
Repositioning FDA-Approved Sulfonamide-Based Drugs as Potential Carbonic Anhydrase Inhibitors in Trypanosoma cruzi: Virtual Screening and In Vitro Studies

 , , , ,
, , , ,  , ,
, ,  and
and 
Abstract
1. Introduction
2. Results
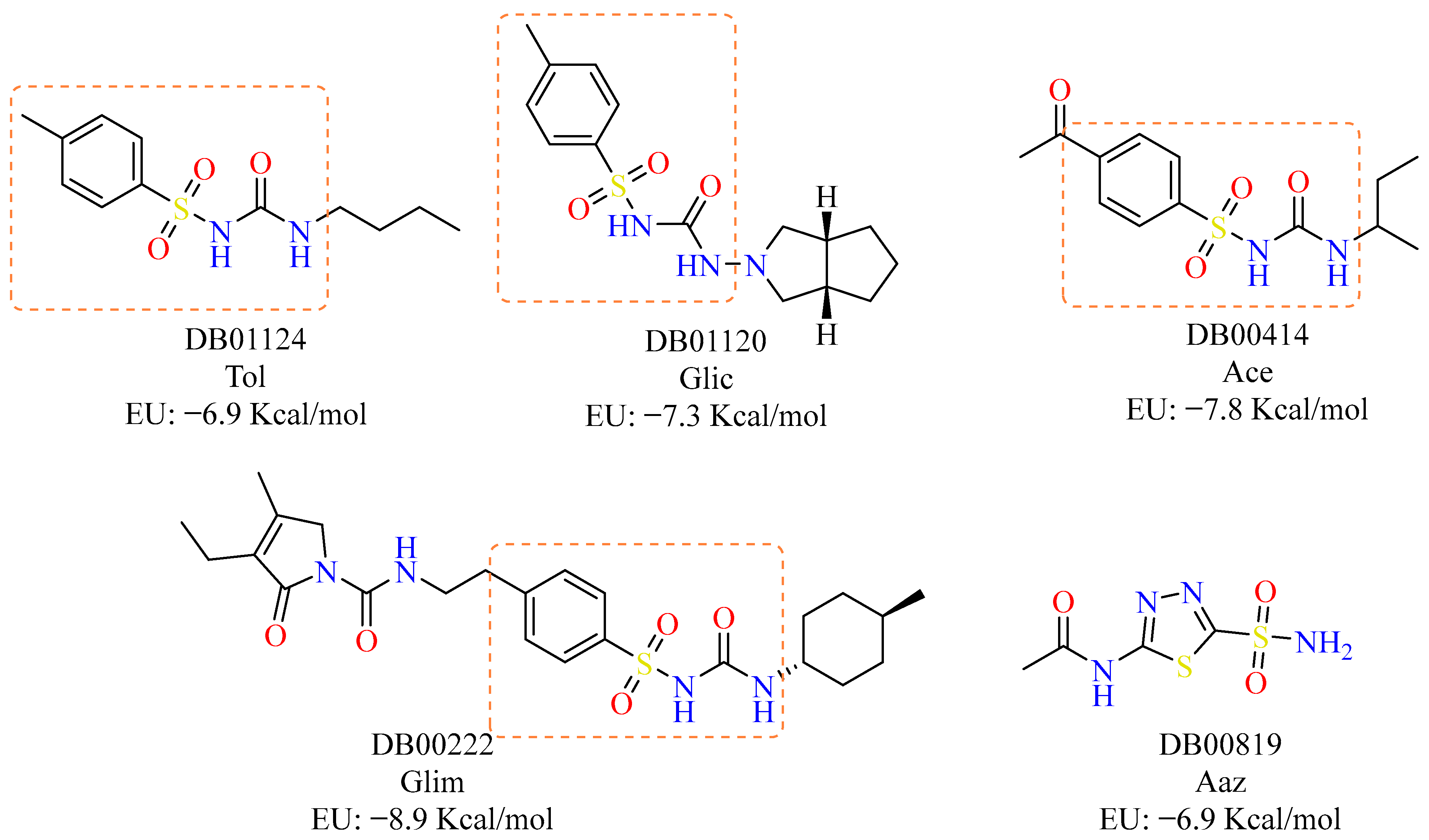
2.1. Virtual Screening
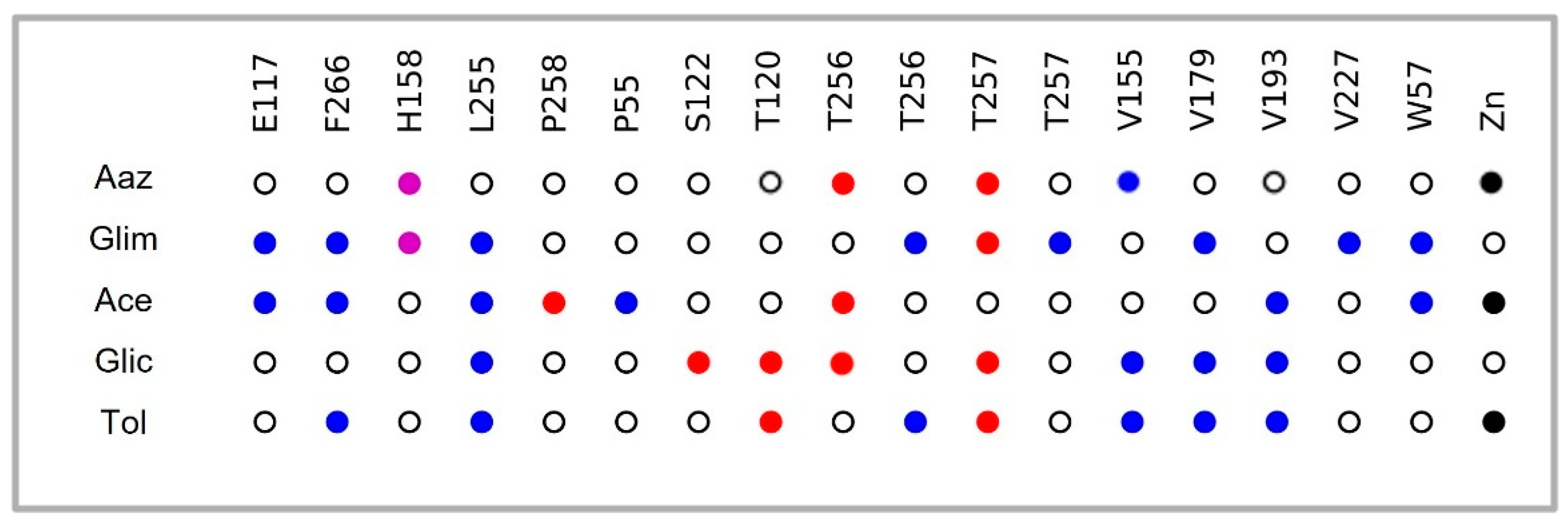
2.2. Interaction Profile Analysis
2.3. Trypanocidal Activity
2.4. α-TcCA Enzymatic Assay
3. Discussion
4. Materials and Methods
4.1. Virtual Screening
Selection of FDA Compounds for Molecular Docking
4.2. Ligand and α-TcCA Receptor Preparation
4.3. Molecular Docking Analysis
4.4. Interaction Analysis
4.5. Trypanocidal Activity
4.6. Ex Vivo Analysis
4.7. Amastigote Assay
4.8. Cytotoxicity in Murine Macrophages
4.9. Carbonic Anhydrase Enzymatic Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| α-TcCA | α-Trypanosoma cruzi Carbonic Anhydrase |
| Aaz | Acetazolamide |
| Ace | Acetohexamide |
| CC50 | Half-maximal cytotoxicity concentration |
| DB | Drug bank |
| BFE | Binding free energy |
| FDA | Food and Drug Administration |
| Glic | Gliclazide |
| Glim | Glimepiride |
| HB | Hydrogen bond |
| HI | Hydrophobic interaction |
| IC50 | Half-maximal inhibitory concentration |
| KI | Inhibitory constant |
| NSAID | Nonsteroidal anti-inflammatory drug |
| PDB | Protein data bank |
| π-c | Pi-cation |
| PLIP | Protein–Ligand Interaction Profile |
| SB | Salt bridge |
| SI | Selectivity index |
| Tol | Tolbutamide |
| Zn | Zinc |
References
- WHO. Available online: https://www.who.int/home (accessed on 17 February 2021).
