



Preclinical and Molecular Docking Insights into the Chemopreventive Role of Fenugreek Seed Extract in a Murine Model of Colorectal Cancer




Abstract

1. Introduction
2. Results
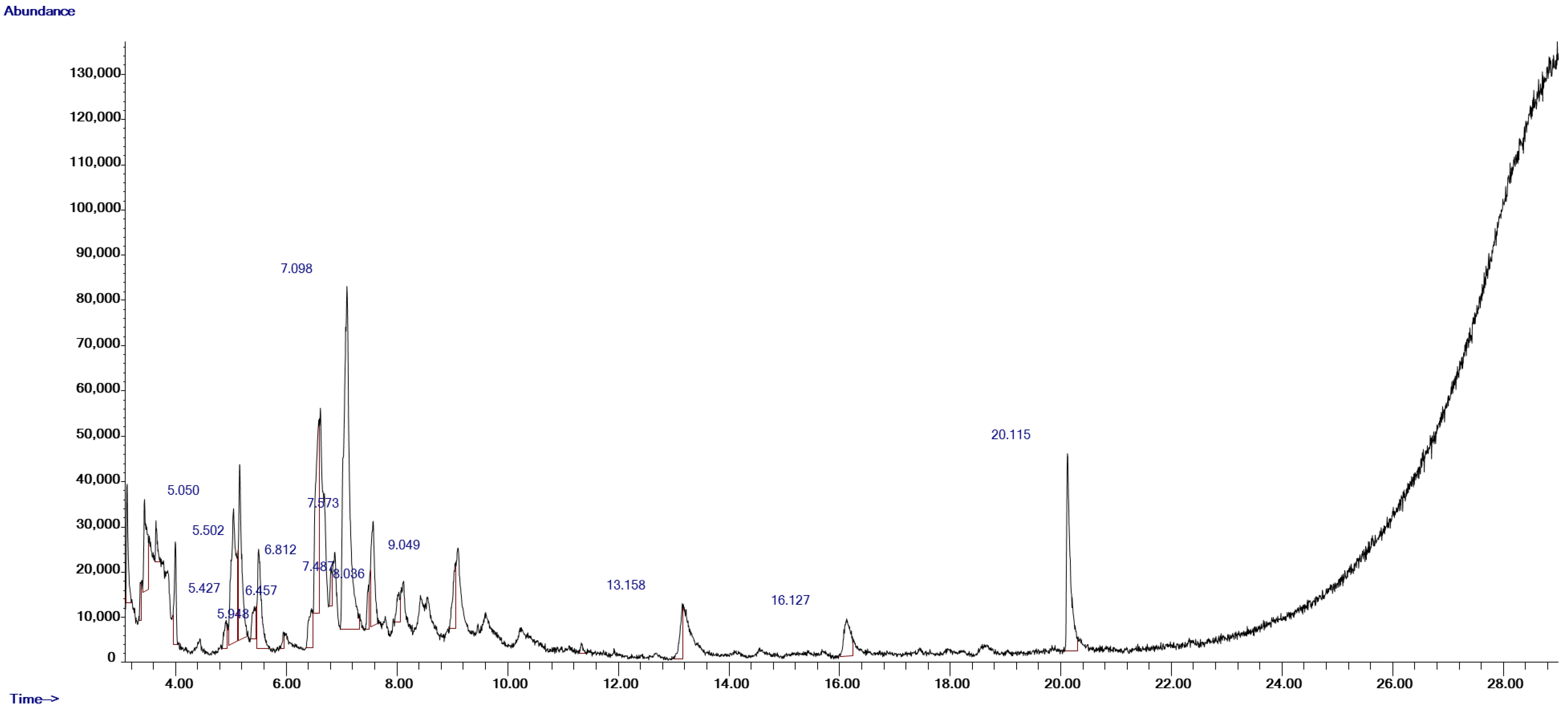
2.1. Yield Percentage of FSE and GC-MS Analysis
2.2. Effect of FSE on AOM-Induced Average Body Weight and Survival Rates
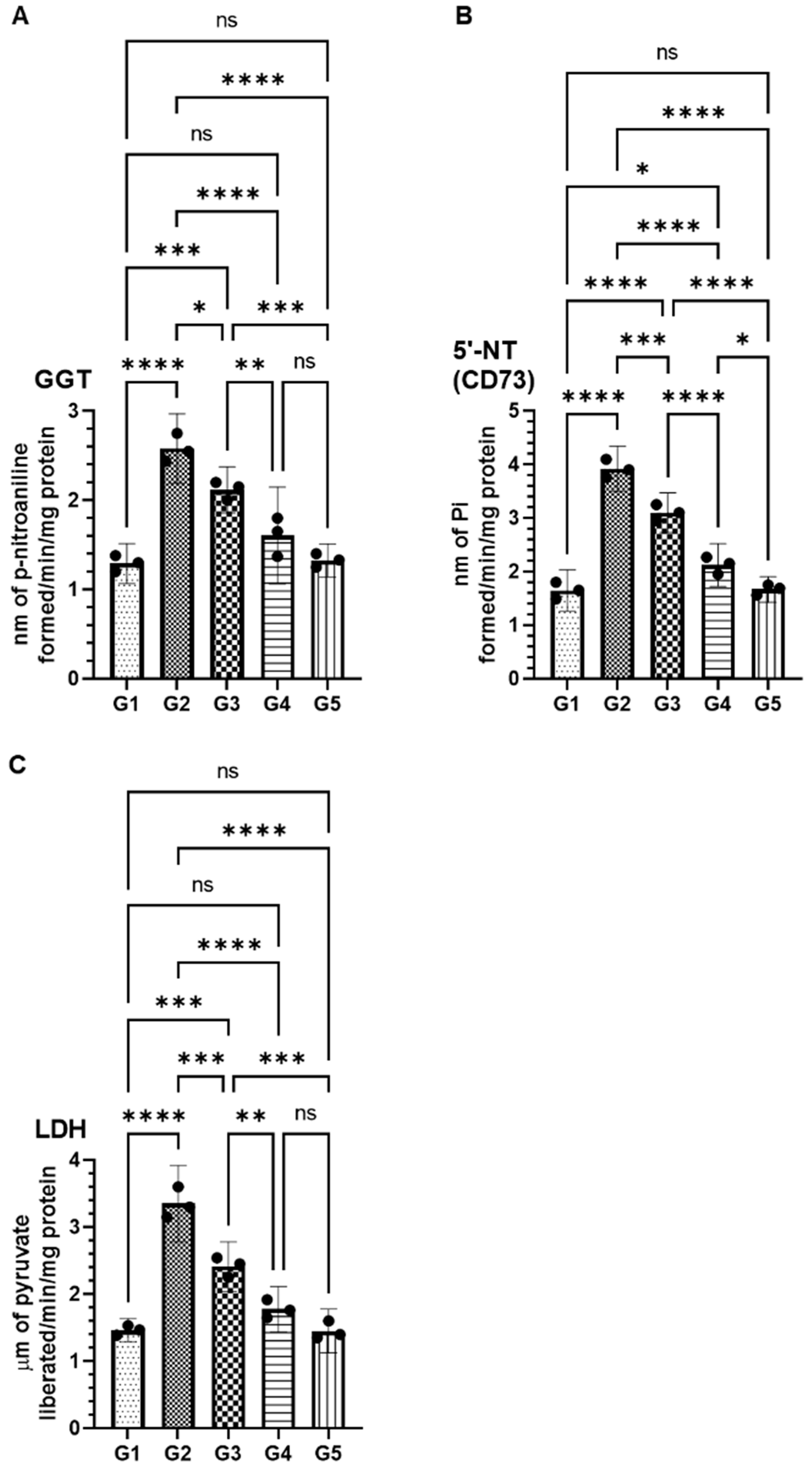
2.3. Effect of FSE on Serum Cancer Marker Enzymes
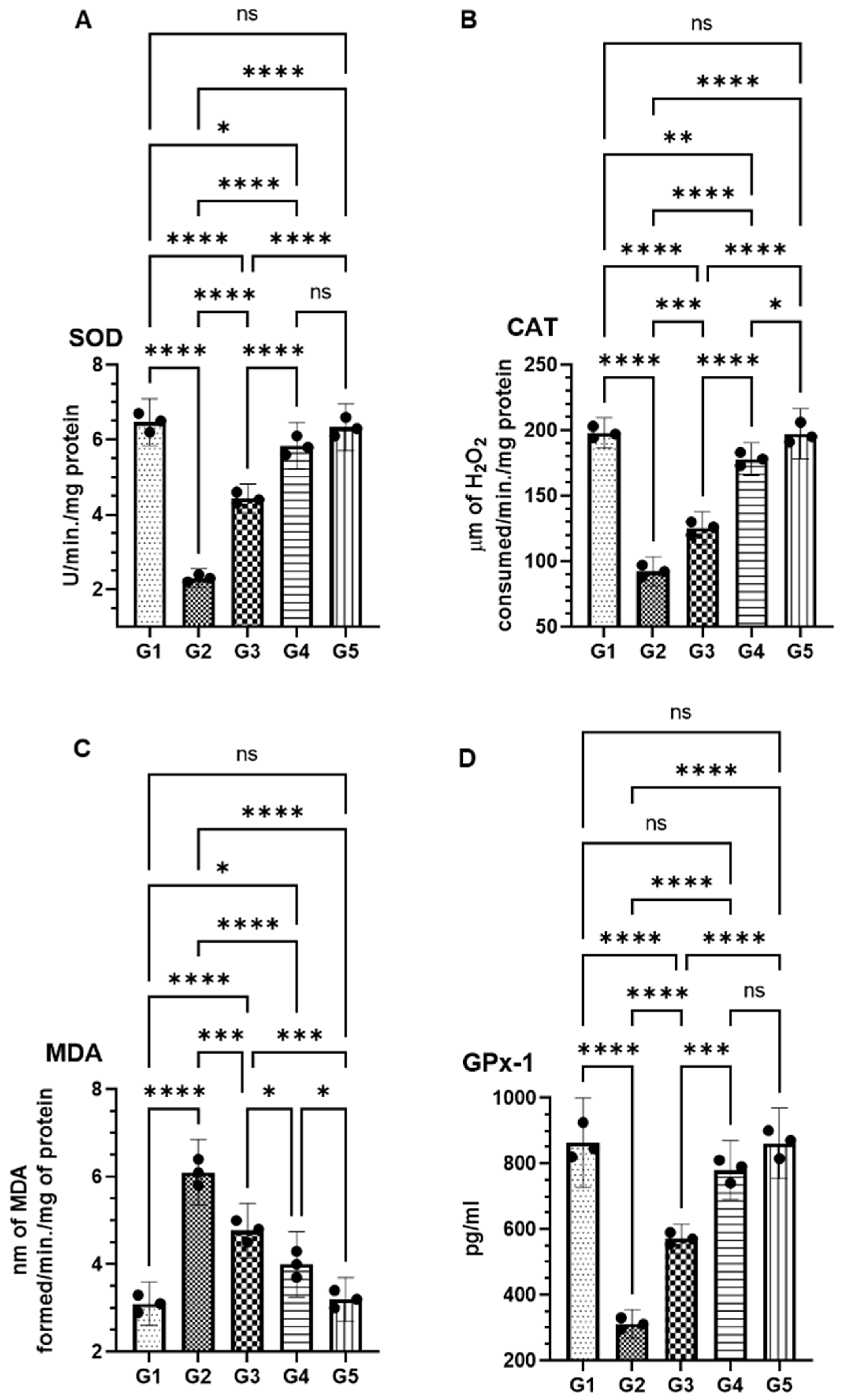
2.4. Modulation of Antioxidant Enzymes by FSE in AOM-Exposed Colonic Tissues
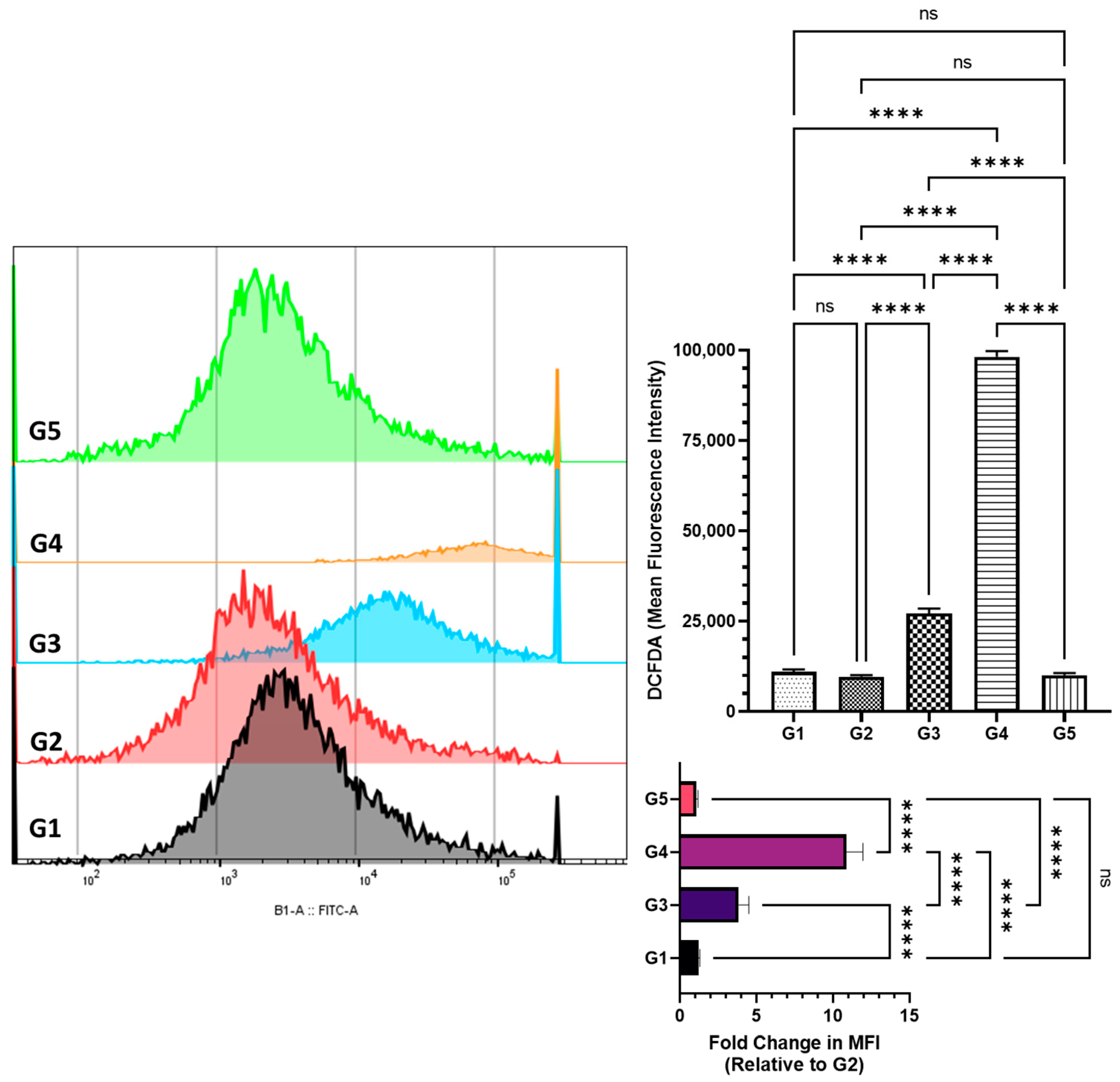
2.5. Assessment of Reactive Oxygen Species in Colonic Cells by DCFDA Flow Cytometry
2.6. Assessment of Apoptosis in Colonic Cells via Annexin V-FITC-PI Flow Cytometry
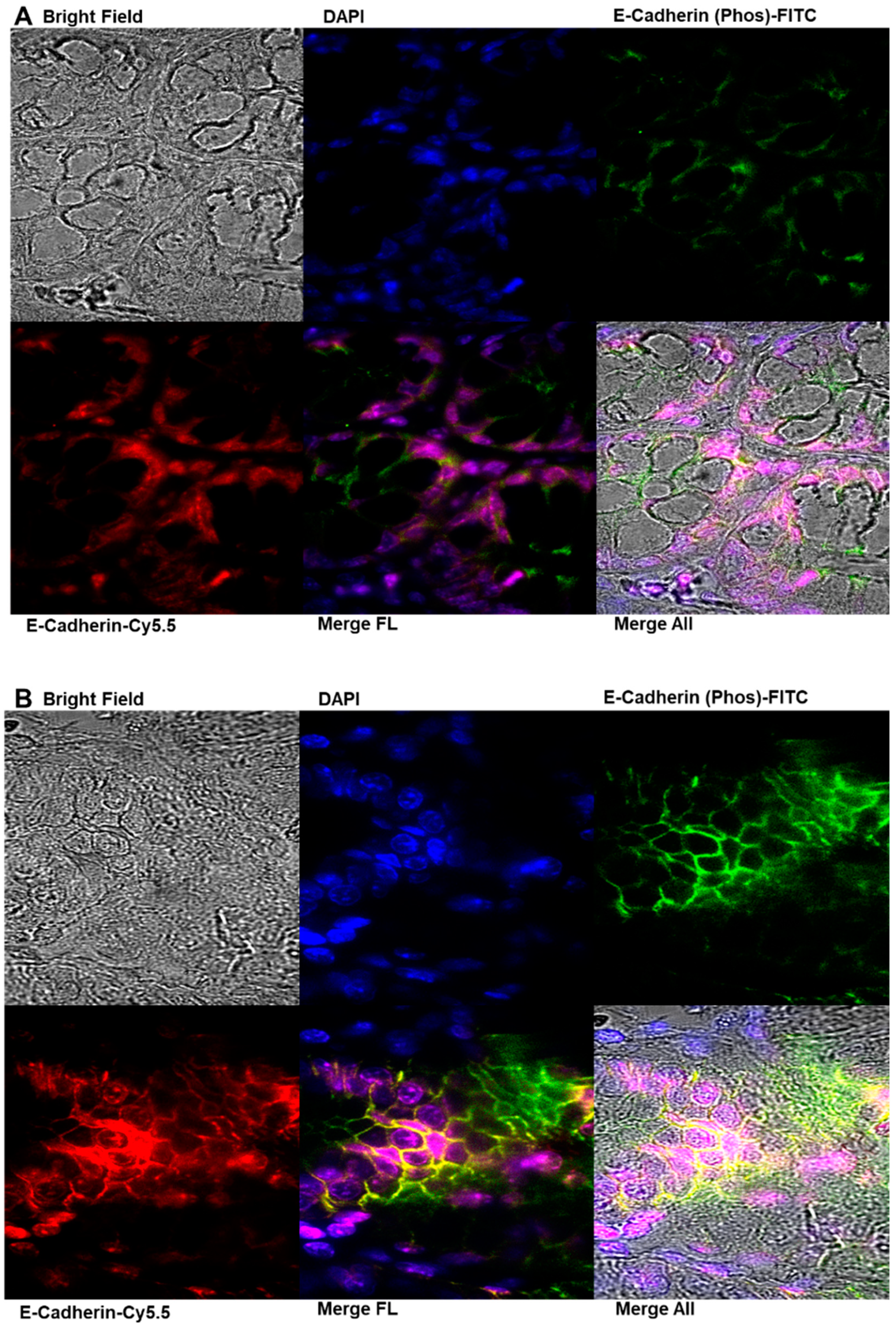
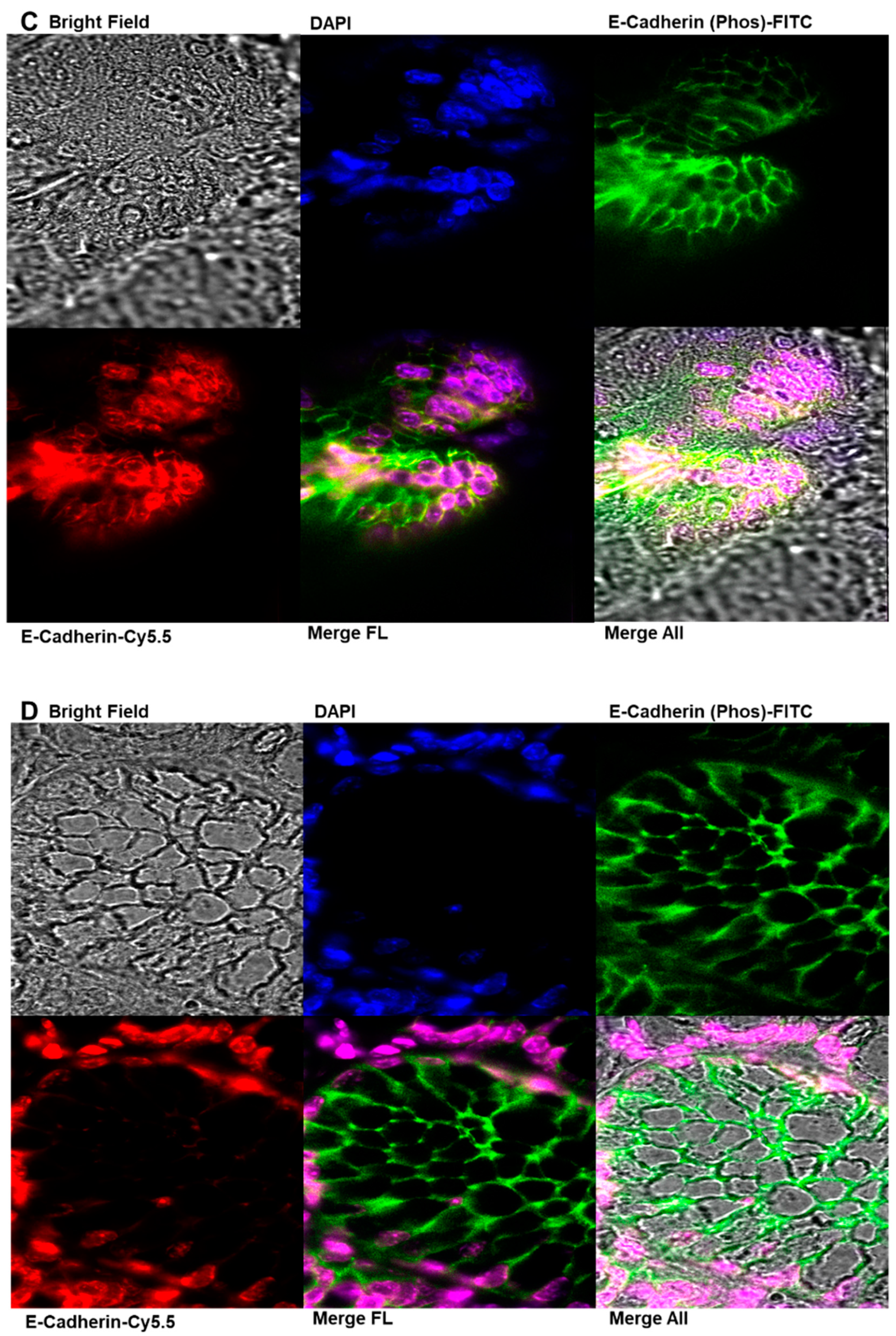
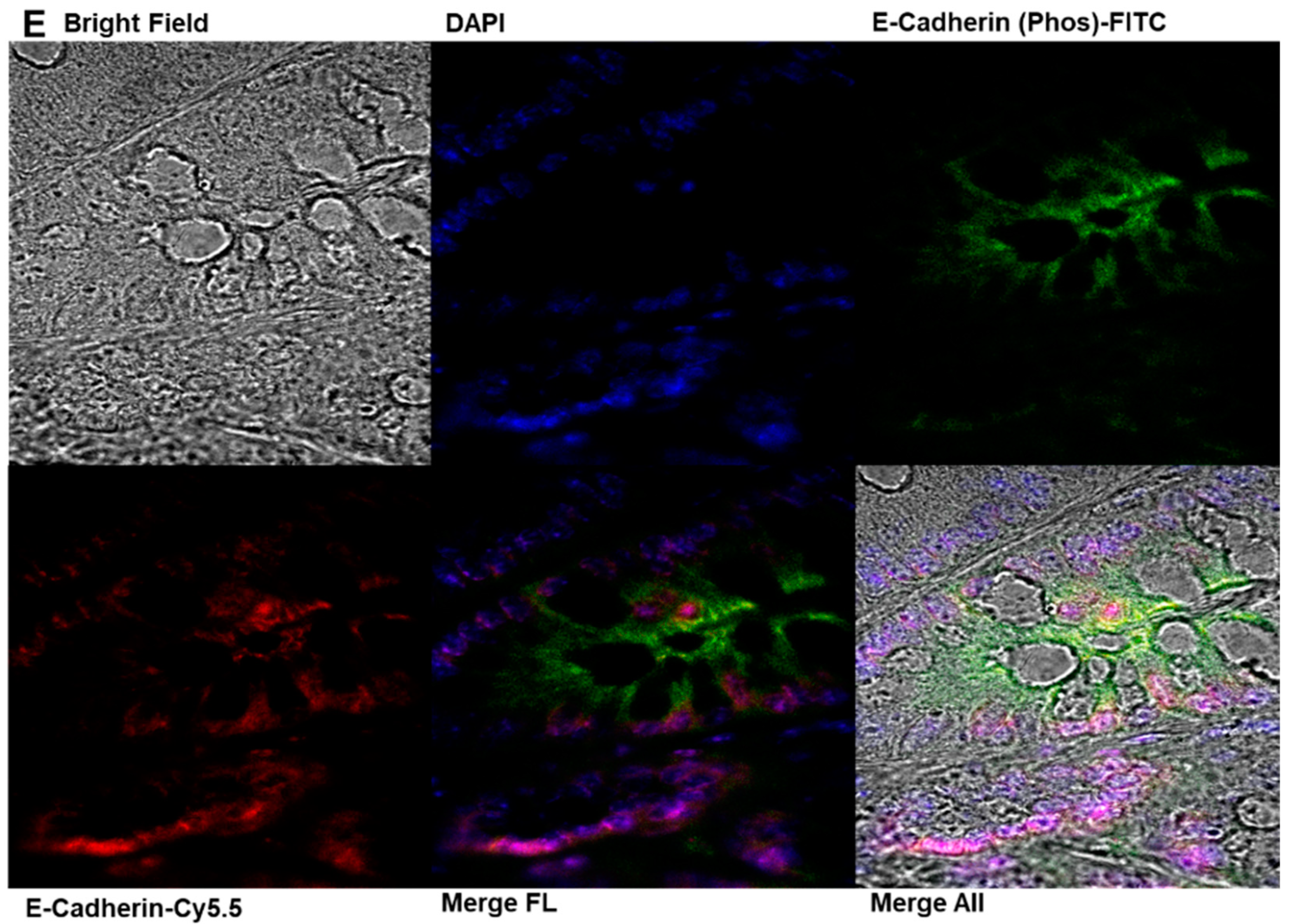
2.7. Immunofluorescence Analysis of E-Cadherin and Phosphorylated E-Cadherin Localization in Colonic Epithelial Tissues
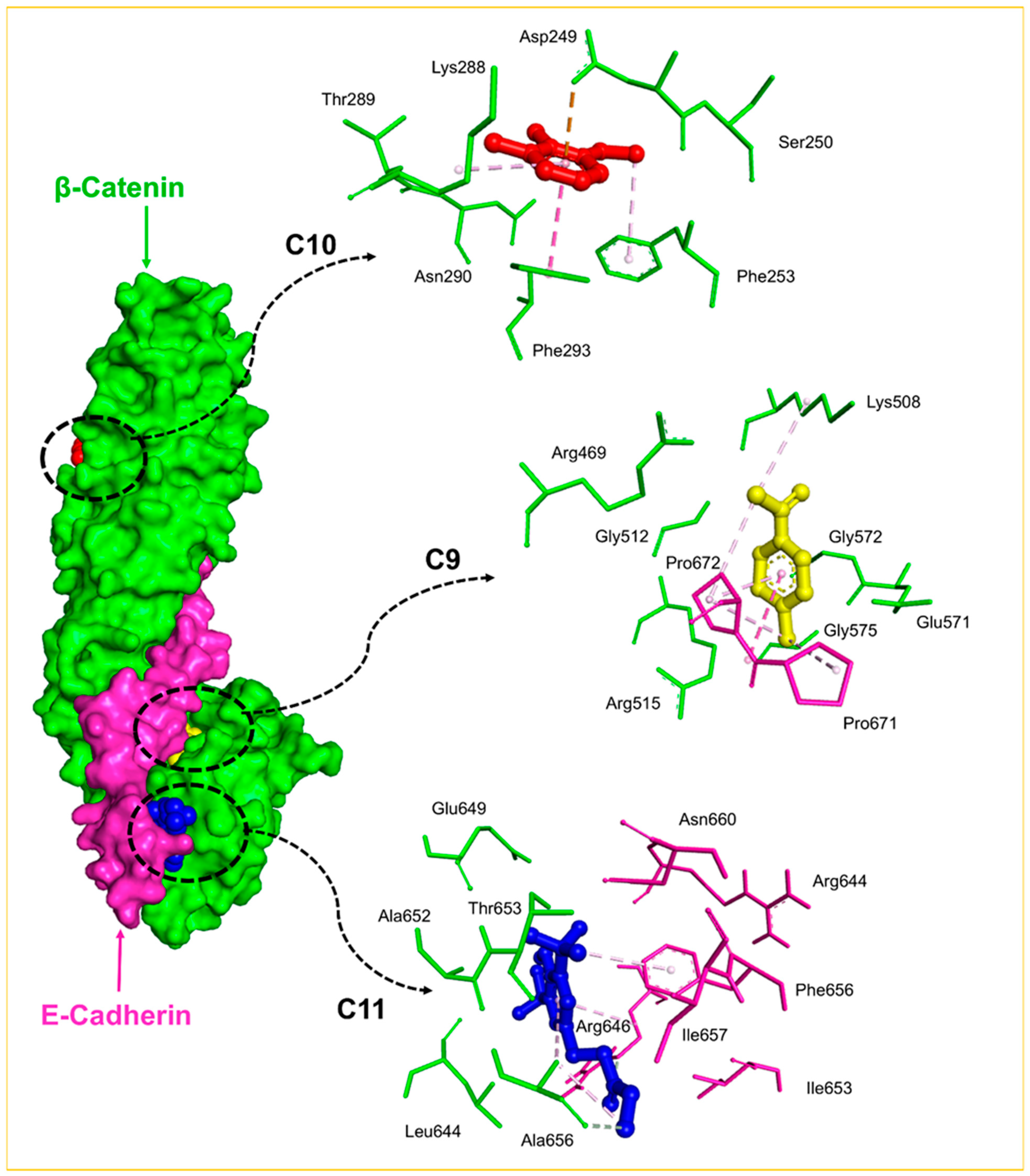
2.8. Molecular Docking Analysis
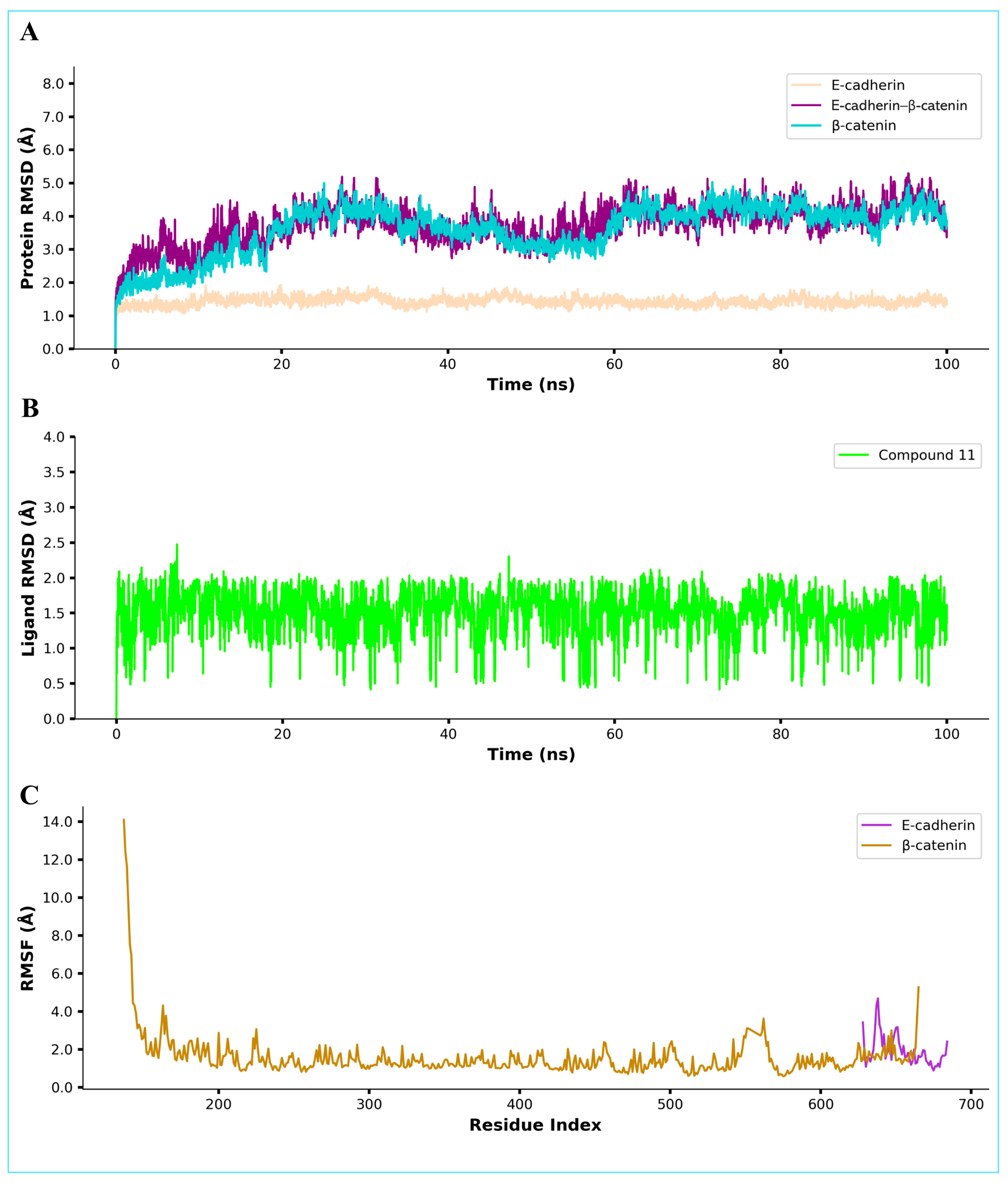
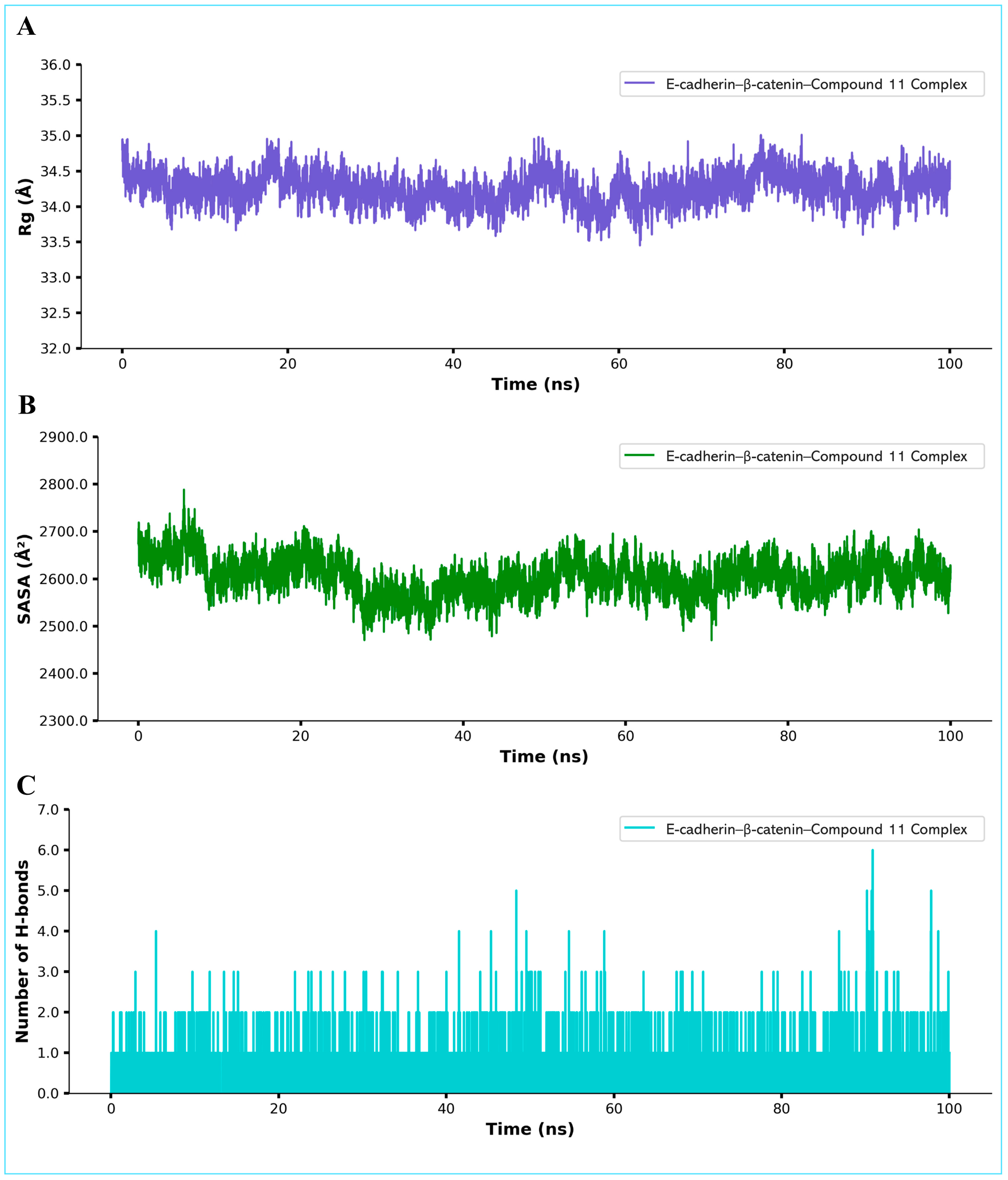
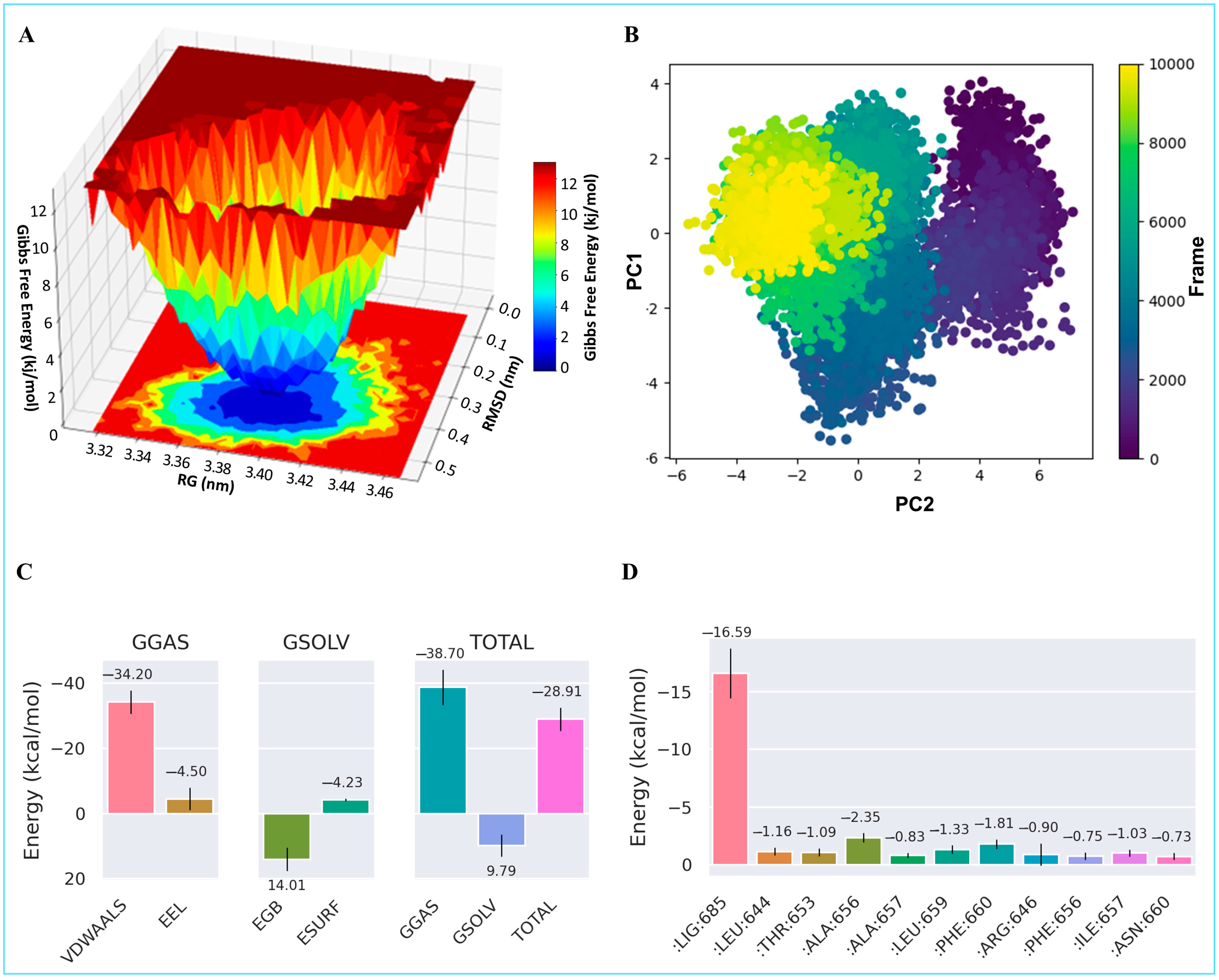
2.9. Molecular Dynamics Simulation
3. Discussions
4. Methods
4.1. Materials
4.2. Methanolic Extraction of Fenugreek Seeds (FSE) Using Cold Maceration Technique
4.3. GC-MS (Gas Chromatography–Mass Spectrometry) Analysis
4.4. Molecular Docking Studies
4.5. Molecular Dynamics Simulation Studies
4.6. MM/GBSA Binding Energy Computations
4.7. In Vivo Studies
4.7.1. Mice
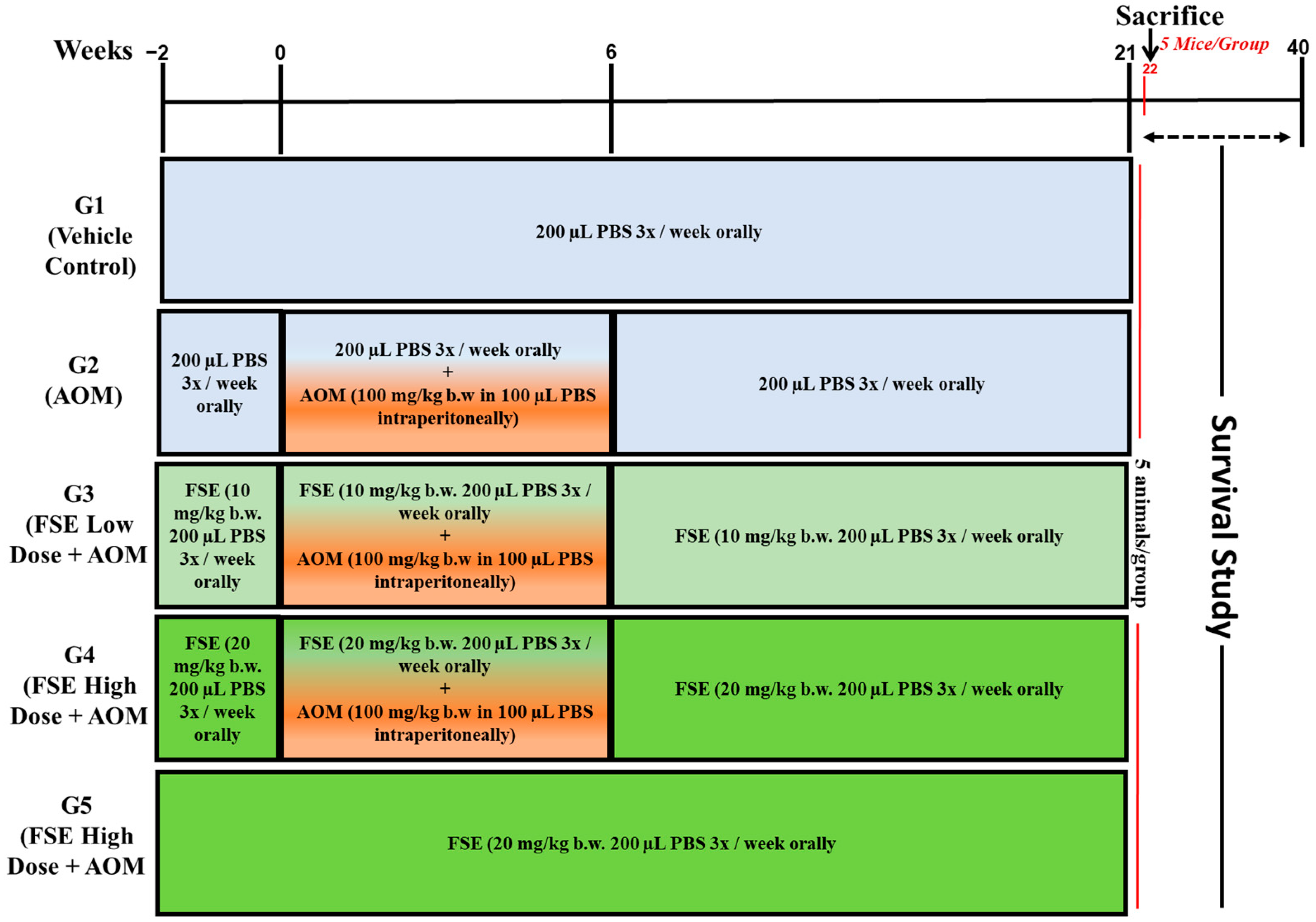
4.7.2. Experimental Design
4.8. Study of Average Body Weight (ABW) and Survival Rate
4.9. Effect of FSE on Serum Cancer Markers
4.10. Effect of FSE on Colonic Antioxidant Enzyme Levels
4.11. Annexin V-FITC/PI Apoptosis Assay and Reactive Oxygen Species (ROS) Detection
4.12. Immunofluorescence Analysis of E-Cadherin and Phosphorylated E-Cadherin Localization
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022, GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Xu, P. Global colorectal cancer burden in 2020 and projections to 2040. Transl. Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef]
- GBD 2019 Colorectal Cancer Collaborators. Global, regional, and national burden of colorectal cancer and its risk factors, 1990-2019, a systematic analysis for the Global Burden of Disease Study 2019. Lancet Gastroenterol. Hepatol. 2022, 7, 627–647. [Google Scholar] [CrossRef]
- Cheng, X.; Zhou, J.; Chen, Y.; Zhao, Y.; Zheng, H.; Wang, Q.; Li, X.; Jiang, S. Patterns and trends of mortality from metastatic colorectal cancer in Shanghai, China from 2005 to 2021, a population-based retrospective analysis. J. Cancer Res. Clin. Oncol. 2024, 150, 68. [Google Scholar] [CrossRef]
