

Identification of Microbial-Based Natural Products as Potential CYP51 Inhibitors for Eumycetoma Treatment: Insights from Molecular Docking, MM-GBSA Calculations, ADMET Analysis, and Molecular Dynamics Simulations

 , , , and
, , , and
Abstract

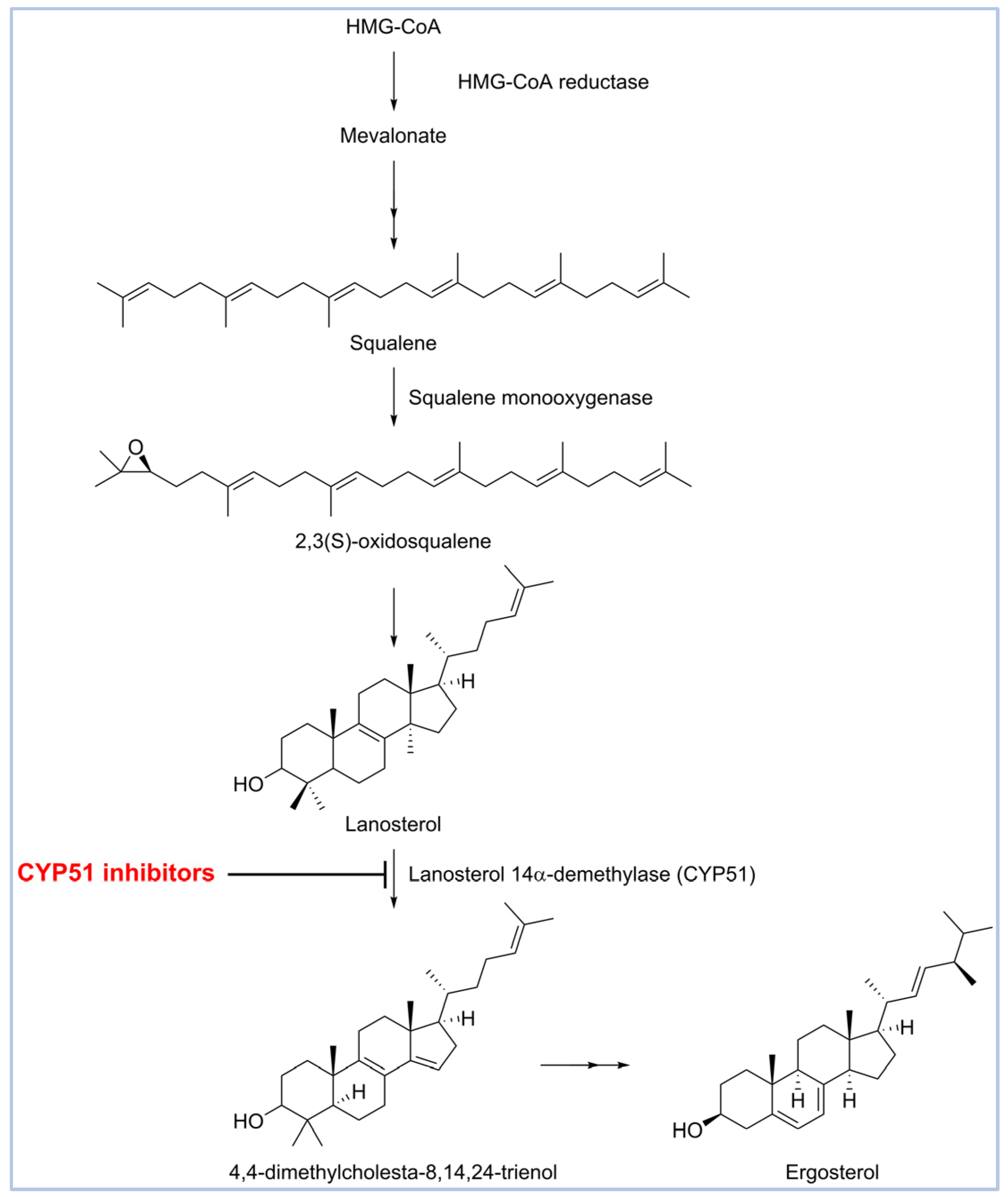
1. Introduction
2. Results and Discussion
2.1. Homology Modeling and Binding Site Identification
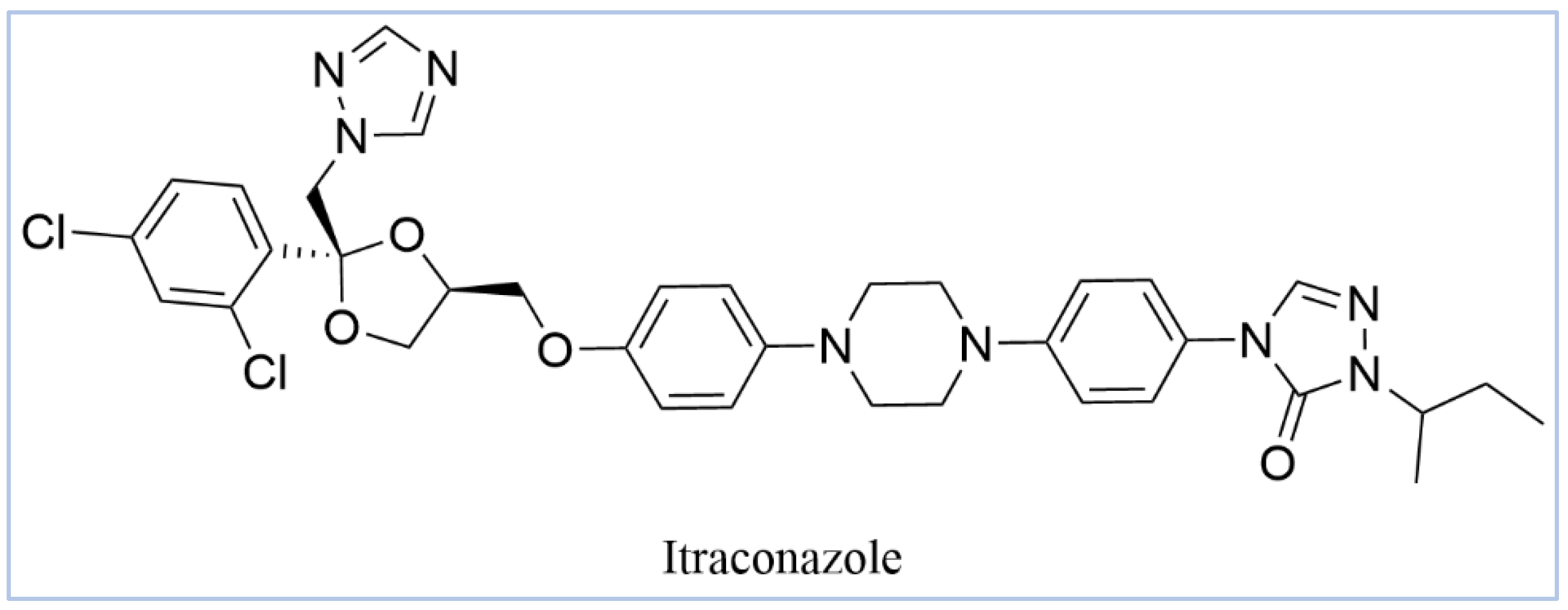
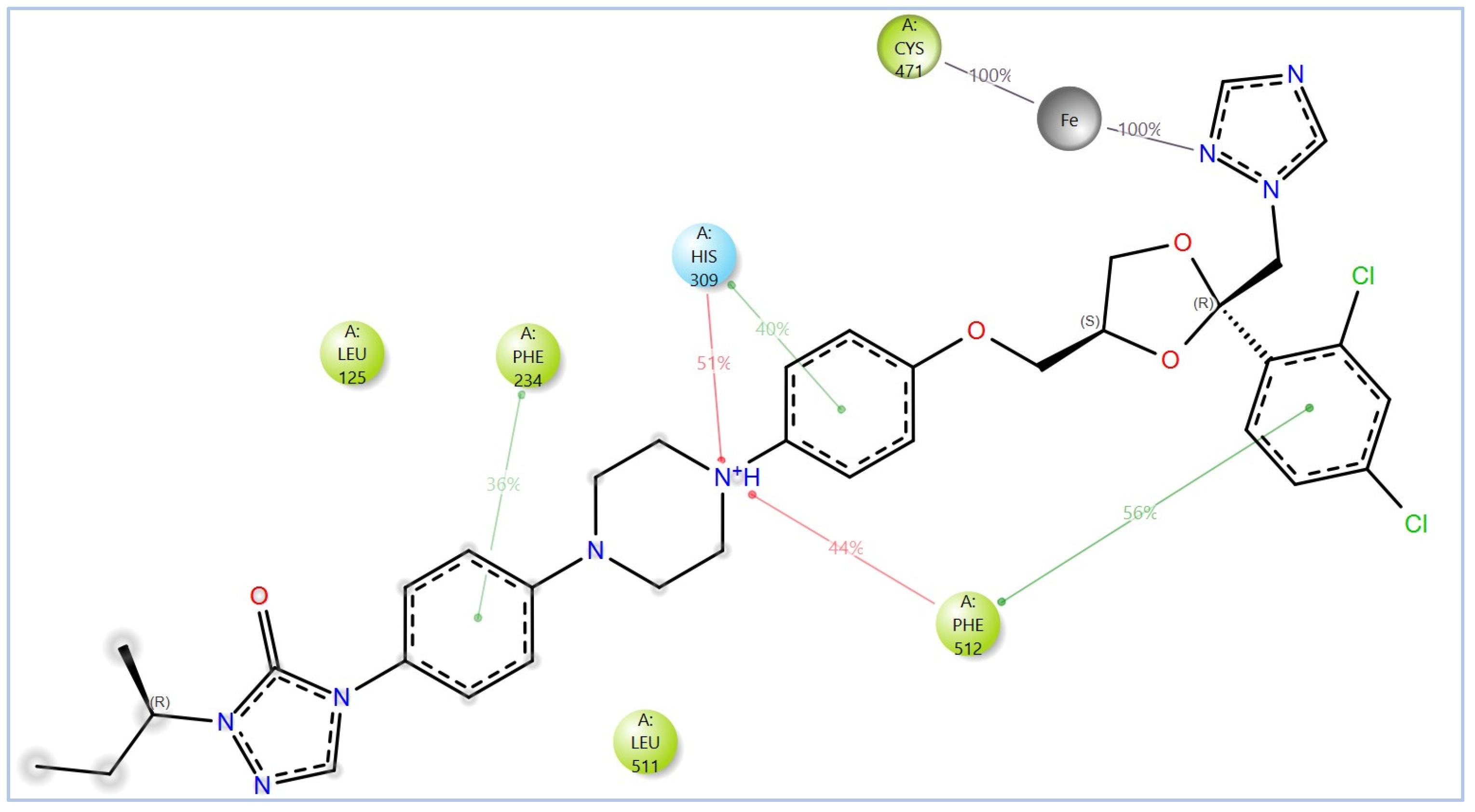
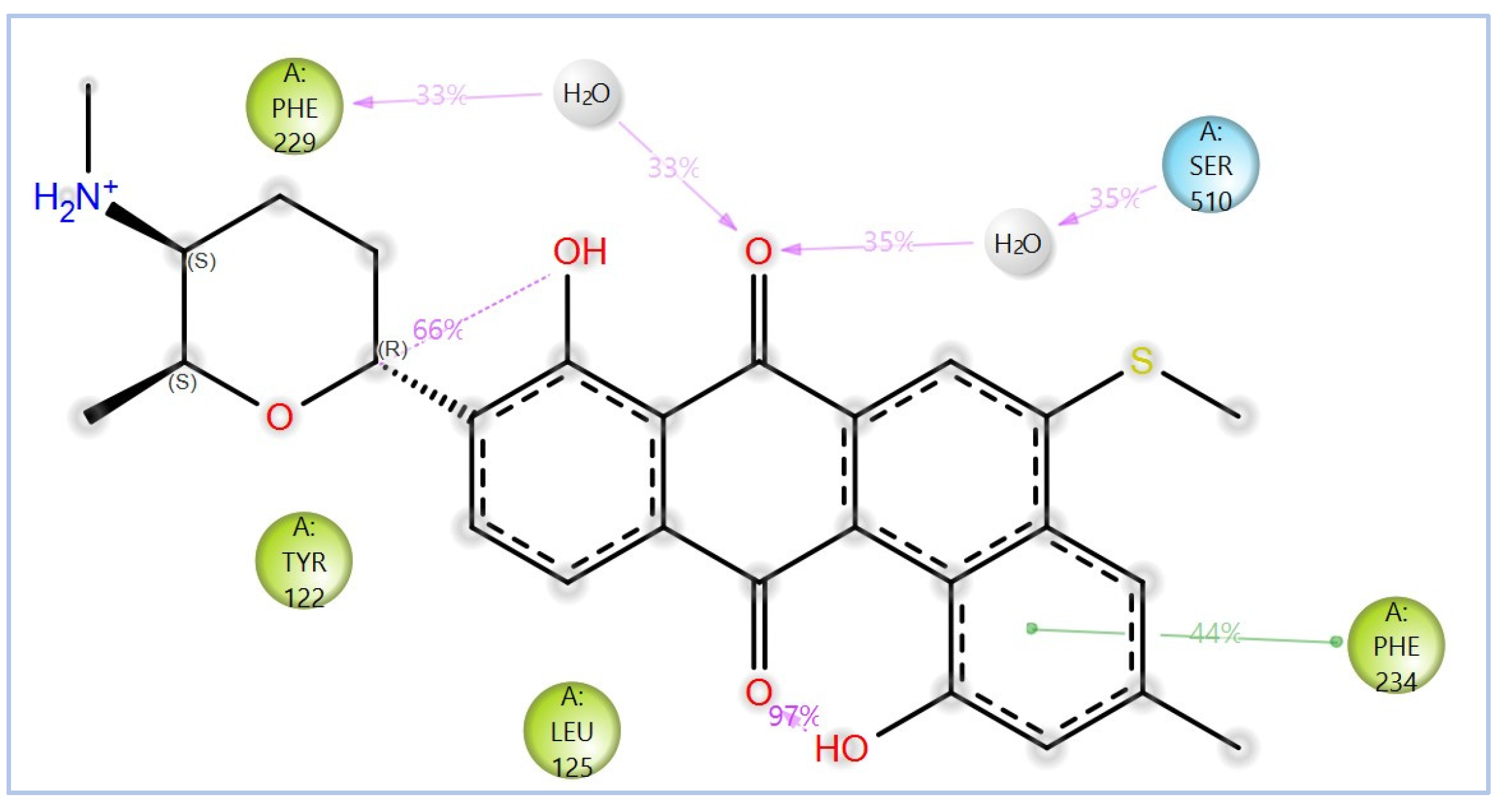
2.2. Virtual Screening, Molecular Docking, and Binding Free Energy Calculations
2.3. In Silico ADMET Profiling
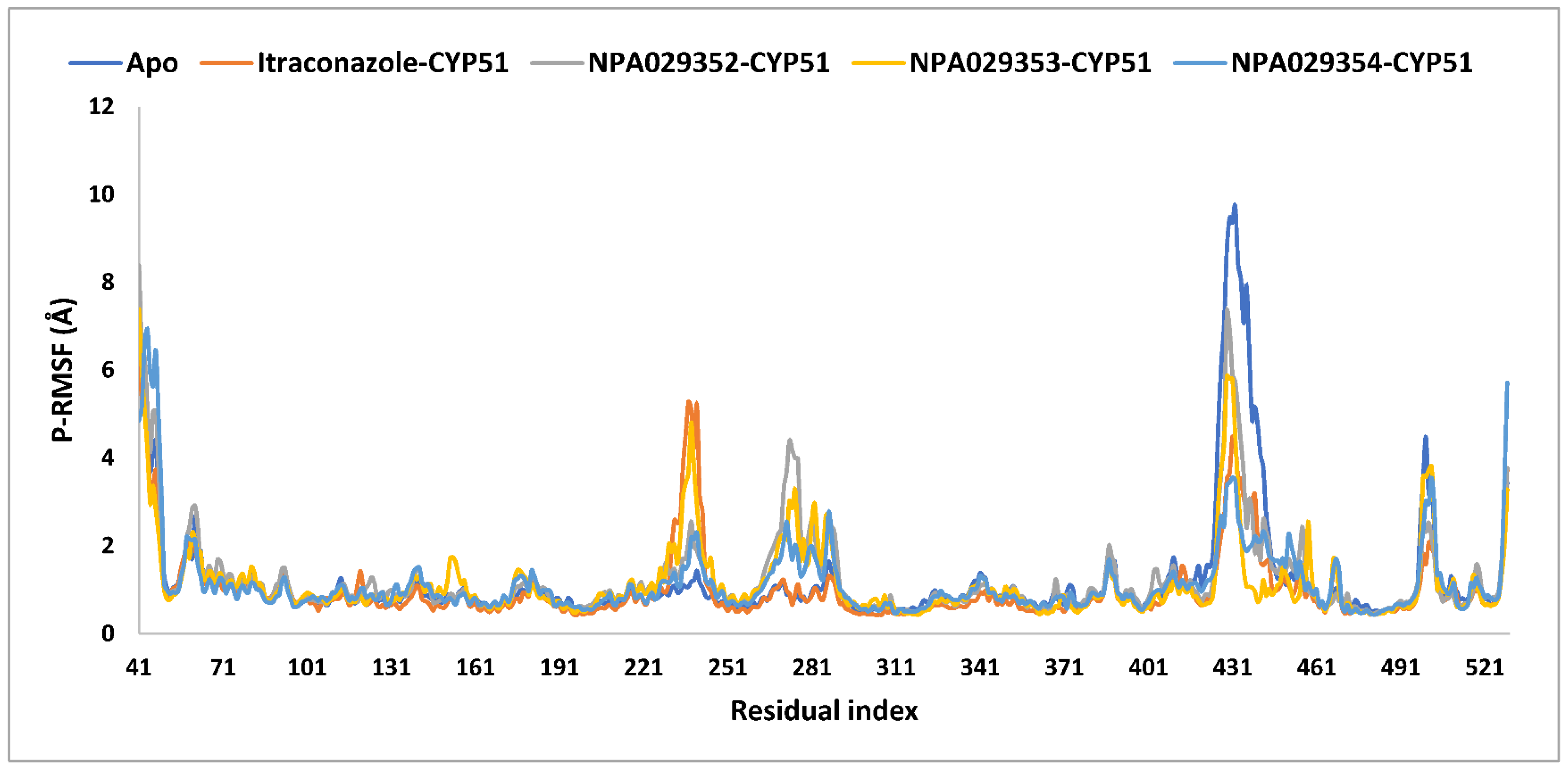
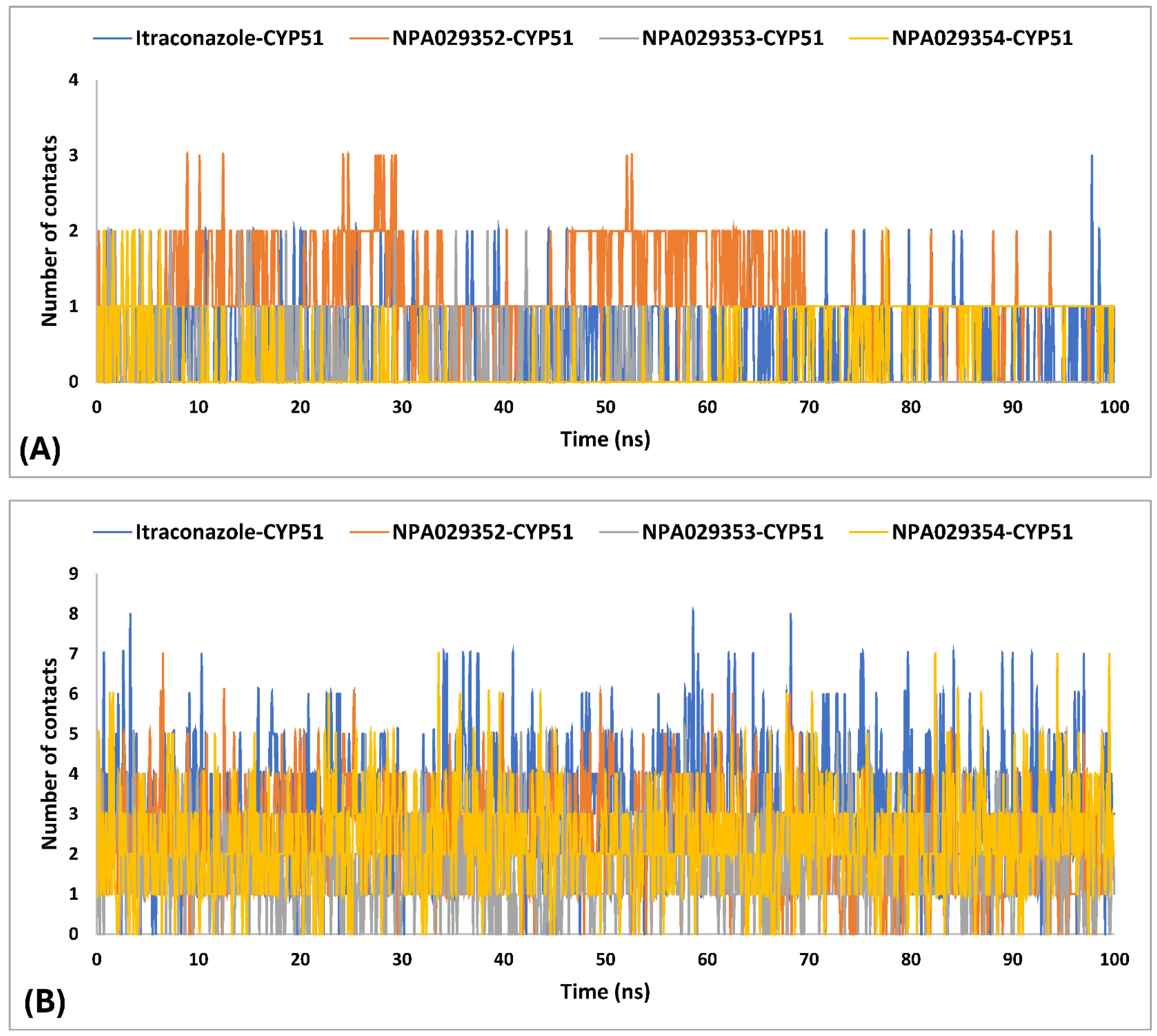
2.4. Molecular Dynamics (MD) Simulations
2.5. Study Limitations and Future Perspectives
3. Material and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Clark, J.E.; Kim, H.Y.; van de Sande, W.W.J.; McMullan, B.; Verweij, P.; Alastruey-Izquierdo, A.; Chakrabarti, A.; Harrison, T.S.; Bongomin, F.; Hay, R.J.; et al. Eumycetoma causative agents: A systematic review to inform the World Health Organization priority list of fungal pathogens. Med. Mycol. 2024, 62, myae044. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, E.E.; van de Sande, W.W.J.; Welsh, O.; Mahgoub, E.S.; Goodfellow, M.; Fahal, A.H. Mycetoma: A unique neglected tropical disease. Lancet Infect. Dis. 2016, 16, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Elkheir, L.Y.M.; Haroun, R.; Mohamed, M.A.; Fahal, A.H. Madurella mycetomatis causing eumycetoma medical treatment: The challenges and prospects. PLoS Negl. Trop. Dis. 2020, 14, e0008307. [Google Scholar] [CrossRef]
- Fahal, A.; Mahgoub, E.L.S.; El Hassan, A.M.; Abdel-Rahman, M.E. Mycetoma in the Sudan: An update from the Mycetoma Research Centre, University of Khartoum, Sudan. PLoS Negl. Trop. Dis. 2015, 9, e0003679. [Google Scholar] [CrossRef]
- Elkheir, L.Y.M.; Delaye, P.-O.; Penichon, M.; Eadie, K.; Mohamed, M.A.; Besson, P.; Chesnay, A.; Desoubeaux, G.; Roger, S.; van de Sande, W.W.J.; et al. Emerging therapeutics: The imidazo[1,2-b]pyridazine scaffold as a novel drug candidate for eumycetoma, a neglected tropical disease. Eur. J. Med. Chem. 2024, 277, 116720. [Google Scholar] [CrossRef]
- Suleiman, S.H.; Wadaella, E.L.S.; Fahal, A.H. The Surgical Treatment of Mycetoma. PLoS Negl. Trop. Dis. 2016, 10, e0004690. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Jagati, A.; Rathod, S.P.; Kalra, K.; Patel, S.; Chaudhari, M. Clinical Features of Mycetoma and the Appropriate Treatment Options. Res. Rep. Trop. Med. 2021, 12, 173–179. [Google Scholar] [CrossRef]
- Efared, B.; Tahiri, L.; Boubacar, M.S.; Atsam-Ebang, G.; Hammas, N.; Hinde, E.F.; Chbani, L. Mycetoma in a non-endemic area: A diagnostic challenge. BMC Clin. Pathol. 2017, 17, 1. [Google Scholar] [CrossRef]
- Emmanuel, P.; Dumre, S.P.; John, S.; Karbwang, J.; Hirayama, K. Mycetoma: A clinical dilemma in resource limited settings. Ann. Clin. Microbiol. Antimicrob. 2018, 17, 35. [Google Scholar] [CrossRef]
- Abd Algaffar, S.O.; Satyal, P.; Ashmawy, N.S.; Verbon, A.; van de Sande, W.W.J.; Khalid, S.A. In Vitro and In Vivo Wide-Spectrum Dual Antimycetomal Activity of Eight Essential Oils Coupled with Chemical Composition and Metabolomic Profiling. Microbiol. Res. 2024, 15, 1280–1297. [Google Scholar] [CrossRef]