- Enriquez, G.F.; Cardinal, M.V.; Orozco, M.M.; Schijman, A.G.; Gürtler, R.E. Detection of Trypanosoma Cruzi Infection in Naturally Infected Dogs and Cats Using Serological, Parasitological and Molecular Methods. Acta Trop. 2013, 126, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, D.E.; Benchimol, M.; Crepaldi, P.H.; de Souza, W. Interactive Multimedia to Teach the Life Cycle of Trypanosoma Cruzi, the Causative Agent of Chagas Disease. PLoS Negl. Trop. Dis. 2012, 6, e1749. [Google Scholar] [CrossRef] [PubMed]
- Martín-Escolano, J.; Marín, C.; Rosales, M.J.; Tsaousis, A.D.; Medina-Carmona, E.; Martín-Escolano, R. An Updated View of the Trypanosoma Cruzi Life Cycle: Intervention Points for an Effective Treatment. ACS Infect. Dis. 2022, 8, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Peña-Callejas, G.; González, J.; Jiménez-Cortés, J.G.; De Fuentes-Vicente, J.A.; Salazar-Schettino, P.M.; Bucio-Torres, M.I.; Cabrera-Bravo, M.; Flores-Villegas, A.L. Chagas Disease: Biology and Transmission of Trypanosoma Cruzi. TIP Rev. Espec. En Cienc. Químico-Biológicas 2022, 25, 1–19. [Google Scholar] [CrossRef]
- Mejía-Jaramillo, A.M.; Ospina-Zapata, H.; Fernandez, G.J.; Triana-Chávez, O. Transcriptomic Analysis of Benznidazole-Resistant Trypanosoma Cruzi Clone Reveals Nitroreductase I-Independent Resistance Mechanisms. PLoS ONE 2025, 20, e0314189. [Google Scholar] [CrossRef]
- Campos, M.C.O.; Leon, L.L.; Taylor, M.C.; Kelly, J.M. Benznidazole-Resistance in Trypanosoma Cruzi: Evidence That Distinct Mechanisms Can Act in Concert. Mol. Biochem. Parasitol. 2014, 193, 17–19. [Google Scholar] [CrossRef]
- Mejia, A.M.; Hall, B.S.; Taylor, M.C.; Gómez-Palacio, A.; Wilkinson, S.R.; Triana-Chávez, O.; Kelly, J.M. Benznidazole-Resistance in Trypanosoma Cruzi Is a Readily Acquired Trait That Can Arise Independently in a Single Population. J. Infect. Dis. 2012, 206, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.R.; Taylor, M.C.; Horn, D.; Kelly, J.M.; Cheeseman, I. A Mechanism for Cross-Resistance to Nifurtimox and Benznidazole in Trypanosomes. Proc. Natl. Acad. Sci. USA 2008, 105, 5022–5027. [Google Scholar] [CrossRef]
- Mosquillo, F.; Scalese, G.; Moreira, R.; Denis, P.A.; Machado, I.; Paulino, M.; Gambino, D.; Pérez-Díaz, L. Platinum and Palladium Organometallic Compounds: Disrupting the Ergosterol Pathway in Trypanosoma Cruzi. ChemBioChem 2023, 24, e202300406. [Google Scholar] [CrossRef]
- Alvarez, G.; Martínez, J.; Aguirre-Lopez, B.; Cabrera, N.; Pérez-Díaz, L.; Gómez-Puyou, M.T.d.; Gómez-Puyou, A.; Pérez-Montfort, R.; Garat, B.; Merlino, A. New Chemotypes as Trypanosoma Cruzi Triosephosphate Isomerase Inhibitors: A Deeper Insight into the Mechanism of Inhibition. J. Enzym. Inhib. Med. Chem. 2014, 29, 198–204. [Google Scholar] [CrossRef]