- Mathew, A.; Baby, B.; Wang, K.; Sirohi, B.; Lei, F.; Chen, Q.; Huang, B. Colorectal cancer incidence in younger adults in India. Gut 2020, 69, 1899–1900. [Google Scholar] [CrossRef] [PubMed]
- Hassanain, M.; Al-Alem, F.; Simoneau, E.; Traiki, T.A.; Alsaif, F.; Alsharabi, A.; Al-Faris, H.; Al-Saleh, K. Colorectal cancer liver metastasis trends in the kingdom of Saudi Arabia. Saudi J. Gastroenterol. 2016, 22, 370. [Google Scholar] [CrossRef]
- Basudan, A.M.; Basuwdan, A.M.; Abudawood, M.; Farzan, R.; Alfhili, M.A. Comprehensive Retrospective Analysis of Colorectal Cancer Incidence Patterns in Saudi Arabia. Life 2023, 13, 2198. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhao, J.; Li, Z.; Sun, J.; Lu, Y.; Zhang, R.; Zhu, Y.; Ding, K.; Rudan, I.; Theodoratou, E.; et al. National and subnational incidence, mortality and associated factors of colorectal cancer in China: A systematic analysis and modelling study. J. Glob. Health 2023, 13, 04096. [Google Scholar] [CrossRef]
- Thanikachalam, K.; Khan, G. Colorectal Cancer and Nutrition. Nutrients 2019, 11, 164. [Google Scholar] [CrossRef]
- Chew, M.H.; Tan, W.S.; Liu, Y.; Cheah, P.Y.; Loi, C.T.; Tang, C.L. Genomics of Hereditary Colorectal Cancer: Lessons Learnt from 25 Years of the Singapore Polyposis Registry. Ann. Acad. Med. Singap. 2015, 44, 290–296. [Google Scholar] [CrossRef]
- Murphy, C.C.; Zaki, T.A. Changing epidemiology of colorectal cancer—Birth cohort effects and emerging risk factors. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Murphy, N.; Moreno, V.; Hughes, D.J.; Vodicka, L.; Vodicka, P.; Aglago, E.K.; Gunter, M.J.; Jenab, M. Lifestyle and dietary environmental factors in colorectal cancer susceptibility. Mol. Asp. Med. 2019, 69, 2–9. [Google Scholar] [CrossRef]
- Yu, L.; Zhang, M.-M.; Hou, J.-G. Molecular and cellular pathways in colorectal cancer: Apoptosis, autophagy and inflammation as key players. Scand. J. Gastroenterol. 2022, 57, 1279–1290. [Google Scholar] [CrossRef]
- Wong, C.C.; Yu, J. Gut microbiota in colorectal cancer development and therapy. Nat. Rev. Clin. Oncol. 2023, 20, 429–452. [Google Scholar] [CrossRef]
- Shi, J.; Sun, Z.; Gao, Z.; Huang, D.; Hong, H.; Gu, J. Radioimmunotherapy in colorectal cancer treatment: Present and future. Front. Immunol. 2023, 14, 1105180. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Ciardiello, D.; Martini, G.; Napolitano, S.; Tabernero, J.; Cervantes, A. Clinical management of metastatic colorectal cancer in the era of precision medicine. CA Cancer J. Clin. 2022, 72, 372–401. [Google Scholar] [CrossRef] [PubMed]
- Zygulska, A.L.; Pierzchalski, P. Novel Diagnostic Biomarkers in Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 852. [Google Scholar] [CrossRef]