- Chandler, D.J.; Bonifaz, A.; van de Sande, W.W.J. An update on the development of novel antifungal agents for eumycetoma. Front. Pharmacol. 2023, 14, 1165273. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Lyons, D.C. The Rise and Fall of Oral Ketoconazole. J. Cutan. Med. Surg. 2015, 19, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, P.V.; Venugopal, T.V. Treatment of eumycetoma with ketoconazole. Australas. J. Dermatol. 1993, 34, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Hussein, S.M.E.; Saeed, A.A.; Fahal, A.H. Mycetoma managment: Therapeutic challenges and the role of pharmacovigilance. PLoS Negl. Trop. Dis. 2025, 19, e0012827. [Google Scholar] [CrossRef]
- Kwizera, R.; Bongomin, F.; Meya, D.B.; Denning, D.W.; Fahal, A.H.; Lukande, R. Mycetoma in Uganda: A neglected tropical disease. PLoS Negl. Trop. Dis. 2020, 14, e0008240. [Google Scholar] [CrossRef]
- Emery, D.; Denning, D.W. The global distribution of actinomycetoma and eumycetoma. PLoS Negl. Trop. Dis. 2020, 14, e0008397. [Google Scholar] [CrossRef]
- Siddig, E.E.; Ahmed, A.; Ali, Y.; Bakhiet, S.M.; Mohamed, N.S.; Ahmed, E.S.; Fahal, A.H. Eumycetoma Medical Treatment: Past, Current Practice, Latest Advances and Perspectives. Microbiol. Res. 2021, 12, 899–906. [Google Scholar] [CrossRef]
- Abdallh, M.G.A.; Hemeda, S.; Elmadani, M.; Ibrahim, B.; Ahmed, A.E.E. Epidemiology, risk factors, and awareness of mycetoma among residents in Eastern Sinnar locality, Sudan, 2021. J. Glob. Health 2025, 15, 04005. [Google Scholar] [CrossRef]
- Hassan, R.; Cano, J.; Fronterre, C.; Bakhiet, S.; Fahal, A.; Deribe, K.; Newport, M. Estimating the burden of mycetoma in Sudan for the period 1991-2018 using a model-based geostatistical approach. PLoS Negl. Trop. Dis. 2022, 16, e0010795. [Google Scholar] [CrossRef]
- Siddig, E.E.; Ahmed, A. The urgent need for developing and implementing a multisectoral One Health strategy for the surveillance, prevention, and control of eumycetoma. IJID One Health 2024, 5, 100048. [Google Scholar] [CrossRef]
- Hernández-Hernández, F.; Méndez-Tovar, L.J. Eumycetoma and Global Warming. In The Impact of Climate Change on Fungal Diseases; Frías-De-León, M.G., Brunner-Mendoza, C., Reyes-Montes, M.d.R., Duarte-Escalante, E., Eds.; Springer International Publishing: Cham, Switzerland, 2022; pp. 99–113. [Google Scholar]
- Mohamed, M.A.; Awadalla, M.K.; Mohamed, M.S.; Elsaman, T.; Eltayib, E.M. Repurposing FDA-Approved Drugs for Eumycetoma Treatment: Homology Modeling and Computational Screening of CYP51 Inhibitors. Int. J. Mol. Sci. 2025, 26, 315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wang, Y.; Wu, A.; Wang, J.; Zhang, J. Strategies of targeting CYP51 for IFIs therapy: Emerging prospects, opportunities and challenges. Eur. J. Med. Chem. 2023, 259, 115658. [Google Scholar] [CrossRef]
- Hargrove, T.Y.; Lamb, D.C.; Wawrzak, Z.; Hull, M.; Kelly, S.L.; Guengerich, F.P.; Lepesheva, G.I. Identification of Potent and Selective Inhibitors of Acanthamoeba: Structural Insights into Sterol 14α-Demethylase as a Key Drug Target. J. Med. Chem. 2024, 67, 7443–7457. [Google Scholar] [CrossRef] [PubMed]
- Hossain, C.M.; Ryan, L.K.; Gera, M.; Choudhuri, S.; Lyle, N.; Ali, K.A.; Diamond, G. Antifungals and Drug Resistance. Encyclopedia 2022, 2, 1722–1737. [Google Scholar] [CrossRef]
- Choi, J.Y.; Podust, L.M.; Roush, W.R. Drug Strategies Targeting CYP51 in Neglected Tropical Diseases. Chem. Rev. 2014, 114, 11242–11271. [Google Scholar] [CrossRef]
- Zhang, J.; Li, L.; Lv, Q.; Yan, L.; Wang, Y.; Jiang, Y. The Fungal CYP51s: Their Functions, Structures, Related Drug Resistance, and Inhibitors. Front. Microbiol. 2019, 10, 691. [Google Scholar] [CrossRef]
- Zhou, S.F.; Zhong, W.Z. Drug Design and Discovery: Principles and Applications. Molecules 2017, 22, 279. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef]
