
Synthesis of Novel Chloro-Benzo [d]imidazole Regioisomers as Selective CB2 Receptor Agonists: Indirect Functional Evaluation and Molecular Insights

 , ,
, ,  and
and
Abstract
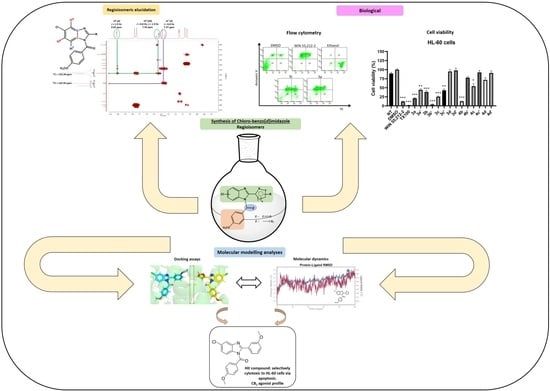
1. Introduction
2. Results and Discussion
2.1. Subsection Design Criteria to Develop CB2 Ligands
2.2. Chemistry
2.3. Biological Evaluations: Cell Viability (MTT) and Flow Cytometry (FC) Experiments
2.4. Docking Simulations
2.5. Molecular Dynamics (MD)
3. Materials and Methods
3.1. Procedure for the Synthesis of 6-Chloro-2-aryl-1H-benzo [d]imidazoles 2(a-c)
3.2. Procedure for the Synthesis of 3-(5-Chloro-1H-benzo [d]imidazol-2-yl)isoxazole (2d)
3.3. Procedure for the Synthesis of (5 or 6)-(Chloro)-2-(aryl)-1H-benzo [d]imidazol-1-yl)(4-methoxyphenyl)methanone 3(a-d) and 3(a’-d’)
3.4. Cell Cultures
3.5. Cell Viability: [MTT-(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)-formazan]
3.6. Flow Cytometry (FC) Experiments
3.7. Molecular Docking Experiments
3.8. Molecular Dynamic (MD) Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, H.-C.; Mackie, K. An Introduction to the Endogenous Cannabinoid System. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a Cannabinoid Receptor and Functional Expression of the Cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular Characterization of a Peripheral Receptor for Cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef]
- Van Sickle, M.D.; Duncan, M.; Kingsley, P.J.; Mouihate, A.; Urbani, P.; Mackie, K.; Stella, N.; Makriyannis, A.; Piomelli, D.; Davison, J.S.; et al. Identification and Functional Characterization of Brainstem Cannabinoid CB2 Receptors. Science 2005, 310, 329–332. [Google Scholar] [CrossRef]
- Palmer, S.L.; Thakur, G.A.; Makriyannis, A. Cannabinergic Ligands. Chem. Phys. Lipids 2002, 121, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Mackie, K. Cannabinoid Receptors as Therapeutic Targets. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 101–122. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.M.; Fowler, C.J. The Endocannabinoid System: Drug Targets, Lead Compounds, and Potential Therapeutic Applications. J. Med. Chem. 2005, 48, 5059–5087. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. The Pharmacology of Cannabinoid Receptors and Their Ligands: An Overview. Int. J. Obes. 2006, 30, 13–18. [Google Scholar] [CrossRef]
- Cassano, T.; Calcagnini, S.; Pace, L.; De Marco, F.; Romano, A.; Gaetani, S. Cannabinoid Receptor 2 Signaling in Neurodegenerative Disorders: From Pathogenesis to a Promising Therapeutic Target. Front. Neurosci. 2017, 11, 30. [Google Scholar] [CrossRef]
- Fonseca, B.; Teixeira, N.; Correia-da-Silva, G. Cannabinoids as Modulators of Cell Death: Clinical Applications and Future Directions. Rev. Physiol. Biochem. Pharmacol. 2017, 173, 63–88. [Google Scholar]
