
Exploring the Protective Effects of Traditional Antidiabetic Medications and Novel Antihyperglycemic Agents in Diabetic Rodent Models

 ,
,  , ,
, ,
Abstract
1. Introduction
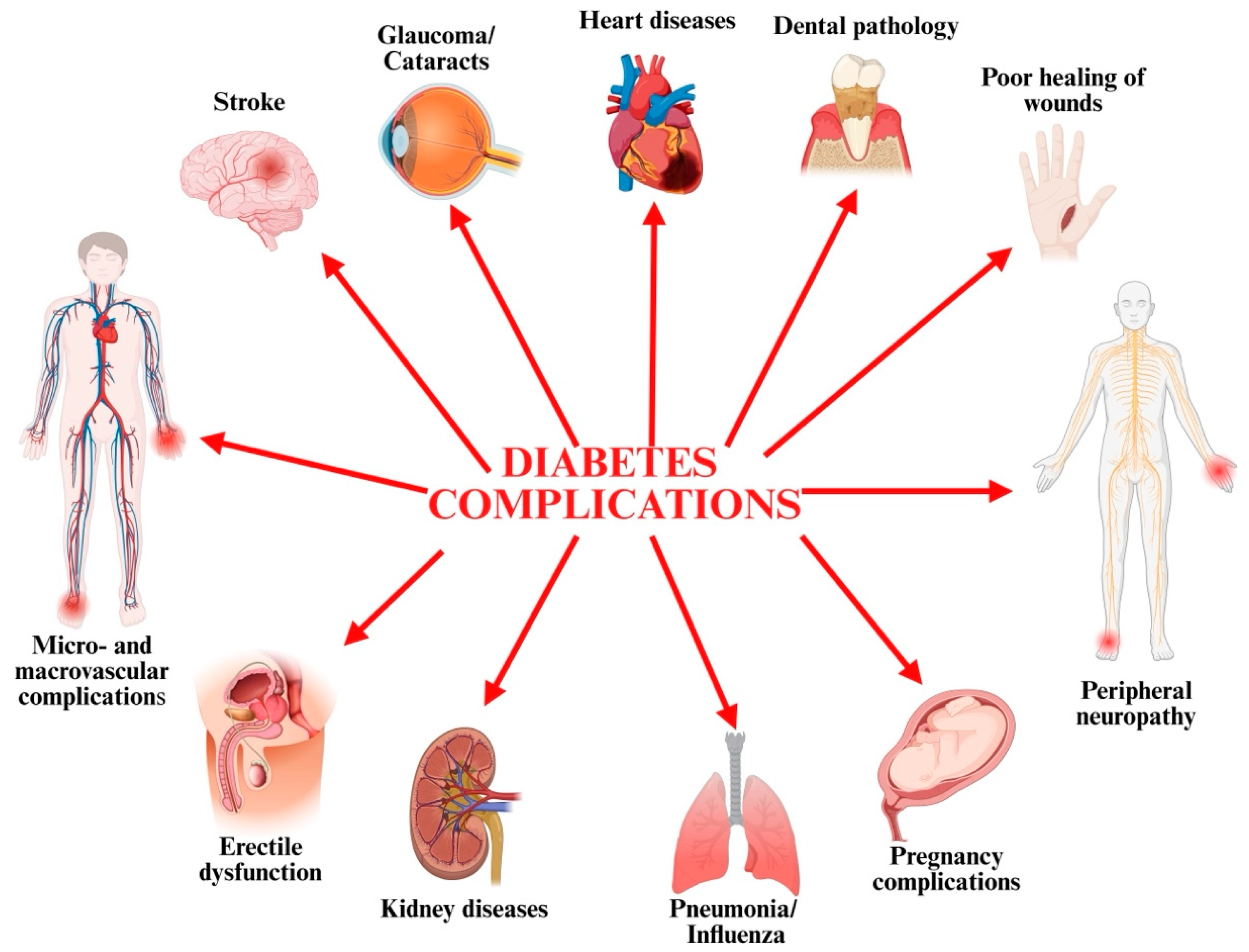
2. Structural Organ Changes in T2D
3. Metformin—Traditional Antidiabetic Agent
4. Novel Antihyperglycemic Agents in the Therapy of T2D
5. Experimental Studies on Organ Protection Provided by Traditional and Novel Antidiabetic Medication
5.1. MET’s Effects
5.2. The Effects of SGLT-2 Inhibitors
5.3. The Effects of GLP-1 Agonists
5.4. TZP’s Effects
6. Limitations
7. Future Perspectives and Clinical Implications
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guo, H.; Wu, H.; Li, Z. The pathogenesis of diabetes. Int. J. Mol. Sci. 2023, 24, 6978. [Google Scholar] [CrossRef]
- Durruty, P.; Sanzana, M.; Sanhueza, L. Pathogenesis of Type 2 Diabetes Mellitus; IntechOpen: London, UK, 2019; pp. 1–14. [Google Scholar]
- Skyler, J.S.; Bakris, G.L.; Bonifacio, E.; Darsow, T.; Eckel, R.H.; Groop, L.; Groop, P.-H.; Handelsman, Y.; Insel, R.A.; Mathieu, C.; et al. Differentiation of diabetes by pathophysiology, natural history, and prognosis. Diabetes 2017, 66, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R. Overview and new insights into the metabolic syndrome: Risk factors and emerging variables in the development of type 2 diabetes and cerebrocardiovascular disease. Medicina 2023, 59, 561. [Google Scholar] [CrossRef]
- Zhao, X.; An, X.; Yang, C.; Sun, W.; Ji, H.; Lian, F. Crucial role and mechanism of insulin resistance in metabolic disease. Front. Endocrinol. 2023, 14, 1149239. [Google Scholar] [CrossRef] [PubMed]
- Baro, B.; Pegu, R.; Hazarika, B.; Sarma, U.; Rabha, G.; Kalita, S. A study of histopathological changes in diabetic and nondiabetic cadaveric pancreas with respect to diabetic status. J. Cardiovasc. Dis. Res. 2024, 15, 3826–3835. [Google Scholar]
- Burns, C.; Francis, N. Type 2 Diabetes-etiology, epidemiology, pathogenesis, treatment. In Metabolic Syndrome; Ahima, R.S., Ed.; Springer: Cham, Switzerland, 2023. [Google Scholar]
- Rojas, A.; Lindner, C.; Schneider, I.; Gonzalez, I.; Uribarri, J. The RAGE axis: A relevant inflammatory hub in human diseases. Biomolecules 2024, 14, 412. [Google Scholar] [CrossRef]
- Pedreanez, A.; Robalino, J.; Tene, D.; Salazar, P. Advanced glycation end products of dietary origin and their association with inflammation in diabetes—A minireview. Endocr. Regul. 2024, 58, 57–67. [Google Scholar] [CrossRef]
- Yang, T.; Qi, F.; Guo, F.; Shao, M.; Song, Y.; Ren, G.; Linlin, Z.; Qin, G.; Zhao, Y. An update on chronic complications of diabetes mellitus: From molecular mechanisms to therapeutic strategies with a focus on metabolic memory. Mol. Med. 2024, 30, 71. [Google Scholar] [CrossRef]
- Liu, H.; Wang, X.; Gao, H.; Yang, C.; Xie, C. Physiological and pathological characteristics of vascular endothelial injury in diabetes and the regulatory mechanism of autophagy. Front. Endocrinol. 2023, 14, 1191426. [Google Scholar] [CrossRef]
- Sanches, J.M.; Na Zhao, L.; Salehi, A.; Wollheim, C.B.; Kaldis, P. Pathophysiology of type 2 diabetes and the impact of altered metabolic interorgan crosstalk. FEBS J. 2023, 290, 620–648. [Google Scholar] [CrossRef]
- Bodhini, D.; Morton, R.W.; Santhakumar, V.; Nakabuye, M.; Pomares-Millan, H.; Clemmensen, C.; Fitzpatrick, S.L.; Guasch-Ferre, M.; Pankow, J.S.; Ried-Larsen, M.; et al. Impact of individual and environmental factors on dietary or lifestyle interventions to prevent type 2 diabetes development: A systematic review. Commun. Med. 2023, 3, 133. [Google Scholar] [CrossRef] [PubMed]
- Gancheva, S.; Jelenik, T.; Álvarez-Hernández, E.; Roden, M. Interorgan metabolic crosstalk in human insulin resistance. Physiol. Rev. 2018, 98, 1371–1415. [Google Scholar] [CrossRef]
- Casado, M.E.; Collado-Pérez, R.; Frago, L.M.; Barrios, V. Recent advances in the knowledge of the mechanisms of leptin physiology and actions in neurological and metabolic pathologies. Int. J. Mol. Sci. 2023, 24, 1422. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.P.; Goodwin, J.E. Loss of endothelial glucocorticoid receptor accelerates organ fibrosis in db/db mice. Am. J. Physiol. Renal. Physiol. 2023, 325, F519–F526. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Liu, S.; Gao, M.; Wang, W.; Chen, K.; Huang, L.; Liu, Y. Diabetic vascular diseases: Molecular mechanisms and therapeutic strategies. Signal Transduct. Target. Ther. 2023, 8, 152. [Google Scholar] [CrossRef]
- Yu, M.G.; Gordin, D.; Fu, J.; Park, K.; Li, Q.; King, G.L. Protective factors and the pathogenesis of complications in diabetes. Endocr. Rev. 2023, 45, 227–252. [Google Scholar] [CrossRef]
- Yan, Z.; Cai, M.; Han, X.; Chen, Q.; Lu, H. The interaction between age and risk factors for diabetes and prediabetes: A community-based cross-sectional study. Diabetes Metab. Syndr. Obes. 2023, 16, 85–93. [Google Scholar] [CrossRef]
- Banday, M.Z.; Sameer, A.S.; Nissar, S. Pathophysiology of diabetes: An overview. Avicenna J. Med. 2020, 10, 174–188. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, W.; Liu, J.; Xie, M.; Liu, Q.; Li, S. Vascular complications of diabetes: A narrative review. Medicine 2023, 102, e35285. [Google Scholar] [CrossRef]