- Mohamed, A.A.E.; Mohamed, K.A.A.; Marwa, S.S.O.; Magdi, A.M.; Mahmoud, M.E.M.; Muzamil, M.A.H.; Malik, S.M.; Mohammed, A.G. Evaluation of antileishmanial activity of valproic acid against Leishmania donovani: An integrated in silico and in vitro study. World J. Pharm. Sci. 2016, 4, 153–159. [Google Scholar]
- Bano, I.; Butt, U.D.; Mohsan, S.A.H. Chapter 25—New challenges in drug discovery. In Novel Platforms for Drug Delivery Applications; Das, S., Thomas, S., Das, P.P., Eds.; Woodhead Publishing: Sawston, UK, 2023; pp. 619–643. [Google Scholar]
- Goupil, L.S.; McKerrow, J.H. Introduction: Drug Discovery and Development for Neglected Diseases. Chem. Rev. 2014, 114, 11131–11137. [Google Scholar] [CrossRef]
- Fahal, A.H.; Ahmed, K.O.; Saeed, A.A.; Elkhawad, A.O.; Bakhiet, S.M. Why the mycetoma patients are still neglected. PLoS Negl. Trop. Dis. 2022, 16, e0010945. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, I.H. Drug discovery for neglected diseases: Molecular target-based and phenotypic approaches. J. Med. Chem. 2013, 56, 7719–7726. [Google Scholar] [CrossRef]
- Weng, H.-B.; Chen, H.-X.; Wang, M.-W. Innovation in neglected tropical disease drug discovery and development. Infect. Dis. Poverty 2018, 7, 67. [Google Scholar] [CrossRef] [PubMed]
- López-López, E.; Barrientos-Salcedo, C.; Prieto-Martínez, F.D.; Medina-Franco, J.L. In silico tools to study molecular targets of neglected diseases: Inhibition of TcSir2rp3, an epigenetic enzyme of Trypanosoma cruzi. Adv. Protein Chem. Struct. Biol. 2020, 122, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Yasuo, N.; Ishida, T.; Sekijima, M. Computer aided drug discovery review for infectious diseases with case study of anti-Chagas project. Parasitol. Int. 2021, 83, 102366. [Google Scholar] [CrossRef]
- Herrera Acevedo, C.; Scotti, L.; Feitosa Alves, M.; Formiga Melo Diniz, M.D.; Scotti, M.T. Computer-Aided Drug Design Using Sesquiterpene Lactones as Sources of New Structures with Potential Activity against Infectious Neglected Diseases. Molecules 2017, 22, 79. [Google Scholar] [CrossRef]
- Adelusi, T.I.; Oyedele, A.-Q.K.; Boyenle, I.D.; Ogunlana, A.T.; Adeyemi, R.O.; Ukachi, C.D.; Idris, M.O.; Olaoba, O.T.; Adedotun, I.O.; Kolawole, O.E.; et al. Molecular modeling in drug discovery. Inform. Med. Unlocked 2022, 29, 100880. [Google Scholar] [CrossRef]
- Gheidari, D.; Mehrdad, M.; karimelahi, Z. Virtual screening, ADMET prediction, molecular docking, and dynamic simulation studies of natural products as BACE1 inhibitors for the management of Alzheimer’s disease. Sci. Rep. 2024, 14, 26431. [Google Scholar] [CrossRef]
- Niazi, S.K.; Mariam, Z. Computer-Aided Drug Design and Drug Discovery: A Prospective Analysis. Pharmaceuticals 2023, 17, 22. [Google Scholar] [CrossRef]
- Ece, A. Computer-aided drug design. BMC Chem. 2023, 17, 26. [Google Scholar] [CrossRef]
- Mushtaq, S.; Abbasi, B.H.; Uzair, B.; Abbasi, R. Natural products as reservoirs of novel therapeutic agents. EXCLI J. 2018, 17, 420–451. [Google Scholar] [CrossRef] [PubMed]
- Smit, S.; Derks Martijn, F.L.; Bervoets, S.; Fahal, A.; van Leeuwen, W.; van Belkum, A.; van de Sande Wendy, W.J. Genome Sequence of Madurella mycetomatis mm55, Isolated from a Human Mycetoma Case in Sudan. Genome Announc. 2016, 4, e00418-16. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.S.; Martin, J.H. x ray crystallography. Mol. Pathol. 2000, 53, 8–14. [Google Scholar] [CrossRef]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Poynton, E.F.; van Santen, J.A.; Pin, M.; Contreras, M.M.; McMann, E.; Parra, J.; Showalter, B.; Zaroubi, L.; Duncan, K.R.; Linington, R.G. The Natural Products Atlas 3.0: Extending the database of microbially derived natural products. Nucleic Acids Res. 2024, 53, D691–D699. [Google Scholar] [CrossRef]
