
Kinin B1 Receptor Agonist Enhances Blood-Brain Barrier Permeability in Healthy and Glioblastoma Environments


 , , and
, , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
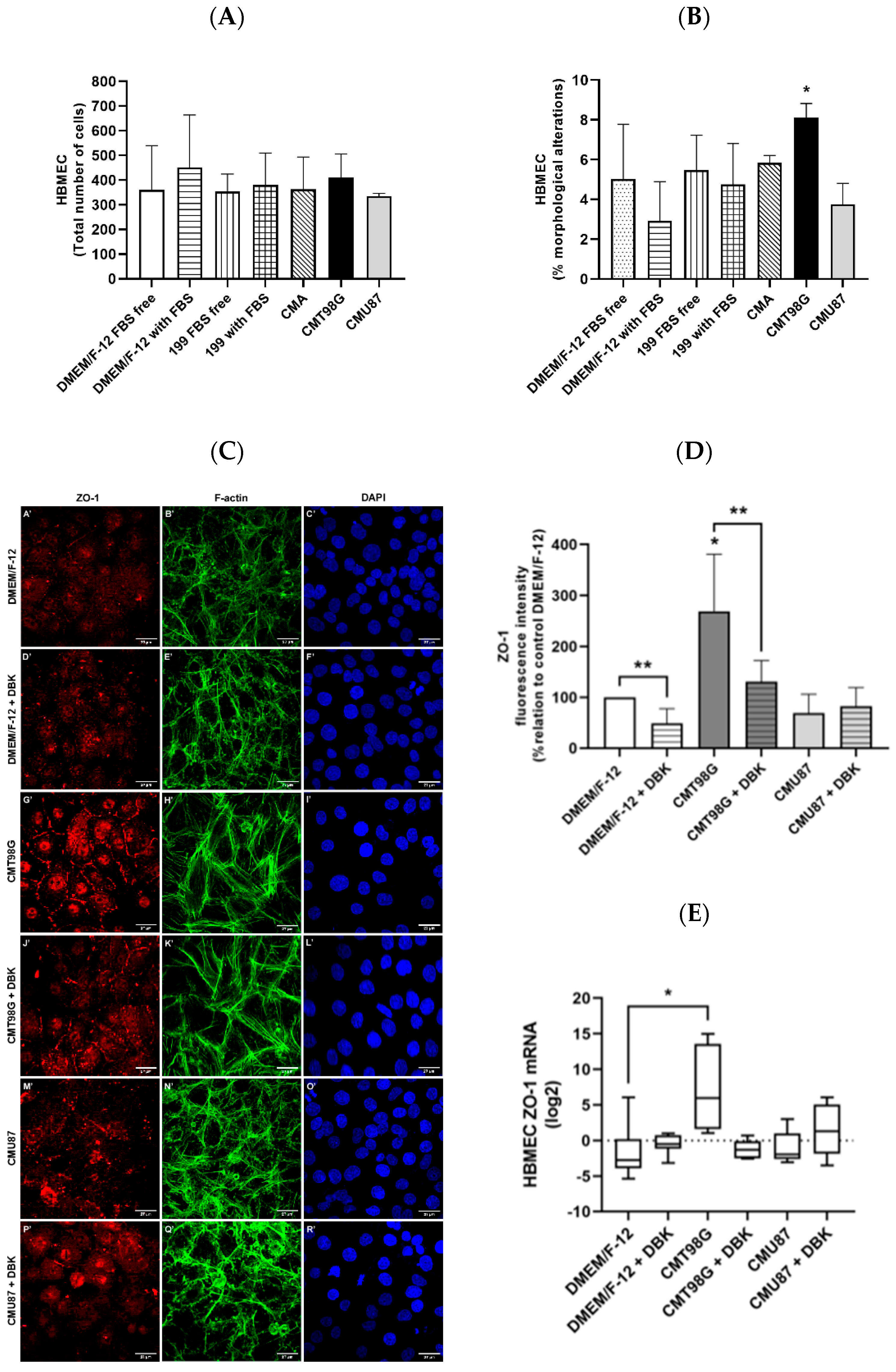
2.1. Effect of T98G- and U87MG-Conditioned Media on Endothelial Cells
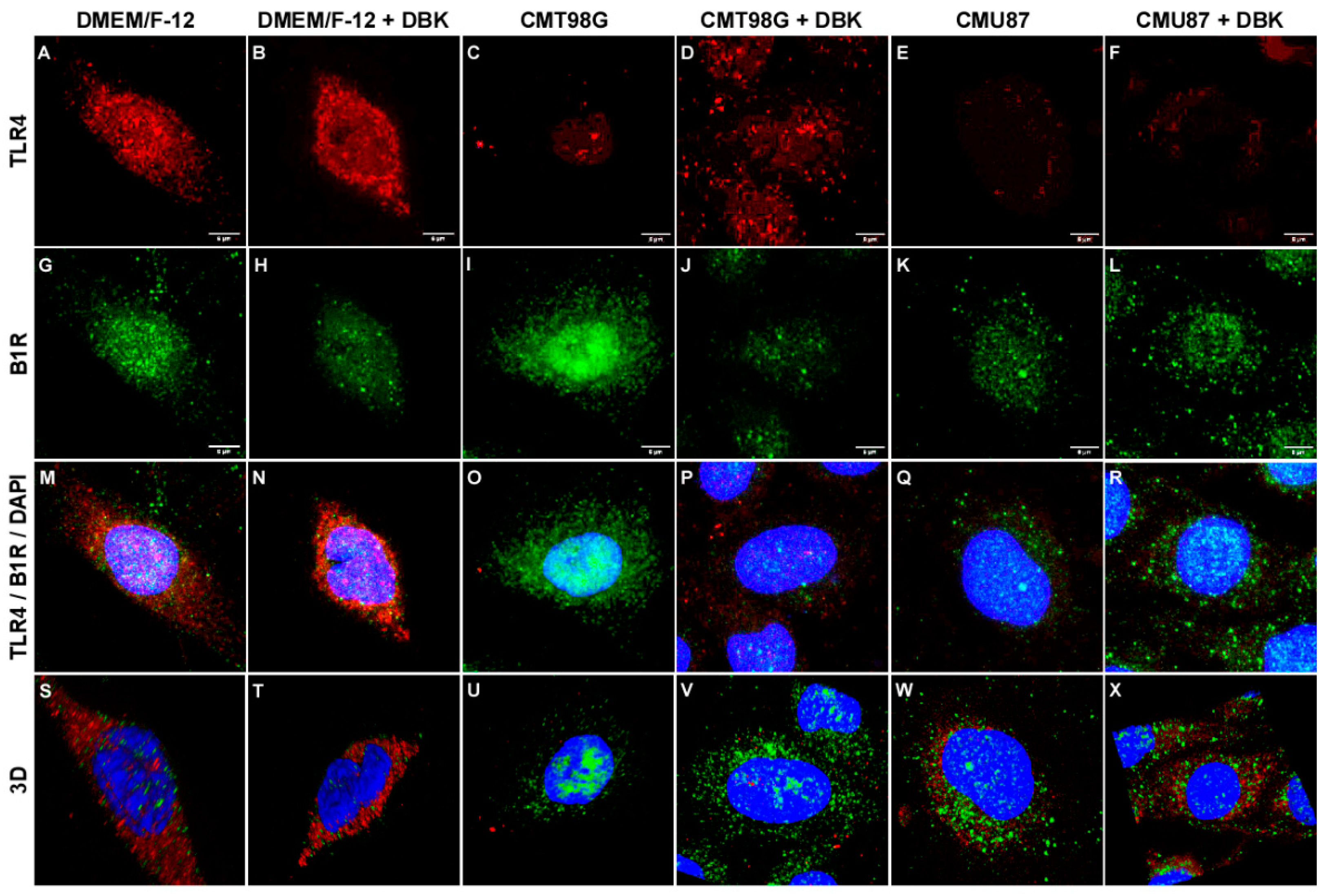
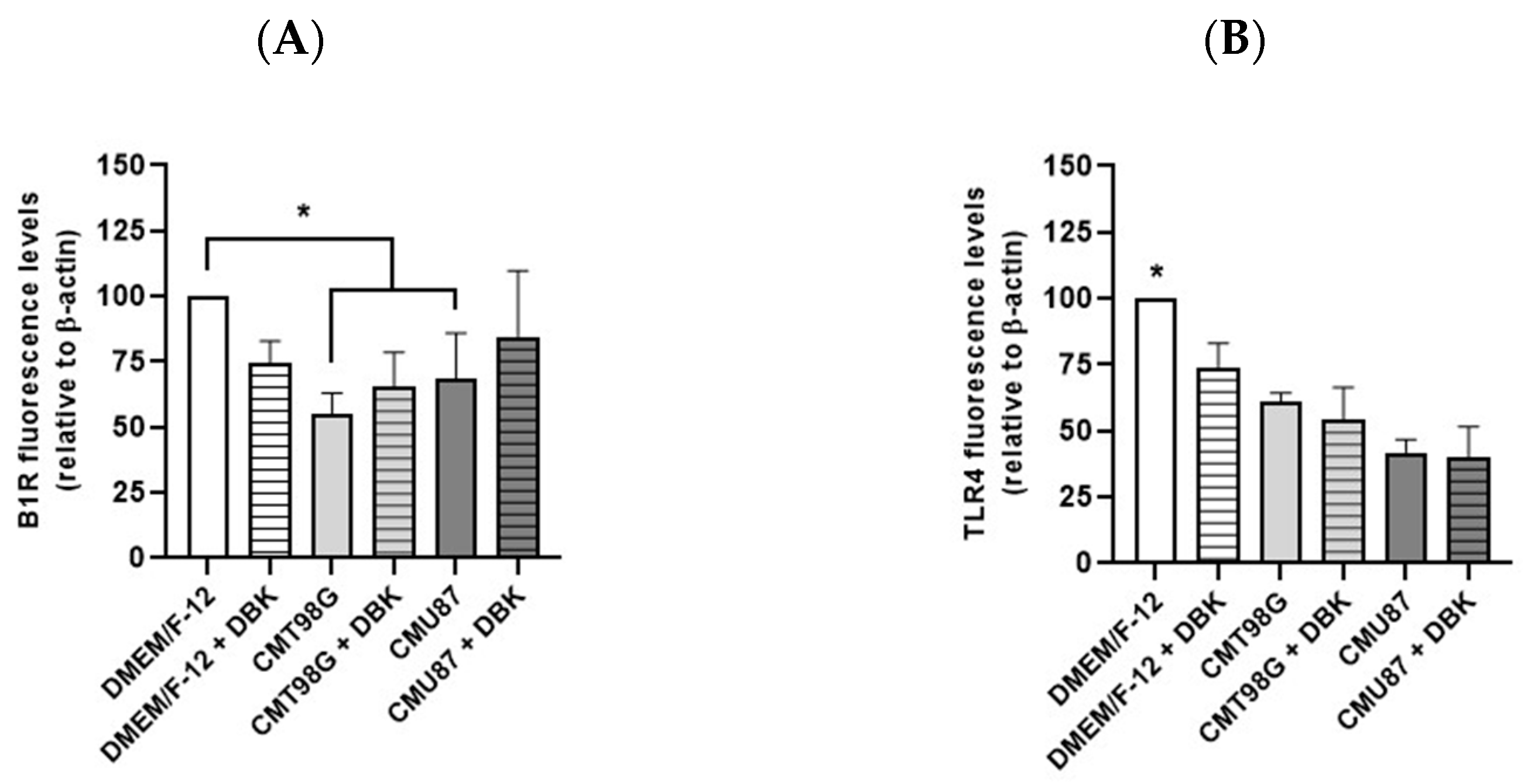
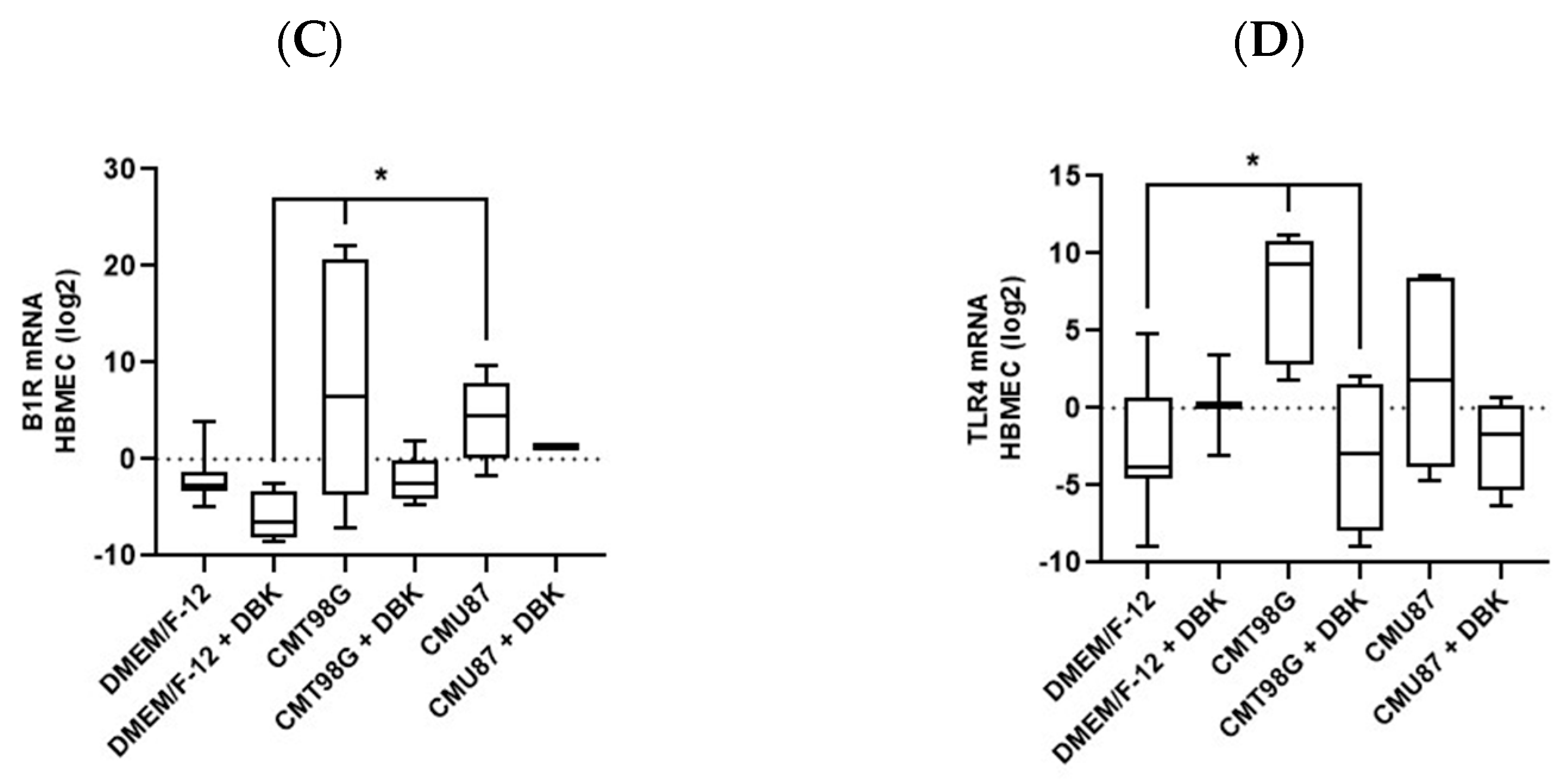
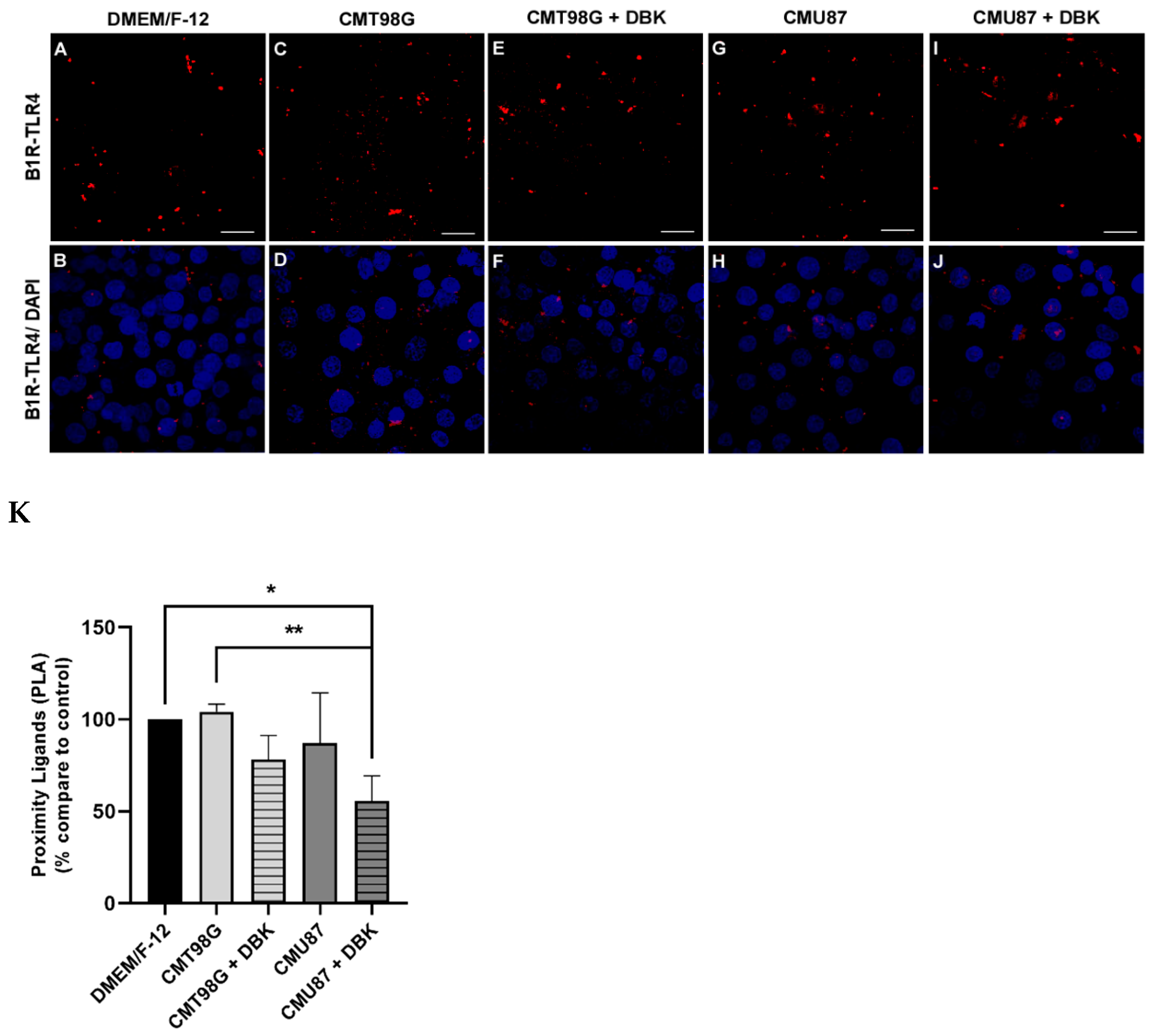
2.2. B1R and TLR4 Analysis in BBB Endothelial Cells Exposed to CMU87 and CMT98G in the Presence or Absence of DBK
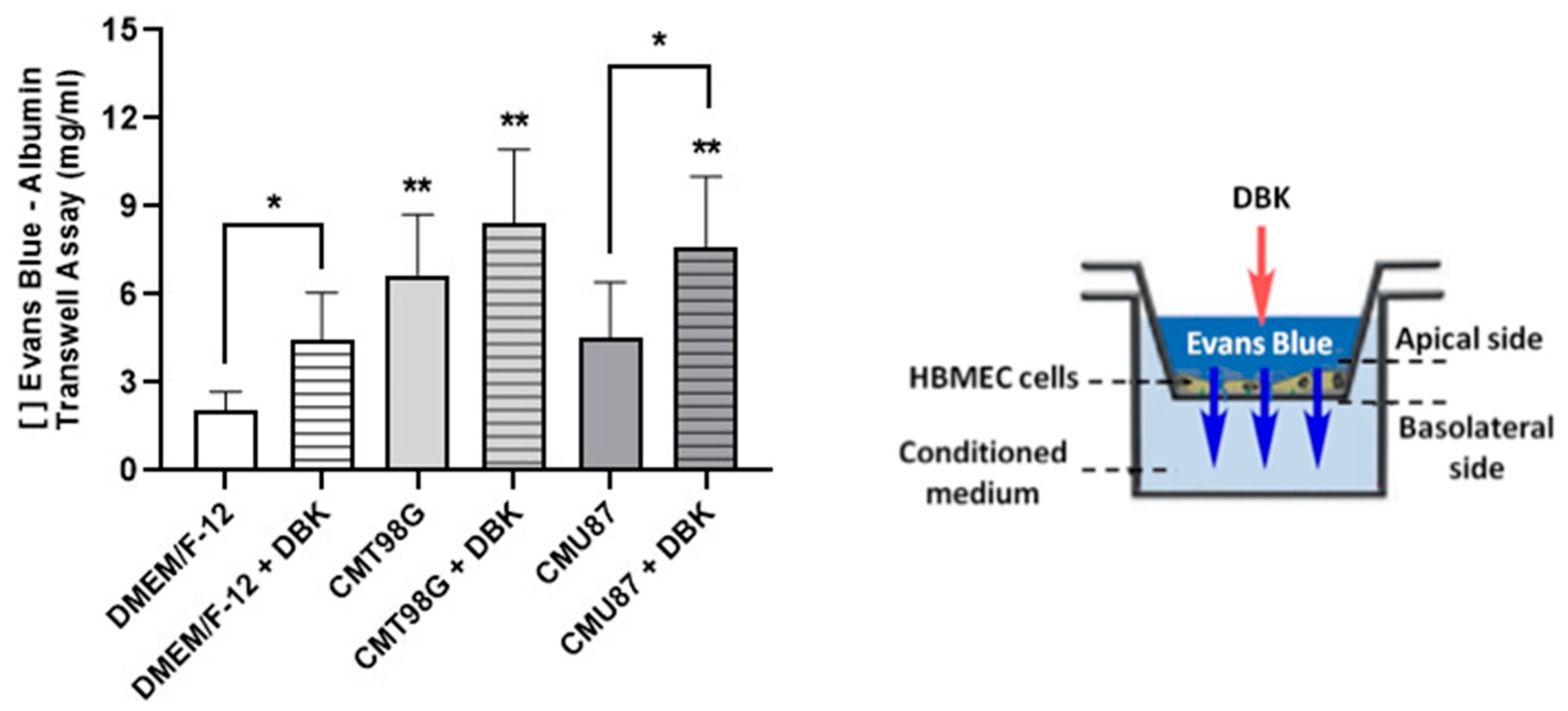
2.3. DBK Promotes Transient Opening of the BBB
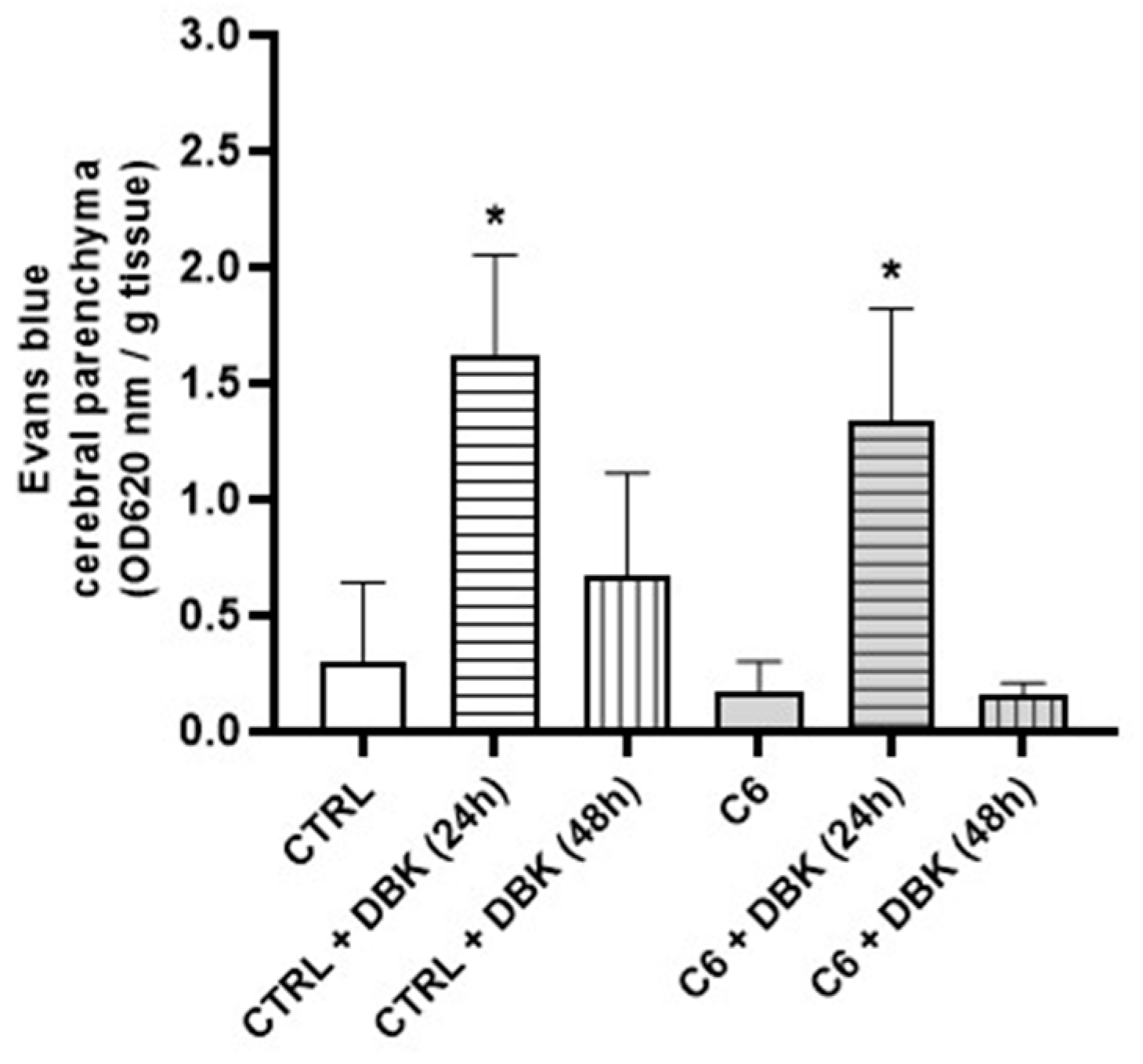
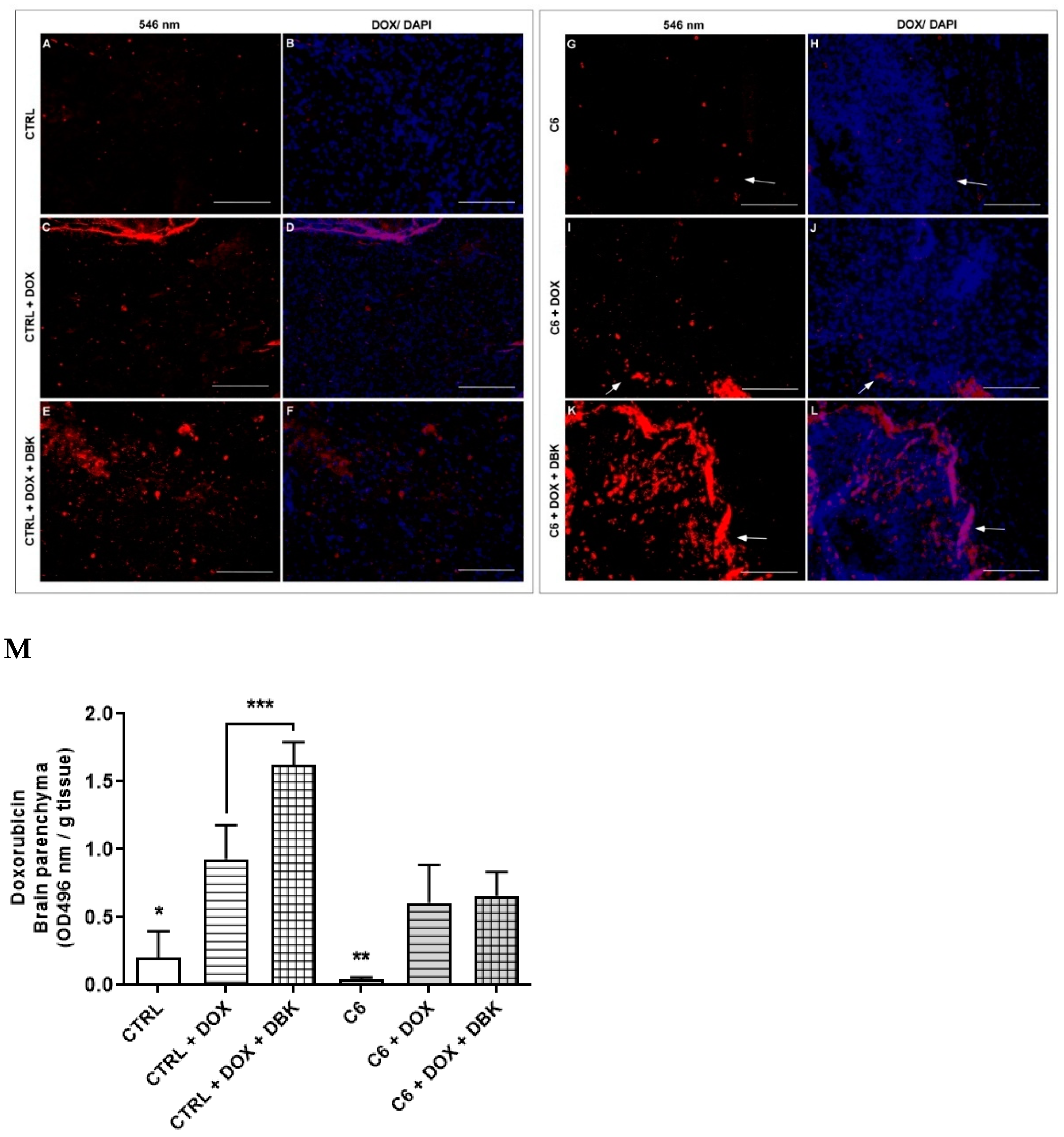
2.4. DBK Effect on DOX Delivery to the Cerebral Parenchyma
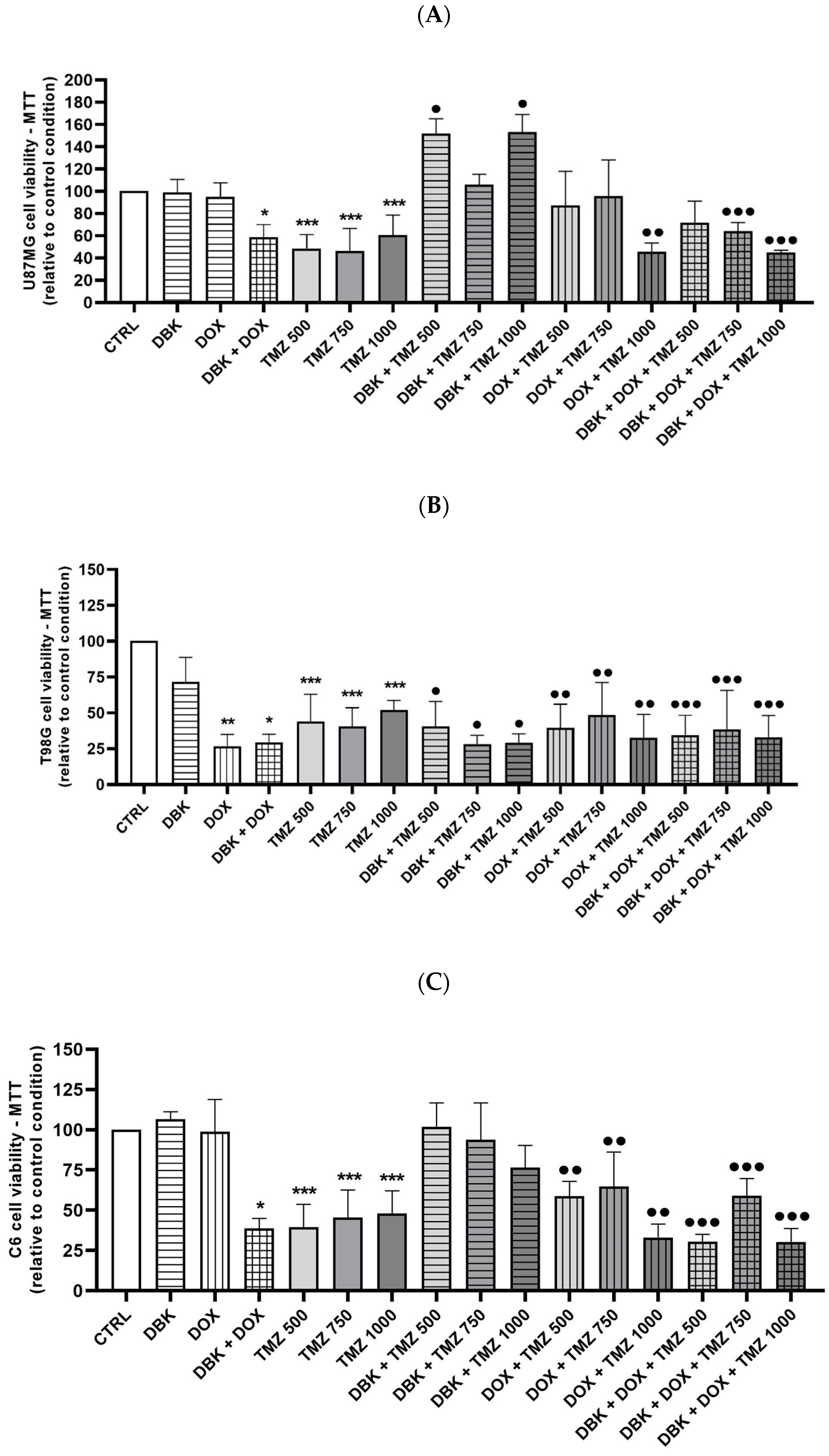
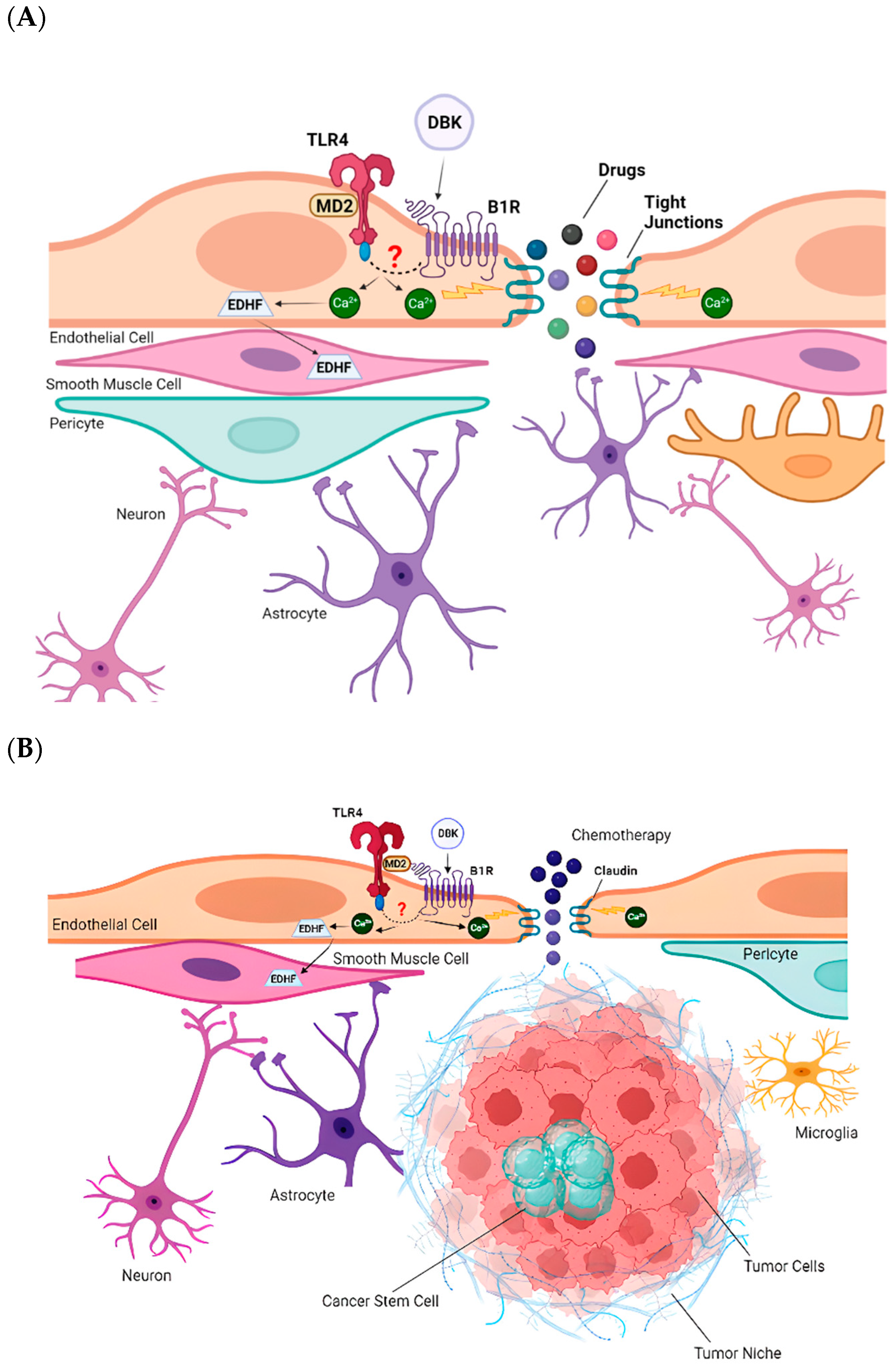
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Maintenance of Cell Lines
4.3. mRNA Expression
4.4. Immunocytochemistry and Immunohistochemistry
4.5. In-Cell Western Assay
4.6. Proximity Ligation Assay
4.7. Paracellular Permeability Assay
4.8. Evaluation of BBB Integrity Using Evans Blue
4.9. Doxorubicin Delivery in Mouse Brain
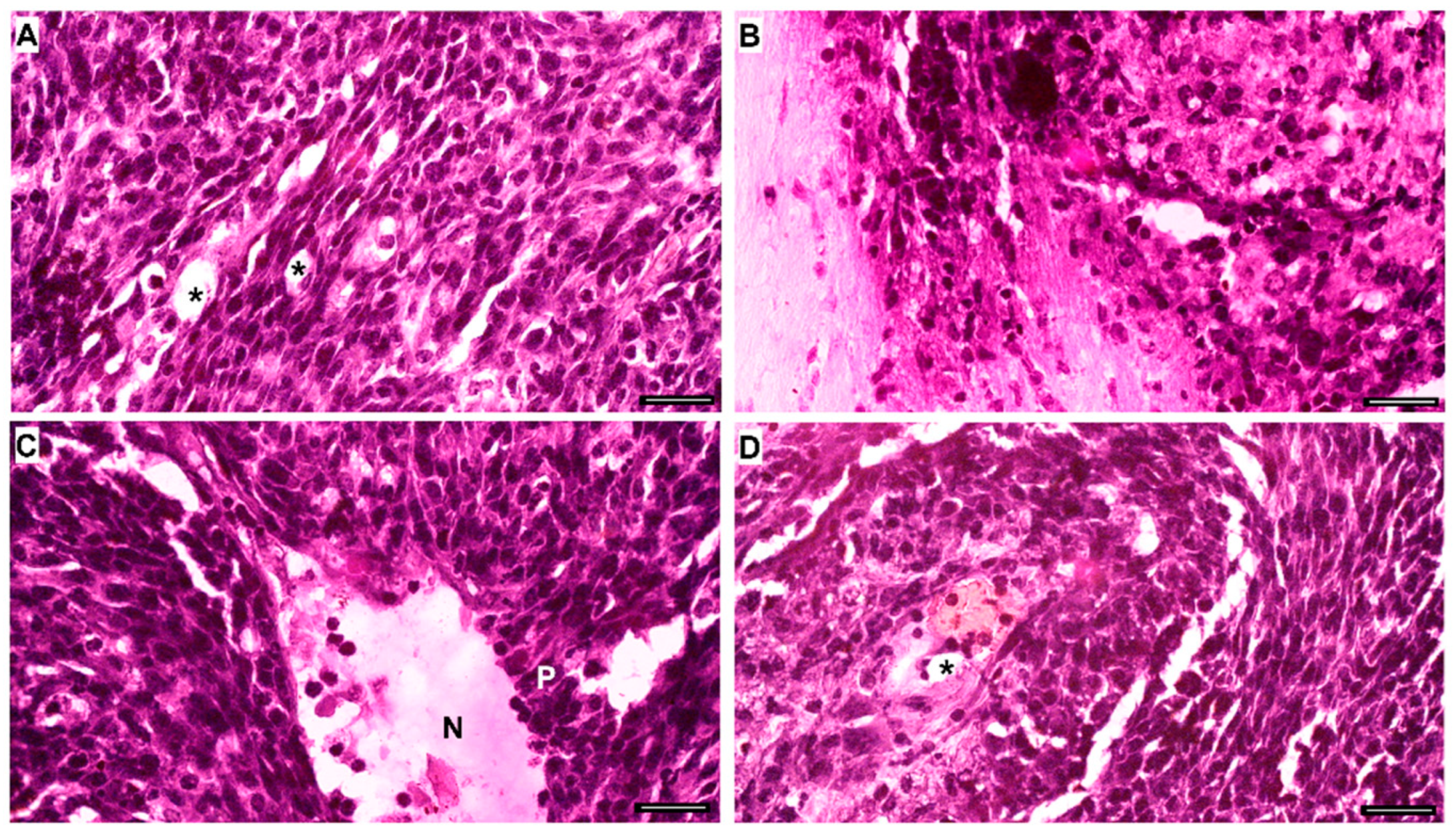
4.10. Histological Staining with Crystal Violet or Hematoxylin and Eosin (H&E)
4.11. Cell Viability Analysis Using MTT Assay
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACM | Astrocyte-conditioned medium |
| BBB | Blood-brain barrier |
| BK | Bradykinin |
| B1R | Kinin B1 receptor |
| B2R | Kinin B2 receptor |
| CMT98G | Conditioned media from glioblastoma cell line T98G |
| CMU87 | Conditioned media from glioblastoma cell line U87MG |
| CNS | Central nervous system |
| CTRL | In vivo experimental control group |
| C6 | Glial cell line isolated from the brain of rat with glioma |
| DAPI | Nuclear staining 4′,6-Diamidino-2-phenylindole |
| DBK | des-Arg9-bradykinin |
| DMEM/F-12 | Dulbecco’s modified Eagle’s medium F12 |
| DMSO | Dimethyl sulfoxide |
| DOX | Doxorubicin |
| EB | Evans blue dye |
| EDHF | Endothelium-derived hyperpolarizing Factor |
| FBS | Fetal bovine serum |
| HBMECs | Human brain microvascular endothelial cells |
| LPS | Lipopolysaccharide |
| Lys-BK | Lys-bradykinin |
| Lys-DBK | Lys-des-Arg9-bradykinin |
| mRNA | Messenger ribonucleic acid |
| NF-κB | Nuclear factor kappa B |
| PFA | Paraformaldehyde |
| PLA | Proximity ligation assay |
| RT-qPCR | Reverse transcription quantitative real-time polymerase chain reaction |
| TLR4 | Toll-like Receptor 4 |
| TMZ | Temozolomide |
| T98G | Human cell line derived from glioblastoma tumor |
| U87MG | Human cell line derived from glioblastoma tumor |
References
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation–associated diseases in organs. Oncotarget 2017, 9, 7204–7218. [Google Scholar] [CrossRef]