- Wilson, V. An overview of complications associated with type 1 and type 2 diabetes. Nurs. Stand. 2023, 38, 77–82. [Google Scholar] [CrossRef]
- Taylor, S.I.; Yazdi, Z.S.; Beitelshees, A.L. Pharmacological treatment of hyperglycemia in type 2 diabetes. J. Clin. Investig. 2021, 131, e142243. [Google Scholar] [CrossRef]
- ElSayed, N.A.; Aleppo, G.; Aroda, V.R.; Bannuru, R.R.; Brown, F.M.; Bruemmer, D.; Collins, B.S.; Hilliard, M.E.; Isaacs, D.; Johnson, E.L.; et al. 9. Pharmacologic approaches to glycemic treatment: Standards of care in diabetes—2023. Diabetes Care 2023, 46 (Suppl. S1), S140–S157. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J. Clinical pharmacology of antidiabetic drugs: What can be expected of their use? Presse Med. 2023, 52, 104158. [Google Scholar] [CrossRef] [PubMed]
- DeMarsilis, A.; Reddy, N.; Boutari, C.; Filippaios, A.; Sternthal, E.; Katsiki, N.; Mantzoros, C. Pharmacotherapy of type 2 diabetes: An update and future directions. Metabolism 2022, 137, 155332. [Google Scholar] [CrossRef]
- Aronne, L.J.; Sattar, N.; Horn, D.B.; Bays, H.E.; Wharton, S.; Lin, W.-Y.; Ahmad, N.N.; Zhang, S.; Liao, R.; Bunck, M.C.; et al. Continued treatment with tirzepatide for maintenance of weight reduction in adults with obesity: The SURMOUNT-4 randomized clinical trial. JAMA 2024, 331, 38–48. [Google Scholar] [CrossRef]
- Lisco, G.; Disoteo, O.E.; De Geronimo, V.; De Tullio, A.; Giagulli, V.A.; Guastamacchia, E.; De Pergola, G.; Jirillo, E.; Triggiani, V. Is tirzepatide the new game changer in type 2 diabetes? Endocrines 2024, 5, 72–86. [Google Scholar] [CrossRef]
- Ma, J.; Liu, M.; Wang, R.; Du, L.; Ji, L. Efficacy and safety of tirzepatide in people with type 2 diabetes by baseline body mass index: An exploratory subgroup analysis of SURPASS-AP-Combo. Diabetes Obes. Metab. 2024, 26, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Kaplan, L.M.; Frias, J.P.; Brouwers, B.; Wu, Q.; Thomas, M.K.; Harris, C.; Schloot, N.C.; Du, Y.; Mather, K.J.; et al. Triple hormone receptor agonist retatrutide for metabolic dysfunction-associated steatotic liver disease: A randomized phase 2a trial. Nat. Med. 2024, 30, 2037–2048. [Google Scholar] [CrossRef]
- Giri, B.; Dey, S.; Das, T.; Sarkar, M.; Banerjee, J.; Dash, S.K. Chronic hyperglycemia mediated physiological alteration and metabolic distortion leads to organ dysfunction, infection, cancer progression and other pathophysiological consequences: An update on glucose toxicity. Biomed. Pharmacother. 2018, 107, 306–328. [Google Scholar] [CrossRef]
- Hu, Q.; Chen, Y.; Deng, X.; Li, Y.; Ma, X.; Zeng, J.; Zhao, Y. Diabetic nephropathy: Focusing on pathological signals, clinical treatment, and dietary regulation. Biomed. Pharmacother. 2023, 159, 114252. [Google Scholar] [CrossRef]
- Kim, Y.H.; Saha, M.K.; Hu, Y.; Kumar, S.; Poulton, C.J.; Hogan, S.L.; Nachman, P.; Jennette, J.C.; Nast, C.C.; Mottl, A.K. Impact of diabetic lesions on pathology, treatment, and outcomes of glomerular diseases. Kidney 2023, 4, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Srivastava, S.; Singh, M.R.; Singh, D. Mechanistic insight into diabetic wounds: Pathogenesis, molecular targets and treatment strategies to pace wound healing. Biomed. Pharmacother. 2019, 112, 108615. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-S.; Lee, J.-S.; Park, J.-H.; Lee, E.-Y.; Moon, J.-S.; Lee, S.-K.; Lee, J.-S.; Kim, J.-H.; Kim, H.-S. Identification of novel biomarker for early detection of diabetic nephropathy. Biomedicines 2021, 9, 457. [Google Scholar] [CrossRef] [PubMed]
- Saleh, S.; Hanna, G.; El-Nabi, S.H.; El-Domiaty, H.; Shabaan, A.; Ewida, S.F. Dapagliflozin, a sodium glucose cotransporter 2 inhibitors, protects cardiovascular function in type-2 diabetic murine model. J. Genet. 2020, 99, 46. [Google Scholar] [CrossRef]
- Huo, J.-L.; Feng, Q.; Pan, S.; Fu, W.-J.; Liu, Z.; Liu, Z. Diabetic cardiomyopathy: Early diagnostic biomarkers, pathogenetic mechanisms, and therapeutic interventions. Cell Death Discov. 2023, 9, 256. [Google Scholar] [CrossRef]
- Cusi, K. Nonalcoholic fatty liver disease in diabetes: A call to action. Diabetes Spectr. 2024, 37, 5–7. [Google Scholar] [CrossRef]
- Dutta, B.J.; Singh, S.; Seksaria, S.; Das Gupta, G.; Singh, A. Inside the diabetic brain: Insulin resistance and molecular mechanism associated with cognitive impairment and its possible therapeutic strategies. Pharmacol. Res. 2022, 182, 106358. [Google Scholar] [CrossRef]
- Sugandh, F.; Chandio, M.; Raveena, F.; Kumar, L.; Karishma, F.; Khuwaja, S.; Memon, U.A.; Bai, K.; Kashif, M.; Varrassi, G.; et al. Advances in the management of diabetes mellitus: A focus on personalized medicine. Cureus 2023, 15, e43697. [Google Scholar] [CrossRef]
- Kannan, S.; Chellappan, D.K.; Kow, C.S.; Ramachandram, D.S.; Pandey, M.; Mayuren, J.; Dua, K.; Candasamy, M. Transform diabetes care with precision medicine. Health Sci. Rep. 2023, 6, e1642. [Google Scholar] [CrossRef]