- Infante, J.R.; Reid, T.R.; Cohn, A.L.; Edenfield, W.J.; Cescon, T.P.; Hamm, J.T.; Malik, I.A.; Rado, T.A.; McGee, P.J.; Richards, D.A.; et al. Axitinib and/or bevacizumab with modified FOLFOX-6 as first-line therapy for metastatic colorectal cancer: A randomized phase 2 study. Cancer 2013, 119, 2555–2563. [Google Scholar] [CrossRef]
- Chen, G.; Gong, T.; Wang, Z.; Wang, Z.; Lin, X.; Chen, S.; Sun, C.; Zhao, W.; Kong, Y.; Ai, H.; et al. Colorectal cancer organoid models uncover oxaliplatin-resistant mechanisms at single cell resolution. Cell. Oncol. 2022, 45, 1155–1167. [Google Scholar] [CrossRef]
- Fyfe, I. Mutations linked to chemotherapy resistance in colorectal cancer. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 269. [Google Scholar] [CrossRef]
- Islam, M.R.; Akash, S.; Rahman, M.M.; Nowrin, F.T.; Akter, T.; Shohag, S.; Rauf, A.; Aljohani, A.S.M.; Simal-Gandara, J. Colon cancer and colorectal cancer: Prevention and treatment by potential natural products. Chem. Biol. Interact. 2022, 368, 110170. [Google Scholar] [CrossRef]
- Rahman, H.S. Preclinical Drug Discovery in Colorectal Cancer: A Focus on Natural Compounds. Curr. Drug Targets 2021, 22, 977–997. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Brockmueller, A.; Ruiz de Porras, V.; Shakibaei, M. Curcumin and its anti-colorectal cancer potential: From mechanisms of action to autophagy. Phytother. Res. 2024, 38, 3525–3551. [Google Scholar] [CrossRef]
- Pricci, M.; Girardi, B.; Giorgio, F.; Losurdo, G.; Ierardi, E.; Di Leo, A. Curcumin and Colorectal Cancer: From Basic to Clinical Evidences. Int. J. Mol. Sci. 2020, 21, 2364. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhu, Q.; Wang, S.; Shi, C. Epigallocatechin-3-gallate inhibits the formation of neutrophil extracellular traps and suppresses the migration and invasion of colon cancer cells by regulating STAT3/CXCL8 pathway. Mol. Cell. Biochem. 2023, 478, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Brockmueller, A.; Buhrmann, C.; Moravejolahkami, A.R.; Shakibaei, M. Resveratrol and p53, How are they involved in CRC plasticity and apoptosis? J. Adv. Res. 2024, 66, 181–195. [Google Scholar] [CrossRef]
- Alrumaihi, F. Exploring the Chemopreventive Potential of Artemisia annua Methanolic Extract in Colorectal Cancer Induced by Azoxymethane in Mice. Pharmaceuticals 2024, 18, 34. [Google Scholar] [CrossRef]
- Alrumaihi, F.; Khan, M.A.; Babiker, A.Y.; Alsaweed, M.; Azam, F.; Allemailem, K.S.; Almatroudi, A.A.; Ahamad, S.R.; AlSuhaymi, N.; Alsugoor, M.H.; et al. The Effect of Liposomal Diallyl Disulfide and Oxaliplatin on Proliferation of Colorectal Cancer Cells: In Vitro and In Silico Analysis. Pharmaceutics 2022, 14, 236. [Google Scholar] [CrossRef]