- Bossi, F.; Peerschke, E.I.; Ghebrehiwet, B.; Tedesco, F. Cross-talk between the complement and the kinin system in vascular permeability. Immunol. Lett. 2011, 140, 7–13. [Google Scholar] [CrossRef] [PubMed]
- e Silva, M.R.; Beraldo, W.T.; Rosenfeld, G. Bradykinin, a hypotensive and smooth muscle stimulating factor released from plasma globulin by snake venoms and by trypsin. Am. J. Physiol. Content 1949, 156, 261–273. [Google Scholar] [CrossRef]
- Bhoola, K.D.; Figueroa, C.D.; Worthy, K. Bioregulation of kinins: Kallikreins, kininogens, and kininases. Pharmacol. Rev. 1992, 44, 1–80. [Google Scholar] [CrossRef] [PubMed]
- Regoli, D.; Barabé, J. Pharmacology of bradykinin and related kinins. Pharmacol. Rev. 1980, 32, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Lora, I. Kinin-mediated inflammation in neurodegenerative disorders. Neurochem. Int. 2012, 61, 72–78. [Google Scholar] [CrossRef]
- Hamid, S.; Rhaleb, I.A.; Kassem, K.M.; Rhaleb, N.-E. Role of kinins in hypertension and heart failure. Pharmaceuticals 2020, 13, 347. [Google Scholar] [CrossRef]
- Marceau, F.; Hess, J.F.; Bachvarov, D.R. The B1 receptors for kinins. Pharmacol. Rev. 1998, 50, 357–386. [Google Scholar] [CrossRef]
- da Costa, P.L.; Sirois, P.; Tannock, I.F.; Chammas, R. The role of kinin receptors in cancer and therapeutic opportunities. Cancer Lett. 2014, 345, 27–38. [Google Scholar] [CrossRef]
- Campos, M.M.; Souza, G.E.P.; Calixto, J.B. In vivo B1 kinin-receptor upregulation. Evidence for involvement of protein kinases and nuclear factor kappaB pathways. Br. J. Pharmacol. 1999, 127, 1851–1859. [Google Scholar] [CrossRef]
- Sabourin, T.; Guay, K.; Houle, S.; Bouthillier, J.; Bachvarov, D.R.; Adam, A.; Marceau, F. Absence of ligand-induced regulation of kinin receptor expression in the rabbit. Br. J. Pharmacol. 2001, 133, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Cayla, C.; Todiras, M.; Iliescu, R.; Saul, V.V.; Gross, V.; Pilz, B.; Chai, G.; Merino, V.F.; Pesquero, J.B.; Baltatu, O.C.; et al. Mice deficient for both kinin receptors are normotensive and protected from endotoxin-induced hypotension. FASEB J. 2007, 21, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Schremmer-Danninger, E.; Öffner, A.; Siebeck, M.; Roscher, A.A. B1 bradykinin receptors and carboxypeptidase M are both upregulated in the aorta of pigs after LPS infusion. Biochem. Biophys. Res. Commun. 1998, 243, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, R.; Cabrini, D.A.; Ferreira, J.; Fernandes, E.S.; Mori, M.A.; Pesquero, J.B.; Bader, M.; Avellar, M.C.; Campos, M.M.; Calixto, J.B. Bradykinin B1 receptor expression induced by tissue damage in the rat portal vein: A critical role for mitogen-activated protein kinase and nuclear factor-kappaB signaling pathways. Circ. Res. 2004, 94, 1375–1382. [Google Scholar] [CrossRef]