- Raj, G.M.; Mathaiyan, J. Precision medicine in diabetes-Finally some light at the end of the tunnel? Br. J. Clin. Pharmacol. 2021, 87, 2625–2628. [Google Scholar] [CrossRef]
- Ahmad, E.; Sargeant, J.A.; Zaccardi, F.; Khunti, K.; Webb, D.R.; Davies, M.J. Where does metformin stand in modern day management of type 2 diabetes? Pharmaceuticals 2020, 13, 427. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.J. Metformin: Therapeutic profile in the treatment of type 2 diabetes. Diabetes Obes. Metab. 2024, 26 (Suppl. S3), 3–19. [Google Scholar] [CrossRef]
- Zhu, H.; Jia, Z.; Li, Y.R.; Danelisen, I. Molecular mechanisms of action of metformin: Latest advances and therapeutic implications. Clin. Exp. Med. 2023, 23, 2941–2951. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.; Williams, K.; Oroszi, T. Metformin—Pharmacokinetic and Pharmacodynamics Journey Through the Body. Pharmacol. Pharm. 2024, 15, 466–477. [Google Scholar] [CrossRef]
- Cheng, M.; Ren, L.; Jia, X.; Wang, J.; Cong, B. Understanding the action mechanisms of metformin in the gastrointestinal tract. Front. Pharmacol. 2024, 15, 1347047. [Google Scholar] [CrossRef] [PubMed]
- Buczyńska, A.; Sidorkiewicz, I.; Krętowski, A.J.; Adamska, A. Examining the clinical relevance of metformin as an antioxidant intervention. Front. Pharmacol. 2024, 15, 1330797. [Google Scholar] [CrossRef]
- Wang, Y.-W.; He, S.-J.; Feng, X.; Cheng, J.; Luo, Y.-T.; Tian, L.; Huang, Q. Metformin: A review of its potential indications. Drug Des. Dev. Ther. 2017, 11, 2421–2429. [Google Scholar] [CrossRef]
- Corcoran, C.; Jacobs, T.F. Metformin. In StatPearls [Internet]; [Updated 2023 Aug 17]; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Andraos, J.; Smith, S.R.; Tran, A.; Pham, D.Q. Narrative review of data supporting alternate first-line therapies over metformin in type 2 diabetes. J. Diabetes Metab. Disord. 2024, 23, 385–394. [Google Scholar] [CrossRef]
- Han, J.; Li, Y.; Liu, X.; Zhou, T.; Sun, H.; Edwards, P.; Gao, H.; Yu, F.-S.; Qiao, X. Metformin suppresses retinal angiogenesis and inflammation in vitro and in vivo. PLoS ONE 2018, 13, e0193031. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kim, M.; Choi, M.Y.; Lee, D.H.; Roh, G.S.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Kim, S.-J.; Yoo, J.-M.; et al. Metformin protects against retinal cell death in diabetic mice. Biochem. Biophys. Res. Commun. 2017, 492, 397–403. [Google Scholar] [CrossRef]
- Nahar, N.; Mohamed, S.; Mustapha, N.M.; Lau, S.; Ishak, N.I.M.; Umran, N.S. Metformin attenuated histopathological ocular deteriorations in a streptozotocin-induced hyperglycemic rat model. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2021, 394, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Oubaha, M.; Miloudi, K.; Dejda, A.; Guber, V.; Mawambo, G.; Germain, M.-A.; Bourdel, G.; Popovic, N.; Rezende, F.A.; Kaufman, R.J.; et al. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci. Transl. Med. 2016, 8, 362ra144. [Google Scholar] [CrossRef]
- Xu, L.; Kong, L.; Wang, J.; Ash, J.D. Stimulation of AMPK prevents degeneration of photoreceptors and the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2018, 115, 10475–10480. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Zhang, C.; Liu, D.; Wu, J.; Tian, H.; Lu, L.; Xu, G.-T.; Liu, F.; Zhang, J. Metformin protects ARPE-19 cells from glyoxal-induced oxidative stress. Oxid. Med. Cell. Longev. 2020, 2020, 1740943. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, L.; Jiang, Y.; Silva, M.; Zhen, X.; Zheng, W. Protective effect of metformin against hydrogen peroxide-induced oxidative damage in human retinal pigment epithelial (RPE) cells by enhancing autophagy through activation of AMPK pathway. Oxidative Med. Cell. Longev. 2020, 2020, 2524174. [Google Scholar] [CrossRef] [PubMed]
- Yi, Q.-Y.; Deng, G.; Chen, N.; Bai, Z.-S.; Yuan, J.-S.; Wu, G.-H.; Wang, Y.-W.; Wu, S.-J. Metformin inhibits development of diabetic retinopathy through inducing alternative splicing of VEGF-A. Am. J. Transl. Res. 2016, 8, 3947–3954. [Google Scholar] [PubMed]
- Zhang, Y.; Chen, F.; Wang, L. Metformin inhibits development of diabetic retinopathy through microRNA-497a-5p. Am. J. Transl. Res. 2017, 9, 5558–5566. [Google Scholar] [PubMed]
- Luodan, A.; Zou, T.; He, J.; Chen, X.; Sun, D.; Fan, X.; Xu, H. Rescue of retinal degeneration in rd1 mice by intravitreally injected metformin. Front. Mol. Neurosci. 2019, 12, 102. [Google Scholar] [CrossRef]
- Ying, Y.; Ueta, T.; Jiang, S.; Lin, H.; Wang, Y.; Vavvas, D.; Wen, R.; Chen, Y.-G.; Luo, Z. Metformin inhibits ALK1-mediated angiogenesis via activation of AMPK. Oncotarget 2017, 8, 32794–32806. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Xiao, J.; Xie, B.; Barba, H.; Boachie-Mensah, M.; Shah, R.N.; Nadeem, U.; Spedale, M.; Dylla, N.; Lin, H.; et al. Oral metformin inhibits choroidal neovascularization by modulating the gut-retina axis. Investig. Opthalmol. Vis. Sci. 2023, 64, 21. [Google Scholar] [CrossRef]
- Xiao, J.F.; Luo, W.; Mani, A.; Barba, H.; Solanki, A.; Droho, S.; Lavine, J.A.; Skondra, D. Intravitreal Metformin Protects Against Choroidal Neovascularization and Light-Induced Retinal Degeneration. Int. J. Mol. Sci. 2024, 25, 11357. [Google Scholar] [CrossRef]
