
MicroRNAs as Sensitizers of Tyrosine Kinase Inhibitor Resistance in Cancer: Small Molecule Partnerships



Abstract

1. Introduction
1.1. MicroRNAs
1.2. Protein Kinases
1.3. Tyrosine Kinase Inhibitors (TKIs) and Mechanism of Resistance
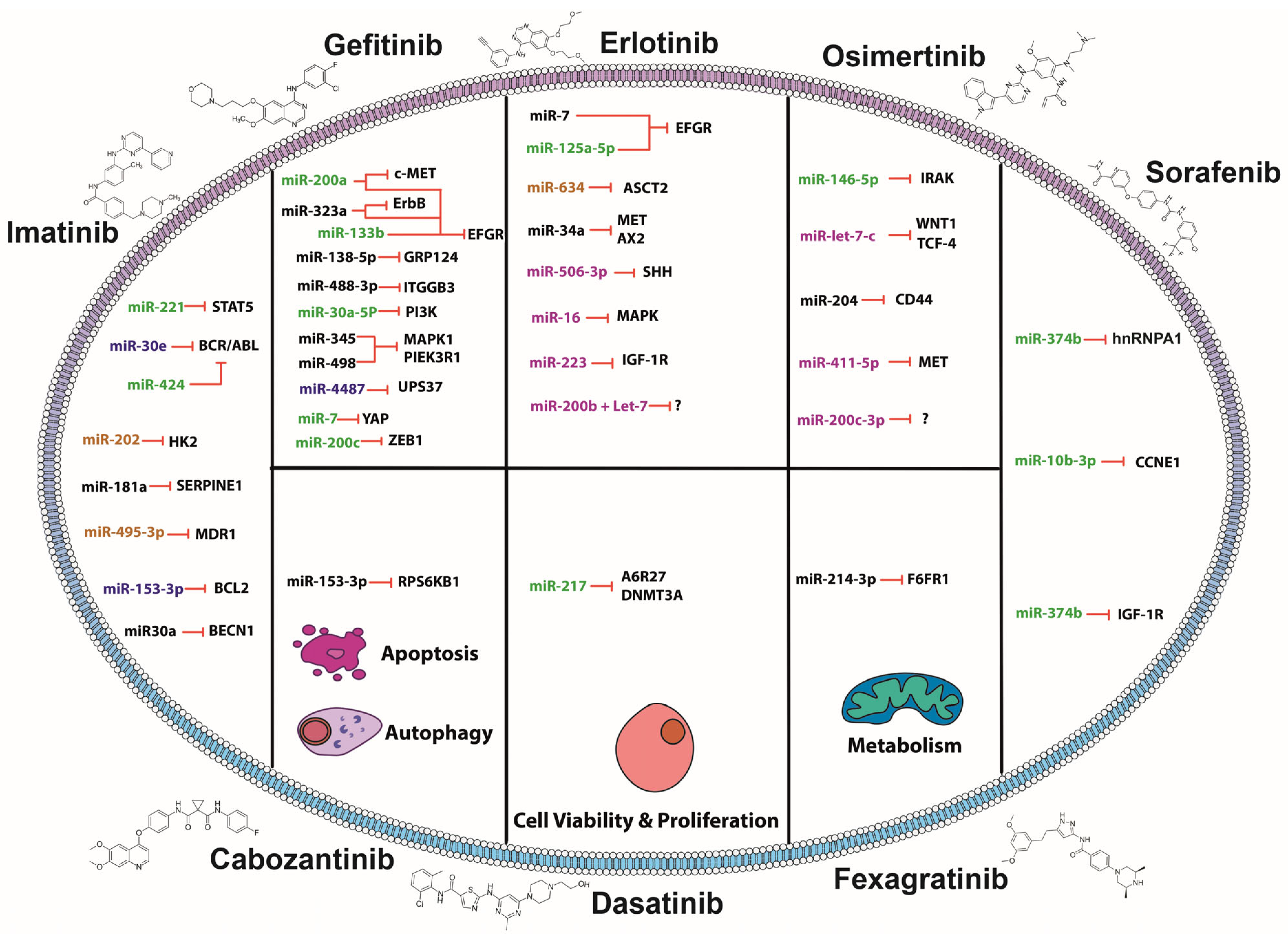
2. MiRNAs and TKIs: Association of Small Molecules in Overcoming Resistance
2.1. MiRNAs Sensitizing to Imatinib
2.2. MiRNAs Sensitizing to Gefitinib
2.3. MiRNAs Sensitizing to Erlotinib
2.4. MiRNAs Sensitizing to Osimertinib
2.5. MiRNAs Sensitizing to Sorafenib
2.6. MiRNAs Sensitizing to Fexagratinib
2.7. MiRNAs Sensitizing to Dasatinib
2.8. MiRNAs Sensitizing to Cabozantinib
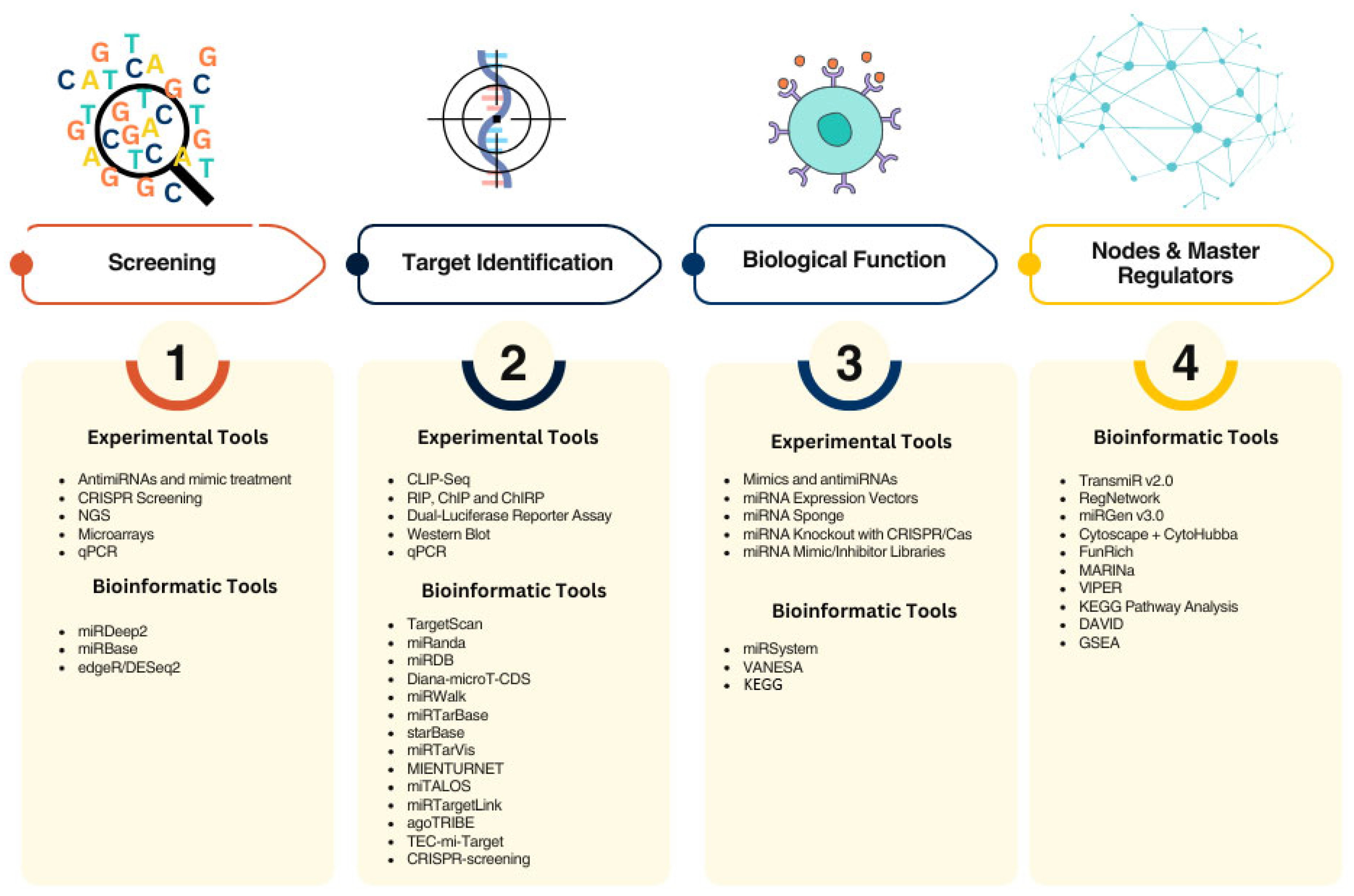
3. Methodological and Clinical Challenges for the Use of miRNAs as Sensitizers of TKIs
4. Significance/Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Available online: https://www.mirbase.org/ (accessed on 14 February 2025).
