
Antibacterial Prodrugs to Overcome Bacterial Antimicrobial Resistance




Abstract
1. Introduction
1.1. Bacteria and Bacterial AMR
1.2. Antibacterial Prodrugs
2. Advancements in Antibacterial Prodrug Applications
2.1. Antituberculosis Prodrugs
2.2. Prodrugs against Gram-Negative Bacteria
2.3. Prodrugs against Gram-Positive Bacteria
2.4. Prodrugs against Gram-Negative and Gram-Positive Bacteria
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Paneri, M.; Sevta, P. Overview of Antimicrobial Resistance: An Emerging Silent Pandemic. Glob. J. Med. Pharm. Biomed. Update 2023, 18, 11–13. [Google Scholar] [CrossRef]
- Chiş, A.A.; Rus, L.L.; Morgovan, C.; Arseniu, A.M.; Frum, A.; Vonica-Ţincu, A.L.; Gligor, F.G.; Mureşan, M.L.; Dobrea, C.M. Microbial Resistance to Antibiotics and Effective Antibiotherapy. Biomedicines 2022, 10, 1121. [Google Scholar] [CrossRef]
- Tang, K.W.K.; Millar, B.C.; Moore, J.E. Antimicrobial resistance (AMR). Br. J. Biomed. Sci. 2023, 80, 11387. [Google Scholar] [CrossRef]
- Lemke, M.; DeSalle, R. The Next Generation of Microbial Ecology and Its Importance in Environmental Sustainability. Microb. Ecol. 2023, 83, 781–795. [Google Scholar] [CrossRef]
- Jubeh, B.; Breijyeh, Z.; Karaman, R. Resistance of Gram-Positive Bacteria to Current Antibacterial Agents and Overcoming Approaches. Molecules 2020, 25, 2888. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Jubeh, B.; Karaman, R. Resistance of Gram-Negative Bacteria to Current Antibacterial Agents and Approaches to Resolve It. Molecules 2020, 25, 1340. [Google Scholar] [CrossRef]
- World Health Organization. 2021 Antibacterial Agents in Clinical and Preclinical Development: An Overview and Analysis; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- Breijyeh, Z.; Karaman, R. Design and Synthesis of Novel Antimicrobial Agents. Antibiotics 2023, 12, 628. [Google Scholar] [CrossRef]
- Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Memesh, R.; Yasir, M.; Ledder, R.G.; Zowawi, H.; McBain, A.J.; Azhar, E.I. An update on the prevalence of colistin and carbapenem-resistant Gram-negative bacteria in aquaculture: An emerging threat to public health. J. Appl. Microbiol. 2024, 135, lxad288. [Google Scholar] [CrossRef]