- Palazuelos, J.; Aguado, T.; Egia, A.; Mechoulam, R.; Guzmán, M.; Galve-Roperh, I. Non-Psychoactive CB2 Cannabinoid Agonists Stimulate Neural Progenitor Proliferation. FASEB J. 2006, 20, 2405–2407. [Google Scholar] [CrossRef] [PubMed]
- Malan, T.P.; Ibrahim, M.M.; Lai, J.; Vanderah, T.W.; Makriyannis, A.; Porreca, F. CB2 Cannabinoid Receptor Agonists: Pain Relief without Psychoactive Effects? Curr. Opin. Pharmacol. 2003, 3, 62–67. [Google Scholar] [CrossRef]
- Fernández-Ruiz, J.; Romero, J.; Velasco, G.; Tolón, R.M.; Ramos, J.A.; Guzmán, M. Cannabinoid CB2 Receptor: A New Target for Controlling Neural Cell Survival? Trends Pharmacol. Sci. 2007, 28, 39–45. [Google Scholar] [CrossRef]
- Ge, H.; Ji, B.; Fang, J.; Wang, J.; Li, J.; Wang, J. Discovery of Potent and Selective CB2 Agonists Utilizing a Function-Based Computational Screening Protocol. ACS Chem. Neurosci. 2023, 14, 3941–3958. [Google Scholar] [CrossRef]
- Gioé-Gallo, C.; Ortigueira, S.; Prieto-Díaz, R.; Contino, M.; Azuaje, J.; Perrone, M.G.; Riganti, C.; Alberga, D.; Mangiatordi, G.F.; Andújar-Arias, A.; et al. Conformational Restriction of Designer Drugs Reveals Subtype-Selective and Biased CB2 Agonists with Neuroprotective Effects. J. Med. Chem. 2025, 68, 17103–17129. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Sun, H.; Ji, J.; Chen, H.; Cheng, J.; Hu, Z.; Gong, X.; Liu, Q.; Peng, S.; Suo, J.; et al. Identification of Cannabigerol-Derived Dual CB2 Receptor Agonists and TRPM8 Antagonists with Anti-Inflammatory and Analgesic Activities. J. Med. Chem. 2025, 68, 13335–13357. [Google Scholar] [CrossRef] [PubMed]
- Aghazadeh Tabrizi, M.; Baraldi, P.G.; Borea, P.A.; Varani, K. Medicinal Chemistry, Pharmacology, and Potential Therapeutic Benefits of Cannabinoid CB2 Receptor Agonists. Chem. Rev. 2016, 116, 519–560. [Google Scholar] [CrossRef]
- Huffman, J.W.; Yu, S.; Showalter, V.; Abood, M.E.; Wiley, J.L.; Compton, D.R.; Martin, B.R.; Bramblett, R.D.; Reggio, P.H. Synthesis and Pharmacology of a Very Potent Cannabinoid Lacking a Phenolic Hydroxyl with High Affinity for the CB2 Receptor. J. Med. Chem. 1996, 39, 3875–3877. [Google Scholar] [CrossRef]
- Ross, R.A.; Brockie, H.C.; Stevenson, L.A.; Murphy, V.L.; Templeton, F.; Makriyannis, A.; Pertwee, R.G. Agonist-Inverse Agonist Characterization at CB1 and CB2 Cannabinoid Receptors of L759633, L759656, and AM630. Br. J. Pharmacol. 1999, 126, 665–672. [Google Scholar] [CrossRef]
- Smoum, R.; Baraghithy, S.; Chourasia, M.; Breuer, A.; Mussai, N.; Attar-Namdar, M.; Kogan, N.M.; Raphael, B.; Bolognini, D.; Cascio, M.G.; et al. CB2 Cannabinoid Receptor Agonist Enantiomers HU-433 and HU-308: An Inverse Relationship between Binding Affinity and Biological Potency. Proc. Natl. Acad. Sci. USA 2015, 112, 8774–8779. [Google Scholar] [CrossRef]
- Marini, P.; Cascio, M.-G.; King, A.; Pertwee, R.G.; Ross, R.A. Characterization of Cannabinoid Receptor Ligands in Tissues Natively Expressing Cannabinoid CB2 Receptors. Br. J. Pharmacol. 2013, 169, 887–899. [Google Scholar] [CrossRef]