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans Heterochronic Gene Lin-4 Encodes Small RNAs with Antisense Complementarity to Lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA Sequences to Function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- MLA Style: Press Release. NobelPrize.Org. Nobel Prize Outreach AB 2024. 2025. Available online: https://www.nobelprize.org/prizes/medicine/2024/press-release/ (accessed on 14 February 2025).
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef]
- Liu, W.; Wang, X. Prediction of Functional microRNA Targets by Integrative Modeling of microRNA Binding and Target Expression Data. Genome Biol. 2019, 20, 18. [Google Scholar] [CrossRef]
- Riolo, G.; Cantara, S.; Marzocchi, C.; Ricci, C. miRNA Targets: From Prediction Tools to Experimental Validation. Methods Protoc. 2020, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.C.; Bovolenta, L.A.; Alves, L.; Figueiredo, L.; Ribeiro, A.O.; Campos, V.F.; Lemke, N.; Pinhal, D. Understanding the Modus Operandi of MicroRNA Regulatory Clusters. Cells 2019, 8, 1103. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The Role of MicroRNAs in Human Cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA Expression Profiles Classify Human Cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef]
- Bautista-Sánchez, D.; Arriaga-Canon, C.; Pedroza-Torres, A.; De La Rosa-Velázquez, I.A.; González-Barrios, R.; Contreras-Espinosa, L.; Montiel-Manríquez, R.; Castro-Hernández, C.; Fragoso-Ontiveros, V.; Álvarez-Gómez, R.M.; et al. The Promising Role of miR-21 as a Cancer Biomarker and Its Importance in RNA-Based Therapeutics. Mol. Ther. Nucleic Acids 2020, 20, 409–420. [Google Scholar] [CrossRef]
- Dasgupta, I.; Chatterjee, A. Recent Advances in miRNA Delivery Systems. Methods Protoc. 2021, 4, 10. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour Heterogeneity and Resistance to Cancer Therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Syed, Y.Y. Givosiran: A Review in Acute Hepatic Porphyria. Drugs 2021, 81, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Abdipourbozorgbaghi, M.; Vancura, A.; Radpour, R.; Haefliger, S. Circulating miRNA Panels as a Novel Non-Invasive Diagnostic, Prognostic, and Potential Predictive Biomarkers in Non-Small Cell Lung Cancer (NSCLC). Br. J. Cancer 2024, 131, 1350–1362. [Google Scholar] [CrossRef]
- MacDonagh, L.; Gallagher, M.F.; Ffrench, B.; Gasch, C.; Gray, S.G.; Reidy, M.; Nicholson, S.; Leonard, N.; Ryan, R.; Young, V.; et al. MicroRNA Expression Profiling and Biomarker Validation in Treatment-Naïve and Drug Resistant Non-Small Cell Lung Cancer. Transl. Lung Cancer Res. 2021, 10, 1773–1791. [Google Scholar] [CrossRef]
- Pavlíková, L.; Šereš, M.; Breier, A.; Sulová, Z. The Roles of microRNAs in Cancer Multidrug Resistance. Cancers 2022, 14, 1090. [Google Scholar] [CrossRef]
- Zhong, S.; Ma, T.; Zhang, X.; Lv, M.; Chen, L.; Tang, J.; Zhao, J. MicroRNA Expression Profiling and Bioinformatics Analysis of Dysregulated microRNAs in Vinorelbine-Resistant Breast Cancer Cells. Gene 2015, 556, 113–118. [Google Scholar] [CrossRef]
- Zhu, M.; Huang, Z.; Zhu, D.; Zhou, X.; Shan, X.; Qi, L.-W.; Wu, L.; Cheng, W.; Zhu, J.; Zhang, L.; et al. A Panel of microRNA Signature in Serum for Colorectal Cancer Diagnosis. Oncotarget 2017, 8, 17081–17091. [Google Scholar] [CrossRef]
- Chen, Q.; Xia, H.-W.; Ge, X.-J.; Zhang, Y.-C.; Tang, Q.-L.; Bi, F. Serum miR-19a Predicts Resistance to FOLFOX Chemotherapy in Advanced Colorectal Cancer Cases. Asian Pac. J. Cancer Prev. 2013, 14, 7421–7426. [Google Scholar] [CrossRef]
- Joshi, D.; Chandrakala, S.; Korgaonkar, S.; Ghosh, K.; Vundinti, B.R. Down-Regulation of miR-199b Associated with Imatinib Drug Resistance in 9q34.1 Deleted BCR/ABL Positive CML Patients. Gene 2014, 542, 109–112. [Google Scholar] [CrossRef]
- Ye, X.; Bai, W.; Zhu, H.; Zhang, X.; Chen, Y.; Wang, L.; Yang, A.; Zhao, J.; Jia, L. MiR-221 Promotes Trastuzumab-Resistance and Metastasis in HER2-Positive Breast Cancers by Targeting PTEN. BMB Rep. 2014, 47, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Liang, G.; Lin, Q.; Fang, X.; Luo, Q.; Cen, Y.; Mehrpour, M.; Hamai, A.; Liu, Z.; Shi, Y.; et al. circCDYL2 Promotes Trastuzumab Resistance via Sustaining HER2 Downstream Signaling in Breast Cancer. Mol. Cancer 2022, 21, 8. [Google Scholar] [CrossRef]
- Bortoletto, A.S.; Parchem, R.J. KRAS Hijacks the miRNA Regulatory Pathway in Cancer. Cancer Res. 2023, 83, 1563–1572. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.I. Roles of MicroRNAs in Disease Biology. JMA J. 2023, 6, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-Targeted Cancer Therapies: Progress, Challenges and Future Directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Cicenas, J.; Zalyte, E.; Bairoch, A.; Gaudet, P. Kinases and Cancer. Cancers 2018, 10, 63. [Google Scholar] [CrossRef]
- Huang, L.; Jiang, S.; Shi, Y. Tyrosine Kinase Inhibitors for Solid Tumors in the Past 20 Years (2001–2020). J. Hematol. Oncol. 2020, 13, 143. [Google Scholar] [CrossRef]
- Schlessinger, J.; Lemmon, M.A. SH2 and PTB Domains in Tyrosine Kinase Signaling. Sci. STKE 2003, 2003, RE12. [Google Scholar] [CrossRef]
- Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef]