- Wang, X.; Liang, X.; Huang, S.; Wei, M.; Xu, Y.; Chen, X.; Miao, Y.; Zong, R.; Lin, X.; Li, S.; et al. Metformin inhibits pathological retinal neovascularization but promotes retinal fibrosis in experimental neovascular age-related macular degeneration. Front. Pharmacol. 2025, 16, 1547492. [Google Scholar] [CrossRef] [PubMed]
- Hasan, I.; Rashid, T.; Jaikaransingh, V.; Heilig, C.; Abdel-Rahman, E.M.; Awad, A.S. SGLT2 inhibitors: Beyond glycemic control. J. Clin. Transl. Endocrinol. 2024, 35, 100335. [Google Scholar] [CrossRef] [PubMed]
- Yankah, R.K.; Anku, E.K.; Eligar, V. Sodium-glucose cotransporter-2 inhibitors and cardiovascular protection among patients with type 2 diabetes mellitus: A systematic review. J. Diabetes Res. 2024, 2024, 9985836. [Google Scholar] [CrossRef]
- Seidu, S.; Alabraba, V.; Davies, S.; Newland-Jones, P.; Fernando, K.; Bain, S.C.; Diggle, J.; Evans, M.; James, J.; Kanumilli, N.; et al. SGLT2 Inhibitors—The new standard of care for cardiovascular, renal and metabolic protection in type 2 diabetes: A narrative review. Diabetes Ther. 2024, 15, 1099–1124. [Google Scholar] [CrossRef] [PubMed]
- Ciardullo, S.; Morieri, M.L.; Daniele, G.; Fiorentino, T.V.; Mezza, T.; Tricò, D.; Consoli, A.; Del Prato, S.; Giorgino, F.; Piro, S.; et al. GLP1-GIP receptor co-agonists: A promising evolution in the treatment of type 2 diabetes. Acta Diabetol. 2024, 61, 941–950. [Google Scholar] [CrossRef]
- Pescariu, S.A.; Elagez, A.; Nallapati, B.; Bratosin, F.; Bucur, A.; Negru, A.; Gaita, L.; Citu, I.M.; Popa, Z.L.; Barata, P.I. Examining the impact of ertugliflozin on cardiovascular outcomes in patients with diabetes and metabolic syndrome: A systematic review of clinical trials. Pharmaceuticals 2024, 17, 929. [Google Scholar] [CrossRef]
- Cheng, Q.; Zou, S.; Feng, C.; Xu, C.; Zhao, Y.; Shi, X.; Sun, M. Effect of ertugliflozin on renal function and cardiovascular outcomes in patients with type 2 diabetes mellitus: A systematic review and meta-analysis. Medicine 2023, 102, e33198. [Google Scholar] [CrossRef]
- Collins, L.; Costello, R.A. Glucagon-like peptide-1 receptor agonists. In StatPearls [Internet]; [Updated 2024 Feb 29]; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Mann, J.F.E.; Rossing, P.; Bakris, G.; Belmar, N.; Bosch-Traberg, H.; Busch, R.; Charytan, D.M.; Hadjadj, S.; Gillard, P.; Górriz, J.L.; et al. Effects of semaglutide with and without concomitant SGLT2 inhibitor use in participants with type 2 diabetes and chronic kidney disease in the FLOW trial. Nat. Med. 2024, 30, 2849–2856. [Google Scholar] [CrossRef]
- Zheng, Z.; Zong, Y.; Ma, Y.; Tian, Y.; Pang, Y.; Zhang, C.; Gao, J. Glucagon-like peptide-1 receptor: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2024, 9, 234. [Google Scholar] [CrossRef]
- Ilias, I.; Zabuliene, L.; Rizzo, M. GLP-1 receptor agonists in diabetes and weight loss: The double-edged sword of innovation and risks. Front. Clin. Diabetes Healthc. 2025, 5, 1530811. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Zhang, R.; Yao, Y.; Wu, Q.; Zhang, J. Tirzepatide as a novel effective and safe strategy for treating obesity: A systematic review and meta-analysis of randomized controlled trials. Front. Public Health 2024, 12, 1277113. [Google Scholar] [CrossRef] [PubMed]
- Mahar, M.U.; Mahmud, O.; Ahmed, S.; Qureshi, S.A.; Kakar, W.G.; Fatima, S.S. The effects of tirzepatide on lipid profile: A systematic review and meta-analysis of randomized controlled trials. J. Obes. Metab. Syndr. 2024, 33, 348–359. [Google Scholar] [CrossRef]
- France, N.L.; Syed, Y.Y. Tirzepatide: A review in type 2 diabetes. Drugs 2024, 84, 227–238. [Google Scholar] [CrossRef]
- Fontanella, R.A.; Ghosh, P.; Pesapane, A.; Taktaz, F.; Puocci, A.; Franzese, M.; Feliciano, M.F.; Tortorella, G.; Scisciola, L.; Sommella, E.; et al. Tirzepatide prevents neurodegeneration through multiple molecular pathways. J. Transl. Med. 2024, 22, 114. [Google Scholar] [CrossRef]
- Almuttairi, R.S. The effects of metformin treatment on diabetic albino rats’ pancreas, liver, and kidney histology. Arch. Razi Inst. 2023, 78, 459–463. [Google Scholar] [CrossRef]
- Salem, A.A.M. Biochemical and histopathological evaluation of the effect of metformin and metformin nanoparticles against alloxan-induced diabetes in Rats. Benha Veter. Med. J. 2022, 42, 39–45. [Google Scholar] [CrossRef]
- Mobasher, M.A.; El-Tantawi, H.G.; El-Said, K.S. Metformin ameliorates oxidative stress induced by diabetes mellitus and hepatocellular carcinoma in rats. Rep. Biochem. Mol. Biol. 2020, 9, 115–128. [Google Scholar] [CrossRef]
- Dallak, M.; Bin-Jaliah, I.; Al-Hashem, F.; Kamar, S.S.; Abdel Kader, D.H.; Amin, S.N.; Haidara, M.A.; Al-Ani, B. Metformin pretreatment ameliorates diabetic nephropathy induced by a combination of high fat diet and streptozotocin in rats. Int. J. Morphol. 2018, 36, 969–974. [Google Scholar] [CrossRef]
- Zhang, S.; Xu, H.; Yu, X.; Wu, Y.; Sui, D. Metformin ameliorates diabetic nephropathy in a rat model of low-dose streptozotocin-induced diabetes. Exp. Ther. Med. 2017, 14, 383–390. [Google Scholar] [CrossRef]