- Aung, M.M.; Griffin, G.; Huffman, J.W.; Wu, M.; Keel, C.; Yang, B.; Showalter, V.M.; Abood, M.E.; Martin, B.R. Influence of the N-1 Alkyl Chain Length of Cannabimimetic Indoles upon CB(1) and CB(2) Receptor Binding. Drug Alcohol Depend. 2000, 60, 133–140. [Google Scholar] [CrossRef]
- Ibrahim, M.M.; Deng, H.; Zvonok, A.; Cockayne, D.A.; Kwan, J.; Mata, H.P.; Vanderah, T.W.; Lai, J.; Porreca, F.; Makriyannis, A.; et al. Activation of CB2 Cannabinoid Receptors by AM1241 Inhibits Experimental Neuropathic Pain: Pain Inhibition by Receptors Not Present in the CNS. Proc. Natl. Acad. Sci. USA 2003, 100, 10529–10533. [Google Scholar] [CrossRef]
- Huffman, J.W.; Marriott, K.-S.C. Recent Advances in the Development of Selective Ligands for the Cannabinoid CB2 Receptor. Curr. Top. Med. Chem. 2008, 8, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Romero-Parra, J.; Mella-Raipán, J.; Palmieri, V.; Allarà, M.; Torres, M.J.; Pessoa-Mahana, H.; Iturriaga-Vásquez, P.; Escobar, R.; Faúndez, M.; Di Marzo, V.; et al. Synthesis, Binding Assays, Cytotoxic Activity and Docking Studies of Benzimidazole and Benzothiophene Derivatives with Selective Affinity for the CB2 Cannabinoid Receptor. Eur. J. Med. Chem. 2016, 124, 17–35. [Google Scholar] [CrossRef]
- Tebbe, M.J.; Spitzer, W.A.; Victor, F.; Miller, S.C.; Lee, C.C.; Sattelberg, T.R.; McKinney, E.; Tang, J.C. Antirhino/Enteroviral Vinylacetylene Benzimidazoles: A Study of Their Activity and Oral Plasma Levels in Mice. J. Med. Chem. 1997, 40, 3937–3946. [Google Scholar] [CrossRef]
- Trivedi, R.; De, S.K.; Gibbs, R.A. A Convenient One-Pot Synthesis of 2-Substituted Benzimidazoles. J. Mol. Catal. A Chem. 2006, 245, 8–11. [Google Scholar] [CrossRef]
- Jaya Preethi, P.; Karthikeyan, E.; Lohita, M.; Goutham Teja, P.; Subhash, M.; Shaheena, P.; Prashanth, Y.; Sai Nandhu, K. Benzimidazole: An Important Scaffold in Drug Discovery. Asian J. Pharm. Tech. 2015, 5, 138–152. [Google Scholar] [CrossRef]
- Guo, Y.; Hou, X.; Fang, H. Recent Applications of Benzimidazole as a Privileged Scaffold in Drug Discovery. Mini Rev. Med. Chem. 2021, 21, 1367–1379. [Google Scholar] [CrossRef] [PubMed]
- Nanda, K.K.; Henze, D.A.; Della Penna, K.; Desai, R.; Leitl, M.; Lemaire, W.; White, R.B.; Yeh, S.; Brouillette, J.N.; Hartman, G.D.; et al. Benzimidazole CB2 Agonists: Design, Synthesis and SAR. Bioorg. Med. Chem. Lett. 2014, 24, 1218–1221. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.; Owen, D.R.; Harding, D.; Kon-I, K.; Lewis, M.L.; Mason, H.J.; Matsumizu, M.; Mukaiyama, T.; Rodriguez-Lens, M.; Shima, A.; et al. Optimisation of a Novel Series of Selective CNS Penetrant CB(2) Agonists. Bioorg. Med. Chem. Lett. 2011, 21, 4284–4287. [Google Scholar] [CrossRef] [PubMed]
- Pagé, D.; Balaux, E.; Boisvert, L.; Liu, Z.; Milburn, C.; Tremblay, M.; Wei, Z.; Woo, S.; Luo, X.; Cheng, Y.-X.; et al. Novel Benzimidazole Derivatives as Selective CB2 Agonists. Bioorg. Med. Chem. Lett. 2008, 18, 3695–3700. [Google Scholar] [CrossRef]