- Subbaiah, M.A.M.; Rautio, J.; Meanwell, N.M. Prodrugs as empowering tools in drug discovery and development: Recent strategic applications of drug delivery solutions to mitigate challenges associated with lead compounds and drug candidates. Chem. Soc. Rev. 2024, 53, 2099–2210. [Google Scholar] [CrossRef]
- Rautio, J.; Meanwell, N.A.; Di, L.; Hageman, M.J. The expanding role of prodrugs in contemporary drug design and development. Nat. Rev. Drug Discov. 2018, 17, 559–589. [Google Scholar] [CrossRef]
- Jubeh, B.; Breijyeh, Z.; Karaman, R. Antibacterial Prodrugs to Overcome Bacterial Resistance. Molecules 2020, 25, 1543. [Google Scholar] [CrossRef]
- Maria, C.; de Matos, A.M.; Rauter, A.P. Recent antibacterial carbohydrate-based prodrugs, drugs and delivery systems to overcome antimicrobial resistance. Curr. Opin. Chem. Biol. 2024, 78, 102419. [Google Scholar] [CrossRef]
- Gordon, S.V.; Parish, T. Microbe Profile: Mycobacterium tuberculosis: Humanity’s deadly microbial foe. Microbiol. 2018, 164, 437–439. [Google Scholar] [CrossRef]
- Chetty, S.; Armstrong, T.; Kharkwal, S.S.; Drewe, W.C.; Matteis, C.I.D.; Evangelopoulos, D.; Bhakta, S.; Thomas, N.R. New InhA Inhibitors Based on Expanded Triclosan and Di-Triclosan Analogues to Develop a New Treatment for Tuberculosis. Pharmaceuticals 2021, 14, 361. [Google Scholar] [CrossRef]
- Arun, K.B.; Madhavan, A.; Abraham, B.; Balaji, M.; Sivakumar, K.C.; Nisha, P.; Kumar, R. Acetylation of Isoniazid Is a Novel Mechanism of Isoniazid Resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2021, 65, 10–1128. [Google Scholar] [CrossRef]
- Safari, J.B.; Mona, L.B.; Sekaleli, B.T.; Avudi, B.K.-N.; Isamura, B.K.; Mukubwa, G.K.; Salami, S.A.; Mbinze, J.K.; Lobb, K.A.; Krause, R.W.M.; et al. Inclusion complexation and liposomal encapsulation of an isoniazid hydrazone derivative in cyclodextrin for pH-dependent controlled release. J. Drug Deliv. Sci. Technol. 2023, 81, 104302. [Google Scholar] [CrossRef]
- Flipo, M.; Frita, R.; Bourotte, M.; Martínez-Martínez, M.S.; Boesche, M.; Boyle, G.W.; Derimanov, G.; Drewes, G.; Gamallo, P.; Ghidelli-Disse, S.; et al. The small-molecule SMARt751 reverses Mycobacterium tuberculosis resistance to ethionamide in acute and chronic mouse models of tuberculosis. Sci. Transl. Med. 2022, 14, eaaz6280. [Google Scholar] [CrossRef]
