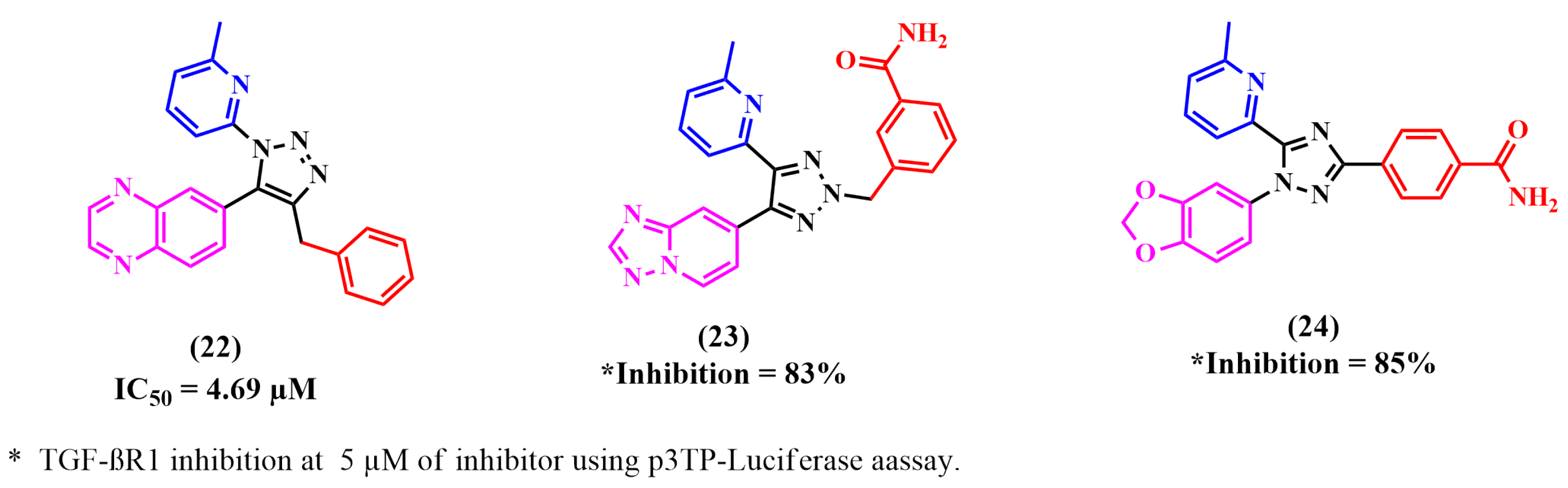
8.4. Triazole Derivatives
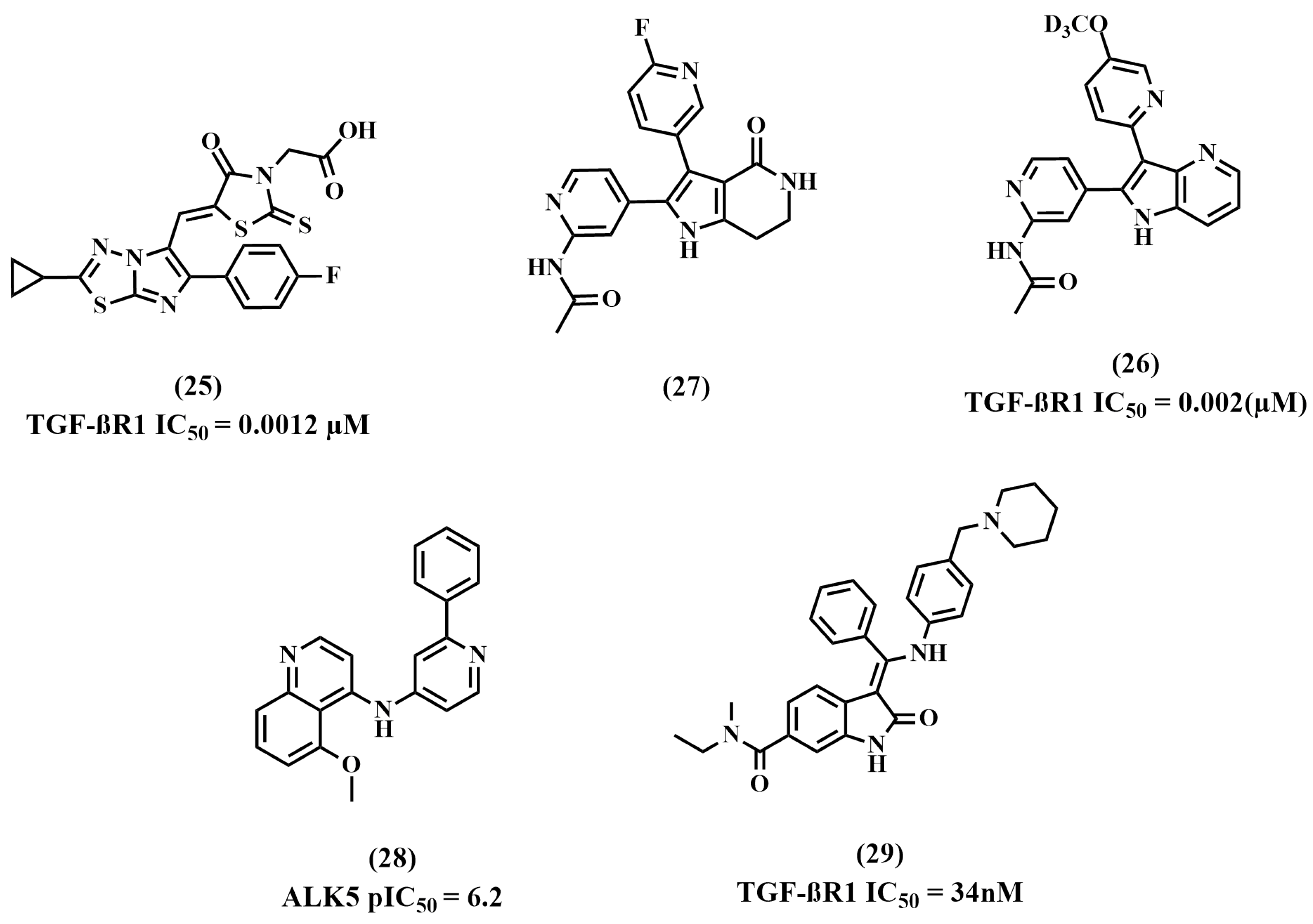
8.5. Miscellaneous
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALK5 | Activin receptor-like kinase 5 |
| ASO | Antisense oligonucleotide |
| ATP | Adenosine triphosphate |
| CDK | Cyclin-dependent kinase |
| CN | Carbonitrile |
| C-H | Carbon–hydrogen |
| CTLs | Cytotoxic T lymphocytes |
| DNA | Deoxyribonucleic acid |
| EMT | Epithelial–mesenchymal transition |
| EW-7197 | TGF-βR1 kinase inhibitor (compound 9) |
| GS | Glycine-serine-rich |
| H-bond | Hydrogen bond |
| IC50 | Half maximal inhibitory concentration |
| IN-1130 | TGF-βR1 kinase inhibitor (compound 7) |
| MAPK | Mitogen-activated protein kinase |
| MOE | Molecular operating environment |
| NHLF | Normal human lung fibroblast |
| NK | Natural killer (cells) |
| PD-1 | Programmed cell death protein 1 |
| PDB | Protein Data Bank |
| R1, R2, R3 | Variable substituent groups in a molecule |
| RNA | Ribonucleic acid |
| SAR | Structure–activity relationship |
| SBE | SMAD binding element |
| SMAD | Homolog of mothers against decapentaplegic (a family of signaling proteins) |
| SMI | Small molecule inhibitor |
| sTGF-βR3 | Soluble transforming growth factor beta receptor type 3 |
| TGF-β | Transforming growth factor beta |
| TGF-βR1 | Transforming growth factor beta receptor type 1 |
| TGF-βR2 | Transforming growth factor beta receptor type 2 |
| TGF-βR3 | Transforming growth factor beta receptor type 3 |
References
- Hinck, A.P. Structural studies of the TGF-βs and their receptors-Insights into evolution of the TGF-β superfamily. FEBS Lett. 2012, 586, 1860–1870. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β family: Context-dependent roles in cell and tissue physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef] [PubMed]
- Burgess, D.J. Keeping one step ahead. Nat. Rev. Drug Discov. 2012, 11, 108. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Fan, T.; Xiao, C.; Tian, H.; Zheng, Y.; Li, C.; He, J. TGF-β signaling in health, disease, and therapeutics. Signal Transduct. Target. Ther. 2024, 9, 61. [Google Scholar] [CrossRef]
- Giarratana, A.O.; Prendergast, C.M.; Salvatore, M.M.; Capaccione, K.M. TGF-β signaling: Critical nexus of fibrogenesis and cancer. J. Transl. Med. 2024, 22, 594. [Google Scholar] [CrossRef]
- Song, N.; Zhou, J.; Cao, B.; Zhao, Y.; Yu, Y.; Lei, H.; Luo, Y. TGF-β’s role in skeletal muscle injury repair: Mechanism and research advances. J. Pract. Med. 2024, 40, 721–726. [Google Scholar] [CrossRef]
- Goumans, M.J.; Zwijsen, A.; ten Dijke, P.; Bailly, S. Bone morphogenetic proteins in vascular homeostasis and disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a031989. [Google Scholar] [CrossRef]
- Adu-Amankwaah, J.; You, Q.; Liu, X.; Jiang, J.; Yang, D.; Liu, K.; Yuan, J.; Wang, Y.; Hu, Q.; Tan, R. Pulmonary Hypertension: Molecular Mechanisms and Clinical Studies. MedComm 2025, 6, e70134. [Google Scholar] [CrossRef]