- Gocek, E.; Moulas, A.N.; Studzinski, G.P. Non-Receptor Protein Tyrosine Kinases Signaling Pathways in Normal and Cancer Cells. Crit. Rev. Clin. Lab. Sci. 2014, 51, 125–137. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.M.; Lin, S.F. The Protein Tyrosine Kinase Family of the Human Genome. Oncogene 2000, 19, 5548–5557. [Google Scholar] [CrossRef] [PubMed]
- Siveen, K.S.; Prabhu, K.S.; Achkar, I.W.; Kuttikrishnan, S.; Shyam, S.; Khan, A.Q.; Merhi, M.; Dermime, S.; Uddin, S. Role of Non Receptor Tyrosine Kinases in Hematological Malignances and Its Targeting by Natural Products. Mol. Cancer 2018, 17, 31. [Google Scholar] [CrossRef]
- Ebrahimi, N.; Fardi, E.; Ghaderi, H.; Palizdar, S.; Khorram, R.; Vafadar, R.; Ghanaatian, M.; Rezaei-Tazangi, F.; Baziyar, P.; Ahmadi, A.; et al. Receptor Tyrosine Kinase Inhibitors in Cancer. Cell. Mol. Life Sci. 2023, 80, 104. [Google Scholar] [CrossRef] [PubMed]
- Steeghs, N.; Nortier, J.W.R.; Gelderblom, H. Small Molecule Tyrosine Kinase Inhibitors in the Treatment of Solid Tumors: An Update of Recent Developments. Ann. Surg. Oncol. 2007, 14, 942–953. [Google Scholar] [CrossRef] [PubMed]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jérusalem, G.; Longuespée, R.; Sounni, N.E. Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef]
- Chen, K.-K.; Du, T.-F.; Wu, K.-S.; Yang, W. First-Line Treatment Strategies for Newly Diagnosed Chronic Myeloid Leukemia: A Network Meta-Analysis. Cancer Manag. Res. 2018, 10, 3891–3910. [Google Scholar] [CrossRef]
- Walz, C.; Sattler, M. Novel Targeted Therapies to Overcome Imatinib Mesylate Resistance in Chronic Myeloid Leukemia (CML). Crit. Rev. Oncol. Hematol. 2006, 57, 145–164. [Google Scholar] [CrossRef]
- Martínez-Castillo, M.; Gómez-Romero, L.; Tovar, H.; Olarte-Carrillo, I.; García-Laguna, A.; Barranco-Lampón, G.; De la Cruz-Rosas, A.; Martínez-Tovar, A.; Hernández-Zavala, A.; Córdova, E.J. Genetic Alterations in the BCR-ABL1 Fusion Gene Related to Imatinib Resistance in Chronic Myeloid Leukemia. Leuk. Res. 2023, 131, 107325. [Google Scholar] [CrossRef]
- Lindström, H.J.G.; Friedman, R. Rotating between Ponatinib and Imatinib Temporarily Increases the Efficacy of Imatinib as Shown in a Chronic Myeloid Leukaemia Model. Sci. Rep. 2022, 12, 5164. [Google Scholar] [CrossRef]
- He, C.; Wang, Z.; Yu, J.; Mao, S.; Xiang, X. Current Drug Resistance Mechanisms and Treatment Options in Gastrointestinal Stromal Tumors: Summary and Update. Curr. Treat. Options Oncol. 2024, 25, 1390–1405. [Google Scholar] [CrossRef]
- Wu, J.-Y.; Wu, S.-G.; Yang, C.-H.; Chang, Y.-L.; Chang, Y.-C.; Hsu, Y.-C.; Shih, J.-Y.; Yang, P.-C. Comparison of Gefitinib and Erlotinib in Advanced NSCLC and the Effect of EGFR Mutations. Lung Cancer 2011, 72, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, C.; Gregorc, V.; Karachaliou, N.; Rosell, R.; Santarpia, M. Mechanisms of Resistance to Osimertinib. J. Thorac. Dis. 2020, 12, 2851–2858. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Chen, Z.; Zhang, W.; Cheng, Y.; Zhang, B.; Wu, F.; Wang, Q.; Wang, S.; Rong, D.; Reiter, F.P.; et al. The Mechanisms of Sorafenib Resistance in Hepatocellular Carcinoma: Theoretical Basis and Therapeutic Aspects. Signal Transduct. Target. Ther. 2020, 5, 87. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Yan, T.; Liu, F.; Liu, Q.; Zhao, J.; Xiong, H.; Jiang, S. Link of Sorafenib Resistance with the Tumor Microenvironment in Hepatocellular Carcinoma: Mechanistic Insights. Front. Pharmacol. 2022, 13, 991052. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, Q.; Jia, R.; Xiang, S.; Xu, L. A Comprehensive Review of the Relationship between Autophagy and Sorafenib-Resistance in Hepatocellular Carcinoma: Ferroptosis Is Noteworthy. Front. Cell Dev. Biol. 2023, 11, 1156383. [Google Scholar] [CrossRef]
- Ezzoukhry, Z.; Louandre, C.; Trécherel, E.; Godin, C.; Chauffert, B.; Dupont, S.; Diouf, M.; Barbare, J.-C.; Mazière, J.-C.; Galmiche, A. EGFR Activation Is a Potential Determinant of Primary Resistance of Hepatocellular Carcinoma Cells to Sorafenib. Int. J. Cancer 2012, 131, 2961–2969. [Google Scholar] [CrossRef]
- Saka, H.; Kitagawa, C.; Kogure, Y.; Takahashi, Y.; Fujikawa, K.; Sagawa, T.; Iwasa, S.; Takahashi, N.; Fukao, T.; Tchinou, C.; et al. Safety, Tolerability and Pharmacokinetics of the Fibroblast Growth Factor Receptor Inhibitor AZD4547 in Japanese Patients with Advanced Solid Tumours: A Phase I Study. Investig. New Drugs 2017, 35, 451–462. [Google Scholar] [CrossRef]
- Picca, A.; Di Stefano, A.L.; Savatovsky, J.; Ducray, F.; Chinot, O.; Moyal, E.C.-J.; Augereau, P.; Le Rhun, E.; Schmitt, Y.; Rousseaux, N.; et al. TARGET: A Phase I/II Open-Label Multicenter Study to Assess Safety and Efficacy of Fexagratinib in Patients with Relapsed/Refractory FGFR Fusion-Positive Glioma. Neurooncol. Adv. 2024, 6, vdae068. [Google Scholar] [CrossRef]
- Aggarwal, C.; Redman, M.W.; Lara, P.N.; Borghaei, H.; Hoffman, P.; Bradley, J.D.; Newman, A.J.; Feldman, M.J.; Minichiello, K.; Miao, J.; et al. SWOG S1400D (NCT02965378), a Phase II Study of the Fibroblast Growth Factor Receptor Inhibitor AZD4547 in Previously Treated Patients with Fibroblast Growth Factor Pathway-Activated Stage IV Squamous Cell Lung Cancer (Lung-MAP Substudy). J. Thorac. Oncol. 2019, 14, 1847–1852. [Google Scholar] [CrossRef]
- Chae, Y.K.; Hong, F.; Vaklavas, C.; Cheng, H.H.; Hammerman, P.; Mitchell, E.P.; Zwiebel, J.A.; Ivy, S.P.; Gray, R.J.; Li, S.; et al. Phase II Study of AZD4547 in Patients with Tumors Harboring Aberrations in the FGFR Pathway: Results From the NCI-MATCH Trial (EAY131) Subprotocol W. J. Clin. Oncol. 2020, 38, 2407–2417. [Google Scholar] [CrossRef]