- Ryckmans, T.; Edwards, M.P.; Horne, V.A.; Correia, A.M.; Owen, D.R.; Thompson, L.R.; Tran, I.; Tutt, M.F.; Young, T. Rapid Assessment of a Novel Series of Selective CB(2) Agonists Using Parallel Synthesis Protocols: A Lipophilic Efficiency (LipE) Analysis. Bioorg. Med. Chem. Lett. 2009, 19, 4406–4409. [Google Scholar] [CrossRef] [PubMed]
- Gijsen, H.J.M.; De Cleyn, M.A.J.; Surkyn, M.; Van Lommen, G.R.E.; Verbist, B.M.P.; Nijsen, M.J.M.A.; Meert, T.; Wauwe, J.V.; Aerssens, J. 5-Sulfonyl-Benzimidazoles as Selective CB2 Agonists-Part 2. Bioorg. Med. Chem. Lett. 2012, 22, 547–552. [Google Scholar] [CrossRef]
- Tonelli, M.; Cichero, E.; Mahmoud, A.M.; Rabbito, A.; Tasso, B.; Fossa, P.; Ligresti, A. Exploring the Effectiveness of Novel Benzimidazoles as CB2 Ligands: Synthesis, Biological Evaluation, Molecular Docking Studies and ADMET Prediction. MedChemComm 2018, 9, 2045–2054. [Google Scholar] [CrossRef] [PubMed]
- Nimczick, M.; Pemp, D.; Darras, F.H.; Chen, X.; Heilmann, J.; Decker, M. Synthesis and Biological Evaluation of Bivalent Cannabinoid Receptor Ligands Based on hCB2R Selective Benzimidazoles Reveal Unexpected Intrinsic Properties. Bioorg. Med. Chem. 2014, 22, 3938–3946. [Google Scholar] [CrossRef]
- Mella-Raipán, J.A.; Lagos, C.F.; Recabarren-Gajardo, G.; Espinosa-Bustos, C.; Romero-Parra, J.; Pessoa-Mahana, H.; Iturriaga-Vásquez, P.; Pessoa-Mahana, C.D. Design, Synthesis, Binding and Docking-Based 3D-QSAR Studies of 2-Pyridylbenzimidazoles—A New Family of High Affinity CB1 Cannabinoid Ligands. Molecules 2013, 18, 3972–4001. [Google Scholar] [CrossRef]
- Romero-Parra, J.; Chung, H.; Tapia, R.A.; Faúndez, M.; Morales-Verdejo, C.; Lorca, M.; Lagos, C.F.; Di Marzo, V.; David Pessoa-Mahana, C.; Mella, J. Combined CoMFA and CoMSIA 3D-QSAR Study of Benzimidazole and Benzothiophene Derivatives with Selective Affinity for the CB2 Cannabinoid Receptor. Eur. J. Pharm. Sci. 2017, 101, 1–10. [Google Scholar] [CrossRef]
- Lorca, M.; Valdes, Y.; Chung, H.; Romero-Parra, J.; Pessoa-Mahana, C.D.; Mella, J. Three-Dimensional Quantitative Structure-Activity Relationships (3D-QSAR) on a Series of Piperazine-Carboxamides Fatty Acid Amide Hydrolase (FAAH) Inhibitors as a Useful Tool for the Design of New Cannabinoid Ligands. Int. J. Mol. Sci. 2019, 20, 2510. [Google Scholar] [CrossRef]
- Cho, A.Y.H.; Chung, H.; Romero-Parra, J.; Kumar, P.; Allarà, M.; Ligresti, A.; Gallardo-Garrido, C.; Pessoa-Mahana, H.; Faúndez, M.; Pessoa-Mahana, C.D. Motifs in Natural Products as Useful Scaffolds to Obtain Novel Benzo [d]Imidazole-Based Cannabinoid Type 2 (CB2) Receptor Agonists. Int. J. Mol. Sci. 2023, 24, 10918. [Google Scholar] [CrossRef]
- Balenga, N.A.; Martínez-Pinilla, E.; Kargl, J.; Schröder, R.; Peinhaupt, M.; Platzer, W.; Bálint, Z.; Zamarbide, M.; Dopeso-Reyes, I.G.; Ricobaraza, A.; et al. Heteromerization of GPR55 and Cannabinoid CB2 Receptors Modulates Signalling. Br. J. Pharmacol. 2014, 171, 5387–5406. [Google Scholar] [CrossRef] [PubMed]