- Andre, P.; Joshi, S.R.; Briscoe, S.D.; Alexander, M.J.; Li, G.; Kumar, R. Therapeutic Approaches for Treating Pulmonary Arterial Hypertension by Correcting Imbalanced TGF-β Superfamily Signaling. Front. Med. 2022, 8, 814222. [Google Scholar] [CrossRef]
- Marvin, D.L.; Heijboer, R.; ten Dijke, P.; Ritsma, L. TGF-β signaling in liver metastasis. Clin. Transl. Med. 2020, 10, e160. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Katz, L.H.; Likhter, M.; Jogunoori, W.; Belkin, M.; Ohshiro, K.; Mishra, L. TGF-β signaling in liver and gastrointestinal cancers. Cancer Lett. 2016, 379, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Gatza, C.E.; Oh, S.Y.; Blobe, G.C. Roles for the type III TGF-β receptor in human cancer. Cell. Signal. 2010, 22, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, F.; van Dinther, M.; Ten Dijke, P. Determining TGF-β receptor levels in the cell membrane. In TGF-β Signaling; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2016; Volume 1344, pp. 35–47. [Google Scholar] [CrossRef]
- Vander Ark, A.; Cao, J.; Li, X. TGF-β receptors: In and beyond TGF-β signaling. Cell. Signal. 2018, 52, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Larsson, J.; Goumans, M.; Sjöstrand, L.J.; Van Rooijen, M.A.; Ward, D.; Levéen, P.; Xu, X.; ten Dijke, P.; Mummery, C.L.; Karlsson, S. Abnormal angiogenesis but intact hematopoietic potential in TGF-β type I receptor-deficient mice. EMBO J. 2001, 20, 1663–1673. [Google Scholar] [CrossRef]
- Clark, D.A.; Coker, R. Molecules in focus Transforming growth factor-beta (TGF-β). Int. J. Biochem. Cell Biol. 1998, 30, 293–298. [Google Scholar] [CrossRef]
- Huang, F.; Chen, Y.G. Regulation of TGF-β receptor activity. Cell Biosci. 2012, 2, 9. [Google Scholar] [CrossRef]
- Oshima, M.; Oshima, H.; Taketo, M.M. TGF-β receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev. Biol. 1996, 179, 297–302. [Google Scholar] [CrossRef]
- Wrana, J.L.; Attisano, L.; Wieser, R.; Ventura, F.; Massagué, J. Mechanism of activation of the TGF-β receptor. Nature 1994, 370, 341–347. [Google Scholar] [CrossRef]
- Galliher, A.J.; Schiemann, W.P. Src phosphorylates Tyr284 in TGF-β type II receptor and regulates TGF-β stimulation of p38 MAPK during breast cancer cell proliferation and invasion. Cancer Res. 2007, 67, 3752–3758. [Google Scholar] [CrossRef]
- Shi, Y.; Massagué, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Miyazono, K.; Ten Dijke, P. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.-H.; Derynck, R. Specificity and versatility in TGF-β signaling through Smads. Annu. Rev. Cell Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.-J.; Valdimarsdottir, G.; Itoh, S.; Lebrin, F.; Larsson, J.; Mummery, C.; Karlsson, S.; Dijke, P.T. Activin receptor-like kinase (ALK) 1 is an antagonistic mediator of lateral TGFβ/ALK5 signaling. Mol. Cell 2003, 12, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.C.; Randall, R.A.; Hill, C.S. Transforming growth factor β-induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage-independent growth. Mol. Cell. Biol. 2008, 28, 6889–6902. [Google Scholar] [CrossRef]