- Coombes, R.C.; Badman, P.D.; Lozano-Kuehne, J.P.; Liu, X.; Macpherson, I.R.; Zubairi, I.; Baird, R.D.; Rosenfeld, N.; Garcia-Corbacho, J.; Cresti, N.; et al. Results of the Phase IIa RADICAL Trial of the FGFR Inhibitor AZD4547 in Endocrine Resistant Breast Cancer. Nat. Commun. 2022, 13, 3246. [Google Scholar] [CrossRef]
- Chen, R.; Chen, B. The Role of Dasatinib in the Management of Chronic Myeloid Leukemia. Drug Des. Dev. Ther. 2015, 9, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Rix, U.; Hantschel, O.; Dürnberger, G.; Remsing Rix, L.L.; Planyavsky, M.; Fernbach, N.V.; Kaupe, I.; Bennett, K.L.; Valent, P.; Colinge, J.; et al. Chemical Proteomic Profiles of the BCR-ABL Inhibitors Imatinib, Nilotinib, and Dasatinib Reveal Novel Kinase and Nonkinase Targets. Blood 2007, 110, 4055–4063. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, T.; Walters, D.K.; Stoffregen, E.P.; Jia, T.; Manley, P.W.; Mestan, J.; Cowan-Jacob, S.W.; Lee, F.Y.; Heinrich, M.C.; Deininger, M.W.N.; et al. In Vitro Activity of Bcr-Abl Inhibitors AMN107 and BMS-354825 against Clinically Relevant Imatinib-Resistant Abl Kinase Domain Mutants. Cancer Res. 2005, 65, 4500–4505. [Google Scholar] [CrossRef]
- Soverini, S.; Martinelli, G.; Colarossi, S.; Gnani, A.; Castagnetti, F.; Rosti, G.; Bosi, C.; Paolini, S.; Rondoni, M.; Piccaluga, P.P.; et al. Presence or the Emergence of a F317L BCR-ABL Mutation May Be Associated with Resistance to Dasatinib in Philadelphia Chromosome-Positive Leukemia. J. Clin. Oncol. 2006, 24, e51–e52. [Google Scholar] [CrossRef] [PubMed]
- Maroto, P.; Porta, C.; Capdevila, J.; Apolo, A.B.; Viteri, S.; Rodriguez-Antona, C.; Martin, L.; Castellano, D. Cabozantinib for the Treatment of Solid Tumors: A Systematic Review. Ther. Adv. Med. Oncol. 2022, 14, 17588359221107112. [Google Scholar] [CrossRef]
- Park, K.Y.; Hefti, H.O.; Liu, P.; Lugo-Cintrón, K.M.; Kerr, S.C.; Beebe, D.J. Immune Cell Mediated Cabozantinib Resistance for Patients with Renal Cell Carcinoma. Integr. Biol. 2021, 13, 259–268. [Google Scholar] [CrossRef]
- Somwar, R.; Smith, R.; Hayashi, T.; Ishizawa, K.; Snyder Charen, A.; Khodos, I.; Mattar, M.; He, J.; Balasubramanian, S.; Stephens, P.; et al. MDM2 Amplification (Amp) to Mediate Cabozantinib Resistance in Patients (Pts) with Advanced RET-Rearranged Lung Cancers. J. Clin. Oncol. 2016, 34, 9068. [Google Scholar] [CrossRef]
- Milojkovic, D.; Apperley, J. Mechanisms of Resistance to Imatinib and Second-Generation Tyrosine Inhibitors in Chronic Myeloid Leukemia. Clin. Cancer Res. 2009, 15, 7519–7527. [Google Scholar] [CrossRef]
- Lupini, L.; Bassi, C.; Ferracin, M.; Bartonicek, N.; D’Abundo, L.; Zagatti, B.; Callegari, E.; Musa, G.; Moshiri, F.; Gramantieri, L.; et al. miR-221 Affects Multiple Cancer Pathways by Modulating the Level of Hundreds Messenger RNAs. Front. Genet. 2013, 4, 64. [Google Scholar] [CrossRef]
- Jiang, X.; Cheng, Y.; Hu, C.; Zhang, A.; Ren, Y.; Xu, X. MicroRNA-221 Sensitizes Chronic Myeloid Leukemia Cells to Imatinib by Targeting STAT5. Leuk. Lymphoma 2019, 60, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Hershkovitz-Rokah, O.; Modai, S.; Pasmanik-Chor, M.; Toren, A.; Shomron, N.; Raanani, P.; Shpilberg, O.; Granot, G. Restoration of miR-424 Suppresses BCR-ABL Activity and Sensitizes CML Cells to Imatinib Treatment. Cancer Lett. 2015, 360, 245–256. [Google Scholar] [CrossRef]
- Yu, Z.; Zhou, X.; Wang, X. Metabolic Reprogramming in Hematologic Malignancies: Advances and Clinical Perspectives. Cancer Res. 2022, 82, 2955–2963. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Li, X.; Feng, J.; Zhang, X. Overexpression of miR-202 Resensitizes Imatinib Resistant Chronic Myeloid Leukemia Cells through Targetting Hexokinase 2. Biosci. Rep. 2018, 38, BSR20171383. [Google Scholar] [CrossRef]
- Rittavee, Y.; Artus, J.; Desterke, C.; Simanic, I.; de Souza, L.E.B.; Riccaldi, S.; Coignard, S.; Ijjeh, Y.; Hugues, P.; Bennaceur-Griscelli, A.; et al. miR-495-3p Sensitizes BCR-ABL1-Expressing Leukemic Cells to Tyrosine Kinase Inhibitors by Targeting Multidrug Resistance 1 Gene in T315I Mutated Cells. Exp. Hematol. 2023, 118, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Jeon, J.; Go, G.; Lee, J.H.; Lee, S.H. The Dual Role of Autophagy in Cancer Development and a Therapeutic Strategy for Cancer by Targeting Autophagy. Int. J. Mol. Sci. 2020, 22, 179. [Google Scholar] [CrossRef]
- Li, Y.-L.; Tang, J.-M.; Chen, X.-Y.; Luo, B.; Liang, G.-H.; Qu, Q.; Lu, Z.-Y. MicroRNA-153-3p Enhances the Sensitivity of Chronic Myeloid Leukemia Cells to Imatinib by Inhibiting B-Cell Lymphoma-2-Mediated Autophagy. Hum. Cell 2020, 33, 610–618. [Google Scholar] [CrossRef]
- Chen, W.; Li, Z.; Liu, H.; Jiang, S.; Wang, G.; Sun, L.; Li, J.; Wang, X.; Yu, S.; Huang, J.; et al. MicroRNA-30a Targets BECLIN-1 to Inactivate Autophagy and Sensitizes Gastrointestinal Stromal Tumor Cells to Imatinib. Cell Death Dis. 2020, 11, 198. [Google Scholar] [CrossRef]
- Zhen, Q.; Liu, J.; Gao, L.; Liu, J.; Wang, R.; Chu, W.; Zhang, Y.; Tan, G.; Zhao, X.; Lv, B. MicroRNA-200a Targets EGFR and c-Met to Inhibit Migration, Invasion, and Gefitinib Resistance in Non-Small Cell Lung Cancer. Cytogenet. Genome Res. 2015, 146, 1–8. [Google Scholar] [CrossRef]