- Khan, A.; Alsahli, M.A.; Aljasir, M.A.; Maswadeh, H.; Mobark, M.A.; Azam, F.; Allemailem, K.S.; Alrumaihi, F.; Alhumaydhi, F.A.; Almatroudi, A.A.; et al. Experimental and Theoretical Insights on Chemopreventive Effect of the Liposomal Thymoquinone Against Benzo[a]pyrene-Induced Lung Cancer in Swiss Albino Mice. J. Inflamm. Res. 2022, 15, 2263–2280. [Google Scholar] [CrossRef]
- Nagulapalli Venkata, K.C.; Swaroop, A.; Bagchi, D.; Bishayee, A. A small plant with big benefits: Fenugreek (Trigonella foenum-graecum Linn.) for disease prevention and health promotion. Mol. Nutr. Food Res. 2017, 61, 1600950. [Google Scholar] [CrossRef]
- Khoja, K.K.; Howes, M.-J.R.; Hider, R.; Sharp, P.A.; Farrell, I.W.; Latunde-Dada, G.O. Cytotoxicity of Fenugreek Sprout and Seed Extracts and Their Bioactive Constituents on MCF-7 Breast Cancer Cells. Nutrients 2022, 14, 784. [Google Scholar] [CrossRef] [PubMed]
- Almalki, D.A.; Naguib, D.M. Anticancer Activity of Aqueous Fenugreek Seed Extract Against Pancreatic Cancer, Histological Evidence. J. Gastrointest. Cancer 2022, 53, 683–686. [Google Scholar] [CrossRef]
- Idris, S.; Mishra, A.; Khushtar, M. Recent Therapeutic Interventions of Fenugreek Seed: A Mechanistic Approach. Drug Res. 2021, 71, 180–192. [Google Scholar] [CrossRef]
- Sur, P.; Das, M.; Gomes, A.; Vedasiromoni, J.R.; Sahu, N.P.; Banerjee, S.; Sharma, R.M.; Ganguly, D.K. Trigonella foenum graecum (fenugreek) seed extract as an antineoplastic agent. Phytother. Res. 2001, 15, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Alrumaihi, F.A.; Khan, M.A.; Allemailem, K.S.; Alsahli, M.A.; Almatroudi, A.; Younus, H.; Alsuhaibani, S.A.; Algahtani, M.; Khan, A. Methanolic Fenugreek Seed Extract Induces p53-Dependent Mitotic Catastrophe in Breast Cancer Cells, Leading to Apoptosis. J. Inflamm. Res. 2021, 14, 1511–1535. [Google Scholar] [CrossRef]
- Khan, A.; Alsahli, M.A.; Aljasir, M.A.; Maswadeh, H.; Mobark, M.A.; Azam, F.; Allemailem, K.S.; Alrumaihi, F.; Alhumaydhi, F.A.; Alwashmi, A.S.S.; et al. Safety, Stability, and Therapeutic Efficacy of Long-Circulating TQ-Incorporated Liposomes: Implication in the Treatment of Lung Cancer. Pharmaceutics 2022, 14, 153. [Google Scholar] [CrossRef]
- Alrumaihi, F.; Khan, M.A.; Babiker, A.Y.; Alsaweed, M.; Azam, F.; Allemailem, K.S.; Almatroudi, A.A.; Ahamad, S.R.; Alsugoor, M.H.; Alharbi, K.N.; et al. Lipid-Based Nanoparticle Formulation of Diallyl Trisulfide Chemosensitizes the Growth Inhibitory Activity of Doxorubicin in Colorectal Cancer Model: A Novel In Vitro, In Vivo and In Silico Analysis. Molecules 2022, 27, 2192. [Google Scholar] [CrossRef]
- Iqbal, M.; Haque, R.; Ahamed, S.; Jafari, S.; Khadeer Ahamed, M.; Abdul Majid, A. Crystal Structures and Cytotoxicity of Ortho-Xylene Linked Bis-benzimidazolium Salts. Med. Chem. 2015, 11, 473–481. [Google Scholar] [CrossRef]