- van Santen, J.A.; Poynton, E.F.; Iskakova, D.; McMann, E.; Alsup, T.A.; Clark, T.N.; Fergusson, C.H.; Fewer, D.P.; Hughes, A.H.; McCadden, C.A.; et al. The Natural Products Atlas 2.0: A database of microbially-derived natural products. Nucleic Acids Res. 2021, 50, D1317–D1323. [Google Scholar] [CrossRef] [PubMed]
- van Santen, J.A.; Jacob, G.; Singh, A.L.; Aniebok, V.; Balunas, M.J.; Bunsko, D.; Neto, F.C.; Castaño-Espriu, L.; Chang, C.; Clark, T.N.; et al. The Natural Products Atlas: An Open Access Knowledge Base for Microbial Natural Products Discovery. ACS Cent. Sci. 2019, 5, 1824–1833. [Google Scholar] [CrossRef] [PubMed]
- Elsaman, T.; Mohamed, M.A. Examining Prenylated Xanthones as Potential Inhibitors Against Ketohexokinase C Isoform for the Treatment of Fructose-Driven Metabolic Disorders: An Integrated Computational Approach. Pharmaceuticals 2025, 18, 126. [Google Scholar] [CrossRef]
- Elsaman, T.; Mohamed, M.A.; Mohamed, M.S.; Eltayib, E.M.; Abdalla, A.E. Microbial-based natural products as potential inhibitors targeting DNA gyrase B of Mycobacterium tuberculosis: An in silico study. Front. Chem. 2025, 13, 1524607. [Google Scholar] [CrossRef]
- Fahal, A.H.; Ahmed, E.S.; Bakhiet, S.M.; Bakhiet, O.E.; Fahal, L.A.; Mohamed, A.A.; Mohamedelamin, E.S.W.; Bahar, M.E.N.; Attalla, H.Y.; Siddig, E.E.; et al. Two dose levels of once-weekly fosravuconazole versus daily itraconazole in combination with surgery in patients with eumycetoma in Sudan: A randomised, double-blind, phase 2, proof-of-concept superiority trial. Lancet Infect. Dis. 2024, 24, 1254–1265. [Google Scholar] [CrossRef]
- Adelusi, T.I.; Bolaji, O.Q.; Ojo, T.O.; Adegun, I.P.; Adebodun, S. Molecular Mechanics with Generalized Born Surface Area (MMGBSA) Calculations and Docking Studies Unravel some Antimalarial Compounds Using Heme O Synthase as Therapeutic Target. ChemistrySelect 2023, 8, e202303686. [Google Scholar] [CrossRef]
- Mohamed, M.A.; Elsaman, T.; Mohamed, M.S.; Eltayib, E.M. Computational investigations of flavonoids as ALDH isoform inhibitors for treatment of cancer. SAR QSAR Environ. Res. 2024, 35, 837–875. [Google Scholar] [CrossRef]
- Mohamed, M.A.; Elsaman, T.; Elderdery, A.Y.; Alsrhani, A.; Ghanem, H.B.; Alruwaili, M.M.; Hamza, S.M.A.; Mekki, S.E.I.; Alotaibi, H.A.; Mills, J. Unveiling the Anticancer Potential: Computational Exploration of Nitrogenated Derivatives of (+)-Pancratistatin as Topoisomerase I Inhibitors. Int. J. Mol. Sci. 2024, 25, 779. [Google Scholar] [CrossRef] [PubMed]
- van de Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.A.; Alanazi, A.F.; Alanazi, W.A.; Elsaman, T.; Mohamed, M.S.; Eltayib, E.M. Repurposing of eluxadoline as a SARS-CoV-2 main protease inhibitor: E-Pharmacophore based virtual screening, molecular docking, MM-GBSA calculations, and molecular dynamics simulations studies. J. Appl. Pharm. Sci. 2025, 15, 102–110. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef]