- Hill, C.S. Nucleocytoplasmic shuttling of Smad proteins. Cell Res. 2009, 19, 36–46. [Google Scholar] [CrossRef]
- Baba, A.B.; Rah, B.; Bhat, G.R.; Mushtaq, I.; Parveen, S.; Hassan, R.; Hameed Zargar, M.; Afroze, D. Transforming Growth Factor-Beta (TGF-β) Signaling in Cancer-A Betrayal Within. Front. Pharmacol. 2022, 13, 791272. [Google Scholar] [CrossRef]
- Mukherjee, P.; Winter, S.L.; Alexandrow, M.G. Cell cycle arrest by transforming growth factor β1 near G1/S is mediated by acute abrogation of prereplication complex activation involving an Rb-MCM interaction. Mol. Cell. Biol. 2010, 30, 845–856. [Google Scholar] [CrossRef]
- Katz, L.H.; Li, Y.; Chen, J.S.; Muñoz, N.M.; Majumdar, A.; Chen, J.; Mishra, L. Targeting TGF-β signaling in cancer. Expert Opin. Ther. Targets 2013, 17, 743–760. [Google Scholar] [CrossRef]
- Yoshida, K.; Matsuzaki, K.; Murata, M.; Yamaguchi, T.; Suwa, K.; Okazaki, K. Clinico-pathological importance of TGF-β/phospho-smad signaling during human hepatic fibrocarcinogenesis. Cancers 2018, 10, 183. [Google Scholar] [CrossRef]
- Zhang, Y.; Alexander, P.B.; Wang, X.-F. TGF-β family signaling in the control of cell proliferation and survival. Cold Spring Harb. Perspect. Biol. 2017, 9, a022145. [Google Scholar] [CrossRef]
- Schrantz, N.; Bourgeade, M.-F.; Mouhamad, S.; Leca, G.; Sharma, S.; Vazquez, A. p38-mediated regulation of an Fas-associated death domain protein-independent pathway leading to caspase-8 activation during TGFβ-induced apoptosis in human Burkitt lymphoma B cells BL41. Mol. Biol. Cell 2001, 12, 3139–3151. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, S.; Wang, J.; Myeroff, L.; Parsons, R.; Sun, L.; Lutterbaugh, J.; Fan, R.S.; Zborowska, E.; Kinzler, K.W.; Vogelstein, B.; et al. Inactivation of the type II TGF-β receptor in colon cancer cells with microsatellite instability. Science 1995, 268, 1336–1338. [Google Scholar] [CrossRef] [PubMed]
- Izumoto, S.; Arita, N.; Ohnishi, T.; Hiraga, S.; Taki, T.; Tomita, N.; Ohue, M.; Hayakawa, T. Microsatellite instability and mutated type II transforming growth factor-β receptor gene in gliomas. Cancer Lett. 1997, 112, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Myeroff, L.L.; Parsons, R.; Kim, S.J.; Hedrick, L.; Cho, K.R.; Orth, K.; Mathis, M.; Kinzler, K.W.; Lutterbaugh, J.; Park, K.; et al. A transforming growth factor beta receptor type II gene mutation common in colon and gastric but rare in endometrial cancers with microsatellite instability. Cancer Res. 1995, 55, 5545–5547. [Google Scholar]