- Asha, R.N.; Daisy, C.; Bhuvanesh, N.; Nayagam, B.R.D. 2,4-bis(bromomethyl)-1,3,5-trimethylbenzene with 2-mercaptopyridine based derivative: Synthesis, crystal structure, in vitro anticancer activity, DFT, Hirshfeld surface analysis, antioxidant, DNA binding and molecular docking studies. J. Mol. Struct. 2022, 1251, 131981. [Google Scholar] [CrossRef]
- Fatima, I.; Safdar, N.; Akhtar, W.; Ayaz, A.; Ali, S.; Elansary, H.O.; Moussa, I.M.; Zaman, W. Green solvent-based extraction of three Fabaceae species: A potential antioxidant, anti-diabetic, and anti-leishmanial agents. Heliyon 2024, 10, e33668. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, S.S.; Makeen, H.A.; Menachery, S.J.; Moni, S.S. Documentation of bioactive principles of the flower from Caralluma retrospiciens (Ehrenb) and in vitro antibacterial activity—Part B. Arab. J. Chem. 2020, 13, 7370–7377. [Google Scholar] [CrossRef]
- Akbari, S.; Abdurahman, N.H.; Yunus, R.M.; Alara, O.R.; Abayomi, O.O. Extraction, characterization and antioxidant activity of fenugreek (Trigonella-Foenum Graecum) seed oil. Mater. Sci. Energy Technol. 2019, 2, 349–355. [Google Scholar] [CrossRef]
- Gavahian, M.; Bannikoppa, A.M.; Majzoobi, M.; Hsieh, C.-W.; Lin, J.; Farahnaky, A. Fenugreek bioactive compounds: A review of applications and extraction based on emerging technologies. Crit. Rev. Food Sci. Nutr. 2024, 64, 10187–10203. [Google Scholar] [CrossRef]
- Natarajan, T.D.; Ramasamy, J.R.; Palanisamy, K. Nutraceutical potentials of synergic foods: A systematic review. J. Ethn. Foods 2019, 6, 27. [Google Scholar] [CrossRef]
- Petruzzelli, M.; Wagner, E.F. Mechanisms of metabolic dysfunction in cancer-associated cachexia. Genes. Dev. 2016, 30, 489–501. [Google Scholar] [CrossRef]
- Marzo, F.; Milagro, F.I.; Barrenetxe, J.; Díaz, M.T.; Martínez, J.A. Azoxymethane-Induced Colorectal Cancer Mice Treated with a Polyphenol-Rich Apple Extract Show Less Neoplastic Lesions and Signs of Cachexia. Foods 2021, 10, 863. [Google Scholar] [CrossRef]
- Corti, A.; Franzini, M.; Paolicchi, A.; Pompella, A. Gamma-glutamyltransferase of Cancer Cells at the Crossroads of Tumor Progression, Drug Resistance and Drug Targeting. Anticancer Res. 2010, 30, 1169–1181. [Google Scholar]
- Kocianova, E.; Piatrikova, V.; Golias, T. Revisiting the Warburg Effect with Focus on Lactate. Cancers 2022, 14, 6028. [Google Scholar] [CrossRef]
- Jurisic, V.; Radenkovic, S.; Konjevic, G. The Actual Role of LDH as Tumor Marker, Biochemical and Clinical Aspects. In Advances in Cancer Biomarkers; Springer: Dordrecht, The Netherlands, 2015; pp. 115–124. [Google Scholar] [CrossRef]