- Zhang, Y.; Liang, S.; Xiao, B.; Hu, J.; Pang, Y.; Liu, Y.; Yang, J.; Ao, J.; Wei, L.; Luo, X. MiR-323a Regulates ErbB3/EGFR and Blocks Gefitinib Resistance Acquisition in Colorectal Cancer. Cell Death Dis. 2022, 13, 256. [Google Scholar] [CrossRef]
- Liu, L.; Shao, X.; Gao, W.; Zhang, Z.; Liu, P.; Wang, R.; Huang, P.; Yin, Y.; Shu, Y. MicroRNA-133b Inhibits the Growth of Non-Small-Cell Lung Cancer by Targeting the Epidermal Growth Factor Receptor. FEBS J. 2012, 279, 3800–3812. [Google Scholar] [CrossRef] [PubMed]
- Fatima, M.; Kakar, S.J.; Adnan, F.; Khan, K.; Mian, A.A.; Khan, D. AXL Receptor Tyrosine Kinase: A Possible Therapeutic Target in Acute Promyelocytic Leukemia. BMC Cancer 2021, 21, 713. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xia, H.; Zhuang, Z.; Miao, L.; Chen, X.; Cai, H. Axl-Altered microRNAs Regulate Tumorigenicity and Gefitinib Resistance in Lung Cancer. Cell Death Dis. 2014, 5, e1227. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Kim, S.H.; Yang, S.H.; Kim, M.S. miR-4487 Enhances Gefitinib-Mediated Ubiquitination and Autophagic Degradation of EGFR in Non-Small Cell Lung Cancer Cells by Targeting USP37. Cancer Res. Treat. 2022, 54, 445–457. [Google Scholar] [CrossRef]
- Gao, Y.; Fan, X.; Li, W.; Ping, W.; Deng, Y.; Fu, X. miR-138-5p Reverses Gefitinib Resistance in Non-Small Cell Lung Cancer Cells via Negatively Regulating G Protein-Coupled Receptor 124. Biochem. Biophys. Res. Commun. 2014, 446, 179–186. [Google Scholar] [CrossRef]
- Zhu, X.; Tao, X.; Lu, W.; Ding, Y.; Tang, Y. Blockade of Integrin Β3 Signals to Reverse the Stem-like Phenotype and Drug Resistance in Melanoma. Cancer Chemother. Pharmacol. 2019, 83, 615–624. [Google Scholar] [CrossRef]
- Gilgan, M.W.; Farquharson, T.E. Paper Chromatographic Separation of Alpha-Ecdysone, Ecdysterone, Inokosterone, Makisterone A and Ponasterone A. Steroids 1973, 22, 365–372. [Google Scholar] [CrossRef]
- Yue, J.; Lv, D.; Wang, C.; Li, L.; Zhao, Q.; Chen, H.; Xu, L. Epigenetic Silencing of miR-483-3p Promotes Acquired Gefitinib Resistance and EMT in EGFR-Mutant NSCLC by Targeting Integrin Β3. Oncogene 2018, 37, 4300–4312. [Google Scholar] [CrossRef]
- Wang, F.; Meng, F.; Wong, S.C.C.; Cho, W.C.S.; Yang, S.; Chan, L.W.C. Combination Therapy of Gefitinib and miR-30a-5p May Overcome Acquired Drug Resistance through Regulating the PI3K/AKT Pathway in Non-Small Cell Lung Cancer. Ther. Adv. Respir. Dis. 2020, 14, 1753466620915156. [Google Scholar] [CrossRef]
- Lu, M.; Liu, B.; Xiong, H.; Wu, F.; Hu, C.; Liu, P. Trans-3,5,4′-Trimethoxystilbene Reduced Gefitinib Resistance in NSCLCs via Suppressing MAPK/Akt/Bcl-2 Pathway by Upregulation of miR-345 and miR-498. J. Cell. Mol. Med. 2019, 23, 2431–2441. [Google Scholar] [CrossRef]
- Wang, Y.-S.; Wang, Y.-H.; Xia, H.-P.; Zhou, S.-W.; Schmid-Bindert, G.; Zhou, C.-C. MicroRNA-214 Regulates the Acquired Resistance to Gefitinib via the PTEN/AKT Pathway in EGFR-Mutant Cell Lines. Asian Pac. J. Cancer Prev. 2012, 13, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Qian, Z.; Xu, X.; Zhang, C.; Niu, Y.; Wang, Z.; Sun, J.; Zhang, X.; Yu, Y. Exosomes-Transmitted miR-7 Reverses Gefitinib Resistance by Targeting YAP in Non-Small-Cell Lung Cancer. Pharmacol. Res. 2021, 165, 105442. [Google Scholar] [CrossRef]
- Zhou, G.; Zhang, F.; Guo, Y.; Huang, J.; Xie, Y.; Yue, S.; Chen, M.; Jiang, H.; Li, M. miR-200c Enhances Sensitivity of Drug-Resistant Non-Small Cell Lung Cancer to Gefitinib by Suppression of PI3K/Akt Signaling Pathway and Inhibites Cell Migration via Targeting ZEB1. Biomed. Pharmacother. 2017, 85, 113–119. [Google Scholar] [CrossRef]
- Lin, C.-C.; Wu, C.-Y.; Tseng, J.T.; Hung, C.-H.; Wu, S.-Y.; Huang, Y.-T.; Chang, W.-Y.; Su, P.-L.; Su, W.-C. Extracellular Vesicle miR-200c Enhances Gefitinib Sensitivity in Heterogeneous EGFR-Mutant NSCLC. Biomedicines 2021, 9, 243. [Google Scholar] [CrossRef] [PubMed]
- Inoue, J.; Fujiwara, K.; Hamamoto, H.; Kobayashi, K.; Inazawa, J. Improving the Efficacy of EGFR Inhibitors by Topical Treatment of Cutaneous Squamous Cell Carcinoma with miR-634 Ointment. Mol. Ther. Oncolytics 2020, 19, 294–307. [Google Scholar] [CrossRef]
- Kalinowski, F.C.; Giles, K.M.; Candy, P.A.; Ali, A.; Ganda, C.; Epis, M.R.; Webster, R.J.; Leedman, P.J. Regulation of Epidermal Growth Factor Receptor Signaling and Erlotinib Sensitivity in Head and Neck Cancer Cells by miR-7. PLoS ONE 2012, 7, e47067. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Kelnar, K.; Bader, A.G. In-Depth Analysis Shows Synergy between Erlotinib and miR-34a. PLoS ONE 2014, 9, e89105. [Google Scholar] [CrossRef]
- Haque, I.; Kawsar, H.I.; Motes, H.; Sharma, M.; Banerjee, S.; Banerjee, S.K.; Godwin, A.K.; Huang, C.H. Downregulation of miR-506-3p Facilitates EGFR-TKI Resistance through Induction of Sonic Hedgehog Signaling in Non-Small-Cell Lung Cancer Cell Lines. Int. J. Mol. Sci. 2020, 21, 9307. [Google Scholar] [CrossRef]
- Fanini, F.; Bandini, E.; Plousiou, M.; Carloni, S.; Wise, P.; Neviani, P.; Murtadha, M.; Foca, F.; Fabbri, F.; Vannini, I.; et al. MicroRNA-16 Restores Sensitivity to Tyrosine Kinase Inhibitors and Outperforms MEK Inhibitors in KRAS-Mutated Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2021, 22, 13357. [Google Scholar] [CrossRef]