- Dawood, A.F.; Alzamil, N.M.; Hewett, P.W.; Momenah, M.A.; Dallak, M.; Kamar, S.S.; Kader, D.H.A.; Yassin, H.; Haidara, M.A.; Maarouf, A.; et al. Metformin protects against diabetic cardiomyopathy: An association between desmin-sarcomere injury and the iNOS/mTOR/TIMP-1 fibrosis axis. Biomedicines 2022, 10, 984. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, M.; Zhang, Y.; Cai, F.; Jiang, B.; Zha, W.; Yu, W. Metformin ameliorates diabetic cardiomyopathy by activating the PK2/PKR pathway. Front. Physiol. 2020, 11, 425. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.; Chen, Z.; Kim, K.T.; Sun, J.; Xue, M.; Chen, G.; Li, S.; Shen, Y.; Zhu, Z.; Wang, X.; et al. Metformin alleviates hyperglycemia-induced endothelial impairment by downregulating autophagy via the Hedgehog pathway. Autophagy 2019, 15, 843–870. [Google Scholar] [CrossRef]
- Yan, Y.; Li, T.; Li, Z.; He, M.; Wang, D.; Xu, Y.; Yang, X.; Bai, Y.; Lao, Y.; Zhang, Z.; et al. Metformin suppresses the progress of diabetes-accelerated atherosclerosis by inhibition of vascular smooth muscle cell migration through AMPK-Pdlim5 pathway. Front. Cardiovasc. Med. 2021, 8, 690627. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Yamanaka, Y.; Tokuda, K.; Sato, N.; Tajima, T.; Tsukamoto, M.; Suzuki, H.; Kawasaki, M.; Nakamura, E.; Sakai, A. Effects of metformin on knee joint capsule fibrosis in a diabetic mouse model. Bone Jt. Res. 2024, 13, 321–331. [Google Scholar] [CrossRef]
- Han, J.-X.; Luo, L.-L.; Wang, Y.-C.; Miyagishi, M.; Kasim, V.; Wu, S.-R. SGLT2 inhibitor empagliflozin promotes revascularization in diabetic mouse hindlimb ischemia by inhibiting ferroptosis. Acta Pharmacol. Sin. 2023, 44, 1161–1174. [Google Scholar] [CrossRef]
- Matthews, J.R.; Schlaich, M.P.; Rakoczy, E.P.; Matthews, V.B.; Herat, L.Y. The effect of SGLT2 inhibition on diabetic kidney disease in a model of diabetic retinopathy. Biomedicines 2022, 10, 522. [Google Scholar] [CrossRef]
- Farias, R.S.; Silva-Aguiar, R.P.; Teixeira, D.E.; Gomes, C.P.; Pinheiro, A.A.S.; Peruchetti, D.B.; Caruso-Neves, C. Inhibition of SGLT2 co-transporter by dapagliflozin ameliorates tubular proteinuria and tubule-interstitial injury at the early stage of diabetic kidney disease. Eur. J. Pharmacol. 2023, 942, 175521. [Google Scholar] [CrossRef]
- Dia, B.; Alkhansa, S.; Njeim, R.; Al Moussawi, S.; Farhat, T.; Haddad, A.; Riachi, M.E.; Nawfal, R.; Azar, W.S.; Eid, A.A. SGLT2 inhibitor-dapagliflozin attenuates diabetes-induced renal injury by regulating inflammation through a CYP4A/20-HETE signaling mechanism. Pharmaceutics 2023, 15, 965. [Google Scholar] [CrossRef]
- Kim, H.K.; Kim, Y.; Kim, R.-H.; Lee, S.; Yang, Y.; Park, H.; Jeon, N.; Lee, M.; Lee, Y.-H.; Cha, B.-S.; et al. Effects of ertugliflozin on podocyte in high-fat diet–induced diabetic kidney disease model, in vivo and in vitro. Diabetes 2022, 71 (Suppl. S1), 385. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, Z.; Wang, J.; Zhang, X.; Yu, X.; Li, Y. Empagliflozin protects diabetic pancreatic tissue from damage by inhibiting the activation of the NLRP3/caspase-1/GSDMD pathway in pancreatic β cells: In vitro and in vivo studies. Bioengineered 2021, 12, 9356–9366. [Google Scholar] [CrossRef] [PubMed]
- Croteau, D.; Luptak, I.; Chambers, J.M.; Hobai, I.; Panagia, M.; Pimentel, D.R.; Siwik, D.A.; Qin, F.; Colucci, W.S. Effects of sodium-glucose linked transporter 2 inhibition with ertugliflozin on mitochondrial function, energetics, and metabolic gene expression in the presence and absence of diabetes mellitus in mice. J. Am. Heart Assoc. 2021, 10, e019995. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Dong, J.; Liu, H.; Chen, H.; Yu, H.; Ye, S.; Yu, S.; Li, Y.; Qiu, L.; Song, N.; et al. Design and discovery of a highly potent ultralong-acting GLP-1 and glucagon co-agonist for attenuating renal fibrosis. Acta Pharm. Sin. B 2024, 14, 1283–1301. [Google Scholar] [CrossRef] [PubMed]
- Iwai, S.; Kaji, K.; Nishimura, N.; Kubo, T.; Tomooka, F.; Shibamoto, A.; Suzuki, J.; Tsuji, Y.; Fujinaga, Y.; Kitagawa, K.; et al. Glucagon-like peptide-1 receptor agonist, semaglutide attenuates chronic liver disease-induced skeletal muscle atrophy in diabetic mice. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1869, 166770. [Google Scholar] [CrossRef]
- Soto-Catalán, M.; Opazo-Ríos, L.; Quiceno, H.; Lázaro, I.; Moreno, J.A.; Gómez-Guerrero, C.; Egido, J.; Mas-Fontao, S. Semaglutide improves liver steatosis and de novo lipogenesis markers in obese and type-2-diabetic mice with metabolic-dysfunction-associated steatotic liver disease. Int. J. Mol. Sci. 2024, 25, 2961. [Google Scholar] [CrossRef]
- Li, R.; Ye, Z.; She, D.; Fang, P.; Zong, G.; Hu, K.; Kong, D.; Xu, W.; Li, L.; Zhou, Y.; et al. Semaglutide may alleviate hepatic steatosis in T2DM combined with NFALD mice via miR-5120/ABHD6. Drug Des. Dev. Ther. 2022, 16, 3557–3572. [Google Scholar] [CrossRef]
- Schwasinger-Schmidt, T.; Robbins, D.C.; Williams, S.J.; Novikova, L.; Stehno-Bittel, L. Long-term liraglutide treatment is associated with increased insulin content and secretion in β-cells, and a loss of α-cells in ZDF rats. Pharmacol. Res. 2013, 76, 58–66. [Google Scholar] [CrossRef]