- Lian, W.; Jiang, D.; Lin, W.; Jiang, M.; Zhang, Y.; Wang, H.; Zhao, L. Dual role of CD73 as a signaling molecule and adenosine-generating enzyme in colorectal cancer progression and immune evasion. Int. J. Biol. Sci. 2024, 20, 137–151. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Pham-Huy, L.A.; He, H.; Pham-Huy, C. Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. 2008, 4, 89–96. [Google Scholar]
- Kensara, O.A.; El-Shemi, A.G.; Mohamed, A.M.; Refaat, B.; Idris, S.; Ahmad, J. Thymoquinone subdues tumor growth and potentiates the chemopreventive effect of 5-fluorouracil on the early stages of colorectal carcinogenesis in rats. Drug Des. Dev. Ther. 2016, 10, 2239–2253. [Google Scholar] [CrossRef]
- Aloliqi, A.A. Therapeutic Potential of 6-Gingerol in Prevention of Colon Cancer Induced by Azoxymethane through the Modulation of Antioxidant Potential and Inflammation. Curr. Issues Mol. Biol. 2022, 44, 6218–6228. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Wang, R.; Weng, S.; Xu, H.; Zhang, Y.; Chen, S.; Liu, S.; Ba, Y.; Zhou, Z.; Luo, p.; et al. Multifaceted role of redox pattern in the tumor immune microenvironment regarding autophagy and apoptosis. Mol. Cancer 2023, 22, 130. [Google Scholar] [CrossRef]
- Marengo, B.; Nitti, M.; Furfaro, A.L.; Colla, R.; Ciucis, C.D.; Marinari, U.M.; Pronzato, M.A.; Traverso, N.; Domenicotti, C. Redox Homeostasis and Cellular Antioxidant Systems: Crucial Players in Cancer Growth and Therapy. Oxid. Med. Cell. Longev. 2016, 2016, 6235641. [Google Scholar] [CrossRef]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Huber, A.H.; Weis, W.I. The Structure of the β-Catenin/E-Cadherin Complex and the Molecular Basis of Diverse Ligand Recognition by β-Catenin. Cell 2001, 105, 391–402. [Google Scholar] [CrossRef]
- Chen, T.; Karedla, N.; Enderlein, J. Observation of E-cadherin adherens junction dynamics with metal-induced energy transfer imaging and spectroscopy. Commun. Biol. 2024, 7, 1596. [Google Scholar] [CrossRef]
- Loh, C.-Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef]
- Yu, W.; Yang, L.; Li, T.; Zhang, Y. Cadherin Signaling in Cancer: Its Functions and Role as a Therapeutic Target. Front. Oncol. 2019, 9, 989. [Google Scholar] [CrossRef]
- Du, W.; Liu, X.; Fan, G.; Zhao, X.; Sun, Y.; Wang, T.; Zhao, R.; Wang, G.; Zhao, C.; Zhu, Y.; et al. From cell membrane to the nucleus: An emerging role of E-cadherin in gene transcriptional regulation. J. Cell. Mol. Med. 2014, 18, 1712–1719. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Gao, Z.; Bao, Y.; Chen, L.; Huang, Y.; Liu, Y.; Dong, Q.; Wei, X. Wnt/β-catenin signaling pathway in carcinogenesis and cancer therapy. J. Hematol. Oncol. 2024, 17, 46. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Pang, K.M.; Evans, M.; Hay, E.D. Overexpression of β-Catenin Induces Apoptosis Independent of Its Transactivation Function with LEF-1 or the Involvement of Major G1 Cell Cycle Regulators. Mol. Biol. Cell 2000, 11, 3509–3523. [Google Scholar] [CrossRef]