- Han, J.; Zhao, F.; Zhang, J.; Zhu, H.; Ma, H.; Li, X.; Peng, L.; Sun, J.; Chen, Z. miR-223 Reverses the Resistance of EGFR-TKIs through IGF1R/PI3K/Akt Signaling Pathway. Int. J. Oncol. 2016, 48, 1855–1867. [Google Scholar] [CrossRef]
- Zhao, F.-Y.; Han, J.; Chen, X.-W.; Wang, J.; Wang, X.-D.; Sun, J.-G.; Chen, Z.-T. miR-223 Enhances the Sensitivity of Non-Small Cell Lung Cancer Cells to Erlotinib by Targeting the Insulin-like Growth Factor-1 Receptor. Int. J. Mol. Med. 2016, 38, 183–191. [Google Scholar] [CrossRef]
- Amri, J.; Molaee, N.; Karami, H. Up-Regulation of MiRNA-125a-5p Inhibits Cell Proliferation and Increases EGFR-TKI Induced Apoptosis in Lung Cancer Cells. Asian Pac. J. Cancer Prev. 2019, 20, 3361–3367. [Google Scholar] [CrossRef]
- Ahmad, A.; Maitah, M.Y.; Ginnebaugh, K.R.; Li, Y.; Bao, B.; Gadgeel, S.M.; Sarkar, F.H. Inhibition of Hedgehog Signaling Sensitizes NSCLC Cells to Standard Therapies through Modulation of EMT-Regulating miRNAs. J. Hematol. Oncol. 2013, 6, 77. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Guerrero, A.; Kelnar, K.; Peltier, H.J.; Bader, A.G. Synergy between next Generation EGFR Tyrosine Kinase Inhibitors and miR-34a in the Inhibition of Non-Small Cell Lung Cancer. Lung Cancer 2017, 108, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-N.; Tsai, M.-F.; Wu, S.-G.; Chang, T.-H.; Tsai, T.-H.; Gow, C.-H.; Wang, H.-Y.; Shih, J.-Y. miR-146b-5p Enhances the Sensitivity of NSCLC to EGFR Tyrosine Kinase Inhibitors by Regulating the IRAK1/NF-κB Pathway. Mol. Ther. Nucleic Acids 2020, 22, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-F.; Shen, W.-Z.; Jin, X.; Ren, P.; Zhang, J. Let-7c Regulated Epithelial-Mesenchymal Transition Leads to Osimertinib Resistance in NSCLC Cells with EGFR T790M Mutations. Sci. Rep. 2020, 10, 11236. [Google Scholar] [CrossRef]
- Wu, S.-G.; Chang, T.-H.; Tsai, M.-F.; Liu, Y.-N.; Huang, Y.-L.; Hsu, C.-L.; Jheng, H.-N.; Shih, J.-Y. miR-204 Suppresses Cancer Stemness and Enhances Osimertinib Sensitivity in Non-Small Cell Lung Cancer by Targeting CD44. Mol. Ther. Nucleic Acids 2024, 35, 102091. [Google Scholar] [CrossRef]
- Romano, G.; Le, P.; Nigita, G.; Saviana, M.; Micalo, L.; Lovat, F.; Del Valle Morales, D.; Li, H.; Nana-Sinkam, P.; Acunzo, M. A-to-I Edited miR-411-5p Targets MET and Promotes TKI Response in NSCLC-Resistant Cells. Oncogene 2023, 42, 1597–1606. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Liu, Y.-N.; Wu, S.-G.; Hsu, C.-L.; Chang, T.-H.; Tsai, M.-F.; Lin, Y.-T.; Shih, J.-Y. MiR-200c-3p Suppression Is Associated with Development of Acquired Resistance to Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitors in EGFR Mutant Non-Small Cell Lung Cancer via a Mediating Epithelial-to-Mesenchymal Transition (EMT) Process. Cancer Biomark. 2020, 28, 351–363. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, H.; Hong, H.; Zhang, Z. MiR-374b Re-Sensitizes Hepatocellular Carcinoma Cells to Sorafenib Therapy by Antagonizing PKM2-Mediated Glycolysis Pathway. Am. J. Cancer Res. 2019, 9, 765–778. [Google Scholar]
- Shao, Y.-Y.; Chen, P.-S.; Lin, L.-I.; Lee, B.-S.; Ling, A.; Cheng, A.-L.; Hsu, C.; Ou, D.-L. Low miR-10b-3p Associated with Sorafenib Resistance in Hepatocellular Carcinoma. Br. J. Cancer 2022, 126, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Huang, J.; Ma, L.; Shan, J.; Shen, J.; Yang, Z.; Liu, L.; Luo, Y.; Yao, C.; Qian, C. MicroRNA-122 Confers Sorafenib Resistance to Hepatocellular Carcinoma Cells by Targeting IGF-1R to Regulate RAS/RAF/ERK Signaling Pathways. Cancer Lett. 2016, 371, 171–181. [Google Scholar] [CrossRef]
- Yang, Y.; Li, Z.; Yuan, H.; Ji, W.; Wang, K.; Lu, T.; Yu, Y.; Zeng, Q.; Li, F.; Xia, W.; et al. Reciprocal Regulatory Mechanism between miR-214-3p and FGFR1 in FGFR1-Amplified Lung Cancer. Oncogenesis 2019, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, C.; Ikezoe, T.; Yang, J.; Nobumoto, A.; Tsuda, M.; Yokoyama, A. Downregulation of miR-217 Correlates with Resistance of Ph(+) Leukemia Cells to ABL Tyrosine Kinase Inhibitors. Cancer Sci. 2014, 105, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Yang, J.; Wang, X.; Xu, K.; Ikezoe, T. miR-217 Sensitizes Chronic Myelogenous Leukemia Cells to Tyrosine Kinase Inhibitors by Targeting pro-Oncogenic Anterior Gradient 2. Exp. Hematol. 2018, 68, 80–88.e2. [Google Scholar] [CrossRef]
- Joo, L.J.S.; Weiss, J.; Gill, A.J.; Clifton-Bligh, R.; Brahmbhatt, H.; MacDiarmid, J.A.; Gild, M.L.; Robinson, B.G.; Zhao, J.T.; Sidhu, S.B. RET Kinase-Regulated MicroRNA-153-3p Improves Therapeutic Efficacy in Medullary Thyroid Carcinoma. Thyroid 2019, 29, 830–844. [Google Scholar] [CrossRef]
- Fu, Z.; Li, G.; Li, Z.; Wang, Y.; Zhao, Y.; Zheng, S.; Ye, H.; Luo, Y.; Zhao, X.; Wei, L.; et al. Endogenous miRNA Sponge LincRNA-ROR Promotes Proliferation, Invasion and Stem Cell-like Phenotype of Pancreatic Cancer Cells. Cell Death Discov. 2017, 3, 17004. [Google Scholar] [CrossRef]
- Rama, A.R.; Quiñonero, F.; Mesas, C.; Melguizo, C.; Prados, J. Synthetic Circular miR-21 Sponge as Tool for Lung Cancer Treatment. Int. J. Mol. Sci. 2022, 23, 2963. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, S.; Wang, H.; Cao, J.; Huang, X.; Chen, Z.; Xu, P.; Sun, G.; Xu, J.; Lv, J.; et al. Circular RNA circNRIP1 Acts as a microRNA-149-5p Sponge to Promote Gastric Cancer Progression via the AKT1/mTOR Pathway. Mol. Cancer 2019, 18, 20. [Google Scholar] [CrossRef]