- Friggeri, L.; Hargrove, T.Y.; Wawrzak, Z.; Blobaum, A.L.; Rachakonda, G.; Lindsley, C.W.; Villalta, F.; Nes, W.D.; Botta, M.; Guengerich, F.P.; et al. Sterol 14α-Demethylase Structure-Based Design of VNI ((R)- N-(1-(2,4-Dichlorophenyl)-2-(1 H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide)) Derivatives To Target Fungal Infections: Synthesis, Biological Evaluation, and Crystallographic Analysis. J. Med. Chem. 2018, 61, 5679–5691. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G.; Spronk, C. YASARA–yet another scientific artificial reality application. YASARA Org 2013, 993, 51–78. [Google Scholar]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Hargrove, T.Y.; Wawrzak, Z.; Guengerich, F.P.; Lepesheva, G.I. A requirement for an active proton delivery network supports a compound I-mediated C–C bond cleavage in CYP51 catalysis. J. Biol. Chem. 2020, 295, 9998–10007. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Eadie, K.; Schippers, M.; Fahal, A.; Laleu, B.; Verbon, A.; van de Sande, W.W.J. Novel Compound MMV1804559 from the Global Health Priority Box Exhibits In Vitro and In Vivo Activity against Madurella mycetomatis. Int. J. Mol. Sci. 2024, 25, 6227. [Google Scholar] [CrossRef]
- Scolding, P.; Fahal, A.; Yotsu, R.R. Drug therapy for Mycetoma. Cochrane Database Syst. Rev. 2018, 2018, CD013082. [Google Scholar] [CrossRef]
- Lim, W.; Verbon, A.; van de Sande, W. Identifying novel drugs with new modes of action for neglected tropical fungal skin diseases (fungal skinNTDs) using an Open Source Drug discovery approach. Expert. Opin. Drug Discov. 2022, 17, 641–659. [Google Scholar] [CrossRef]
- Elsaman, T.; Ahmad, I.; Eltayib, E.M.; Suliman Mohamed, M.; Yusuf, O.; Saeed, M.; Patel, H.; Mohamed, M.A. Flavonostilbenes natural hybrids from Rhamnoneuron balansae as potential antitumors targeting ALDH1A1: Molecular docking, ADMET, MM-GBSA calculations and molecular dynamics studies. J. Biomol. Struct. Dyn. 2024, 42, 3249–3266. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [PubMed]
- Sgobba, M.; Caporuscio, F.; Anighoro, A.; Portioli, C.; Rastelli, G. Application of a post-docking procedure based on MM-PBSA and MM-GBSA on single and multiple protein conformations. Eur. J. Med. Chem. 2012, 58, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert. Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Hoffer, L.; Muller, C.; Roche, P.; Morelli, X. Chemistry-driven Hit-to-lead Optimization Guided by Structure-based Approaches. Mol. Inform. 2018, 37, e1800059. [Google Scholar] [CrossRef]
- Albanese, S.K.; Chodera, J.D.; Volkamer, A.; Keng, S.; Abel, R.; Wang, L. Is Structure-Based Drug Design Ready for Selectivity Optimization? J. Chem. Inf. Model. 2020, 60, 6211–6227. [Google Scholar] [CrossRef]
- Ferreira, L.L.G.; Andricopulo, A.D. ADMET modeling approaches in drug discovery. Drug Discov. Today 2019, 24, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.S. Preclinical pharmacokinetics: An approach towards safer and efficacious drugs. Curr. Drug Metab. 2006, 7, 165–182. [Google Scholar] [CrossRef]
- Wu, F.; Zhou, Y.; Li, L.; Shen, X.; Chen, G.; Wang, X.; Liang, X.; Tan, M.; Huang, Z. Computational Approaches in Preclinical Studies on Drug Discovery and Development. Front. Chem. 2020, 8, 726. [Google Scholar] [CrossRef]
- Ioakimidis, L.; Thoukydidis, L.; Mirza, A.; Naeem, S.; Reynisson, J. Benchmarking the Reliability of QikProp. Correlation between Experimental and Predicted Values. QSAR Comb. Sci. 2008, 27, 445–456. [Google Scholar] [CrossRef]
- Ntie-Kang, F.; Lifongo, L.L.; Mbah, J.A.; Owono Owono, L.C.; Megnassan, E.; Mbaze, L.M.; Judson, P.N.; Sippl, W.; Efange, S.M. In silico drug metabolism and pharmacokinetic profiles of natural products from medicinal plants in the Congo basin. In Silico Pharmacol. 2013, 1, 12. [Google Scholar] [CrossRef] [PubMed]
- Ntie-Kang, F.; Mbah, J.A.; Lifongo, L.L.; Owono Owono, L.C.; Megnassan, E.; Meva’a Mbaze, L.; Judson, P.N.; Sippl, W.; Efange, S.M. Assessing the pharmacokinetic profile of the CamMedNP natural products database: An in silico approach. Org. Med. Chem. Lett. 2013, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef]