- Wu, G.S.; Ding, Z. Caspase 9 is required for p53-dependent apoptosis and chemosensitivity in a human ovarian cancer cell line. Oncogene 2002, 21, 1205020. [Google Scholar] [CrossRef] [PubMed]
- Roger, L.; Jullien, L.; Gire, V.; Roux, P. Gain of oncogenic function of p53 mutants regulates E-cadherin expression uncoupled from cell invasion in colon cancer cells. J. Cell Sci. 2010, 123, 1295–1305. [Google Scholar] [CrossRef]
- Cheng, Y.; Mao, M.; Lu, Y. The biology of YAP in programmed cell death. Biomark. Res. 2022, 10, 34. [Google Scholar] [CrossRef]
- Wen, Z.; Wang, Y.; Qi, S.; Ma, M.; Li, J.; Yu, F.-X. Regulation of TP73 transcription by Hippo-YAP signaling. Biochem. Biophys. Res. Commun. 2020, 531, 96–104. [Google Scholar] [CrossRef]
- Julien, S.; Puig, I.; Caretti, E.; Bonaventure, J.; Nelles, L.; van Roy, F.; Dargemont, C.; de Herreros, A.G.; Bellacosa, A.; Larue, L. Activation of NF-κB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene 2007, 26, 7445–7456. [Google Scholar] [CrossRef]
- Wang, T.; Li, X. E-Cadherin. In Encyclopedia of Signaling Molecules; Springer: New York, NY, USA, 2016; pp. 1–11. [Google Scholar] [CrossRef]
- Chiou, Y.S.; Tsai, M.L.; Nagabhushanam, K.; Wang, Y.J.; Wu, C.H.; Ho, C.T.; Pan, M.H. Pterostilbene Is More Potent than Resveratrol in Preventing Azoxymethane (AOM)-Induced Colon Tumorigenesis via Activation of the NF-E2-Related Factor 2 (Nrf2)-Mediated Antioxidant Signaling Pathway. J. Agric. Food Chem. 2011, 59, 2725–2733. [Google Scholar] [CrossRef]
- Hao, J.; Dai, X.; Gao, J.; Li, Y.; Hou, Z.; Chang, Z.; Wang, Y. Curcumin suppresses colorectal tumorigenesis via the Wnt/β-catenin signaling pathway by downregulating Axin2. Oncol. Lett. 2021, 21, 186. [Google Scholar] [CrossRef]
- Rebersek, M. Gut microbiome and its role in colorectal cancer. BMC Cancer 2021, 21, 1325. [Google Scholar] [CrossRef]
- Song, M.; Chan, A.T.; Sun, J. Influence of the Gut Microbiome, Diet, and Environment on Risk of Colorectal Cancer. Gastroenterology 2020, 158, 322–340. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4, Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Lundborg, M.; Lindahl, E. Automatic GROMACS Topology Generation and Comparisons of Force Fields for Solvation Free Energy Calculations. J. Phys. Chem. B 2015, 119, 810–823. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Eisenhaber, F.; Lijnzaad, P.; Argos, P.; Sander, C.; Scharf, M. The double cubic lattice method: Efficient approaches to numerical integration of surface area and volume and to dot surface contouring of molecular assemblies. J. Comput. Chem. 1995, 16, 273–284. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Number | RT (min) | Scan Number (#) | Area (Ab·s) | Baseline Height (Ab) | Absolute Height (Ab) | Peak Width 50% (min) | Hit Number | Hit Name | Quality | Mol Weight (amu) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 5.05 | 343 | 182,804 | 29,865 | 34,215 | 0.16 | 1 | Benzene, 1,3-dimethyl- | 94 | 106.078 |