- Chen, L.-N.; Lyu, J.; Yang, X.-F.; Ji, W.-J.; Yuan, B.-X.; Chen, M.-X.; Ma, X.; Wang, B. Liraglutide ameliorates glycometabolism and insulin resistance through the upregulation of GLUT4 in diabetic KKAy mice. Int. J. Mol. Med. 2013, 32, 892–900. [Google Scholar] [CrossRef]
- Alobaid, S.M.; Alshahrani, R.M.; Alonazi, A.S.; Alrasheed, N.M.; Alamin, M.A.; Alshammari, T.K.; Bin Dayel, A.F.; Elnagar, D.M.; Alotaibi, R.R.; Almuthnabi, L.A.; et al. Liraglutide attenuates diabetic cardiomyopathy via the ILK/PI3K/AKT/PTEN signaling pathway in rats with streptozotocin-induced type 2 diabetes mellitus. Pharmaceuticals 2024, 17, 374. [Google Scholar] [CrossRef]
- Martos-Guillami, N.; Vergara, A.; Llorens-Cebrià, C.; Motto, A.E.; Martínez-Díaz, I.; Gonçalves, F.; Garcias-Ramis, M.M.; Allo-Urzainqui, E.; Narváez, A.; Bermejo, S.; et al. SGLT2i and GLP1-RA exert additive cardiorenal protection with a RAS blocker in uninephrectomized db/db mice. Front. Pharmacol. 2024, 15, 1415879. [Google Scholar] [CrossRef]
- Tian, Y.; Tian, R.; Juan, H.; Guo, Y.; Yan, P.; Cheng, Y.; Li, R.; Wang, B. GLP-1/GIP dual agonist tirzepatide normalizes diabetic nephropathy via PI3K/AKT mediated suppression of oxidative stress. Int. Immunopharmacol. 2025, 146, 113877. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, Y.; Kimura, T.; Dan, K.; Iwamoto, H.; Sanada, J.; Fushimi, Y.; Katakura, Y.; Shimoda, M.; Yamasaki, Y.; Nogami, Y.; et al. Tirzepatide, a dual glucose-dependent insulinotropic polypeptide/glucagon-like peptide 1 receptor agonist, exhibits favourable effects on pancreatic β-cells and hepatic steatosis in obese type 2 diabetic db/db mice. Diabetes Obes. Metab. 2024, 26, 5982–5994. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Liu, W.; Jiang, X.; Huang, Y.; Zong, L.; Ding, H.; Shen, X.; Sun, Y.; Feng, X.; Li, X.; et al. Molecular dynamics-guided optimization of BGM0504 enhances dual-target agonism for combating diabetes and obesity. Sci. Rep. 2024, 14, 16680. [Google Scholar] [CrossRef] [PubMed]
- Jeong, B.-K.; Choi, W.-I.; Choi, W.; Moon, J.; Lee, W.H.; Choi, C.; Choi, I.Y.; Lee, S.-H.; Kim, J.K.; Ju, Y.S.; et al. A male mouse model for metabolic dysfunction-associated steatotic liver disease and hepatocellular carcinoma. Nat. Commun. 2024, 15, 6506. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, Y.; Zhou, Y.; Deng, J.; Wu, L. Tirzepatide alleviates oxidative stress and inflammation in diabetic nephropathy via IL-17 signaling pathway. Mol. Cell. Biochem. 2025, 480, 1241–1254. [Google Scholar] [CrossRef]

{kind=link}
| Article | Animals | Study Design | Pathology Model | Biochemical, Histological, Immunohistochemical Tests | Organs | Drug (Dose, Duration of Drug Exposure) | Other Drugs |
|---|---|---|---|---|---|---|---|
| Almuttairi, 2023 [80] | Albino rats | In vivo, randomized | Alloxan-induced T2D | Markers: None reported. Histology: H&E | Pancreas Liver Kidney | MET (1000 mg/kg daily, oral gavage, 4 weeks) | - |
| Salem, 2022 [81] | Albino rats | In vivo, randomized | Alloxan-induced T2D | Markers: Glucose, AST, ALT, ALP, Urea, Creatinine, Uric acid. Histology: H&E | Pancreas Liver Kidney | MET (45 mg/kg/day, oral, 28 days). MET nanoparticles (45 mg/kg/day, oral, 28 days) | - |
| Mobasher et al., 2020 [82] | Sprague Dawley albino rats | In vivo, randomized | STZ-induced DM | Markers: SOD, CAT, GSH, MDA, ALT, AST, ALP, arginase, albumin, total bilirubin, TP, PCNA, caspase-3. Histology: H&E, ICH (PCNA, caspase-3). | Liver | MET (150 mg/kg, oral gavage, every other day for 100 days) | - |
| Dallak et al., 2018 [83] | Albino rats | In vivo, randomized | HFD + STZ-induced T2DN | Markers: Glucose, urea, creatinine, TNF-α, hs-CRP. Histology: H&E, PAS. | Kidney | MET (200 mg/kg/day, oral gavage, started 2 weeks before STZ and continued for 12 weeks) | - |
| Zhang et al., 2017 [84] | Wistar rats | In vivo, randomized | HFD + low-dose STZ-induced T2DN | Markers: TGF-β1, CTGF, SOD, MDA, GSH-Px, FBG, HDL-c, LDL-c, TG, TC, BUN, SCr, UAER. Histology: H&E, ICH, TEM. | Kidney | MET (70 mg/kg/day, oral gavage, 13 weeks) | Captopril (10 mg/kg/day, oral gavage, 13 weeks) |
| Dawood et al., 2022 [85] | Wistar rats | In vivo, randomized | HCFD + STZ-induced T2D-induced DCM | Markers: Desmin, iNOS, AMPK, mTOR, TIMP-1, collagen. Histology: H&E, Masson’s trichrome, TEM, ICH, Western blot, qPCR, ECG. | Heart (left ventricle) | MET (200 mg/kg/day, oral, started day 1, continued for 12 weeks) | - |
| Yang et al., 2020 [86] | Male C57BL/6J mice | In vivo and in vitro, randomized | STZ-induced DCM | Markers: LDH, AST, CK, TC, TG, Bax, Bcl-2, PK2, PKR1, PKR2, p-AKT, AKT, p-GSK3β, GSK3β. Histology: H&E, Masson’s trichrome, TUNEL, ICH, electron microscopy, RT-qPCR, Western blot. | Heart | MET (250 mg/kg/day, oral in drinking water, 16 weeks). | - |
| Niu et al., 2019 [87] | db/db mice | In vivo, randomized | TMM | Markers: PECAM1/CD31, GLI1, PTCH1, SMO, HHIP, LC3-II, CASP3, CASP8, CASP9, BECN1, BNIP3, BCL2. Histology: Immunofluorescence, electron microscopy, Western blot, TUNEL assay, luciferase assay. | Retina Aorta | MET (300 mg/kg daily, i.p. injection, 4 weeks) | - |