- Aguilera, E.; Varela, J.; Serna, E.; Torres, S.; Yaluff, G.; Bilbao, N.V.d.; Cerecetto, H.; Alvarez, G.; González, M. Looking for Combination of Benznidazole and Trypanosoma Cruzi-Triosephosphate Isomerase Inhibitors for Chagas Disease Treatment. Mem. Inst. Oswaldo Cruz 2018, 113, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Nardy, A.F.F.R.; Freire-de-Lima, C.G.; Pérez, A.R.; Morrot, A. Role of Trypanosoma Cruzi Trans-Sialidase on the Escape from Host Immune Surveillance. Front. Microbiol. 2016, 7, 348. [Google Scholar] [CrossRef] [PubMed]
- Nunes, J.A.; da Silva Santos-Júnior, P.F.; Gomes, M.C.; Ferreira, L.A.S.; Padilha, E.K.A.; Teixeira, T.R.; Stanger, E.J.; Kaur, Y.; da Silva, E.B.; Costa, C.A.C.B. Nanomolar Activity of Coumarin-3-Thiosemicarbazones Targeting Trypanosoma Cruzi Cruzain and the T. Brucei Cathepsin L-like Protease. Eur. J. Med. Chem. 2025, 283, 117109. [Google Scholar] [CrossRef]
- Beltran-Hortelano, I.; Alcolea, V.; Font, M.; Pérez-Silanes, S. Examination of Multiple Trypanosoma Cruzi Targets in a New Drug Discovery Approach for Chagas Disease. Bioorg. Med. Chem. 2022, 58, 116577. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Pérez, E.; Mendez-Alvarez, D.; Juarez-Saldivar, A.; Rodriguez-Moreno, A.; De Alba-Alvarado, M.; Gonzalez-Gonzalez, A.; Vazquez, K.; Martinez-Vazquez, A.V.; Nogueda-Torres, B.; Lara-Ramírez, E.E.; et al. A Computational Approach Using α-Carbonic Anhydrase to Find Anti-Trypanosoma Cruzi Agents. Med. Chem. 2024, 20, 46–60. [Google Scholar] [CrossRef]
- Mann, T.; Keilin, D. Sulphanilamide as a Specific Inhibitor of Carbonic Anhydrase. Nature 1940, 146, 164–165. [Google Scholar] [CrossRef]
- Hoff, E.; Zou, D.; Schiza, S.; Demir, Y.; Grote, L.; Bouloukaki, I.; Beydemir, Ş.; Eskandari, D.; Stenlöf, K.; Hedner, J. Carbonic Anhydrase, Obstructive Sleep Apnea and Hypertension: Effects of Intervention. J. Sleep Res. 2020, 29, e12956. [Google Scholar] [CrossRef]
- Swenson, E.R. New Insights into Carbonic Anhydrase Inhibition, Vasodilation, and Treatment of Hypertensive-Related Diseases. Curr. Hypertens. Rep. 2014, 16, 467. [Google Scholar] [CrossRef]
- Carta, F.; Supuran, C.T. Diuretics with Carbonic Anhydrase Inhibitory Action: A Patent and Literature Review (2005–2013). Expert Opin. Ther. Pat. 2013, 23, 681–691. [Google Scholar] [CrossRef]
- Khokhlov, A.L.; Yaichkov, I.I.; Korsakov, M.K.; Shetnev, A.A.; Ivanovskiy, S.A.; Alexeev, M.A.; Gasilina, O.A.; Volkhin, N.N.; Petukhov, S.S. Identification and Synthesis of Metabolites of the New Antiglaucoma Drug. Res. Results Pharmacol. 2024, 10, 53–66. [Google Scholar] [CrossRef]
- Supuran, C.T.; Scozzafava, A.; Casini, A. Carbonic Anhydrase Inhibitors. Med. Res. Rev. 2003, 23, 146–189. [Google Scholar] [CrossRef] [PubMed]