- Parsons, R.; Myeroff, L.L.; Liu, B.; Willson, J.K.V.; Markowitz, S.D.; Kinzler, K.W.; Vogelstein, B. Microsatellite instability and mutations of the transforming growth factor β type II receptor gene in colorectal cancer. Cancer Res. 1995, 55, 5548–5550. [Google Scholar]
- Schutte, M.; Hruban, R.H.; Hedrick, L.; Cho, K.R.; Nadasdy, G.M.; Weinstein, C.L.; Bova, G.S.; Isaacs, W.B.; Cairns, P.; Nawroz, H.; et al. DPC4 gene in various tumor types. Cancer Res. 1996, 56, 2527–2530. [Google Scholar]
- Hahn, S.A.; Schutte, M.; Shamsul Hoque, A.T.M.; Moskaluk, C.A.; Da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21. 1. Science 1996, 271, 350–353. [Google Scholar] [CrossRef]
- Vincent, F.; Hagiwara, K.; Ke, Y.; Stoner, G.D.; Demetrick, D.J.; Bennett, W.P. Mutation analysis of the transforming growth factor β type II receptor in sporadic human cancers of the pancreas, liver, and breast. Biochem. Biophys. Res. Commun. 1996, 223, 561–564. [Google Scholar] [CrossRef]
- Jones, E.; Pu, H.; Kyprianou, N. Targeting TGF-β in prostate cancer: Therapeutic possibilities during tumor progression. Expert Opin. Ther. Targets 2009, 13, 227–234. [Google Scholar] [CrossRef]
- Gillani, S.R.; Mahar, S.K.; Badar, Q.; Sardar, A.; Soomro, I.A. Exploring the Molecular Mechanism of Cancer Metastasis Focus on Epithelial Mesenchymal Transition (EMT). Indus J. Biosci. Res. 2025, 3, 425–437. [Google Scholar] [CrossRef]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-β and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.A.; Massagué, J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Chung, C.L.; Hu, T.H.; Chen, J.J.; Liu, P.F.; Chen, C.L. Recent progress in TGF-β inhibitors for cancer therapy. Biomed. Pharmacother. 2021, 134, 111046. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A. TGFβ-directed therapeutics: 2020. Pharmacol. Ther. 2021, 217, 107666. [Google Scholar] [CrossRef]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I Study of GC1008 (Fresolimumab): A Human Anti-Transforming Growth Factor-Beta (TGFβ) Monoclonal Antibody in Patients with Advanced Malignant Melanoma or Renal Cell Carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef]
- Chen, Y.; Di, C.; Zhang, X.; Wang, J.; Wang, F.; Yan, J.; Xu, C.; Zhang, J.; Zhang, Q.; Li, H.; et al. Transforming growth factor β signaling pathway: A promising therapeutic target for cancer. J. Cell. Physiol. 2020, 235, 1903–1914. [Google Scholar] [CrossRef]
- Gellibert, F.; Woolven, J.; Fouchet, M.-H.; Mathews, N.; Goodland, H.; Lovegrove, V.; Laroze, A.; Nguyen, V.L.; Sautet, S.; Wang, R.; et al. Identification of 1, 5-naphthyridine derivatives as a novel series of potent and selective TGF-β type I receptor inhibitors. J. Med. Chem. 2004, 47, 4494–4506. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), 2024.0601; Chemical Computing Group ULC: Montreal, QC, Canada, 2024.