- Butnarasu, C.; Garbero, O.V.; Petrini, P.; Visai, L.; Visentin, S. Permeability Assessment of a High-Throughput Mucosal Platform. Pharmaceutics 2023, 15, 380. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Prasanna, S.; Doerksen, R.J. Topological polar surface area: A useful descriptor in 2D-QSAR. Curr. Med. Chem. 2009, 16, 21–41. [Google Scholar] [CrossRef]
- Asano, D.; Takakusa, H.; Nakai, D. Oral Absorption of Middle-to-Large Molecules and Its Improvement, with a Focus on New Modality Drugs. Pharmaceutics 2024, 16, 47. [Google Scholar] [CrossRef]
- Divyashri, G.; Krishna Murthy, T.P.; Sundareshan, S.; Kamath, P.; Murahari, M.; Saraswathy, G.R.; Sadanandan, B. In silico approach towards the identification of potential inhibitors from Curcuma amada Roxb against H. pylori: ADMET screening and molecular docking studies. Bioimpacts 2021, 11, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Shi, H.; Chen, H.; Zhang, K.; Wang, M.; Qiu, F. Oral Bioavailability Comparison of Artemisinin, Deoxyartemisinin, and 10-Deoxoartemisinin Based on Computer Simulations and Pharmacokinetics in Rats. ACS Omega 2021, 6, 889–899. [Google Scholar] [CrossRef]
- Wanat, K.; Żydek, G.; Hekner, A.; Brzezińska, E. In Silico Plasma Protein Binding Studies of Selected Group of Drugs Using TLC and HPLC Retention Data. Pharmaceuticals 2021, 14, 202. [Google Scholar] [CrossRef] [PubMed]
- Lexa, K.W.; Dolghih, E.; Jacobson, M.P. A structure-based model for predicting serum albumin binding. PLoS ONE 2014, 9, e93323. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Sun, J.; Zhao, Q. Investigating cardiotoxicity related with hERG channel blockers using molecular fingerprints and graph attention mechanism. Comput. Biol. Med. 2023, 153, 106464. [Google Scholar] [CrossRef]
- Masimirembwa, C.M.; Bredberg, U.; Andersson, T.B. Metabolic Stability for Drug Discovery and Development. Clin. Pharmacokinet. 2003, 42, 515–528. [Google Scholar] [CrossRef]
- Twycross, R.; Ross, J.; Kotlinska-Lemieszek, A.; Charlesworth, S.; Mihalyo, M.; Wilcock, A. Variability in Response to Drugs. J. Pain. Symptom Manag. 2015, 49, 293–306. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Zhang, M.-Q.; Wilkinson, B. Drug discovery beyond the ‘rule-of-five’. Curr. Opin. Biotechnol. 2007, 18, 478–488. [Google Scholar] [CrossRef]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Kurczab, R.; Ali, W.; Łażewska, D.; Kotańska, M.; Jastrzębska-Więsek, M.; Satała, G.; Więcek, M.; Lubelska, A.; Latacz, G.; Partyka, A.; et al. Computer-Aided Studies for Novel Arylhydantoin 1,3,5-Triazine Derivatives as 5-HT6 Serotonin Receptor Ligands with Antidepressive-Like, Anxiolytic and Antiobesity Action In Vivo. Molecules 2018, 23, 2529. [Google Scholar] [CrossRef]
- Trösken, E.R.; Adamska, M.; Arand, M.; Zarn, J.A.; Patten, C.; Völkel, W.; Lutz, W.K. Comparison of lanosterol-14α-demethylase (CYP51) of human and Candida albicans for inhibition by different antifungal azoles. Toxicology 2006, 228, 24–32. [Google Scholar] [CrossRef]
- Jäger, M.C.; Joos, F.L.; Winter, D.V.; Odermatt, A. Characterization of the interferences of systemic azole antifungal drugs with adrenal steroid biosynthesis using H295R cells and enzyme activity assays. Curr. Res. Toxicol. 2023, 5, 100119. [Google Scholar] [CrossRef]
- Borjian Boroujeni, M.; Shahbazi Dastjerdeh, M.; Shokrgozar, M.; Rahimi, H.; Omidinia, E. Computational driven molecular dynamics simulation of keratinocyte growth factor behavior at different pH conditions. Inform. Med. Unlocked 2021, 23, 100514. [Google Scholar] [CrossRef]