- Elsayad, K.A.; Elmasry, G.F.; Mahmoud, S.T.; Awadallah, F.M. Sulfonamides as Anticancer Agents: A Brief Review on Sulfonamide Derivatives as Inhibitors of Various Proteins Overexpressed in Cancer. Bioorg. Chem. 2024, 147, 107409. [Google Scholar] [CrossRef] [PubMed]
- Pastorekova, S.; Zatovicova, M.; Pastorek, J. Cancer-Associated Carbonic Anhydrases and Their Inhibition. Curr. Pharm. Des. 2008, 14, 685–698. [Google Scholar] [CrossRef]
- Singh, S.; Lomelino, C.L.; Mboge, M.Y.; Frost, S.C.; McKenna, R. Cancer Drug Development of Carbonic Anhydrase Inhibitors beyond the Active Site. Molecules 2018, 23, 1045. [Google Scholar] [CrossRef]
- Scarim, C.B.; Chelucci, R.C.; Dos Santos, J.L.; Chin, C.M. The Use of Sulfonamide Derivatives in the Treatment of Trypanosomatid Parasites Including Trypanosoma Cruzi, Trypanosoma Brucei, and Leishmania ssp. Med. Chem. 2020, 16, 24–38. [Google Scholar] [CrossRef]
- Pillai, M.; Wu, D. Validation Approaches for Computational Drug Repurposing: A Review. In AMIA Annual Symposium Proceedings; American Medical Informatics Association: Washington, DC, USA, 2024; Volume 2023, p. 559. [Google Scholar]
- Berdigaliyev, N.; Aljofan, M. An Overview of Drug Discovery and Development. Future Med. Chem. 2020, 12, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Busardò, F.P.; Lo Faro, A.F.; Sirignano, A.; Giorgetti, R.; Carlier, J. In Silico, in Vitro, and in Vivo Human Metabolism of Acetazolamide, a Carbonic Anhydrase Inhibitor and Common “Diuretic and Masking Agent” in Doping. Arch. Toxicol. 2022, 96, 1989–2001. [Google Scholar] [CrossRef]
- Beatriz Vermelho, A.; Rodrigues, G.C.; Nocentini, A.; Mansoldo, F.R.P.; Supuran, C.T. Discovery of Novel Drugs for Chagas Disease: Is Carbonic Anhydrase a Target for Antiprotozoal Drugs? Expert Opin. Drug Discov. 2022, 17, 1147–1158. [Google Scholar] [CrossRef]
- Khalifah, R.G. The Carbon Dioxide Hydration Activity of Carbonic Anhydrase: I. Stop-Flow Kinetic Studies on the Native Human Isoenzymes B and C. J. Biol. Chem. 1971, 246, 2561–2573. [Google Scholar] [CrossRef]
- Pinard, M.A.; Mahon, B.; McKenna, R. Probing the Surface of Human Carbonic Anhydrase for Clues towards the Design of Isoform Specific Inhibitors. Biomed Res. Int. 2015, 2015, 453543. [Google Scholar] [CrossRef]
- Ovung, A.; Bhattacharyya, J. Sulfonamide Drugs: Structure, Antibacterial Property, Toxicity, and Biophysical Interactions. Biophys. Rev. 2021, 13, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Llanos, M.A.; Sbaraglini, M.L.; Villalba, M.L.; Ruiz, M.D.; Carrillo, C.; Alba Soto, C.; Talevi, A.; Angeli, A.; Parkkila, S.; Supuran, C.T. A Structure-Based Approach towards the Identification of Novel Antichagasic Compounds: Trypanosoma Cruzi Carbonic Anhydrase Inhibitors. J. Enzym. Inhib. Med. Chem. 2020, 35, 21–30. [Google Scholar] [CrossRef]
- DrugBank. Available online: https://go.drugbank.com/ (accessed on 3 February 2021).