- Huse, M.; Chen, Y.G.; Massagué, J.; Kuriyan, J. Crystal structure of the cytoplasmic domain of the type I TGF beta receptor in complex with FKBP12. Cell 1999, 96, 425–436. [Google Scholar] [CrossRef]
- Wieser, R.; Wrana, J.L.; Massagué, J. GS domain mutations that constitutively activate TβR-I, the downstream signaling component in the TGF-β receptor complex. EMBO J. 1995, 14, 2199–2208. [Google Scholar] [CrossRef]
- Eyers, P.A.; Craxton, M.; Morricel, N.; Cohen, P.; Goedert, M. Conversion of SB 203580-insensitive MAP kinase family members to drug-sensitive forms by a single amino-acid substitution. Chem. Biol. 1998, 5, 321–328. [Google Scholar] [CrossRef]
- Kim, D.-K.; Jang, Y.; Lee, H.S.; Park, H.-J.; Yoo, J. Synthesis and Biological Evaluation of 4(5)-(6-Alkylpyridin-2-yl)imidazoles as Transforming Growth Factor-â Type 1 Receptor Kinase Inhibitors. J. Med. Chem. 2007, 50, 3143–3147. [Google Scholar] [CrossRef]
- Inman, G.J.; Nicolás, F.J.; Callahan, J.F.; Harling, J.D.; Gaster, L.M.; Reith, A.D.; Laping, N.J.; Hill, C.S. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2002, 62, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Callahan, J.F.; Burgess, J.L.; Fornwald, J.A.; Gaster, L.M.; Harling, J.D.; Harrington, F.P.; Heer, J.; Kwon, C.; Lehr, R.; Mathur, A.; et al. Identification of novel inhibitors of the transforming growth factor β1 (TGF-β1) type 1 receptor (ALK5). J. Med. Chem. 2002, 45, 999–1001. [Google Scholar] [CrossRef]
- Yingling, J.M.; Blanchard, K.L.; Sawyer, J.S. Development of TGF-β signalling inhibitors for cancer therapy. Nat. Rev. Drug Discov. 2004, 3, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- DaCosta Byfield, S.; Major, C.; Laping, N.J.; Roberts, A.B. SB-505124 is a selective inhibitor of transforming growth factor-beta type I receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2004, 65, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Gellibert, F.; Fouchet, M.H.; Nguyen, V.L.; Wang, R.; Krysa, G.; de Gouville, A.C.; Huet, S.; Dodic, N. Design of novel quinazoline derivatives and related analogues as potent and selective ALK5 inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 2277–2281. [Google Scholar] [CrossRef]
- Bonafoux, D.; Chuaqui, C.; Boriack-Sjodin, P.A.; Fitch, C.; Hankins, G.; Josiah, S.; Black, C.; Hetu, G.; Ling, L.; Lee, W.-C. 2-Aminoimidazoles inhibitors of TGF-β receptor 1. Bioorg. Med. Chem. Lett. 2009, 19, 912–916. [Google Scholar] [CrossRef]
- Sawyer, J.S.; Beight, D.W.; Britt, K.S.; Anderson, B.D.; Campbell, R.M.; Goodson, T.J.; Herron, D.K.; Li, H.-Y.; McMillen, W.T.; Mort, N.; et al. Synthesis and activity of new aryl- and heteroaryl-substituted 5,6-dihydro-4H-pyrrolo[1,2-b]pyrazole inhibitors of the transforming growth factor-beta type I receptor kinase domain. Bioorg. Med. Chem. Lett. 2004, 14, 3581–3584. [Google Scholar] [CrossRef]
- Tojo, M.; Hamashima, Y.; Hanyu, A.; Kajimoto, T.; Saitoh, M.; Miyazono, K.; Node, M.; Imamura, T. The ALK-5 inhibitor A-83-01 inhibits Smad signaling and epithelial-to-mesenchymal transition by transforming growth factor-beta. Cancer Sci. 2005, 96, 791–800. [Google Scholar] [CrossRef]
- Amada, H.; Sekiguchi, Y.; Ono, N.; Matsunaga, Y.; Koami, T.; Asanuma, H.; Shiozawa, F.; Endo, M.; Ikeda, A.; Aoki, M.; et al. Design, synthesis, and evaluation of novel 4-thiazolylimidazoles as inhibitors of transforming growth factor-β type I receptor kinase. Bioorg. Med. Chem. Lett. 2012, 22, 2024–2029. [Google Scholar] [CrossRef]