- Talley, A.K.; Thurston, A.; Moore, G.; Gupta, V.K.; Satterfield, M.; Manyak, E.; Stokes, S.; Dane, A.; Melnick, D. First-in-Human Evaluation of the Safety, Tolerability, and Pharmacokinetics of SPR720, a Novel Oral Bacterial DNA Gyrase (GyrB) Inhibitor for Mycobacterial Infections. Antimicrob. Agents Chemother. 2021, 65, e0120821. [Google Scholar] [CrossRef]
- Cioetto- Mazzabò, L.; Boldrin, F.; Beauvineau, C.; Speth, M.; Marina, A.; Namouchi, A.; Segafreddo, G.; Cimino, M.; Favre-Rochex, S.; Balasingham, S.; et al. SigH stress response mediates killing of Mycobacterium tuberculosis by activating nitronaphthofuran prodrugs via induction of Mrx2 expression. Nucleic Acids Res. 2023, 51, 144–165. [Google Scholar] [CrossRef]
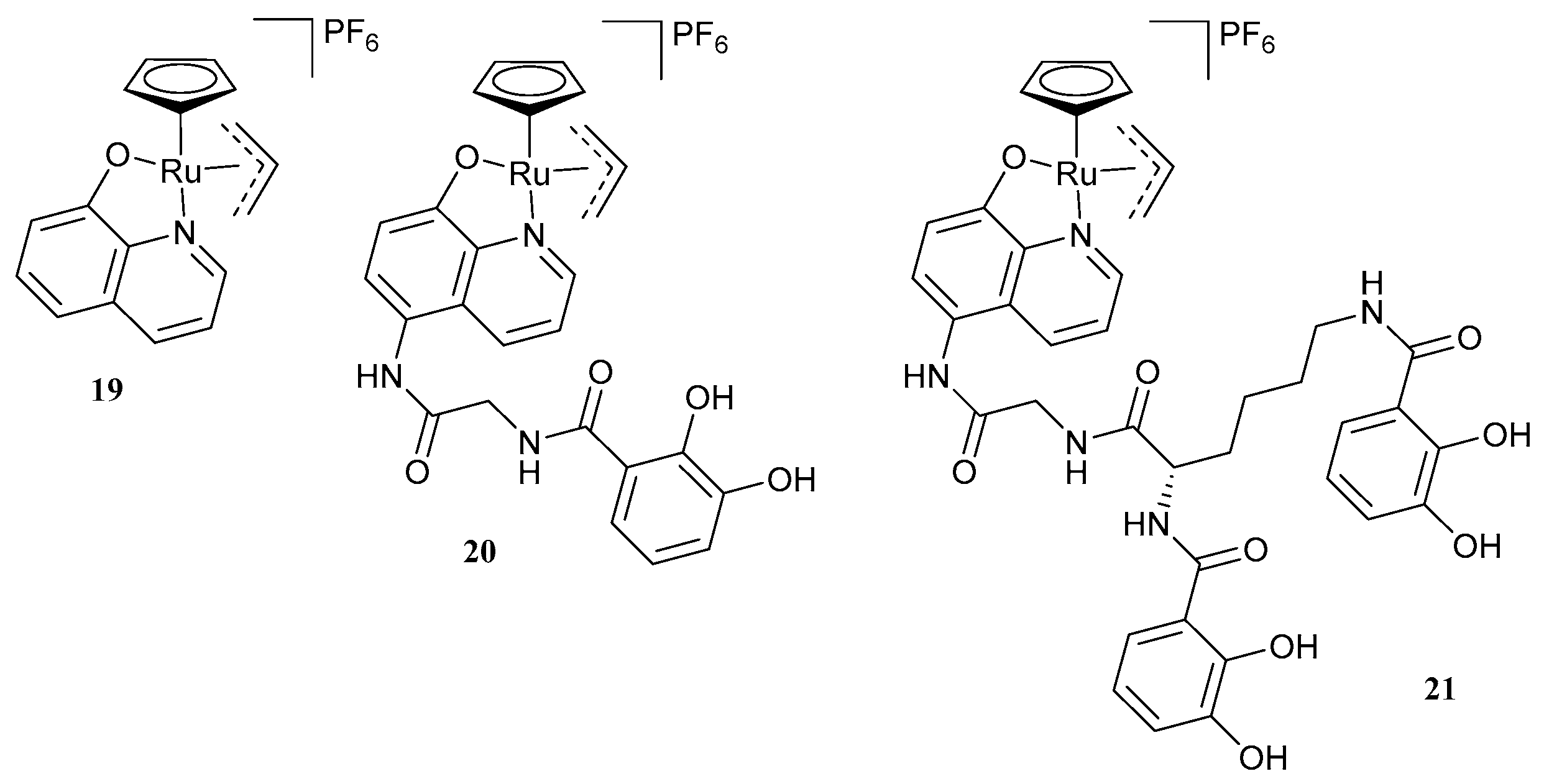
- Meiers, J.; Rox, K.; Titz, A. Lectin-Targeted Prodrugs Activated by Pseudomonas aeruginosa for Self-Destructive Antibiotic Release. J. Med. Chem. 2022, 65, 13988–14014. [Google Scholar] [CrossRef]
- Southwell, J.W.; Herman, R.; Raines, D.J.; Clarke, J.E.; Böswald, I.; Dreher, T.; Gutenthaler, S.M.; Schubert, N.; Seefeldt, J.; Metzler-Nolte, N.; et al. Siderophore-Linked Ruthenium Catalysts for Targeted Allyl Ester Prodrug Activation within Bacterial Cells. Chem. Eur. J. 2023, 29, e202202536. [Google Scholar] [CrossRef]
- Sun, Z.; Su, L.; Cotroneo, N.; Critchley, I.; Pucci, M.; Jain, A.; Palzkill, T. Evaluation of Tebipenem Hydrolysis by β-Lactamases Prevalent in Complicated Urinary Tract Infections. Antimicrob. Agents Chemother. 2022, 66, e0239621. [Google Scholar] [CrossRef]
- Eckburg, P.B.; Muir, L.; Critchley, I.A.; Walpole, S.; Kwak, H.; Phelan, A.-M.; Moore, G.; Jain, A.; Keutzer, T.; Dane, A.; et al. Oral Tebipenem Pivoxil Hydrobromide in Complicated Urinary Tract Infection. N. Eng. J. Med. 2022, 386, 1327–1338. [Google Scholar] [CrossRef]
- Ahmad, M.N.; Dasgupta, C.N.; Chopra, S. Tebipenem pivoxil hydrobromide. Drugs Future 2022, 47, 311–323. [Google Scholar] [CrossRef]