- Farwa, U.; Raza, M.A. Heterocyclic Compounds as a Magic Bullet for Diabetes Mellitus: A Review. RSC Adv. 2022, 12, 22951–22973. [Google Scholar] [CrossRef]
- Osmaniye, D.; Yuva, O.; Sağlık, B.N.; Levent, S.; Ozkay, Y.; Kaplancıklı, Z.A. Design, Synthesis, Evaluation of Biological Activities and Molecular Docking and Dynamic Studies of Novel Acetazolamide Analog Compounds. J. Biomol. Struct. Dyn. 2024, 42, 7243–7256. [Google Scholar] [CrossRef] [PubMed]
- Guzel-Akdemir, Ö.; Akdemir, A.; Pan, P.; Vermelho, A.B.; Parkkila, S.; Scozzafava, A.; Capasso, C.; Supuran, C.T. A Class of Sulfonamides with Strong Inhibitory Action against the α-Carbonic Anhydrase from Trypanosoma Cruzi. J. Med. Chem. 2013, 56, 5773–5781. [Google Scholar] [CrossRef]
- Vela, A.; Coral-Almeida, M.; Sereno, D.; Costales, J.A.; Barnabé, C.; Brenière, S.F. In Vitro Susceptibility of Trypanosoma Cruzi Discrete Typing Units (Dtus) to Benznidazole: A Systematic Review and Meta-Analysis. PLoS Negl. Trop. Dis. 2021, 15, e0009269. [Google Scholar] [CrossRef] [PubMed]
- Juárez-Saldivar, A.; Schroeder, M.; Salentin, S.; Joachim Haupt, V.; Saavedra, E.; Vázquez, C.; Reyes-Espinosa, F.; Herrera-Mayorga, V.; Villalobos-Rocha, J.C.; García-Pérez, C.A.; et al. Computational Drug Repositioning for Chagas Disease Using Protein-Ligand Interaction Profiling. Int. J. Mol. Sci. 2020, 21, 4270. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An Open-Source Program for Chemistry Aware Data Visualization and Analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Bisong, E.; Bisong, E. Google Colaboratory. In Building Machine Learning and Deep Learning Models on Google Cloud Platform: A Comprehensive Guide for Beginners; Apress: Berkeley, CA, USA, 2019; pp. 59–64. [Google Scholar]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An Open-Source Molecular Graphics Tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- McNutt, A.T.; Francoeur, P.; Aggarwal, R.; Masuda, T.; Meli, R.; Ragoza, M.; Sunseri, J.; Koes, D.R. GNINA 1.0: Molecular Docking with Deep Learning. J. Cheminform. 2021, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the Scope of the Protein–Ligand Interaction Profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Protein Data Bank. Available online: https://www.rcsb.org/ (accessed on 20 May 2021).
- Yuan, S.; Chan, H.C.S.; Hu, Z. Using PyMOL as a Platform for Computational Drug Design. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1298. [Google Scholar] [CrossRef]
- Coimbra, J.R.M.; Baptista, S.J.; Dinis, T.C.P.; Silva, M.; Moreira, P.I.; Santos, A.E.; Salvador, J.A.R. Combining Virtual Screening Protocol and in Vitro Evaluation towards the Discovery of BACE1 Inhibitors. Biomolecules 2020, 10, 535. [Google Scholar] [CrossRef]
- Supuran, C.T. Inhibition of Carbonic Anhydrase from Trypanosoma Cruzi for the Management of Chagas Disease: An Underexplored Therapeutic Opportunity. Future Med. Chem. 2016, 8, 311–324. [Google Scholar] [CrossRef]
- PLIP. Available online: https://plip-tool.biotec.tu-dresden.de/plip-web/plip/index (accessed on 1 October 2021).