- Merino, V.F.; Todiras, M.; Campos, L.A.; Saul, V.; Popova, E.; Baltatu, O.C.; Pesquero, J.B.; Bader, M. Increased susceptibility to endotoxic shock in transgenic rats with endothelial overexpression of kinin B(1) receptors. J. Mol. Med. 2008, 86, 791–798. [Google Scholar] [CrossRef]
- Dutra, R.C. Kinin receptors: Key regulators of autoimmunity. Autoimmun. Rev. 2017, 16, 192–207. [Google Scholar] [CrossRef]
- Deepak, K.; Roy, P.K.; Kola, P.; Mukherjee, B.; Mandal, M. An overview of kinin mediated events in cancer progression and therapeutic applications. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188807. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Zong, H.; Parada, L.F.; Baker, S.J. Cell of origin for malignant gliomas and its implication in the therapeutic development. Cold Spring Harb. Perspect. Biol. 2015, 7, a020610. [Google Scholar] [CrossRef]
- Kane, J.R. The role of brain vasculature in glioblastoma. Mol. Neurobiol. 2019, 56, 6645–6653. [Google Scholar] [CrossRef]
- Cruz, J.V.R.; Batista, C.; Afonso, B.d.H.; Alexandre-Moreira, M.S.; Dubois, L.G.; Pontes, B.; Neto, V.M.; Mendes, F.A. Obstacle to glioblastoma treatment two decades after temozolomide. Cancers 2022, 14, 3203. [Google Scholar] [CrossRef] [PubMed]
- Di Nunno, V.; Franceschi, E.; Tosoni, A.; Gatto, L.; Bartolini, S.; Brandes, A.A. Glioblastoma microenvironment: From na inviolable defense to therapeutic chance. Front. Oncol. 2022, 12, 852950. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Aaroe, A.; Liang, J.; Puduvalli, V.K. Tumor microenvironment in glioblastoma: Current and emerging concepts. Neuro-Oncol. Adv. 2023, 5, vdad009. [Google Scholar] [CrossRef]
- Alvarado, A.G.; Thiagarajan, P.S.; Mulkearns-Hubert, E.E.; Silver, D.J.; Hale, J.S.; Alban, T.J.; Turaga, S.M.; Jarrar, A.; Reizes, O.; Longworth, M.S.; et al. Glioblastoma cancer stem cells evade innate immune supression of self-renewal through reduced TLR4 expression. Cell Stem Cell 2017, 20, 450–461. [Google Scholar] [CrossRef]
- Erices, J.I.; Bizama, C.; Niechi, I.; Uribe, D.; Rosales, A.; Fabres, K.; Navarro-Martínez, G.; Torres, Á.; Martín, R.S.; Roa, J.C.; et al. Glioblastoma microenvironment and invasiveness: New insights and therapeutic targets. Int. J. Mol. Sci. 2023, 24, 7047. [Google Scholar] [CrossRef]
- Guevara-Lora, I.; Blonska, B.; Faussner, A.; Kozik, A. Kinin-generating cellular model obtained from human glioblastoma cell line U-373. Acta Biochim. Pol. 2013, 60, 299–305. [Google Scholar] [CrossRef]
- Nicoletti, N.T.; Erig, T.C.; Zanin, R.F.; Pereira, T.C.B.; Bogo, M.R.; Campos, M.M.; Morrone, F.B. Mechanisms involved in kinin-induced glioma cells proliferation: The role of ERK1/2 and PI3K/Akt pathways. J. Neurooncol. 2014, 120, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, N.F.; Sénécal, J.; da Silva, V.D.; Roxo, M.R.; Ferreira, N.P.; de Morais, R.L.T.; Pesquero, J.B.; Campos, M.M.; Couture, R.; Morrone, F.B. Primary role for kinin B1 and B2 receptors in glioma proliferation. Mol. Neurobiol. 2016, 54, 7869–7882. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Van Tellingen, O.; Yetkin-Arik, B.; De Gooijer, M.; Wesseling, P.; Wurdinger, T.; De Vries, H.; De Lange, E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updates 2015, 19, 1–12. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The microenvironmental landscape of brain tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef]