- Barta, T.; Peskova, L.; Hampl, A. miRNAsong: A Web-Based Tool for Generation and Testing of miRNA Sponge Constructs in Silico. Sci. Rep. 2016, 6, 36625. [Google Scholar] [CrossRef]
- Lennox, K.A.; Behlke, M.A. Chemical Modification and Design of Anti-miRNA Oligonucleotides. Gene Ther. 2011, 18, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Ørom, U.A.; Kauppinen, S.; Lund, A.H. LNA-Modified Oligonucleotides Mediate Specific Inhibition of microRNA Function. Gene 2006, 372, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Pagoni, M.; Cava, C.; Sideris, D.C.; Avgeris, M.; Zoumpourlis, V.; Michalopoulos, I.; Drakoulis, N. miRNA-Based Technologies in Cancer Therapy. J. Pers. Med. 2023, 13, 1586. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, L.; Lun, A.T.L.; Baldoni, P.L.; Smyth, G.K. edgeR v4: Powerful Differential Analysis of Sequencing Data with Expanded Functionality and Improved Support for Small Counts and Larger Datasets. Nucleic Acids Res. 2025, 53, gkaf018. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Zhang, L.; Su, W.; Liu, S.; Huang, C.; Ghalandari, B.; Divsalar, A.; Ding, X. Recent Progresses in Electrochemical DNA Biosensors for MicroRNA Detection. Phenomics 2022, 2, 18–32. [Google Scholar] [CrossRef]
- Merk, D.J.; Paul, L.; Tsiami, F.; Hohenthanner, H.; Kouchesfahani, G.M.; Haeusser, L.A.; Walter, B.; Brown, A.; Persky, N.S.; Root, D.E.; et al. CRISPR-Cas9 Screens Reveal Common Essential miRNAs in Human Cancer Cell Lines. Genome Med. 2024, 16, 82. [Google Scholar] [CrossRef]
- Lu, Y.; Chan, Y.-T.; Wu, J.; Feng, Z.; Yuan, H.; Li, Q.; Xing, T.; Xu, L.; Zhang, C.; Tan, H.-Y.; et al. CRISPR/Cas9 Screens Unravel miR-3689a-3p Regulating Sorafenib Resistance in Hepatocellular Carcinoma via Suppressing CCS/SOD1-Dependent Mitochondrial Oxidative Stress. Drug Resist. Updates 2023, 71, 101015. [Google Scholar] [CrossRef]
- Soares, F.; Chen, B.; Lee, J.B.; Ahmed, M.; Ly, D.; Tin, E.; Kang, H.; Zeng, Y.; Akhtar, N.; Minden, M.D.; et al. CRISPR Screen Identifies Genes That Sensitize AML Cells to Double-Negative T-Cell Therapy. Blood 2021, 137, 2171–2181. [Google Scholar] [CrossRef]
- Sun, J.; Warden, A.R.; Ding, X. Recent Advances in Microfluidics for Drug Screening. Biomicrofluidics 2019, 13, 061503. [Google Scholar] [CrossRef]
- McGeary, S.E.; Lin, K.S.; Shi, C.Y.; Pham, T.M.; Bisaria, N.; Kelley, G.M.; Bartel, D.P. The Biochemical Basis of microRNA Targeting Efficacy. Science 2019, 366, eaav1741. [Google Scholar] [CrossRef]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microRNA.Org Resource: Targets and Expression. Nucleic Acids Res. 2008, 36, D149–D153. [Google Scholar] [CrossRef]
- Jung, D.; Kim, B.; Freishtat, R.J.; Giri, M.; Hoffman, E.; Seo, J. miRTarVis: An Interactive Visual Analysis Tool for microRNA-mRNA Expression Profile Data. BMC Proc. 2015, 9, S2. [Google Scholar] [CrossRef]
- Licursi, V.; Conte, F.; Fiscon, G.; Paci, P. MIENTURNET: An Interactive Web Tool for microRNA-Target Enrichment and Network-Based Analysis. BMC Bioinform. 2019, 20, 545. [Google Scholar] [CrossRef]
- Kowarsch, A.; Preusse, M.; Marr, C.; Theis, F.J. miTALOS: Analyzing the Tissue-Specific Regulation of Signaling Pathways by Human and Mouse microRNAs. RNA 2011, 17, 809–819. [Google Scholar] [CrossRef]
- Kern, F.; Aparicio-Puerta, E.; Li, Y.; Fehlmann, T.; Kehl, T.; Wagner, V.; Ray, K.; Ludwig, N.; Lenhof, H.-P.; Meese, E.; et al. miRTargetLink 2.0-Interactive miRNA Target Gene and Target Pathway Networks. Nucleic Acids Res. 2021, 49, W409–W416. [Google Scholar] [CrossRef] [PubMed]
- Sekar, V.; Mármol-Sánchez, E.; Kalogeropoulos, P.; Stanicek, L.; Sagredo, E.A.; Widmark, A.; Doukoumopoulos, E.; Bonath, F.; Biryukova, I.; Friedländer, M.R. Detection of Transcriptome-Wide microRNA-Target Interactions in Single Cells with agoTRIBE. Nat. Biotechnol. 2024, 42, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Wang, Y.; He, Y. TEC-miTarget: Enhancing microRNA Target Prediction Based on Deep Learning of Ribonucleic Acid Sequences. BMC Bioinform. 2024, 25, 159. [Google Scholar] [CrossRef]
- Yang, B.; McJunkin, K. CRISPR Screening Strategies for microRNA Target Identification. FEBS J. 2020, 287, 2914–2922. [Google Scholar] [CrossRef]
- Das, R.P.; Konkimalla, V.B.; Rath, S.N.; Hansa, J.; Jagdeb, M. Elucidation of the Molecular Interaction between miRNAs and the HOXA9 Gene, Involved in Acute Myeloid Leukemia, by the Assistance of Argonaute Protein through a Computational Approach. Genom. Inform. 2015, 13, 45–52. [Google Scholar] [CrossRef]
- Karginov, F.V.; Conaco, C.; Xuan, Z.; Schmidt, B.H.; Parker, J.S.; Mandel, G.; Hannon, G.J. A Biochemical Approach to Identifying microRNA Targets. Proc. Natl. Acad. Sci. USA 2007, 104, 19291–19296. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.P.; Lee, C.Y.; Tsai, M.H.; Chiu, Y.C.; Hsiao, C.K.; Lai, L.C.; Chuang, E.Y. miRSystem: An integrated system for characterizing enriched functions and pathways of microRNA targets. PLoS ONE 2012, 7, e42390. [Google Scholar] [CrossRef]
- Hamzeiy, H.; Suluyayla, R.; Brinkrolf, C.; Janowski, S.J.; Hofestaedt, R.; Allmer, J. Visualization and Analysis of MicroRNAs within KEGG Pathways Using VANESA. J. Integr. Bioinform. 2017, 14, 20160004. [Google Scholar] [CrossRef]
- Alvarez, M.J.; Shen, Y.; Giorgi, F.M.; Lachmann, A.; Ding, B.B.; Ye, B.H.; Califano, A. Functional Characterization of Somatic Mutations in Cancer Using Network-Based Inference of Protein Activity. Nat. Genet. 2016, 48, 838–847. [Google Scholar] [CrossRef]