- Karlowsky, J.A.; Hackel, M.A.; Stone, G.G.; Sahm, D.F. In vitro Activity of Ceftibuten-Avibactam against β-Lactamase-Positive Enterobacterales from the ATLAS Global Surveillance Program. Antimicrob. Agents Chemother. 2023, 67, e0134622. [Google Scholar] [CrossRef]
- Álvaro, E.F.; Vinh, P.V.; Cozar, C.; Willé, D.R.; Urones, B.; Cortés, A.; Price, A.; Hoang, N.T.D.; Thanh, T.H.; McCloskey, M.; et al. The repurposing of Tebipenem pivoxil as alternative therapy for severe gastrointestinal infections caused by extensively drug-resistant Shigella spp. Elife 2022, 15, e69798. [Google Scholar] [CrossRef]
- Huband, M.D.; Fedler, K.A.; Sader, H.S.; Stone, G.G.; Castanheira, M. Determination of MIC Quality Control Ranges for Ceftibuten-Avibactam (Fixed 4 μg/mL), a Novel β-Lactam/β-Lactamase Inhibitor Combination. J. Clin. Microbiol. 2021, 61, e01657-22. [Google Scholar] [CrossRef]
- Sader, H.S.; Carvalhaes, C.G.; Huband, M.D.; Mendes, R.E.; Castanheira, M. Antimicrobial activity of ceftibuten-avibactam against a global collection of Enterobacterales from patients with urinary tract infections (2021). Eur. J. Clin. Microbiol. 2023, 42, 453–459. [Google Scholar] [CrossRef]
- Antraygues, K.; Maingot, M.; Schellhorn, B.; Trebosc, V.; Gitzinger, M.; Deprez, B.; Defert, O.; Dale, G.E.; Bourotte, M.; Lociuro, S.; et al. Design and synthesis of water-soluble prodrugs of rifabutin for intraveneous administration. Eur. J. Med. Chem. 2022, 238, 114515. [Google Scholar] [CrossRef]
- Li, X.; Li, W.; Li, K.; Chen, X.; Wang, C.; Qiao, M.; Hong, W. Albumin-coated pH-responsive dimeric prodrug-based nano-assemblies with high biofilm eradication capacity. Biomater. Sci. 2023, 11, 1031–1041. [Google Scholar] [CrossRef]
- Landa, G.; Alejo, T.; Sauzet, T.; Laroche, J.; Sebastian, V.; Tewes, F.; Arruebo, M. Colistin-loaded aerosolizable particles for the treatment of bacterial respiratory infections. Int. J. Pharm. 2023, 635, 122732. [Google Scholar] [CrossRef]
- Zhao, H.; Zhong, L.-I.; Yang, C.; Tang, N.; He, Y.; He, W.; Zhao, Z.; Wu, C.; Yuan, P.; Yang, Y.Y.; et al. Antibiotic–Polymer Self-Assembled Nanocomplex to Reverse Phenotypic Resistance of Bacteria toward Last-Resort Antibiotic Colistin. ACS Nano 2023, 17, 15411–15423. [Google Scholar] [CrossRef]