- Domínguez-Díaz, L.R.; Ochoa, M.E.; Soto-Castro, D.; Farfán, N.; Morales-Chamorro, M.; Yépez-Mulia, L.; Pérez-Campos, E.; Santillan, R.; Moreno-Rodríguez, A. In Vitro, Ex Vivo and in Vivo Short-Term Screening of DHEA Nitrate Derivatives Activity over Trypanosoma Cruzi Ninoa and TH Strains from Oaxaca State, México. Bioorg. Med. Chem. 2021, 48, 116417. [Google Scholar] [CrossRef] [PubMed]
- Adasme, M.F.; Bolz, S.N.; Adelmann, L.; Salentin, S.; Haupt, V.J.; Moreno-Rodríguez, A.; Nogueda-Torres, B.; Castillo-Campos, V.; Yepez-Mulia, L.; De Fuentes-Vicente, J.A.; et al. Repositioned Drugs for Chagas Disease Unveiled via Structure-Based Drug Repositioning. Int. J. Mol. Sci. 2020, 21, 8809. [Google Scholar] [CrossRef]
- Nocentini, A.; Cadoni, R.; Dumy, P.; Supuran, C.T.; Winum, J.Y. Carbonic Anhydrases from Trypanosoma Cruzi and Leishmania Donovani Chagasi Are Inhibited by Benzoxaboroles. J. Enzym. Inhib. Med. Chem. 2018, 33, 286–289. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| DB ID | BFE (kcal/mol) | DB ID | BFE (kcal/mol) | ||||
|---|---|---|---|---|---|---|---|
| 1 | DB00222 | −8.9 | 24 | DB00580 | −7.4 | ||
| 2 | DB11395 | −8.8 | 25 | DB13773 | −7.3 | ||
| 3 | DB00214 | −8.4 | 26 | DB00436 | −7.3 | ||
| 4 | DB05015 | −8.1 | 27 | DB15861 | −7.3 | ||
| 5 | DB00774 | −8 | 28 | DB01120 | −7.3 | ||
| 6 | DB06268 | −8 | 29 | DB11739 | −7.3 | ||
| 7 | DB00310 | −7.9 | 30 | DB01015 | −7.2 | ||
| 8 | DB08881 | −7.8 | 31 | DB00554 | −7.2 | ||
| 9 | DB00414 | −7.8 | 32 | DB11462 | −7.2 | ||
| 10 | DB14033 | −7.7 | 33 | DB11464 | −7.2 | ||
| 11 | DB08942 | −7.7 | 34 | DB00880 | −7.1 | ||
| 12 | DB00278 | −7.7 | 35 | DB06150 | −7.1 | ||
| 13 | DB00482 | −7.7 | 36 | DB01298 | −7.1 | ||
| 14 | DB11461 | −7.6 | 37 | DB08439 | −7.1 | ||
| 15 | DB00263 | −7.5 | 38 | DB01325 | −7.1 | ||
| 16 | DB00999 | −7.4 | 39 | DB00814 | −7.1 | ||
| 17 | DB00695 | −7.4 | 40 | DB08912 | −7 | ||
| 18 | DB13532 | −7.4 | 41 | DB00606 | −7 | ||
| 19 | DB06729 | −7.4 | 42 | DB01021 | −7 | ||
| 20 | DB09289 | −7.4 | 43 | DB13165 | −7 | ||
| 21 | DB14973 | −7.4 | 44 | DB09215 | −6.9 | ||
| 22 | DB13284 | −7.4 | 45 | DB01124 | −6.9 | ||
| 23 | DB00808 | −7.4 | 46 | DB00819_Aaz | −6.9 | ||
| Hypoglycaemics | NSAIDs | ||||||
| Diuretics | Antihypertensives | ||||||
| Antibacterials | Antidepressants | ||||||
| Vet approved | Histone deacetylase (HDAC), kinase and COX-2 inhibitor | ||||||
| ID | Epimastigote (In Vitro) | Trypomastigote (Ex Vivo) | Amastigote (Ex Vivo) | J774.2 b CC50 (μM ± SD) | |||
|---|---|---|---|---|---|---|---|
| NINOA | A1 | NINOA | A1 | NINOA | A1 | ||
| a IC50 (μM ± SD) | |||||||
| Glic | >200 | 10.7 ± 1.5 | 89.4 ± 0.1 | >200 | >200 | 12.3 ± 0.01 | >200 |
| Glim | 70.2 ± 3.2 | 37.6 ± 1.5 | >200 | >200 | >200 | 50.26 ± 0.01 | 14.2 ± 2.3 |
| Ace | 6.5 ± 2.1 | 52.9 ± 2.7 | 46.5 ± 0.1 | >200 | 46 ± 0.6 | >200 | >200 |
| Tol | 8.5 ± 1.4 | >200 | 9.8 ± 0.1 | 97.7 ± 0.1 | 148.7 ± 0.01 | 44.5 ± 0.01 | >200 |