- Johnson, R.H.; Kho, D.T.; Carroll, S.J.O.; Angel, C.E.; Graham, E.S. The functional and inflammatory response of brain endothelial cells to toll-like receptor agonists. Sci. Rep. 2018, 8, 10102. [Google Scholar] [CrossRef] [PubMed]
- Weiss, N.; Miller, F.; Cazaubon, S.; Couraud, P.-O. The blood-brain barrier in brain homeostasis and neurological diseases. Biochim. Biophys. Acta Biomembr. 2009, 1788, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Tatter, S.B.; Debinski, W. Neurosurgical techniques for disruption of the blood-brain barrier for glioblastoma treatment. Pharmaceutics 2015, 7, 175–187. [Google Scholar] [CrossRef]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The blood-brain barrier and blood-tumour barrier in brain tumours and metastases. Nat. Rev. Cancer 2020, 20, 26–41. [Google Scholar] [CrossRef]
- Steeg, P.S. The blood-tumour barrier in cancer biology and therapy. Nat. Rev. Clin. Oncol. 2021, 18, 696–714. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.S.; Muresanu, D.F.; Castellani, R.J.; Nozari, A.; Lafuente, J.V.; Tian, Z.R.; Sahib, S.; Bryukhovetskiy, I.; Bryukhovetskiy, A.; Buzoianu, A.D.; et al. Phatophysiology of blood-brain barrier in brain tumor. Novel therapeutic advances using nanomedicine. Int. Rev. Neurobiol. 2020, 151, 1–66. [Google Scholar] [CrossRef]
- Allen, B.D.; Limoli, C.L. Breaking barriers: Neurodegenerative repercussions of radiotherapy induced damage on the blood-brain and blood-tumor barrier. Free. Radic. Biol. Med. 2021, 178, 189–201. [Google Scholar] [CrossRef]
- Whelan, R.; Hargaden, G.C.; Knox, A.J.S. Modulating the blood-brain barrier: A comprehensive review. Pharmaceutics 2021, 13, 1980. [Google Scholar] [CrossRef]
- Ningaraj, N.S.; Rao, M.; Hashizume, K.; Asotra, K.; Black, K.L. Regulation of blood-brain tumor barrier permeability by calcium-activated potassium channels. J. Pharmacol. Exp. Ther. 2002, 301, 838–851. [Google Scholar] [CrossRef]
- Prados, M.D.; Schold, S.C., Jr.; Fine, H.A.; Jaeckle, K.; Hochberg, F.; Mechtler, L.; Fetell, M.R.; Phuphanich, S.; Feun, L.; Janus, T.J.; et al. A randomized, double-blind, placebo-controlled, phase 2 study of RMP-7 in combination with carboplatin adminstered intravenously for the treatment of recurrent malignant glioma. Neuro-Oncol. 2003, 5, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Kunigelis, K.E.; Vogelbaum, M.A. Therapeutic delivery to central nervous system. Neurosurg. Clin. 2021, 32, 291–303. [Google Scholar] [CrossRef]