- Kim, D.K.; Jung, S.H.; Lee, H.S.; Dewang, P.M. Synthesis and biological evaluation of benzenesulfonamide-substituted 4-(6-alkylpyridin-2-yl)-5-(quinoxalin-6-yl)imidazoles as transforming growth factor-β type 1 receptor kinase inhibitors. Eur. J. Med. Chem. 2009, 44, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.W.; Kim, Y.K.; Lee, J.Y.; Chang, K.T.; Lee, H.J.; Kim, D.-K.; Sheen, Y.Y. Pharmacokinetics and tissue distribution of 3-((5-(6-methylpyridin-2-yl)-4-(quinoxalin-6-yl)-1 H-imidazol-2-yl) methyl) benzamide; a novel ALK5 inhibitor and a potential anti-fibrosis drug. Xenobiotica 2008, 38, 325–339. [Google Scholar] [CrossRef]
- Kim, D.K.; Lee, Y.I.; Lee, Y.W.; Dewang, P.M.; Sheen, Y.Y.; Kim, Y.W.; Park, H.-J.; Yoo, J.; Lee, H.S.; Kim, Y.-K. Synthesis and biological evaluation of 4(5)-(6-methylpyridin-2-yl)imidazoles and -pyrazoles as transforming growth factor-β type 1 receptor kinase inhibitors. Bioorg. Med. Chem. 2010, 18, 4459–4467. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.H.; Krishnaiah, M.; Sreenu, D.; Subrahmanyam, V.B.; Rao, K.S.; Lee, H.J.; Park, S.-J.; Park, H.-J.; Lee, K.; Sheen, Y.Y.; et al. Discovery of N-((4-([1,2,4]Triazolo[1,5-a]pyridin-6-yl)-5-(6-methylpyridin-2-yl)-1H-imidazol-2-yl)methyl)-2-fluoroaniline (EW-7197): A Highly Potent, Selective, and Orally Bioavailable Inhibitor of TGF-β Type I Receptor Kinase as Cancer Immunotherapeutic. J. Med. Chem. 2014, 57, 4213–4238. [Google Scholar] [CrossRef] [PubMed]
- Krishnaiah, M.; Jin, C.H.; Sheen, Y.Y.; Kim, D.K. Synthesis and biological evaluation of 5-(fluoro-substituted-6-methylpyridin-2-yl)-4-([1,2,4]triazolo[1,5-a]pyridin-6-yl)imidazoles as inhibitors of transforming growth factor-β type I receptor kinase. Bioorg. Med. Chem. Lett. 2015, 25, 5228–5231. [Google Scholar] [CrossRef]
- Guo, Z.; Song, X.; Zhao, L.M.; Piao, M.G.; Quan, J.; Piao, H.R.; Jin, C.H. Synthesis and biological evaluation of novel benzo[c][1,2,5]thiadiazol-5-yl and thieno[3,2-c]-pyridin-2-yl imidazole derivatives as ALK5 inhibitors. Bioorg. Med. Chem. Lett. 2019, 29, 2070–2075. [Google Scholar] [CrossRef]
- Krishnaiah, M.; Jin, C.H.; Sreenu, D.; Subrahmanyam, V.B.; Rao, K.S.; Son, D.H.; Park, H.-J.; Kim, S.W.; Sheen, Y.Y.; Kim, D.-K. Synthesis and biological evaluation of 2-benzylamino-4(5)-(6-methylpyridin-2-yl)-5(4)-([1,2,4]triazolo[1,5-a]-pyridin-6-yl)thiazoles as transforming growth factor-β type 1 receptor kinase inhibitors. Eur. J. Med. Chem. 2012, 57, 74–84. [Google Scholar] [CrossRef]
- Kim, D.K.; Kim, J.; Park, H.J. Synthesis and biological evaluation of novel 2-pyridinyl-[1,2,3]triazoles as inhibitors of transforming growth factor β1 type 1 receptor. Bioorg. Med. Chem. Lett. 2004, 14, 2401–2405. [Google Scholar] [CrossRef]
- Kim, D.K.; Choi, J.H.; An, Y.J.; Lee, H.S. Synthesis and biological evaluation of 5-(pyridin-2-yl)thiazoles as transforming growth factor-β type1 receptor kinase inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 2122–2127. [Google Scholar] [CrossRef]
- Kim, D.K.; Kim, J.; Park, H.J. Design, synthesis, and biological evaluation of novel 2-pyridinyl-[1,2,4] triazoles as inhibitors of transforming growth factor β1 type 1 receptor. Bioorg. Med. Chem. 2004, 12, 2013–2020. [Google Scholar] [CrossRef]
- Du, Y.; Xie, C.; Ravikumar, S.; Orme, J.; Li, L.; Zhou, X.J.; Mohan, C. Heightened Crescentic Glomerulonephritis in Immune Challenged 129sv Mice Is TGF-β/Smad3 Dependent. Int. J. Mol. Sci. 2021, 22, 2059. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Crosby, A.; Yang, X.; Southwood, M.; Upton, P.D.; Kim, D.-K.; Morrell, N.W. Altered bone morphogenetic protein and transforming growth factor-β signaling in rat models of pulmonary hypertension: Potential for activin receptor-like kinase-5 inhibition in prevention and progression of disease. Circulation 2009, 119, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.W.; Kim, Y.K.; Kim, D.-K.; Sheen, Y.Y. Identification of human cytochrome P450 enzymes involved in the metabolism of IN-1130, a novel activin receptor-like kinase-5 (ALK5) inhibitor. Xenobiotica 2008, 38, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.-K.; Piao, S.; Shin, H.-Y.; Choi, M.J.; Zhang, L.W.; Jin, H.-R.; Kim, W.J.; Han, J.-Y.; Hong, S.S.; Park, S.H.; et al. IN-1130, a novel transforming growth factor-β type I receptor kinase (activin receptor-like kinase 5) inhibitor, promotes regression of fibrotic plaque and corrects penile curvature in a rat model of Peyronie’s disease. J. Sex. Med. 2009, 6, 1284–1296. [Google Scholar] [CrossRef]