| Reference drugs | |||||||
| Aaz | 5.5 ± 1.7 | 15.2 ± 2.8 | 198 ± 0.1 | >200 | >200 | 9.6 ± 0.01 | 12.6 ± 4.7 |
| Nfx | 7.1 ± 0.1 | 39.1 ± 0.07 | 156 ± 0.1 | 118.2 ± 0.02 | 70.5 ± 0.6 | 54.8 ± 0.01 | 164.2 ± 0.3 |
| Bzn | 30.3 ± 0.03 | 19.3 ± 0.08 | 167.1 ± 0.03 | 145.3 ± 0.2 | Ne | Ne | 133.9 ± 0.06 |
| Did not exceed drug controls | Exceeded one or two drug controls | ||||||
| a SI | ||||||
|---|---|---|---|---|---|---|
| ID | Epimastigote | Trypomastigote | Amastigote | |||
| NINOA | A1 | NINOA | A1 | NINOA | A1 | |
| Glic | b Nc | >18.7 | >2.2 | b Nc | b Nc | >16.3 |
| Glim | 0.2 | 0.38 | 0.07 | 0.07 | 0.07 | 0.3 |
| Ace | >30.7 | >3.8 | >4.3 | b Nc | >4.34 | b Nc |
| Tol | >23.5 | b Nc | >20.4 | >2.0 | >1.3 | >4.5 |
| Reference drugs | ||||||
| Aaz | 2.3 | 0.8 | 0.06 | 0.06 | 0.06 | 1.3 |
| Nfx | 23.2 | 4.2 | 1.0 | 1.4 | 2.3 | 3 |
| Bzn | 4.4 | 6.9 | 0.8 | 0.9 | c Ne | c Ne |
| Compound | KI (μM) a | ||
|---|---|---|---|
| hCA I | hCA II | TcCA | |
| Glic | >100 | >100 | >100 |
| Glim | >100 | 19.9 | 35.7 |
| Ace | 17.3 | 2.9 | 5.6 |
| Tol | >100 | >100 | >100 |
| Aaz | 0.25 | 0.012 | 0.061 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortiz-Pérez, E.; Moreno-Rodríguez, A.; Delgado-Maldonado, T.; Ortega-Balleza, J.L.; González-González, A.; Paz-González, A.D.; Vázquez, K.; Avalos-Navarro, G.; Giovannuzzi, S.; Supuran, C.T.; et al. Repositioning FDA-Approved Sulfonamide-Based Drugs as Potential Carbonic Anhydrase Inhibitors in Trypanosoma cruzi: Virtual Screening and In Vitro Studies. Pharmaceuticals 2025, 18, 669. https://doi.org/10.3390/ph18050669
Ortiz-Pérez E, Moreno-Rodríguez A, Delgado-Maldonado T, Ortega-Balleza JL, González-González A, Paz-González AD, Vázquez K, Avalos-Navarro G, Giovannuzzi S, Supuran CT, et al. Repositioning FDA-Approved Sulfonamide-Based Drugs as Potential Carbonic Anhydrase Inhibitors in Trypanosoma cruzi: Virtual Screening and In Vitro Studies. Pharmaceuticals. 2025; 18(5):669. https://doi.org/10.3390/ph18050669
Chicago/Turabian StyleOrtiz-Pérez, Eyra, Adriana Moreno-Rodríguez, Timoteo Delgado-Maldonado, Jessica L. Ortega-Balleza, Alonzo González-González, Alma D. Paz-González, Karina Vázquez, Guadalupe Avalos-Navarro, Simone Giovannuzzi, Claudiu T. Supuran, and et al. 2025. "Repositioning FDA-Approved Sulfonamide-Based Drugs as Potential Carbonic Anhydrase Inhibitors in Trypanosoma cruzi: Virtual Screening and In Vitro Studies" Pharmaceuticals 18, no. 5: 669. https://doi.org/10.3390/ph18050669
APA StyleOrtiz-Pérez, E., Moreno-Rodríguez, A., Delgado-Maldonado, T., Ortega-Balleza, J. L., González-González, A., Paz-González, A. D., Vázquez, K., Avalos-Navarro, G., Giovannuzzi, S., Supuran, C. T., & Rivera, G. (2025). Repositioning FDA-Approved Sulfonamide-Based Drugs as Potential Carbonic Anhydrase Inhibitors in Trypanosoma cruzi: Virtual Screening and In Vitro Studies. Pharmaceuticals, 18(5), 669. https://doi.org/10.3390/ph18050669