- Martinez-Gutierrez, A.D.; Cantú de León, D.; Millan-Catalan, O.; Coronel-Hernandez, J.; Campos-Parra, A.D.; Porras-Reyes, F.; Exayana-Alderete, A.; López-Camarillo, C.; Jacobo-Herrera, N.J.; Ramos-Payan, R.; et al. Identification of miRNA Master Regulators in Breast Cancer. Cells 2020, 9, 1610. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.; Cui, Q.; Wang, J.; Zhou, Y. TransmiR v2.0: An Updated Transcription Factor-microRNA Regulation Database. Nucleic Acids Res. 2019, 47, D253–D258. [Google Scholar] [CrossRef]
- Devaraj, S.; Natarajan, J. miRNA-mRNA Network Detects Hub mRNAs and Cancer Specific miRNAs in Lung Cancer. In Silico Biol. 2011, 11, 281–295. [Google Scholar] [CrossRef]
- Joung, J.-G.; Hwang, K.-B.; Nam, J.-W.; Kim, S.-J.; Zhang, B.-T. Discovery of microRNA-mRNA Modules via Population-Based Probabilistic Learning. Bioinformatics 2007, 23, 1141–1147. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Subfamily | Members |
|---|---|
| 1. EGFR (Epidermal growth factor receptor) | EGFR, ERBB2/3/4 (receptor tyrosine-protein kinase erbB-2/3/4) |
| 2. Ins (Insulin) | INSR (insulin receptor), IGFR (insulin-like growth factor 1 receptor) |
| 3. PDGF (Platelet-derived growth factor) | PDGFRα, PDGFRβ (platelet-derived growth factor receptor alpha/beta), M-CSFR (macrophage colony-stimulating factor receptor), KIT (Mast/stem cell growth factor receptor kit), FLT3L (FMS-like tyrosine kinase 3 ligand) |
| 4. VEGF (Vascular endothelial growth factor) | VEGFR1/2/3 (vascular endothelial growth factor receptor 1/2/3) |
| 5. FGFs (Fibroblast growth factor) | FGFR1/2/3/4 (fibroblast growth factor receptor 1/2/3/4) |
| 6. CCK (colon carcinoma kinase) | CCK4 |
| 7. NGF (Beta-nerve growth factor) | TRKA (hig- affinity nerve growth factor receptor), TRKB (BDNF/NT-3 growth factors receptor), TRKC (NT-3 growth factor receptor) |
| 8. HGF (Hepatocyte growth factor) | RON (macrophage-stimulating protein receptor), MET (hepatocyte growth factor receptor) |
| 9. Eph (Ephrin receptors) | EPHA1–6 (ephrin type-A receptor 1–6), EPHB1–6 (ephrin type-B receptor 1–6) |
| 10. AXL (Tyrosine-protein kinase receptor UFO) | AXL, MER (tyrosine-protein kinase mer), TYRO3 (tyrosine-protein kinase receptor TYRO3) |
| 11. TIE (Tyrosine-protein kinase receptor Tie-1) | TIE, TEK (angiopoietin-1 receptor) |
| 12. RYK (Tyrosine-protein kinase RYK) | RYK |
| 13. DDRs (Discoidin Domain Receptor Tyrosine Kinase) | DDR1/2 |
| 14. RET (Proto-oncogene tyrosine-protein kinase receptor Ret) | RET |
| 15. ROS (Proto-oncogene tyrosine-protein kinase ROS) | ROS |
| 16. LTK (Leukocyte tyrosine kinase receptor) | ALK (ALK tyrosine kinase receptor), LTK |
| 17. ROR (Inactive tyrosine-protein kinase transmembrane receptor ROR) | ROR1/2 |
| 18. MuSK (Muscle, skeletal receptor tyrosine-protein kinase) | MuSK |
| 19. LMR (Lemur tyrosine kinase) | AATYK1/2/3 (serine/threonine-protein kinase LMTK1/2/3) |
| 20. Undetermined | RTK106 |
| Subfamily | Members |
|---|---|
| 1. ABL (Tyrosine-protein kinase ABL) | ABL1, ABL2 (ARG) |
| 2. ACK (Activated CDC42 kinase) | TNK1 (non-receptor tyrosine-protein kinase TNK1), TNK2 (ACK1) (tyrosine kinase non-receptor protein 2) |
| 3. CSK (Tyrosine-protein kinase CSK) | CSK, MATK (megakaryocyte-associated tyrosine-protein kinase) |
| 4. FAK (Focal adhesion kinase 1) | PTK2 (FAK), PTK2B (PYK2) (protein-tyrosine kinase 2-beta) |
| 5. FES (Tyrosine-protein kinase Fes/Fps) | FER (tyrosine-protein kinase Fer), FES |
| 6. FRK (Tyrosine-protein kinase FRK) | FRK, PTK6 (BRK) (protein-tyrosine kinase 6), SRMS (tyrosine-protein kinase Srms) |
| 7. JAK (Tyrosine-protein kinase JAK) | JAK1, JAK2, JAK3, TYK2 (non-receptor tyrosine-protein kinase TYK2) |
| 8. SRC-A (Proto-oncogene tyrosine-protein kinase Src) | FGR (tyrosine-protein kinase Fgr), FYN (tyrosine-protein kinase Fyn), SRC, YES1 (tyrosine-protein kinase Yes) |
| 8. SRC-B | BLK (tyrosine-protein kinase Blk), HCK (tyrosine-protein kinase HCK), LCK (tyrosine-protein kinase Lck), LYN (tyrosine-protein kinase Lyn) |
| 9. TEC (Tyrosine-protein kinase Tec) | BTK (tyrosine-protein kinase BTK), ITK (tyrosine-protein kinase ITK/TSK), TEC (tyrosine-protein kinase Tec), TXK (tyrosine-protein kinase TXK) |
| 10. SYK (Tyrosine-protein kinase SYK) | SYK, ZAP70 (tyrosine-protein kinase ZAP-70) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos-Parra, A.D.; Sánchez-Marín, D.; Acevedo-Sánchez, V. MicroRNAs as Sensitizers of Tyrosine Kinase Inhibitor Resistance in Cancer: Small Molecule Partnerships. Pharmaceuticals 2025, 18, 492. https://doi.org/10.3390/ph18040492
Campos-Parra AD, Sánchez-Marín D, Acevedo-Sánchez V. MicroRNAs as Sensitizers of Tyrosine Kinase Inhibitor Resistance in Cancer: Small Molecule Partnerships. Pharmaceuticals. 2025; 18(4):492. https://doi.org/10.3390/ph18040492
Chicago/Turabian StyleCampos-Parra, Alma D., David Sánchez-Marín, and Víctor Acevedo-Sánchez. 2025. "MicroRNAs as Sensitizers of Tyrosine Kinase Inhibitor Resistance in Cancer: Small Molecule Partnerships" Pharmaceuticals 18, no. 4: 492. https://doi.org/10.3390/ph18040492
APA StyleCampos-Parra, A. D., Sánchez-Marín, D., & Acevedo-Sánchez, V. (2025). MicroRNAs as Sensitizers of Tyrosine Kinase Inhibitor Resistance in Cancer: Small Molecule Partnerships. Pharmaceuticals, 18(4), 492. https://doi.org/10.3390/ph18040492