- Oyewusi, H.A.; Wahab, R.A.; Akinyede, K.A.; Albadrani, G.M.; Al-Ghadi, M.Q.; Abdel-Daim, M.M.; Ajiboye, B.O.; Huyop, F. Bioinformatics analysis and molecular dynamics simulations of azoreductases (AzrBmH2) from Bacillus megaterium H2 for the decolorization of commercial dyes. Environ. Sci. Eur. 2024, 36, 31. [Google Scholar] [CrossRef]
- Patil, R.; Das, S.; Stanley, A.; Yadav, L.; Sudhakar, A.; Varma, A.K. Optimized hydrophobic interactions and hydrogen bonding at the target-ligand interface leads the pathways of drug-designing. PLoS ONE 2010, 5, e12029. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Lin, L.; Liu, L.; Yu, Y.; Wang, H. Antifungal activity and molecular mechanisms of mulberrin derivatives against Colletotrichum gloeosporioides for mango storage. Int. J. Food Microbiol. 2022, 378, 109817. [Google Scholar] [CrossRef]
- Agarwal, S.K.; Singh, S.S.; Verma, S.; Kumar, S. Antifungal activity of anthraquinone derivatives from Rheum emodi. J. Ethnopharmacol. 2000, 72, 43–46. [Google Scholar] [CrossRef]
- Masi, M.; Evidente, A. Fungal Bioactive Anthraquinones and Analogues. Toxins 2020, 12, 714. [Google Scholar] [CrossRef]
- Ma, J.; Todd, M.; van de Sande, W.W.J.; Biersack, B. Antifungal Activity of Natural Naphthoquinones and Anthraquinones against Madurella mycetomatis. Chem. Biodivers. 2023, 20, e202300151. [Google Scholar] [CrossRef] [PubMed]
- Pinto, E.; Afonso, C.; Duarte, S.; Vale-Silva, L.; Costa, E.; Sousa, E.; Pinto, M. Antifungal activity of xanthones: Evaluation of their effect on ergosterol biosynthesis by high-performance liquid chromatography. Chem. Biol. Drug Des. 2011, 77, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.C.; Yao, K.; Kaplan, Z.; Chelliah, M.; Leswing, K.; Seekins, S.; Watts, S.; Calkins, D.; Chief Elk, J.; Jerome, S.V.; et al. Epik: pKa and Protonation State Prediction through Machine Learning. J. Chem. Theory Comput. 2023, 19, 2380–2388. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Pandya, V.; Rao, P.; Prajapati, J.; Rawal, R.M.; Goswami, D. Pinpointing top inhibitors for GSK3β from pool of indirubin derivatives using rigorous computational workflow and their validation using molecular dynamics (MD) simulations. Sci. Rep. 2024, 14, 49. [Google Scholar] [CrossRef]
- Klyshko, E.; Kim, J.S.-H.; McGough, L.; Valeeva, V.; Lee, E.; Ranganathan, R.; Rauscher, S. Functional protein dynamics in a crystal. Nat. Commun. 2024, 15, 3244. [Google Scholar] [CrossRef]
- Ivánczi, M.; Balogh, B.; Kis, L.; Mándity, I. Molecular Dynamics Simulations of Drug-Conjugated Cell-Penetrating Peptides. Pharmaceuticals 2023, 16, 1251. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Chemical Structure | Docking Score (kcal/mol) | MM-GBSA (kcal/mol) | |
|---|---|---|---|---|
| ID | Name | |||
| NPA020764 | Octacosamicin A | 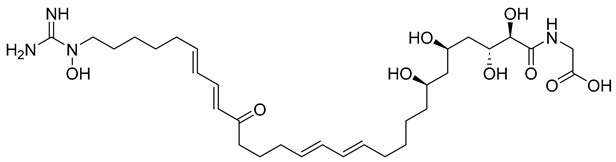 | −11.385 | −76.66 |
| NPA029353 | Monacyclinone H |  | −11.649 | −72.39 |
| NPA029354 | Monacyclinone I | 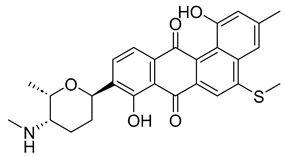 | −11.460 | −65.62 |
| NPA017021 | Monacyclinone E |  | −13.669 | −62.35 |
| NPA026108 | 3′-N-methyl-medermycin |  | −10.299 | −62.10 |
| NPA029352 | Monacyclinone G |  | −11.716 | −58.89 |
| NPA020514 | gamma-iso-Rubromycin | 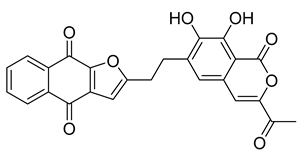 | −10.877 | −55.05 |
| NPA023185 | 2,2′-bis-(7-methyl-1,4,5-trihydroxy-anthracene-9,10-dione) | 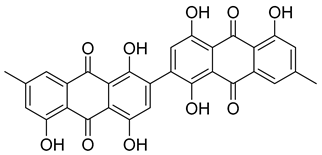 | −14.449 | −54.51 |
| NPA018887 | Roseobacticide K |  | −9.626 | −54.34 |
| - | Itraconazole-ionized |  | −5.952 | −54.07 |
| - | Itraconazole-unionized | 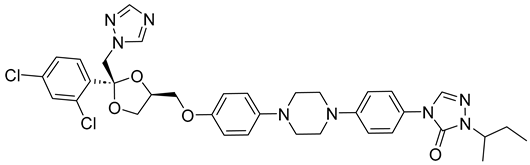 | −6.565 | −36.07 |