| 2 | 5.427 | 409 | 28,045 | 7262 | 12,506 | 0.086 | 1 | 1,3-Cyclopentadiene, 5-(1-methylethylidene)- | 94 | 106.078 |
| 3 | 5.502 | 422 | 103,925 | 22,117 | 25,147 | 0.217 | 1 | o-Xylene | 95 | 106.078 |
| 4 | 5.948 | 500 | 8311 | 3799 | 6860 | 0.063 | 1 | Benzene, (1-methylethyl)- | 43 | 120.094 |
| 5 | 6.457 | 589 | 42,947 | 8712 | 12,028 | 0.126 | 1 | Benzene, propyl- | 80 | 120.094 |
| 6 | 6.812 | 651 | 20,495 | 8476 | 20,988 | 0.057 | 1 | Benzene, 1-ethyl-2-methyl- | 93 | 120.094 |
| 7 | 7.098 | 701 | 549,840 | 75,785 | 83,135 | 0.338 | 1 | Benzene, 1,2,3-trimethyl- | 95 | 120.094 |
| 8 | 7.487 | 769 | 20,667 | 9854 | 17,249 | 0.057 | 1 | Benzene, 1-ethyl-3-methyl- | 90 | 120.094 |
| 9 | 8.036 | 865 | 25,453 | 6644 | 15,658 | 0.097 | 1 | 1,3,8-p-Menthatriene | 52 | 134.11 |
| 10 | 9.049 | 1042 | 51,010 | 14,409 | 22,019 | 0.109 | 1 | Benzene, 1-ethyl-2,3-dimethyl- | 94 | 134.11 |
| 11 | 13.158 | 1760 | 41,098 | 12,250 | 13,143 | 0.154 | 1 | Tridecane | 59 | 184.219 |
| 12 | 16.127 | 2279 | 66,807 | 8267 | 9759 | 0.223 | 1 | Dotriacontane | 72 | 450.516 |
| 13 | 20.115 | 2976 | 206,276 | 43,730 | 46,268 | 0.24 | 1 | Benzenepropanoic acid, 3,5-bis(1,1-dimethylethyl)-4-hydroxy-, methyl ester | 93 | 292.204 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, A.; Allemailem, K.S.; Alradhi, A.E.; Azam, F. Preclinical and Molecular Docking Insights into the Chemopreventive Role of Fenugreek Seed Extract in a Murine Model of Colorectal Cancer. Pharmaceuticals 2025, 18, 490. https://doi.org/10.3390/ph18040490
Khan A, Allemailem KS, Alradhi AE, Azam F. Preclinical and Molecular Docking Insights into the Chemopreventive Role of Fenugreek Seed Extract in a Murine Model of Colorectal Cancer. Pharmaceuticals. 2025; 18(4):490. https://doi.org/10.3390/ph18040490
Chicago/Turabian StyleKhan, Arif, Khaled S. Allemailem, Arwa Essa Alradhi, and Faizul Azam. 2025. "Preclinical and Molecular Docking Insights into the Chemopreventive Role of Fenugreek Seed Extract in a Murine Model of Colorectal Cancer" Pharmaceuticals 18, no. 4: 490. https://doi.org/10.3390/ph18040490
APA StyleKhan, A., Allemailem, K. S., Alradhi, A. E., & Azam, F. (2025). Preclinical and Molecular Docking Insights into the Chemopreventive Role of Fenugreek Seed Extract in a Murine Model of Colorectal Cancer. Pharmaceuticals, 18(4), 490. https://doi.org/10.3390/ph18040490