| Yan et al., 2021 [88] | ApoE−/− mice, Myh11-cre-EGFP mice | In vivo, randomize | STZ + HFD-induced diabetes-accelerated atherosclerosis model | Markers: Pdlim5, AMPK, α-SMA, Oil Red O staining, phosphorylated Pdlim5. Histology: ICH, Western blotting, en face aortic lesion analysis. | Aorta—en face lesion analysis, Oil Red O staining. Carotid artery—wire injury model to study vascular smooth muscle cell (VSMC) migration. | MET (300 mg/kg daily, oral gavage, 8 weeks) | - |
| Naito et al., 2024 [89] | db/db mice, WT mice | In vivo, randomized | TMM | Markers: Col1a1, Col1a2, Col3a1, Acta2, Ccn2, TGF-β1. Histology: H&E, PSR, ICH for TGF-β1, Acta2, Ccn2. | Knee joint capsule Visceral fat (for adiponectin expression analysis) | MET (100 mg/kg daily, i.p. injection, 4, 8 weeks) | - |
| Srivastava and Goodwin, 2023 [16] | db/db mice, db/db; GRECKO mice | In vivo, randomized | TMM | Markers: IL-6, Wnt3a, α-SMA, FSP-1, Axin2. Histology: Masson’s trichrome stain, PSR, ICH, qPCR, cytokine array (IL-1β, IL-6, IFN-γ). | Kidney Heart Liver Adipose tissue | MET (100 mg/kg daily, oral gavage, for 8 weeks) | Wnt inhibitor (LGK974, 5 mg/kg, oral gavage, 6 doses per week for 8 weeks). |
| Article | Animals | Study Design | Pathology Model | Biochemical, Histological, Immunohistochemical Tests | Organs | Drug (Dose, Duration of Drug Exposure) | Other Drugs |
|---|---|---|---|---|---|---|---|
| Han et al., 2023 [90] | C57BL/6 mice | In vivo, randomized | STZ + HFD-induced HLI | Markers: GPX4, PECAM-1, α-SMA, 4-HNE. Histology: Immunofluorescence, Western blot, qRT-PCR, TEM. | Skeletal muscle (gastrocnemius) Hindlimb vasculature | EMPA (10 mg/kg, intramuscular injection, every 3 days for 21 days) | - |
| Matthews et al., 2022 [91] | Kimba and Akimba mice | In vivo, randomized | TMM | Markers: SGLT2, insulin. Histology: H&E, PAS, Masson’s trichrome staining, ICH, Western blot, kidney-to-body weight ratio. | Kidney Pancreas | DAPA, EMPA, CANA (25 mg/kg/day, oral, in drinking water, 8 weeks) | - |
| Farias et al., 2023 [92] | Wistar rats | In vivo, randomized | STZ-induced T2D, early-stage DKD | Markers: Albumin, β2-microglobulin, LDH, γ-GT, megalin. Histology: PAS staining, immunofluorescence, SDS-PAGE, Western blot, renal histomorphometry. | Kidney | DAPA (1 mg/kg/day, oral gavage, started 1 day post-STZ, continued for 8 weeks) | - |
| Dia et al., 2023 [93] | C57BL/6 and FVB/NJ mice | In vivo, randomized | HFD + STZ-induced T2D, DKD | Markers: CYP4A, 20-HETE, MCP-1, IL-1β, IL-6, IL-17, TNFα, fibronectin, collagen IV. Histology: PAS, Masson’s trichrome, Western blot, RT-PCR, ELISA, HPLC, NADPH oxidase assay. | Kidney | DAPA (1.5 mg/kg/day, i.p., 8 weeks). HET0016 (5 mg/kg/day, s.c., 10 weeks) | Insulin (2 IU/day, i.p., 8 weeks). |
| Kim et al., 2022 [94] | C57BL/6J mice | In vivo, randomized | HFD-induced early DKD | Markers: Albuminuria, SGLT2 (mRNA and protein), glomerular volume, mesangial expansion, GBM thickness. Histology: ICH, renal histomorphometry. | Kidney (renal cortex) | ERTU (mixed with HFD, oral, 16 weeks) | - |
| Liu et al., 2021 [95] | db/db mice | In vivo and in vitro, randomized | TMM | Markers: NLRP3, caspase-1, GSDMD. Histology: H&E, ICH, immunofluorescence, Western blot (pancreas and β TC-6 cells). | Pancreas | EMPA (10 mg/kg/day, oral gavage, 6 months) | - |
| Croteau et al., 2021 [96] | C57BL/6J mice | In vivo, randomized | HFHS induced DCM | Markers: HbA1c, glucose, insulin, HOMA-IR, ROS (4-HNE), ATP, phosphocreatine/ATP. Histology: H&E, PRS, ICH, 4-HNE staining, 31P-NMR spectroscopy, RNA-seq. | Heart (left ventricle) | ERTU (0.5 mg/g of diet, oral, 4 months) | - |
| Article | Animals | Study Design | Pathology Model | Biochemical, Histological, Immunohistochemical Tests | Organs | Drug (Dose, Duration of Drug Exposure) | Other Drugs |
|---|---|---|---|---|---|---|---|
| Zhao et al., 2024 [97] | db/db mice, UUO mice, ZDF rats | In vivo, randomized | TMM | Markers: GLP-1R, GCGR, TGF-β1, α-SMA, COL1A1, NF-κB, IL-1β, TNF-α, PGC-1α, PGC-1β. Histology: H&E, Masson’s trichrome, Sirius Red, ICH, Western blot, qPCR, RNA sequencing, mitochondrial OCR assay. | Kidney, Liver, Epididymis adipose tissue | SEMA (120 mg/kg/day, s.c. for 8 weeks) | - |
| Iwai et al., 2023 [98] | Diabetic KK-Ay mice | In vivo, randomized | TMM | Markers: GLP-1R, PGC-1α, PKA, AKT, NF-κB, MSTN, MyoG, MyoD, MuRF-1. Histology: H&E, Sirius Red, Immunofluorescence, Western blot, qRT-PCR, mitochondrial OCR assay. | Liver Gastrocnemius muscle | SEMA (3 nmol/kg, s.c., every 3 days for 6 weeks) | - |
| Soto-Catalán et al., 2024 [99] | BKS db/db mice | In vivo, randomized | TMM | Markers: GLP-1R, ALT, AST, AP, triglycerides, total cholesterol, c-HDL, SCD1, Acaca, Fasn. Histology: H&E, Oil Red O staining, DXA body composition, qPCR, Western blot, lipidomic analysis. | Liver | SEMA (25 µg/kg/week for 2 weeks, then 100 µg/kg/week for 9 weeks, s.c.) | - |