- Guo, C.; Nolan, E.M. Heavy-Metal Trojan Horse: Enterobactin-Directed Delivery of Platinum (IV) Prodrugs to Escherichia coli. J. Am. Chem. Soc. 2022, 144, 12756–12768. [Google Scholar] [CrossRef]
- Rosales-Hurtado, M.; Sannio, F.; Lari, L.; Verdirosa, F.; Feller, G.; Carretero, E.; Vo-Hoang, Y.; Licznar-Fajardo, P.; Docquier, J.-D.; Gavara, L. Zidovudine-β-Lactam Pronucleoside Strategy for Selective Delivery into Gram-Negative Bacteria Triggered by β-Lactamases. ACS Infect. Dis. 2023, 9, 1546–1557. [Google Scholar] [CrossRef]
- Kaul, M.; Ferrer-González, E.; Mark, L.; Parhi, A.K.; LaVoie, E.J.; Pilch, D.S. Combination with a FtsZ inhibitor potentiates the in vivo efficacy of oxacillin against methicillin-resistant Staphylococcus aureus. Med. Chem. Res. 2022, 31, 1705–1715. [Google Scholar] [CrossRef]
- Xiao, T.; Liu, K.; Gao, Q.; Chen, M.; Kim, Y.S.; Jin, S.; Ding, Y.; Huigens, R.W., III. Design, Synthesis, and Evaluation of Carbonate-Linked Halogenated Phenazine-Quinone Prodrugs with Improved Water-Solubility and Potent Antibacterial Profiles. ACS Infect. Dis. 2023, 9, 899–915. [Google Scholar] [CrossRef]
- Liu, K.; Xiao, T.; Yang, H.; Chen, M.; Gao, Q.; Brummel, B.R.; Ding, Y.; Huigens III, R.W. Design, synthesis and evaluation of halogenated phenazine antibacterial prodrugs targeting nitroreductase enzymes for activation. RSC Med. Chem. 2023, 14, 1472–1481. [Google Scholar] [CrossRef]
- Artim, C.M.; Kunala, M.; O’Leary, M.K.; Alabi, C.A. PEGylated Oligothioetheramide Prodrugs Activated by Host Serum Proteases. Chembiochem 2021, 22, 2697–2702. [Google Scholar] [CrossRef]
- Li, D.; Tang, G.; Yao, H.; Zhu, Y.; Shi, C.; Fu, Q.; Yang, F.; Wang, X. Formulation of pH-responsive PEGylated nanoparticles with high drug loading capacity and programmable drug release for enhanced antibacterial activity. Bioact. Mater. 2022, 16, 47–56. [Google Scholar] [CrossRef]
- Dai, X.; Liu, X.; Yang, L.; Yuan, S.; Xu, Q.; Li, Y.; Gao, F. pH-Responsive non-antibiotic polymer prodrugs eradicate intracellular infection by killing bacteria and regulating immune response. Colloids Surf. B 2022, 220, 112889. [Google Scholar] [CrossRef]