- Côté, J.; Savard, M.; Neugebauer, W.; Fortin, D.; Lepage, M.; Gobeil, F. Dual kinin B1 and B2 receptor activation provides enhanced blood-brain barrier permeability and anticancer drug delivery into brain tumors. Cancer Biol. Ther. 2013, 14, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Tan, J.; Wan, L.; Chen, C.; Wu, B.; Ke, X.; Wu, R.; Ran, X. Increase in blood-brain barrier permeability is modulated by tissue kallikrein via activation of bradykinin B1 and B2 receptor-mediated signaling. J. Inflamm. Res. 2021, 14, 4283–4297. [Google Scholar] [CrossRef]
- Sikpa, D.; Whittingstall, L.; Savard, M.; Lebel, R.; Côté, J.; McManus, S.; Chemtob, S.; Fortin, D.; Lepage, M.; Gobeil, F. Pharmacological modulation of blood-brain barrier permeability by kinin analogs in normal and pathologic conditions. Pharmaceuticals 2020, 13, 279. [Google Scholar] [CrossRef] [PubMed]
- Bouthillier, J.; Deblois, D.; Marceau, F. Studies on the induction of pharmacological responses to des-Arg9-bradykinin in vitro and in vivo. Br. J. Pharmacol. 1987, 92, 257–264. [Google Scholar] [CrossRef]
- Pesquero, J.B.; Pesquero, J.L.; Oliveira, S.M.; Roscher, A.A.; Metzger, R.; Ganten, D.; Bader, M. Molecular cloning and functional characterization of a mouse bradykinin B1 receptor gene. Biochem. Biophys. Res. Commun. 1996, 224, 281. [Google Scholar] [CrossRef]
- Qadri, F.; Bader, M. Kinin B1 receptors as a therapeutic target for inflammation. Expert Opin. Ther. Targets 2017, 22, 31–44. [Google Scholar] [CrossRef]
- Han, S.; Wang, C.; Qin, X.; Xia, J.; Wu, A. LPS alters the immuno-phenotype of glioma and glioma stem-like cells and induces in vivo antitumor immunity via TLR4. J. Exp. Clin. Cancer Res. 2017, 36, 83. [Google Scholar] [CrossRef]
- Bachar, O.; Adner, M.; Uddman, R.; Cardell, L.-O. Toll-like receptor stimulation induces airway hyper-responsiveness to bradykinin, an effect mediated by JNK and NF-kappa B signaling pathways. Eur. J. Immunol. 2004, 34, 1196–1207. [Google Scholar] [CrossRef]
- Passos, G.F.; Fernandes, E.S.; Campos, M.M.; Araújo, J.G.V.C.; Pesquero, J.L.; Souza, G.E.P.; Avellar, M.C.W.; Teixeira, M.M.; Calixto, J.B. Kinin B1 receptor up-regulation after lipopolysaccharide administration: Role of proinflammatory cytokines and neutrophil influx. J. Immunol. 2004, 172, 1839–1847. [Google Scholar] [CrossRef] [PubMed]
- Batista, C.; Sales, V.; Merino, V.; Bader, M.; Feres, T.; Pesquero, J. Role of endotelial kinin B1 receptor on the membrane potential of transgenic rat aorta. Physiol. Res. 2022, 71, 477–487. [Google Scholar] [CrossRef]
- Batista, C.; Cruz, J.V.R.; Stipursky, J.; Mendes, F.A.; Pesquero, J.B. Kinin B1 receptor and TLR4 interaction in inflammatory response. Inflamm. Res. 2024, 73, 1459–1476. [Google Scholar] [CrossRef] [PubMed]
- Lawley, T.J.; Kubota, Y. Induction of morphologic differentiation of endothelial cells in culture. J. Investig. Dermatol. 1989, 93 (Suppl. 2), S59–S61. [Google Scholar] [CrossRef]