| Li et al., 2022 [100] | C57BL/6J mice | In vivo, randomized | STZ + HFD-induced T2D and NAFLD | Markers: ABHD6, miR-5120, GLP-1R, CD36, PPARγ, PPARα, ALT, AST, TG, TC, FFA, LDL-C, HDL-C. Histology: H&E, Sirius Red, Oil Red O staining, qPCR, Western blot, ELISA, OGTT, IPITT, dual-luciferase assay. | Liver | SEMA (0.42 mg/kg/week, s.c., for 12 weeks) | - |
| Schwasinger-Schmidt et al., 2013 [101] | ZDF rats | In vivo and in vitro, randomized | TMM | Markers: Insulin, Proinsulin, Glucagon, Somatostatin, Ki-67, CD34. Histology: H&E, ICH, immunofluorescence, electron microscopy, insulin ELISA, islet density, morphology, granule analysis. | Pancreas | LIRA (0.225 µg/g, s.c., twice daily for 9 weeks) | - |
| Chen et al., 2013 [102] | Male KKAy mice and C57BL/6J (C57) mice | In vivo, randomized | TMM | Markers: GLUT4, keletal muscle pyruvate kinase, hexokinase, TEM, RNA isolation, real-time PCR. | Liver, Skeletal muscle | LIRA (250 μg/kg/day, s.c., 6 weeks) | - |
| Alobaid et al., 2024 [103] | Wistar albino rats | In vivo, randomized | STZ + HFD-induced T2D and DCM | Markers: Troponin I, CK-MB, ILK, PI3K, AKT, PTEN, BCL2, BAX, caspase 3, MDA, SOD, GPx. Histology: H&E, TUNEL assay, ICH, ELISA, Western blot. | Heart | LIRA (150 µg/kg, s.c., twice daily for 3 weeks after 3 weeks of vehicle) | - |
| Martos-Guillami et al., 2024 [104] | db/db mice | In vivo, randomized | TMM | Markers: Ccl2, TGF-β1, ACE2, ACE, FABP4, IGFBP4, Myh7, WT1. Histology: H&E, PAS, PRS, IHC, qPCR, Western blot, GFR, albuminuria, CT. | Kidney Heart | SEMA (10 nmol/kg, s.c., twice weekly, 8 weeks) EMPA (10 mg/kg/day, oral gavage, 5 days/week, 8 weeks) | Ramipril (8 mg/kg/day, drinking water, 8 weeks) |
| Article | Animals | Study Design | Pathology Model | Biochemical, Histological, Immunohistochemical Tests | Organs | Drug (Dose, Duration of Drug Exposure) | Other Drugs |
|---|---|---|---|---|---|---|---|
| Tian et al., 2025 [105] | C57BL/6J mice | In vivo, randomized | STZ + HFD-induced T2DN | Markers: PI3K, AKT, p-PI3K, p-AKT, GPX4, SOD, CAT. Histology: H&E, Masson’s trichrome, ICH, Western blot, RNA sequencing, oxidative stress enzyme assays. | Kidney | TZP (10 nmol/kg, i.p., daily for 2 weeks) SEMA (30 nmol/kg, i.p., daily for 2 weeks) | Insulin glargine (0.5 IU/day, s.c., for 2 weeks) |
| Iwamoto et al., 2024 [106] | db/db mice | In vivo, randomized | TMM | Markers: Blood glucose, insulin, glucagon, ALT, AST, LDH, insulin-related genes (Ins1, Ins2, Pdx-1, MafA, NeuroD, Munc18), Chemokine and cytokine genes (Ccl3, Ccl4, Ccl5, Tnf-α, IL-1β, Mcp1). Histology: H&E, Oil Red O, Azan, immunofluorescence, TEM, qPCR, flow cytometry, CT imaging. | Pancreas Liver | TZP (30 nmol/kg, s.c., twice weekly, 4 weeks) SEMA (200 nmol/kg, s.c., twice weekly, 4 weeks) | - |
| Yuan et al., 2024 [107] | db/db mice, STZ + HFD-induced C57BL/6 mice | In vivo, randomized | TMM STZ + HFD-induced | Markers: GLP-1R, GIPR, ALT, AST, TC, TG, LDL, HDL. Histology: H&E, Sirius Red staining, qPCR, Western blot, ELISA, GTT, lipidomic analysis. | Liver | TZP (0.15 mg/kg, s.c., every 3 days, for 31 days) | BGM0504 (0.05, 0.15, 0.5 mg/kg, s.c., every 3 days, for 31 days) |
| Jeong et al., 2024 [108] | C57BL/6J mice | In vivo, randomized | STZ + HFD-induced MASLD (including MASH, fibrosis, HCC) | Markers: ALT, AST, NAS score, fibrosis stage, triglycerides, gene expression via RNA-seq. Histology: H&E, Masson’s trichrome, in vivo liver imaging, ICH, transcriptomics, ATAC-seq. | Liver | TZP (dose not specified, administered for 10–11 weeks at stages 21–32 weeks, 28–38 weeks, 41–52 weeks) | - |
| Yang et al., 2025 [109] | BALB/c mice | In vivo, randomized | STZ-induced T2DN | Markers: BUN, sCr, AGEs, insulin, SOD, CAT, MDA, IL-1β, IL-6, TNF-α, NGAL, podocin, cystatin C, IL-17A, IL-17F, GLP-1R, Bax, Bcl-2. Histology: H&E, PAS, TUNEL, RT-qPCR, Western blot, ELISA, renal histomorphometry. | Kidney | TZP (3 and 10 nmol/kg/day, i.p., 8 weeks) | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tartau, C.G.; Boboc, I.K.S.; Mititelu-Tartau, L.; Bogdan, M.; Buca, B.R.; Pavel, L.L.; Amalinei, C. Exploring the Protective Effects of Traditional Antidiabetic Medications and Novel Antihyperglycemic Agents in Diabetic Rodent Models. Pharmaceuticals 2025, 18, 670. https://doi.org/10.3390/ph18050670
Tartau CG, Boboc IKS, Mititelu-Tartau L, Bogdan M, Buca BR, Pavel LL, Amalinei C. Exploring the Protective Effects of Traditional Antidiabetic Medications and Novel Antihyperglycemic Agents in Diabetic Rodent Models. Pharmaceuticals. 2025; 18(5):670. https://doi.org/10.3390/ph18050670
Chicago/Turabian StyleTartau, Cosmin Gabriel, Ianis Kevyn Stefan Boboc, Liliana Mititelu-Tartau, Maria Bogdan, Beatrice Rozalina Buca, Liliana Lacramioara Pavel, and Cornelia Amalinei. 2025. "Exploring the Protective Effects of Traditional Antidiabetic Medications and Novel Antihyperglycemic Agents in Diabetic Rodent Models" Pharmaceuticals 18, no. 5: 670. https://doi.org/10.3390/ph18050670
APA StyleTartau, C. G., Boboc, I. K. S., Mititelu-Tartau, L., Bogdan, M., Buca, B. R., Pavel, L. L., & Amalinei, C. (2025). Exploring the Protective Effects of Traditional Antidiabetic Medications and Novel Antihyperglycemic Agents in Diabetic Rodent Models. Pharmaceuticals, 18(5), 670. https://doi.org/10.3390/ph18050670