- Yakub, G.; Manolova, N.E.; Rashkov, I.B.; Markova, N.; Toshkova, R.; Georgieva, A.; Mincheva, R.; Toncheva, A.; Raquez, J.-M.; Dubois, P. Pegylated Curcumin Derivative: Water-Soluble Conjugates with Antitumor and Antibacterial Activity. ACS Omega 2022, 7, 36403–36414. [Google Scholar] [CrossRef]
- AbouAitah, K.; Piotrowska, U.; Wojnarowics, J.; Swiderka-Sroda, A.; El-Desoky, A.H.H.; Lojkowski, W. Enhanced Activity and Sustained Release of Protocatechuic Acid, a Natural Antibacterial Agent, from Hybrid Nanoformulations with Zinc Oxide Nanoparticles. Int. J. Mol. Sci. 2022, 22, 5287. [Google Scholar] [CrossRef]
- Alshememry, A.; Alkholief, M.; Kalam, M.A.; Raish, M.; Ali, R.; Alhudaithi, S.S.; Iqbal, M.; Alshamsan, A. Perspectives of Positively Charged Nanocrystals of Tedizolid Phosphate as a Topical Ocular Application in Rabbits. Molecules 2022, 27, 4619. [Google Scholar] [CrossRef]
- McLeod, J.R.; Harvey, P.A.; Detweiler, C.S. An Oral Fluorouracil Prodrug, Capecitabine, Mitigates a Gram-Positive Systemic Infection in Mice. Microbiol. Spectr. 2021, 9, e0027521. [Google Scholar] [CrossRef]
- Negrya, S.D.; Jasko, M.V.; Makarov, D.A.; Karpenko, I.L.; Solyev, P.N.; Chekhov, V.O.; Efremenkova, O.V.; Vasilieva, B.F.; Efimenko, T.A.; Kochetkov, S.N.; et al. Oligoglycol carbonate prodrugs of 5-modified 2′-deoxyuridines: Synthesis and antibacterial activity. Mendeleev Commun. 2022, 32, 433–435. [Google Scholar] [CrossRef]
- Li, Z.; Yang, Y.-J.; Qin, Z.; Li, S.-H.; Bai, L.-X.; Li, J.-Y.; Liu, X.-W. Florfenicol-Polyarginine Conjugates Exhibit Promising Antibacterial Activity Against Resistant Strains. Front. Chem. 2022, 10, 921091. [Google Scholar] [CrossRef]
- Weng, Y.; Chen, H.; Chen, X.; Yang, H.; Chen, C.-H.; Tan, H. Adenosine triphosphate-activated prodrug system for on-demand bacterial inactivation and wound disinfection. Nat. Commun. 2022, 13, 4712. [Google Scholar] [CrossRef]
- Wang, S.; Fang, Y.; Zhang, Z.; Jin, Q.; Ji, J. Bacterial infection microenvironment sensitive prodrug micelles with enhanced photodynamic activities for infection control. Colloids Interface Sci. Commun. 2021, 40, 100354. [Google Scholar] [CrossRef]