- Kim, D.-E.; Oh, K.-H.; Yang, J.-H.; Kwon, S.-K.; Cho, T.-J.; Lee, S.-B.; Nam, H.; Lee, D.-S.; Lee, J.-R.; Lee, G.; et al. The porcine aortic tissue culture system in vitro for stem cell research. Int. J. Stem Cells 2011, 4, 116–122. [Google Scholar] [CrossRef]
- Qu, D.; Wang, L.; Huo, M.; Song, W.; Lau, C.-W.; Xu, J.; Xu, A.; Yao, X.; Chiu, J.-J.; Tian, X.Y.; et al. Focal TLR4 activation mediates disturbed flow-induced endothelial inflammation. Cardiovasc. Res. 2019, 116, 226–236. [Google Scholar] [CrossRef]
- Perrin, S.L.; Samuel, M.S.; Koszyca, B.; Brown, M.P.; Ebert, L.M.; Oksdath, M.; Gomez, G.A. Glioblastoma heterogeneity and the tumour microenvironment: Implications for preclinical research and development of new treatments. Biochem. Soc. Trans. 2019, 47, 625–638. [Google Scholar] [CrossRef]
- Karmakar, S.; Olive, M.F.; Banik, N.L.; Ray, S.K. Intracranial stereotaxic cannulation for development of orthotopic glioblastoma allograft in Sprague-Dawley rats and histoimmunopathological characterization of the brain tumor. Neurochem. Res. 2007, 32, 2235–2242. [Google Scholar] [CrossRef]
- Barth, R.F.; Kaur, B. Rat brain tumor models in experimental neuro-oncology: The C6, 9L, T9, RG2, F98, BT4C, RT-2 and CNS-1 gliomas. J. Neurooncol. 2009, 94, 299–312. [Google Scholar] [CrossRef]
- Lukina, M.; Yashin, K.; Kiseleva, E.E.; Alekseeva, A.; Dudenkova, V.; Zagaynova, E.V.; Bederina, E.; Medyanic, I.; Becker, W.; Mishra, D.; et al. Label-free macroscopic fluorescence lifetime imaging of brain tumors. Front. Oncol. 2021, 11, 666059. [Google Scholar] [CrossRef]
- Bona, N.P.; Pedra, N.S.; Spohr, L.; da Silva dos Santos, F.; Saraiva, J.T.; Carvalho, F.B.; Fernandes, M.C.; Fernandes, A.S.; Saraiva, N.; Filipa, M.; et al. Antitumoral activity of Cecropia pachystachya leaves extract in vitro and in vivo model of rat glioma: Brain and blood effects. Mol. Neurobiol. 2024, 61, 8234–8252. [Google Scholar] [CrossRef]
- Jia, W.; Lu, R.; Martin, T.A.; Jiang, W.G. The role of claudin-5 in blood-brain barrier (BBB) and brain metastases (review). Mol. Med. Rep. 2013, 9, 779–785. [Google Scholar] [CrossRef]
- Prasain, N.; Stevens, T. The actin cytoskeleton in endothelial cell phenotypes. Microvasc. Res. 2009, 77, 53–63. [Google Scholar] [CrossRef]
- Oliveira, M.N.; Pillat, M.M.; Motaln, H.; Ulrich, H.; Lah, T.T. Kinin-B1 receptor stimulation promotes invasion and is involved in cell-cell interaction of co-cultured glioblastoma and mesenchymal stem cells. Sci. Rep. 2018, 8, 1299. [Google Scholar] [CrossRef]
- Belot, N.; Rorive, S.; Doyen, I.; Lefranc, F.; Bruyneel, E.; Dedecker, R.; Micik, S.; Brotchi, J.; Decaestecker, C.; Salmon, I.; et al. Molecular characterization of cell substratum attachments in human glial tumors relates to prognostic features. Glia 2001, 36, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef]