- Ciaglia, E.; Torelli, G.; Pisanti, S.; Picardi, P.; D’Alessandro, A.; Laezza, C.; Malfitano, A.M.; Fiore, D.; Pagano Zottola, A.C.; Proto, M.C.; et al. Cannabinoid Receptor CB1 Regulates STAT3 Activity and Its Expression Dictates the Responsiveness to SR141716 Treatment in Human Glioma Patients’ Cells. Oncotarget 2015, 6, 15464–15481. [Google Scholar] [CrossRef]
- Romero, J.; Lastres-Becker, I.; de Miguel, R.; Berrendero, F.; Ramos, J.A.; Fernández-Ruiz, J. The Endogenous Cannabinoid System and the Basal Ganglia: Biochemical, Pharmacological, and Therapeutic Aspects. Pharmacol. Ther. 2002, 95, 137–152. [Google Scholar] [CrossRef]
- Guzmán, M. Cannabinoids: Potential Anticancer Agents. Nat. Rev. Cancer 2003, 3, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Cudaback, E.; Marrs, W.; Moeller, T.; Stella, N. The Expression Level of CB1 and CB2 Receptors Determines Their Efficacy at Inducing Apoptosis in Astrocytomas. PLoS ONE 2010, 5, e8702. [Google Scholar] [CrossRef]
- Downer, E.; Boland, B.; Fogarty, M.; Campbell, V. Δ9-Tetrahydrocannabinol Induces the Apoptotic Pathway in Cultured Cortical Neurones via Activation of the CB1 Receptor. Neuroreport 2001, 12, 3973–3978. [Google Scholar] [CrossRef]
- Maccarrone, M.; Finazzi-Agró, A. The Endocannabinoid System, Anandamide and the Regulation of Mammalian Cell Apoptosis. Cell Death Differ. 2003, 10, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Campbell, V.A. Tetrahydrocannabinol-Induced Apoptosis of Cultured Cortical Neurones Is Associated with Cytochrome c Release and Caspase-3 Activation. Neuropharmacology 2001, 40, 702–709. [Google Scholar] [CrossRef]
- Rieder, S.A.; Chauhan, A.; Singh, U.; Nagarkatti, M.; Nagarkatti, P. Cannabinoid-Induced Apoptosis in Immune Cells as a Pathway to Immunosuppression. Immunobiology 2010, 215, 598–605. [Google Scholar] [CrossRef]
- Compton, D.R.; Gold, L.H.; Ward, S.J.; Balster, R.L.; Martin, B.R. Aminoalkylindole Analogs: Cannabimimetic Activity of a Class of Compounds Structurally Distinct from Delta 9-Tetrahydrocannabinol. J. Pharmacol. Exp. Ther. 1992, 263, 1118–1126. [Google Scholar] [CrossRef]
- Xing, C.; Zhuang, Y.; Xu, T.-H.; Feng, Z.; Zhou, X.E.; Chen, M.; Wang, L.; Meng, X.; Xue, Y.; Wang, J.; et al. Cryo-EM Structure of the Human Cannabinoid Receptor CB2-Gi Signaling Complex. Cell 2020, 180, 645–654.e13. [Google Scholar] [CrossRef] [PubMed]
- Elderfield, R.C.; McCarthy, J.R. The Reaction of O-Phenylenediamines with Carbonyl Compounds. J. Am. Chem. Soc. 1951, 73, 975–984. [Google Scholar] [CrossRef]
- Ghosh, P.; Subba, R. MgCl2·6H2O Catalyzed Highly Efficient Synthesis of 2-Substituted-1H-Benzimidazoles. Tetrahedron Lett. 2015, 56, 2691–2694. [Google Scholar] [CrossRef]