- Cojocaru, E.; Ghitman, J.; Pircalabioru, G.G.; Zaharia, A.; Iovu, H.; Sarbu, A. Electrospun/3D-Printed Bicomponent Scaffold Co-Loaded with a Prodrug and a Drug with Antibacterial and Immunomodulatory Properties. Polymers 2023, 15, 2854. [Google Scholar] [CrossRef]
- Lu, Y.; Shan, P.; Lu, W.; Yin, X.; Liu, H.; Lian, X.; Jin, J.; Qi, Y.; Li, Z.; Li, Z. ROS-Responsive and Self-Amplifying polymeric prodrug for accelerating infected wound healing. J. Chem. Eng. 2023, 463, 142311. [Google Scholar] [CrossRef]
- Grassiri, B.; Mezzetta, A.; Maisetta, G.; Migone, C.; Fabiano, A.; Esin, S.; Guazzelli, L.; Zambito, Y.; Batoni, G.; Piras, A.M. Betaine- and l-Carnitine-Based Ionic Liquids as Solubilising and Stabilising Agents for the Formulation of Antimicrobial Eye Drops Containing Diacerein. Int. J. Mol. Sci. 2023, 24, 2714. [Google Scholar] [CrossRef]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. Nr. | Structure | Antibacterial Activity | Ref. | ||
|---|---|---|---|---|---|
| Tuberculosis | GNB | GPB | |||
| 1. Isoniazid |  | X | - | - | [16] |
| 2. INH-HB | 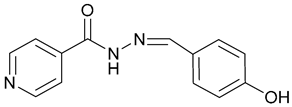 | X | - | - | [18] |
| 4. Ethionamide |  | X | - | - | [19] |
| 5.SMARt751 | 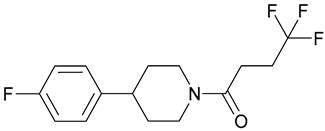 | ||||
| 6. SPR720 | 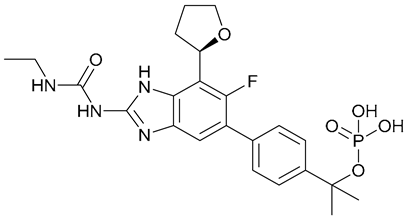 | X | - | - | [20] |
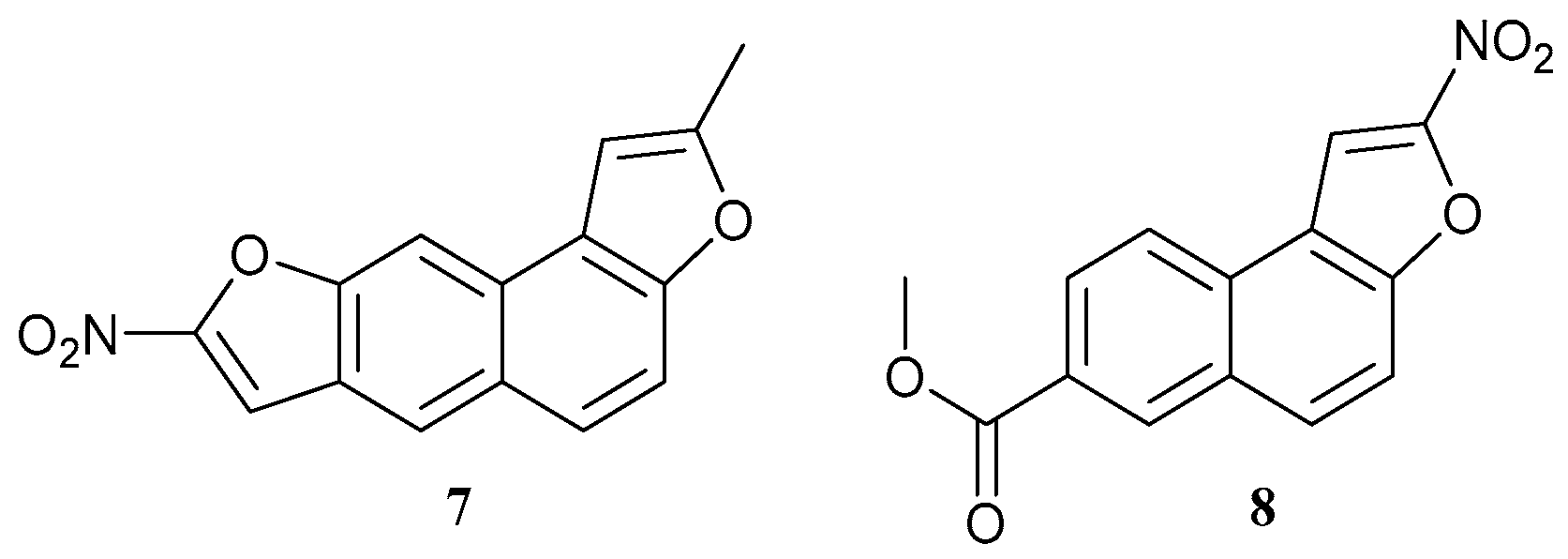
| 7 | 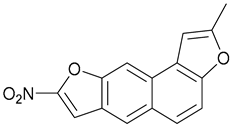 | X | - | - | [21] |
| 8 | 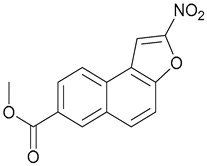 | ||||
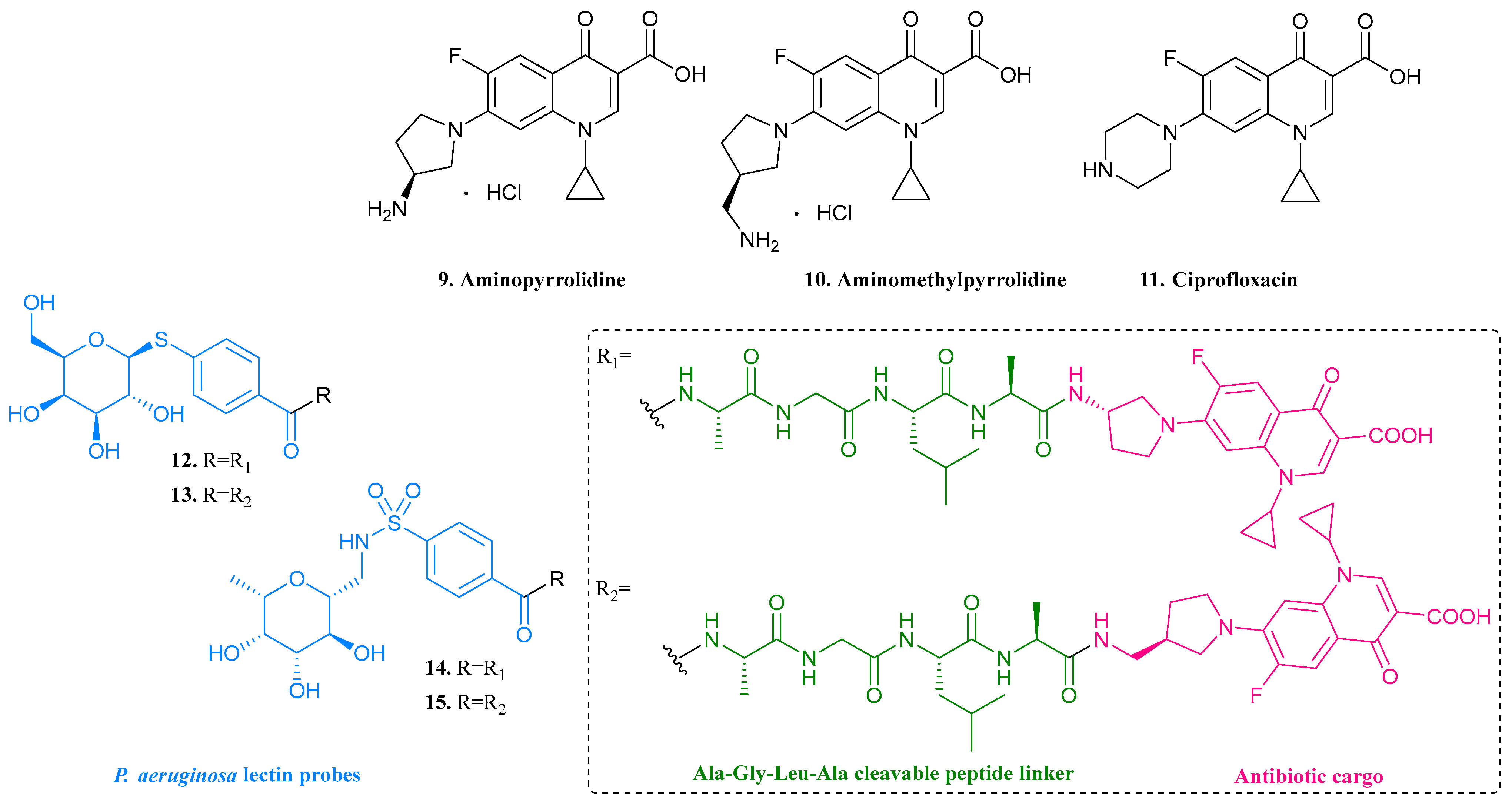
| 9. Aminopyrro- lidine | 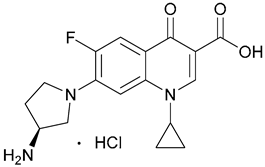 | - | X | - | [22] |
| 10. Aminomethylpyrro-lidine | 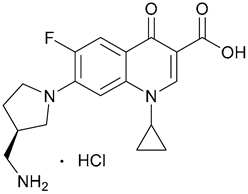 | ||||
| 11. Ciprofloxacin | 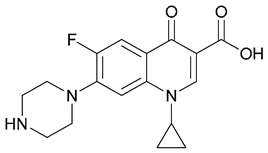 | ||||
| 12 |  | - | X | - | [22] |
| 13 |  | ||||
| 14 |  | ||||
| 15 | 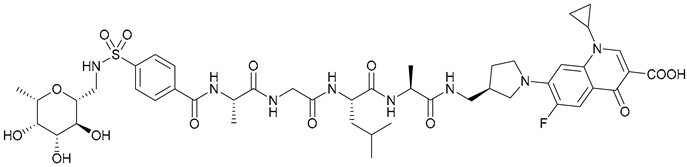 | ||||
| 16. Moxifloxacin | 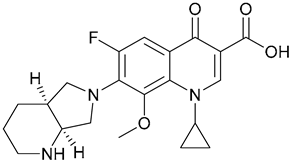 | - | X | - | [23] |
| 17. N-Moxi | 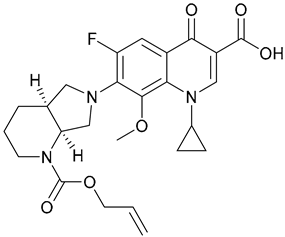 | ||||
| 18. C-Moxi |  | ||||
| 22. TBP-PI-HBr | 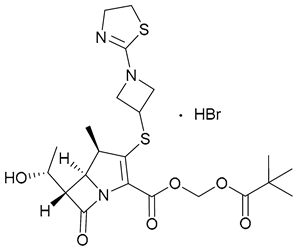 | - | X | - | [24,25,26,27,28] |
| 23. Ceftibuten | 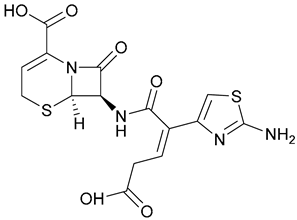 | - | X | - | [29,30] |
| 24. Avibactam |  | ||||
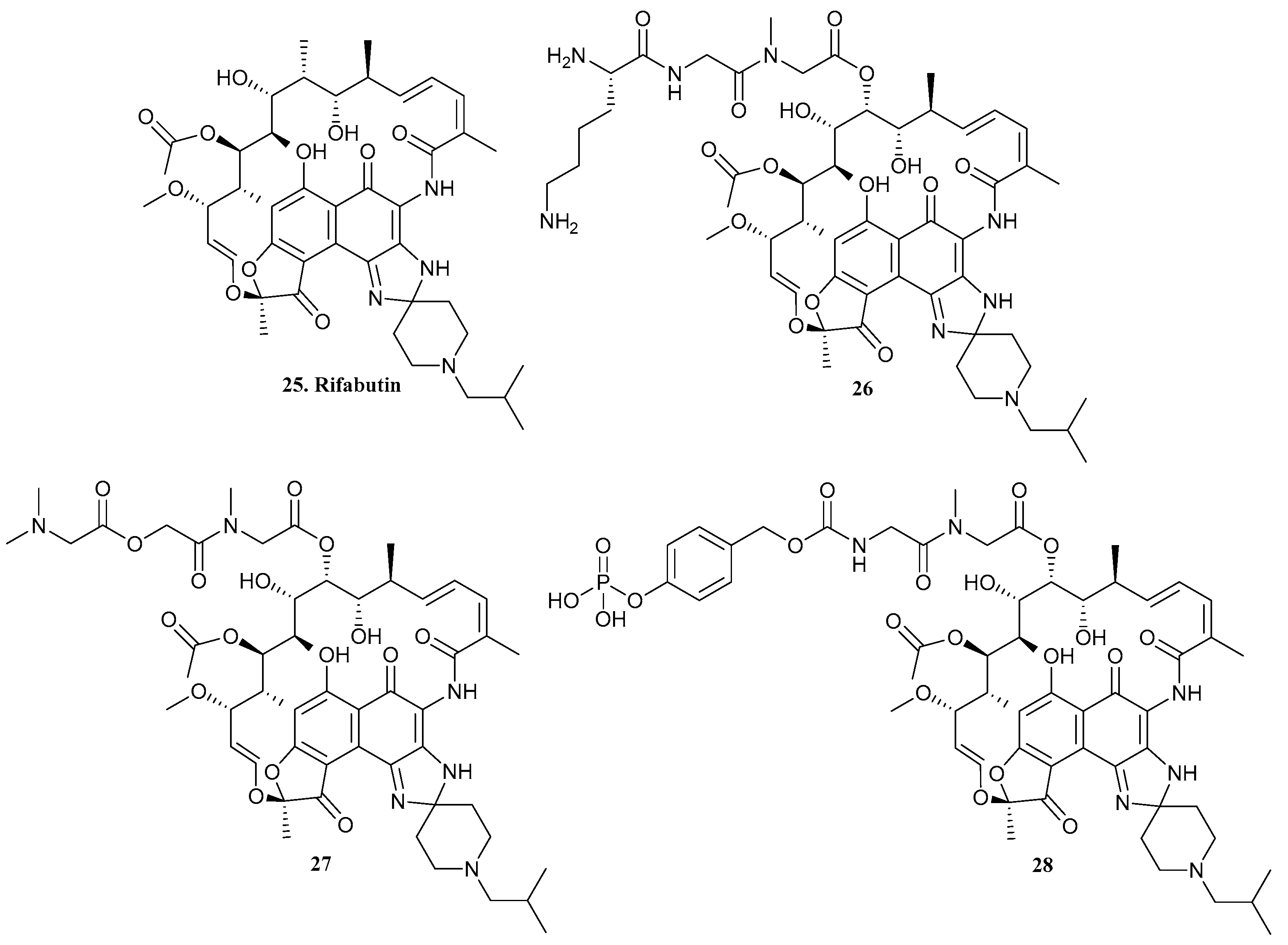