- Dewang, P.M.; Kim, D.K. Synthesis and biological evaluation of 2-pyridyl-substituted pyrazoles and imidazoles as transforming growth factor-β type 1 receptor kinase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 4228–4232. [Google Scholar] [CrossRef]
- Jin, C.H.; Krishnaiah, M.; Sreenu, D.; Rao, K.S.; Subrahmanyam, V.B.; Park, C.Y.; Son, J.-Y.; Sheen, Y.Y.; Kim, D.-K. Synthesis and biological evaluation of 1-substituted-3(5)-(6-methylpyridin-2-yl)-4-(quinolin-6-yl)pyrazoles as transforming growth factor-β type 1 receptor kinase inhibitors. Bioorg. Med. Chem. 2011, 19, 2633–2640. [Google Scholar] [CrossRef]
- Jin, C.H.; Sreenu, D.; Krishnaiah, M.; Subrahmanyam, V.B.; Rao, K.S.; Nagendra Mohan, A.V.; Park, C.-Y.; Son, J.-Y.; Son, D.-H.; Park, H.-J.; et al. Synthesis and biological evaluation of 1-substituted-3(5)-(6-methylpyridin-2-yl)-4-(quinoxalin-6-yl)pyrazoles as transforming growth factor-β type 1 receptor kinase inhibitors. Eur. J. Med. Chem. 2011, 46, 3917–3925. [Google Scholar] [CrossRef]
- Jin, C.H.; Krishnaiah, M.; Sreenu, D.; Subrahmanyam, V.B.; Rao, K.S.; Mohan, A.V.N.; Park, C.-Y.; Son, J.-Y.; Sheen, Y.Y.; Kim, D.-K. Synthesis and biological evaluation of 1-substituted-3-(6-methylpyridin-2-yl)-4-([1,2,4triazolo[1,5-apyridin-6-yl)pyrazoles as transforming growth factor-β type 1 receptor kinase inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 6049–6053. [Google Scholar] [CrossRef]
- Jin, C.H.; Krishnaiah, M.; Sreenu, D.; Subrahmanyam, V.B.; Park, H.J.; Park, S.J.; Sheen, Y.Y.; Kim, D.-K. 4-([1,2,4]Triazolo[1,5-a]pyridin-6-yl)-5(3)-(6-methylpyridin-2-yl)imidazole and -pyrazole derivatives as potent and selective inhibitors of transforming growth factor-β type I receptor kinase. Bioorg. Med. Chem. 2014, 22, 2724–2732. [Google Scholar] [CrossRef]
- Xu, G.; Zhang, Y.; Wang, H.; Guo, Z.; Wang, X.; Li, X.; Chang, S.; Sun, T.; Yu, Z.; Xu, T.; et al. Synthesis and biological evaluation of 4-(pyridin-4-oxy)-3-(3,3-difluorocyclobutyl)-pyrazole derivatives as novel potent transforming growth factor-β type 1 receptor inhibitors. Eur. J. Med. Chem. 2020, 198, 112354. [Google Scholar] [CrossRef]
- Tan, B.; Zhang, X.; Quan, X.; Zheng, G.; Li, X.; Zhao, L.; Li, W.; Li, B. Design, synthesis and biological activity evaluation of novel 4-((1-cyclopropyl-3-(tetrahydro-2H-pyran-4-yl)-1H-pyrazol-4-yl) oxy) pyridine-2-yl) amino derivatives as potent transforming growth factor-β (TGF-β) type I receptor inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 127339. [Google Scholar] [CrossRef] [PubMed]
- Meldal, M.; Tornøe, C.W. Cu-catalyzed azide− alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Park, Y.; Hah, J.M.; Ryu, J.S. Synthesis and biological evaluation of 1-(6-methylpyridin-2-yl)-5-(quinoxalin-6-yl)-1,2,3-triazoles as transforming growth factor-β type 1 receptor kinase inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, T.F.; Seibel, G.L.; Kassis, S.; Laydon, J.T.; Blumenthal, M.J.; Lee, J.C.; Lee, D.; Boehm, J.C.; Fier-Thompson, S.M.; Abt, J.W.; et al. Regulation of stress-induced cytokine production by pyridinylimidazoles; inhibition of CSBP kinase. Bioorg. Med. Chem. 1997, 5, 49–64. [Google Scholar] [CrossRef]