- Hein, D.; Alheim, R.J.; Leavitt, J. The Use of Polyphosphoric Acid in the Synthesis of 2-Aryl-and 2-Alkyl-Substituted Benzimidazoles, Benzoxazoles and Benzothiazoles1. J. Am. Chem. Soc. 1957, 79, 427–429. [Google Scholar] [CrossRef]
- Pertwee, R.G. Pharmacological Actions of Cannabinoids. In Cannabinoids; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2005; Volume 168, pp. 1–51. [Google Scholar]
- An, D.; Peigneur, S.; Hendrickx, L.A.; Tytgat, J. Targeting Cannabinoid Receptors: Current Status and Prospects of Natural Products. Int. J. Mol. Sci. 2020, 21, 5064. [Google Scholar] [CrossRef]
- Darzynkiewicz, Z.; Bruno, S.; Del Bino, G.; Gorczyca, W.; Hotz, M.A.; Lassota, P.; Traganos, F. Features of Apoptotic Cells Measured by Flow Cytometry. Cytometry 1992, 13, 795–808. [Google Scholar] [CrossRef]
- Pozarowski, P.; Grabarek, J.; Darzynkiewicz, Z. Flow Cytometry of Apoptosis. Curr. Protoc. Cell Biol. 2004, 18, 18. [Google Scholar]
- Kepp, O.; Galluzzi, L.; Lipinski, M.; Yuan, J.; Kroemer, G. Cell Death Assays for Drug Discovery. Nat. Rev. Drug Discov. 2011, 10, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, M.; Turnbull, T.; Sebastian, S.; Kempson, I. The MTT Assay: Utility, Limitations, Pitfalls, and Interpretation in Bulk and Single-Cell Analysis. Int. J. Mol. Sci. 2021, 22, 12827. [Google Scholar] [CrossRef]
- Stasiulewicz, A.; Lesniak, A.; Bujalska-Zadrożny, M.; Pawiński, T.; Sulkowska, J. Identification of novel CB2 ligands through virtual screening and in vitro evaluation. J. Chem. Inf. Model. 2023, 63, 1012–1027. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, M.I. Synthesis and in Vitro α-Chymotrypsin Inhibitory Activity of 6-Chlorobenzimidazole Derivatives. Bioorg. Med. Chem. 2016, 24, 3387–3395. [Google Scholar]
- Jung, M.H.; Park, J.M.; Lee, I.-Y.C.; Ahn, M. Synthesis of 2-(1-Methyl-1,2,5,6-Tetrahydropyridin-3-Yl)Benzimidazoles. J. Heterocycl. Chem. 2003, 40, 37–44. [Google Scholar] [CrossRef]
- Pham, E.C.; Thi Le, T.V.; Truong, T.N. Design, Synthesis, Bio-Evaluation, and in Silico Studies of Some N-Substituted 6-(Chloro/Nitro)-1H-Benzimidazole Derivatives as Antimicrobial and Anticancer Agents. RSC Adv. 2022, 12, 21621–21646. [Google Scholar] [CrossRef] [PubMed]
- Mosorov, V. The Lambert-Beer Law in Time Domain Form and Its Application. Appl. Radiat. Isot. 2017, 128, 1–5. [Google Scholar] [CrossRef]
- Schrödinger Release 2018-2: Maestro; Version 11.8; Schrödinger, LLC: New York, NY, USA, 2018.
- Rose, P.W.; Prlić, A.; Bi, C.; Bluhm, W.F.; Christie, C.H.; Dutta, S.; Green, R.K.; Goodsell, D.S.; Westbrook, J.D.; Woo, J.; et al. The RCSB Protein Data Bank: Views of Structural Biology for Basic and Applied Research and Education. Nucleic Acids Res. 2015, 43, D345–D356. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W. PyMol: An Open-Source Molecular Graphics Tool. CCP4 Newsletter on Protein Crystallography. 2002. Available online: https://mozart.gmb.bio.br/uploads/6/4/3/6/64362047/pymol.pdf#page=44 (accessed on 19 October 2025).