| Molecule | Stars | mol_MW | SASA | donorHB | accptHB | QPlogPo/w | QPlogS | QPlogHERG | QPPCaco | QPlogBB | #metab | QPlogKhsa | % Oral Absorption | PSA | Rule of Five | Rule of Three |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NPA017021 | 0 | 497.54 | 769.51 | 1 | 8.95 | 1.10 | −5.01 | −4.20 | 2.35 | −2.15 | 8 | 0.25 | 40.04 | 166.75 | 0 | 2 |
| NPA018887 | 1 | 522.58 | 691.09 | 1 | 6.75 | 4.89 | −6.03 | −5.43 | 594.45 | −0.64 | 1 | 0.86 | 92.32 | 98.02 | 1 | 1 |
| NPA020514 | 0 | 444.39 | 732.41 | 1 | 9.5 | 1.71 | −5.04 | −6.41 | 17.05 | −2.95 | 5 | −0.04 | 59.01 | 172.60 | 0 | 1 |
| NPA020764 | 12 | 624.77 | 1218.33 | 8.25 | 15.75 | 1.54 | −4.94 | −4.09 | 0.03 | −8.82 | 10 | −1.14 | 0 | 278.04 | 3 | 2 |
| NPA023185 | 4 | 538.46 | 799.47 | 0 | 6.5 | 3.26 | −6.82 | −6.29 | 2.36 | −4.07 | 8 | 0.80 | 26.84 | 219.93 | 2 | 3 |
| NPA026108 | 2 | 443.45 | 701.58 | 2 | 13.35 | −0.20 | −2.70 | −5.66 | 21.16 | −1.42 | 9 | −0.56 | 49.45 | 161.75 | 0 | 2 |
| NPA029352 | 0 | 433.50 | 699.01 | 2 | 7.7 | 3.11 | −4.47 | −6.18 | 242.72 | −0.26 | 6 | 0.50 | 87.87 | 83.35 | 0 | 0 |
| NPA029353 | 0 | 491.60 | 769.96 | 1 | 8.7 | 3.53 | −5.26 | −6.15 | 128.76 | −0.59 | 6 | 0.62 | 85.38 | 98.81 | 0 | 0 |
| NPA029354 | 0 | 477.57 | 782.85 | 2 | 8.2 | 3.53 | −5.72 | −6.55 | 114.73 | −0.71 | 6 | 0.68 | 84.49 | 105.81 | 0 | 1 |
| CYP51 Complex | Apo | Itraconazole | NPA029352 | NPA029353 | NPA029354 |
|---|---|---|---|---|---|
| PL-RMSD (Å) | |||||
| average | 3.1 | 3.2 | 3.1 | 3.1 | 2.7 |
| maximum | 4.2 | 3.7 | 4.1 | 3.8 | 3.5 |
| minimum | 1.5 | 1.5 | 1.2 | 1.4 | 1.3 |
| P-RMSF (Å) | |||||
| average | 1.2 | 1.0 | 1.2 | 1.2 | 1.1 |
| maximum | 9.7 | 6.3 | 8.4 | 7.4 | 6.9 |
| minimum | 0.5 | 0.4 | 0.4 | 0.4 | 0.4 |
| H-bond contacts | |||||
| average | - | 0.5 | 1.2 | 0.2 | 0.4 |
| maximum | - | 3.0 | 3.0 | 2.0 | 2.0 |
| minimum | - | 0.0 | 0.0 | 0.0 | 0.0 |
| Hydrophobic contacts | |||||
| average | - | 3.1 | 2.4 | 1.7 | 2.3 |
| maximum | - | 8.0 | 7.0 | 6.0 | 7.0 |
| minimum | - | 0.0 | 0.0 | 0.0 | 0.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elsaman, T.; Awadalla, M.K.A.; Mohamed, M.S.; Eltayib, E.M.; Mohamed, M.A. Identification of Microbial-Based Natural Products as Potential CYP51 Inhibitors for Eumycetoma Treatment: Insights from Molecular Docking, MM-GBSA Calculations, ADMET Analysis, and Molecular Dynamics Simulations. Pharmaceuticals 2025, 18, 598. https://doi.org/10.3390/ph18040598
Elsaman T, Awadalla MKA, Mohamed MS, Eltayib EM, Mohamed MA. Identification of Microbial-Based Natural Products as Potential CYP51 Inhibitors for Eumycetoma Treatment: Insights from Molecular Docking, MM-GBSA Calculations, ADMET Analysis, and Molecular Dynamics Simulations. Pharmaceuticals. 2025; 18(4):598. https://doi.org/10.3390/ph18040598
Chicago/Turabian StyleElsaman, Tilal, Mohamed Khalid Alhaj Awadalla, Malik Suliman Mohamed, Eyman Mohamed Eltayib, and Magdi Awadalla Mohamed. 2025. "Identification of Microbial-Based Natural Products as Potential CYP51 Inhibitors for Eumycetoma Treatment: Insights from Molecular Docking, MM-GBSA Calculations, ADMET Analysis, and Molecular Dynamics Simulations" Pharmaceuticals 18, no. 4: 598. https://doi.org/10.3390/ph18040598
APA StyleElsaman, T., Awadalla, M. K. A., Mohamed, M. S., Eltayib, E. M., & Mohamed, M. A. (2025). Identification of Microbial-Based Natural Products as Potential CYP51 Inhibitors for Eumycetoma Treatment: Insights from Molecular Docking, MM-GBSA Calculations, ADMET Analysis, and Molecular Dynamics Simulations. Pharmaceuticals, 18(4), 598. https://doi.org/10.3390/ph18040598