- Patel, H.M.; Sing, B.; Bhardwaj, V.; Palkar, M.; Shaikh, M.S.; Rane, R.; Alwan, W.S.; Gadad, A.K.; Noolvi, M.N.; Karpoormath, R. Design, synthesis and evaluation of small molecule imidazo[2,1-b][1,3,4]thiadiazoles as inhibitors of transforming growth factor-β type-I receptor kinase (ALK5). Eur. J. Med. Chem. 2015, 93, 599–613. [Google Scholar] [CrossRef]
- Goldberg, F.W.; Ward, R.A.; Powell, S.J.; Debreczeni, J.E.; Norman, R.A.; Roberts, N.J.; Dishington, A.P.; Gingell, H.J.; Wickson, K.F.; Roberts, A.L. Rapid generation of a high quality lead for transforming growth factor-beta (TGF-β) type I receptor (ALK5). J. Med. Chem. 2009, 52, 7901–7905. [Google Scholar] [CrossRef]
- Harikrishnan, L.S.; Warrier, J.; Tebben, A.J.; Tonukunuru, G.; Madduri, S.R.; Baligar, V.; Mannoori, R.; Seshadri, B.; Rahaman, H.; Arunachalam, P.; et al. Heterobicyclic inhibitors of transforming growth factor beta receptor I (TGFβRI). Bioorg. Med. Chem. 2018, 26, 1026–1034. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, Y.; Tebben, A.J.; Sheriff, S.; Ruzanov, M.; Fereshteh, M.P.; Fan, Y.; Lippy, J.; Swanson, J.; Ho, C.-P.; et al. Discovery of 4-Azaindole Inhibitors of TGFβRI as Immuno-oncology Agents. ACS Med. Chem. Lett. 2018, 9, 1117–1122. [Google Scholar] [CrossRef]
- Sabat, M.; Wang, H.; Scorah, N.; Lawson, J.D.; Atienza, J.; Kamran, R.; Hixon, M.S.; Dougan, D.R. Design, synthesis and optimization of 7-substituted-pyrazolo[4,3-b]pyridine ALK5 (activin receptor-like kinase 5) inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 1955–1961. [Google Scholar] [CrossRef]
- Chaudhary, N.I.; Roth, G.J.; Hilberg, F.; Müller-Quernheim, J.; Prasse, A.; Zissel, G.; Schnapp, A.; Park, J.E. Inhibition of PDGF, VEGF and FGF signalling attenuates fibrosis. Eur. Respir. J. 2007, 29, 976–985. [Google Scholar] [CrossRef]
- Roth, G.J.; Heckel, A.; Colbatzky, F.; Handschuh, S.; Kley, J.; Lehmann-Lintz, T.; Lotz, R.; Tontsch-Grunt, U.; Walter, R.; Hilberg, F. Design, synthesis, and evaluation of indolinones as triple angiokinase inhibitors and the discovery of a highly specific 6-methoxycarbonyl-substituted indolinone (BIBF 1120). J. Med. Chem. 2009, 52, 4466–4480. [Google Scholar] [CrossRef]
- Roth, G.J.; Heckel, A.; Brandl, T.; Grauert, M.; Hoerer, S.; Kley, J.T.; Schnapp, G.; Baum, P.; Mennerich, D.; Schnapp, A.; et al. Design, synthesis, and evaluation of indolinones as inhibitors of the transforming growth factor β receptor I (TGFβRI). J. Med. Chem. 2010, 53, 7287–7295. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babiker, N.A.; Nadeem, S.; Abu Kariem, H.; Abdul Hameed, A.; Negmeldin, A.T.; El-labbad, E.M. Medicinal Chemistry Strategies in Targeting TGF-βR1 Kinase Domain: Unveiling Insights into Inhibitor Structure–Activity Relationship (SAR). Pharmaceuticals 2025, 18, 716. https://doi.org/10.3390/ph18050716
Babiker NA, Nadeem S, Abu Kariem H, Abdul Hameed A, Negmeldin AT, El-labbad EM. Medicinal Chemistry Strategies in Targeting TGF-βR1 Kinase Domain: Unveiling Insights into Inhibitor Structure–Activity Relationship (SAR). Pharmaceuticals. 2025; 18(5):716. https://doi.org/10.3390/ph18050716
Chicago/Turabian StyleBabiker, Nusaiba A., Soam Nadeem, Hasan Abu Kariem, Afra Abdul Hameed, Ahmed T. Negmeldin, and Eman M. El-labbad. 2025. "Medicinal Chemistry Strategies in Targeting TGF-βR1 Kinase Domain: Unveiling Insights into Inhibitor Structure–Activity Relationship (SAR)" Pharmaceuticals 18, no. 5: 716. https://doi.org/10.3390/ph18050716
APA StyleBabiker, N. A., Nadeem, S., Abu Kariem, H., Abdul Hameed, A., Negmeldin, A. T., & El-labbad, E. M. (2025). Medicinal Chemistry Strategies in Targeting TGF-βR1 Kinase Domain: Unveiling Insights into Inhibitor Structure–Activity Relationship (SAR). Pharmaceuticals, 18(5), 716. https://doi.org/10.3390/ph18050716