- Liu, Y.-S.; Hsu, J.-W.; Lin, H.-Y.; Lai, S.-W.; Huang, B.-R.; Tsai, C.-F.; Lu, D.-Y. Bradykinin B1 receptor contributes to interleukin-8 production and glioblastoma migration through interaction of STAT3 and SP-1. Neuropharmacology 2019, 144, 143–154. [Google Scholar] [CrossRef]
- Liu, Y.; Hashizume, K.; Chen, Z.; Samoto, K.; Ningaraj, N.; Asotra, K.; Black, K.L. Correlation between bradykinin-induced blood-tumor barrier permeability and B2 receptor expression in experimental brain tumors. Neurol. Res. 2001, 23, 379–387. [Google Scholar] [CrossRef]
- Calixto, J.B.; Medeiros, R.; Fernandes, E.S.; Ferreira, J.; A Cabrini, D.; Campos, M.M. Kinin B1 receptors: Key G-protein-coupled receptors and their role in inflammatory and painful processes. Br. J. Pharmacol. 2004, 143, 803–818. [Google Scholar] [CrossRef]
- Loiola, R.A.; Reis, F.C.; Kawamoto, E.M.; Scavone, C.; Abdalla, D.S.; Fernandes, L.; Pesquero, J.B. Role of vascular Kinin B1 and B2 receptors in endothelial nitric oxide metabolismo. Peptides 2011, 32, 1700–1705. [Google Scholar] [CrossRef]
- Merino, V.F.; Silva, J.A.; Araújo, R.C.; Avellar, C.W.; Bascands, J.-L.; Schanstra, J.P.; Paiva, A.C.M.; Bader, M.; Pesquero, J.B. Molecular structure and transcriptional regulation by nuclear factor-kappaB of the mouse kinin B1 receptor gene. Biol. Chem. 2005, 386, 515–522. [Google Scholar] [CrossRef]
- Xiao, C.; Ghosh, S. NF-κB, an evolutionarily conserved mediator of immune and inflammatory responses. Adv. Exp. Med. Biol. 2005, 560, 541–545. [Google Scholar] [CrossRef]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.-W. NF-kappaB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef]
- Sun, J.; Sui, X.; Bradbury, A.; Zeldin, D.C.; Conte, M.S.; Liao, J.K. Inhibition of vascular smooth muscle cell migration by cytochrome P450 epoxygenase-derived eicosanoids. Circ. Res. 2002, 90, 1020–1027. [Google Scholar] [CrossRef]
- Wang, F.; Ji, S.; Wang, M.; Liu, L.; Li, Q.; Jiang, F.; Cen, J.; Ji, B. HMGB1 promoted P-glycoprotein at the blood-brain barrier in MCAO rats via TLR4/NF-κB signaling pathway. Eur. J. Pharmacol. 2020, 880, 173189. [Google Scholar] [CrossRef]
- Liebner, S.; Fischmann, A.; Rascher, G.; Duffner, F.; Grote, E.-H.; Kalbacher, H.; Wolburg, H. Claudin-1 and claudin-5 expression and tigh junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropathol. 2000, 100, 323–331. [Google Scholar] [CrossRef]
- Chao, H.-H.; Chen, P.-Y.; Hao, W.-R.; Chiang, W.-P.; Cheng, T.-H.; Loh, S.-H.; Leung, Y.-M.; Liu, J.-C.; Chen, J.-J.; Sung, L.-C. Lipopolysaccharide pretreatment increases protease-activated receptor-2 expression and monocyte chemoattractant protein-1 secretion in vascular endothelial cells. J. Biomed. Sci. 2017, 24, 85. [Google Scholar] [CrossRef]
- Ando, Y.; Okada, H.; Takemura, G.; Suzuki, K.; Takada, C.; Tomita, H.; Zaikokuji, R.; Hotta, Y.; Miyazaki, N.; Yano, H.; et al. Brain-specific ultrastructure of capillary endothelial glycocalyx and its possible contribution for blood brain barrier. Sci. Rep. 2018, 8, 17523. [Google Scholar] [CrossRef]