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cell Viability Percentage (%) HEK293 | Cell Viability Percentage (%) U-87MG | Cell Viability Percentage (%) HL-60 | Selectivity Index% Cytotoxicity HL-60/% Cytotoxicity U-87MG |
|---|---|---|---|---|
| 3a | 60.87 ± 12 | 72.37 ± 20 | 21.14 ± 6 | 0.29 |
| 3a’ | 5.38 ± 13 | 6.71 ± 3 | 44.29 ± 11 | 6.6 |
| 3b | 72.71 ± 24 | 84.78 ± 26 | 40.94 ± 16 | 0.48 |
| 3b’ | 74.71 ± 19 | 78.94 ± 13 | 3.88 ± 6 | 0.05 |
| 3c | 84.44 ± 15 | 91.29 ± 28 | 25.42 ± 7 | 0.28 |
| 3c’ | 63.41 ± 30 | 73.91 ± 11 | 43.04 ± 14 | 0.58 |
| 3d | 75.39 ± 30 | 68.23 ± 14 | 94.88 ± 20 | 1.39 |
| 3d’ | 71.71 ± 23 | 95.21 ± 23 | 95.58 ± 23 | 1.00 |
| 4b | 103.12 ± 21 | 101.6 ± 27 | 12.34 ± 3 | 0.12 |
| 4b’ | 77.7 ± 34 | 93.93 ± 25 | 77.9 ± 11 | 0.83 |
| 4c | 115.07 ± 22 | 92.94 ± 16 | 54.07 ± 28 | 0.58 |
| 4c’ | 95.5 ± 32 | 105.92 ± 28 | 92.43 ± 21 | 0.87 |
| 4d | 105.7 ± 45 | 118.86 ± 33 | 68.8 ± 27 | 0.58 |
| 4d’ | 77.66 ± 19 | 92.87 ± 12 | 87.98 ± 23 | 0.95 |
| WIN-55,212-2 | 89.6 ± 23 | 36.21 ± 12 | 12.21 ± 4 | 0.34 |
| Compound | Binding Energy (kcal/mol) |
|---|---|
| 3a | −10.640 |
| 3c | −10.059 |
| 3b’ | −9.632 |
| 4b | −9.822 |
| AM630 | −10.681 |
| WIN-55,212-2 | −11.081 |
| Compound 3a | Compound 3c | ||
|---|---|---|---|
| Time (ns) | Energy (kcal/mol) | Time (ns) | Energy (kcal/mol) |
| 220 | −86.23 | 180 | −73.89 |
| 240 | −78.15 | 200 | −75.46 |
| 260 | −77.30 | 220 | −78.51 |
| 280 | −81.65 | 240 | −74.00 |
| 300 | −78.49 | 260 | −85.65 |
| 320 | −84.81 | 280 | −74.22 |
| 340 | −78.02 | 300 | −74.70 |
| 360 | −87.13 | 320 | −74.32 |
| 380 | −79.08 | 340 | −70.93 |
| Average | −81.21 | Average | −75.74 |
| Std. Dev. | 3.88 | Std. Dev. | 4.20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salazar, V.Z.; Ravanal, R.B.; Soto-Flores, J.; Sabadini, G.; González, J.V.; Mella, J.; Romero-Parra, J. Synthesis of Novel Chloro-Benzo [d]imidazole Regioisomers as Selective CB2 Receptor Agonists: Indirect Functional Evaluation and Molecular Insights. Pharmaceuticals 2025, 18, 1599. https://doi.org/10.3390/ph18111599
Salazar VZ, Ravanal RB, Soto-Flores J, Sabadini G, González JV, Mella J, Romero-Parra J. Synthesis of Novel Chloro-Benzo [d]imidazole Regioisomers as Selective CB2 Receptor Agonists: Indirect Functional Evaluation and Molecular Insights. Pharmaceuticals. 2025; 18(11):1599. https://doi.org/10.3390/ph18111599
Chicago/Turabian StyleSalazar, Valeria Zuñiga, Renato Burgos Ravanal, Jonathan Soto-Flores, Gianfranco Sabadini, José Vicente González, Jaime Mella, and Javier Romero-Parra. 2025. "Synthesis of Novel Chloro-Benzo [d]imidazole Regioisomers as Selective CB2 Receptor Agonists: Indirect Functional Evaluation and Molecular Insights" Pharmaceuticals 18, no. 11: 1599. https://doi.org/10.3390/ph18111599
APA StyleSalazar, V. Z., Ravanal, R. B., Soto-Flores, J., Sabadini, G., González, J. V., Mella, J., & Romero-Parra, J. (2025). Synthesis of Novel Chloro-Benzo [d]imidazole Regioisomers as Selective CB2 Receptor Agonists: Indirect Functional Evaluation and Molecular Insights. Pharmaceuticals, 18(11), 1599. https://doi.org/10.3390/ph18111599