- Liu, L.; Xue, Y.; Liu, Y.; Wang, Y. Bradykinin increases blood-tumor barrier permeability by down-regulating the expression levels of ZO-1, occludin, and claudin-5 and rearranging actin cytoskeleton. J. Neurosci. Res. 2008, 86, 1153–1168. [Google Scholar] [CrossRef]
- Ifuku, M.; Fäber, K.; Okuno, Y.; Yamakawa, Y.; Miyamoto, T.; Nolte, C.; Merino, V.F.; Kita, S.; Iwamoyo, T.; Komuro, I.; et al. Bradykinin-induced microglial migration mediated by B1-bradykinin receptors depends on Ca2+ influx via reverse-mode activity of the Na+/Ca2+ exchanger. J. Neurosci. 2007, 27, 13065–13073. [Google Scholar] [CrossRef]
- Su, B.; Wang, R.; Xie, Z.; Ruan, H.; Li, J.; Xie, C.; Lu, W.; Wang, J.; Wang, D.; Liu, M. Effect of retro-inverso isomer of bradykinin on size-dependent penetration of blood-brain tumor barrier. Small 2018, 14, 1702331. [Google Scholar] [CrossRef]
- Goldim, M.P.S.; Della Giustina, A.; Petronilho, F. Using Evans blue dye to determine blood-brain barrier integrity in rodents. Curr. Protoc. Immunol. 2019, 126, e83. [Google Scholar] [CrossRef]
- Radu, M.; Chernoff, J. An in vivo assay to test blood vessel permeability. J. Vis. Exp. 2013, 73, e50062. [Google Scholar] [CrossRef]
- Wick, M.J.; Harral, J.W.; Loomis, Z.L.; Dempsey, E.C. An optimized Evans blue protocol to assess vascular leak in the mouse. J. Vis. Exp. 2018, 139, 57037. [Google Scholar] [CrossRef]
- Du, K.; Xia, Q.; Heng, H.; Feng, F. Temozolomide-doxorubicin conjugate as a double intercalating agent and delivery by apoferritin for glioblastoma chemotherapy. ACS Appl. Mater. Interfaces 2020, 12, 34599–34609. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batista, C.; Cruz, J.V.R.; Siqueira, M.; Pesquero, J.B.; Stipursky, J.; Mendes, F.d.A. Kinin B1 Receptor Agonist Enhances Blood-Brain Barrier Permeability in Healthy and Glioblastoma Environments. Pharmaceuticals 2025, 18, 591. https://doi.org/10.3390/ph18040591
Batista C, Cruz JVR, Siqueira M, Pesquero JB, Stipursky J, Mendes FdA. Kinin B1 Receptor Agonist Enhances Blood-Brain Barrier Permeability in Healthy and Glioblastoma Environments. Pharmaceuticals. 2025; 18(4):591. https://doi.org/10.3390/ph18040591
Chicago/Turabian StyleBatista, Carolina, João Victor Roza Cruz, Michele Siqueira, João Bosco Pesquero, Joice Stipursky, and Fabio de Almeida Mendes. 2025. "Kinin B1 Receptor Agonist Enhances Blood-Brain Barrier Permeability in Healthy and Glioblastoma Environments" Pharmaceuticals 18, no. 4: 591. https://doi.org/10.3390/ph18040591
APA StyleBatista, C., Cruz, J. V. R., Siqueira, M., Pesquero, J. B., Stipursky, J., & Mendes, F. d. A. (2025). Kinin B1 Receptor Agonist Enhances Blood-Brain Barrier Permeability in Healthy and Glioblastoma Environments. Pharmaceuticals, 18(4), 591. https://doi.org/10.3390/ph18040591