| 25. Rifabutin | 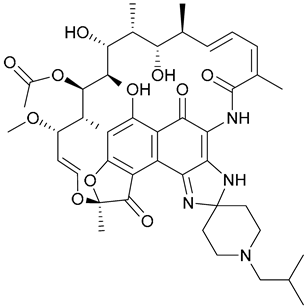 | - | X | - | [31] |
| 26 | 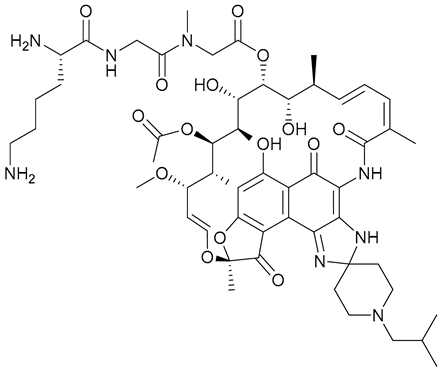 | ||||
| 27 |  | ||||
| 28 | 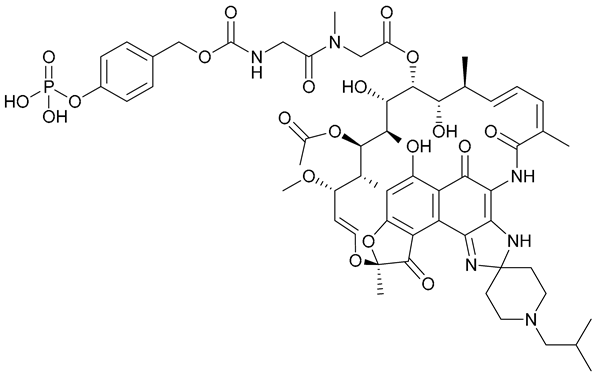 | ||||
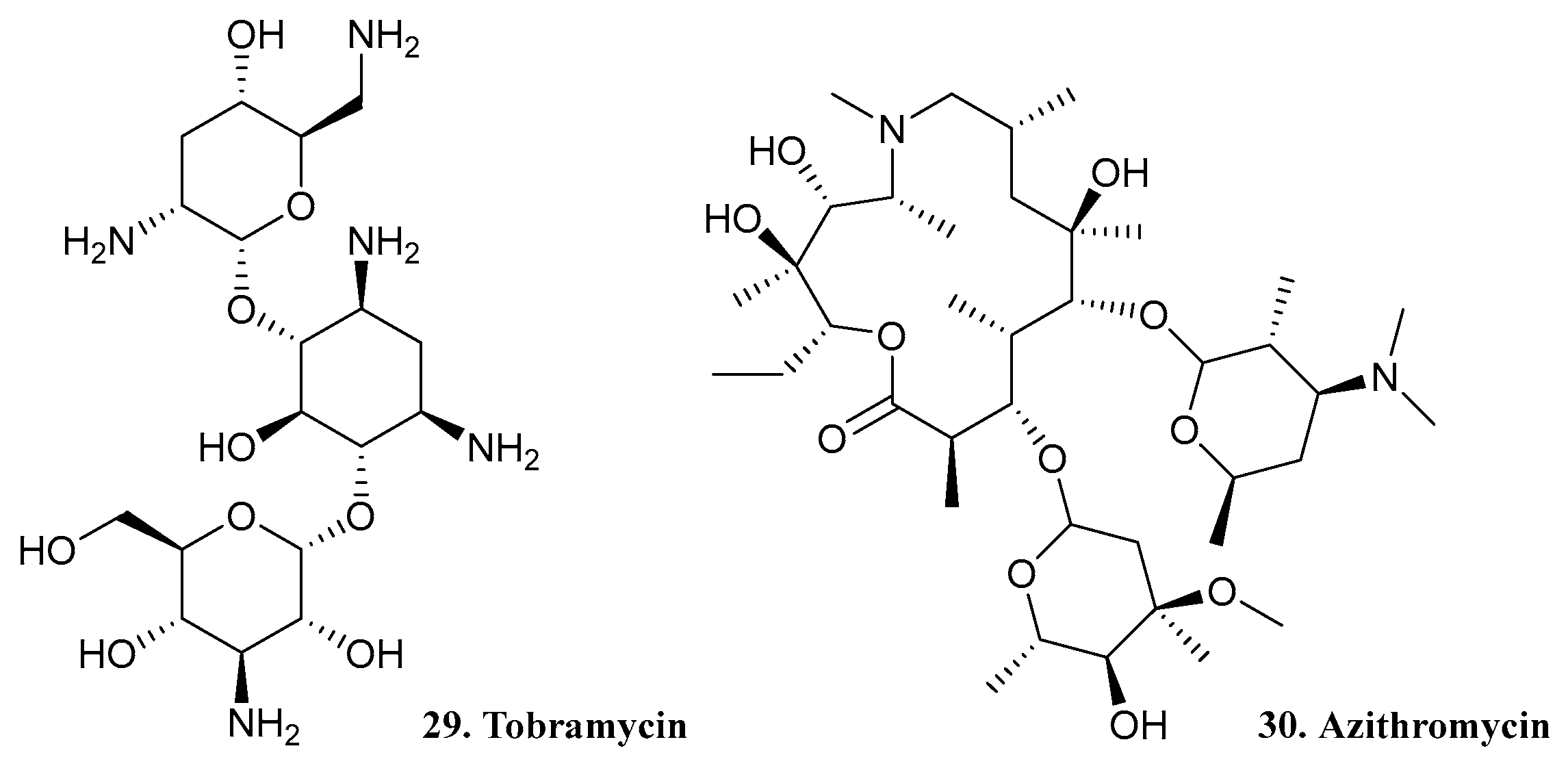
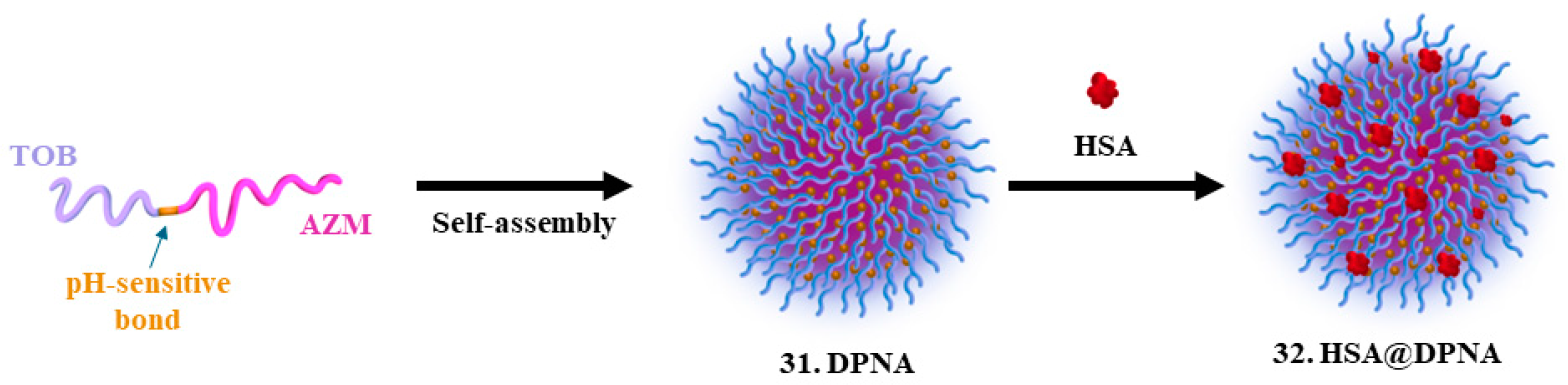
| 29. Tobramycin | 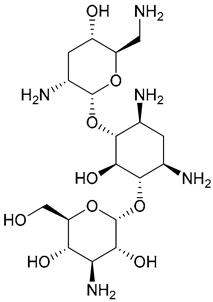 | - | X | - | [32] |
| 30. Azithromycin | 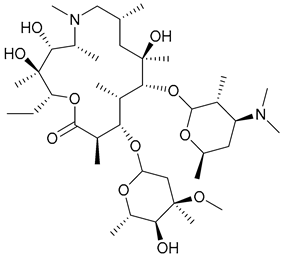 | ||||
| 33. Colistin methanesulfonate | 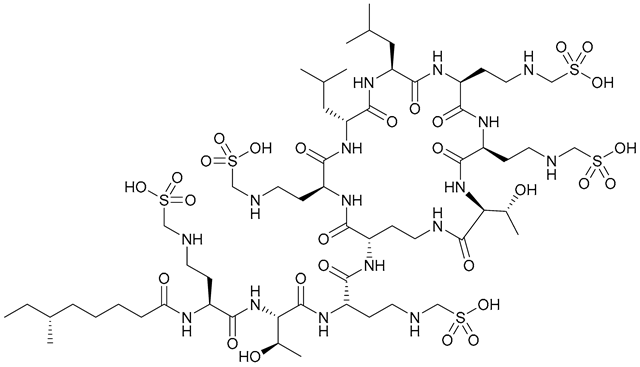 | - | X | - | [34] |
| 34. pEt_20 | 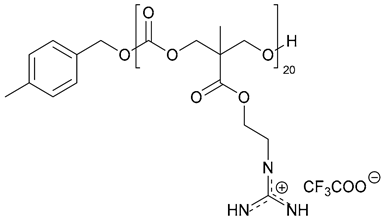 | ||||
| 38, 39. l-EP/d-EP |  | - | X | - | [35] |
| 40. Zidovudine | 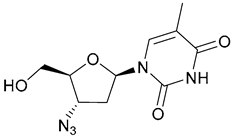 | - | X | - | [36] |
| 41 |  | ||||
| 42 | 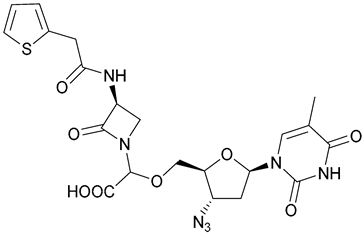 | ||||
| 43. Oxacillin | 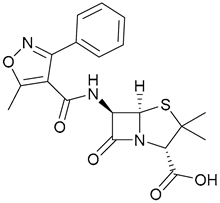 | - | - | X | [37] |
| 44. TXA709 | 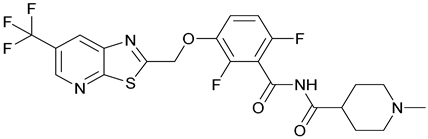 | ||||
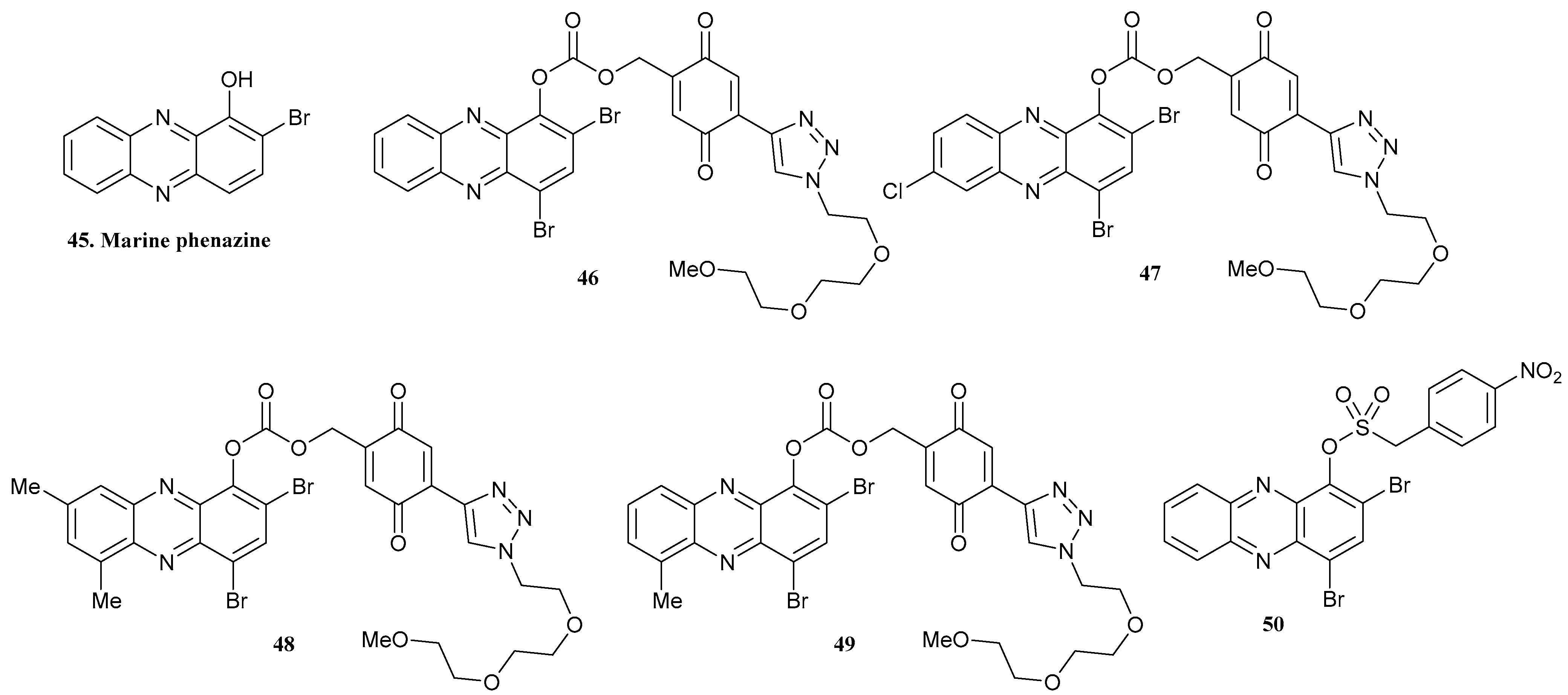
| 45. Marine Phenazine | 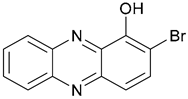 | - | - | X | [38] |
| 46 | 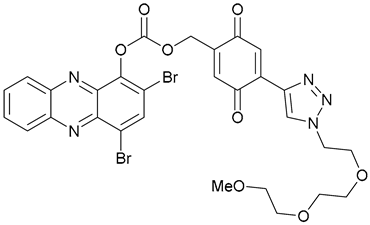 | ||||
| 47 | 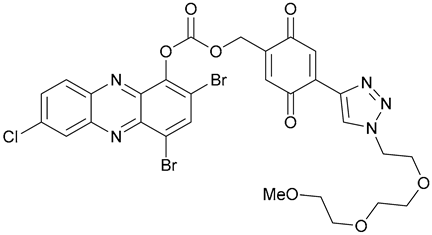 | ||||
| 48 |  | ||||
| 49 | 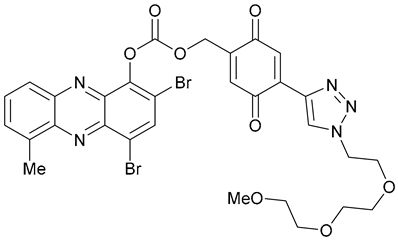 | ||||
| 50 | 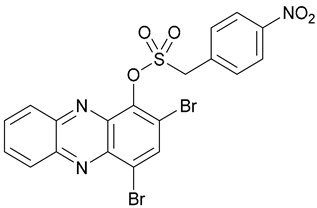 | - | - | X | [39] |
| 54. Vancomycin | 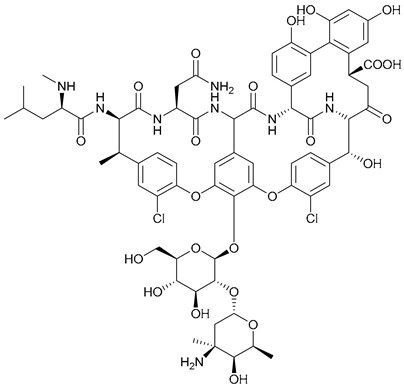 | - | - | X | [41] |
| 55. PEG-Schiff -Van | 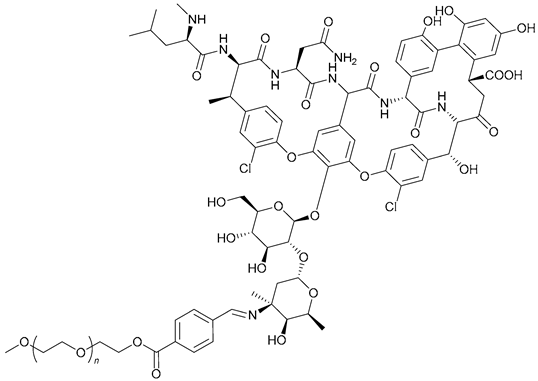 | ||||
| 57. PEG-b-PCAE | 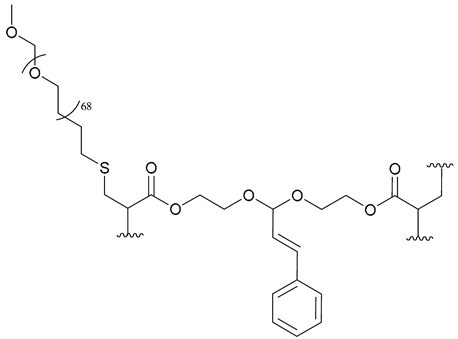 | - | - | X | [42] |
| 58. Curcumin | 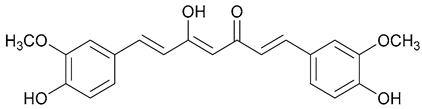 | - | - | X | [43] |
| 59. PEG600-Curc | 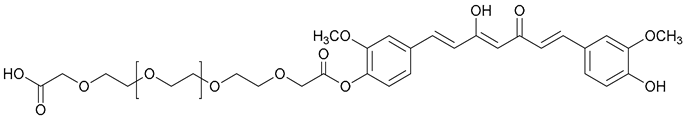 | ||||
| 60. Protocatechuic acid |  | - | - | X | [44] |
| 61. Tedizolid phosphate | 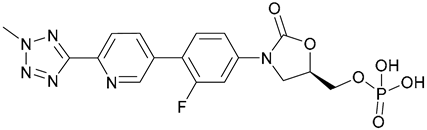 | - | - | X | [45] |
| 62. Capecitabine | 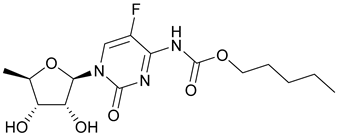 | - | - | X | [46] |
| 63 |  | - | - | X | [47] |
| 64 | 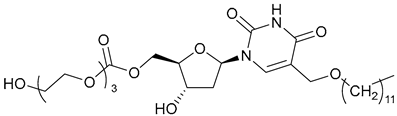 | ||||
| 65 (n = 1); 66 (n = 2); 67 (n = 3) | 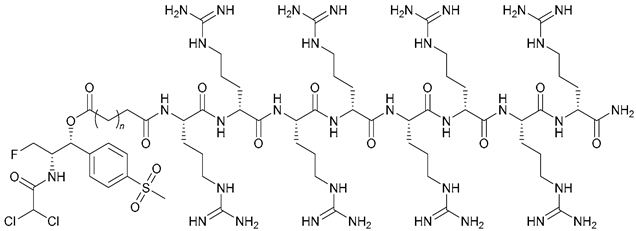 | - | X | X | [48] |
| 68. HiZP |  | - | X | X | [49] |
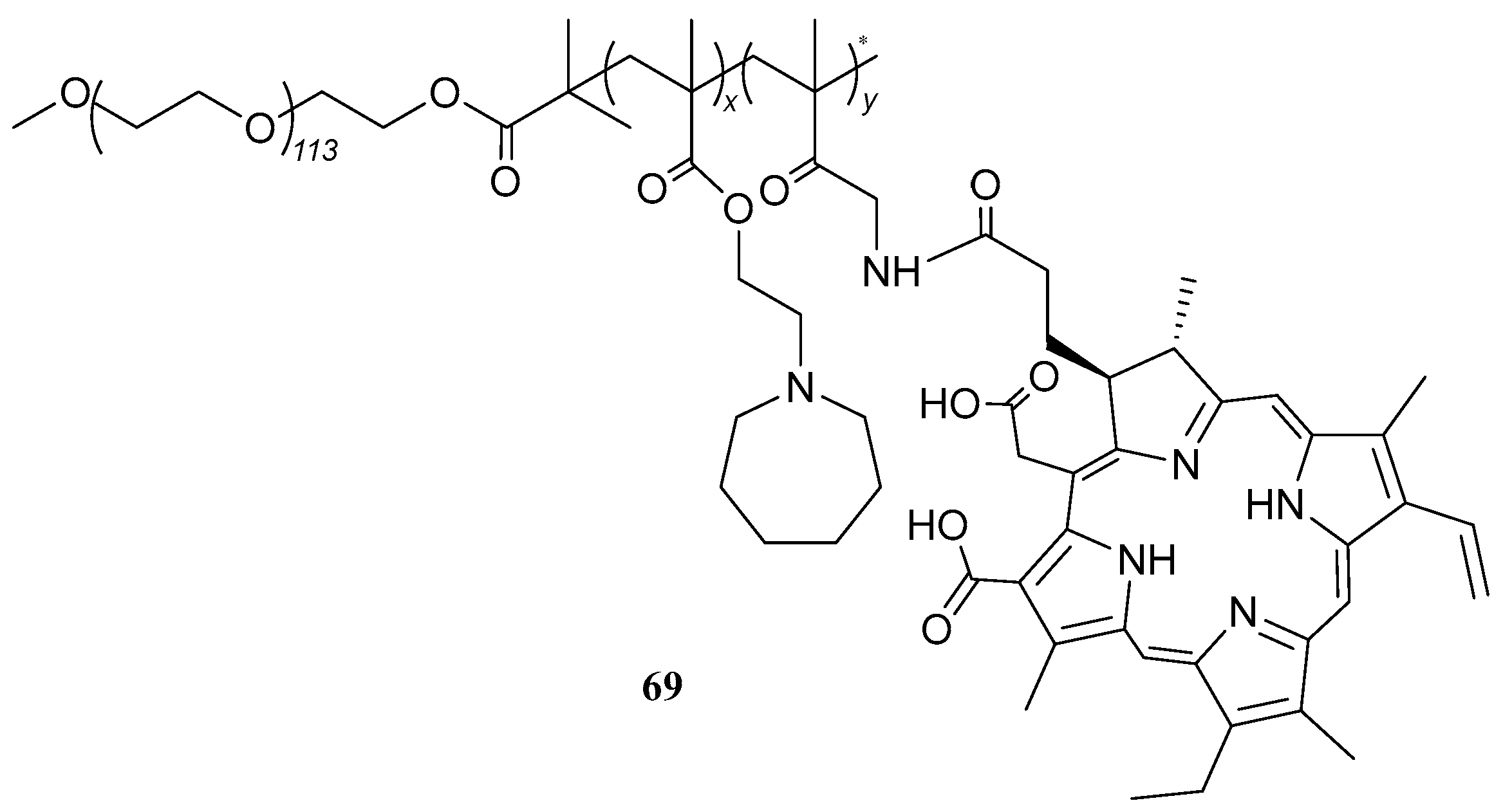
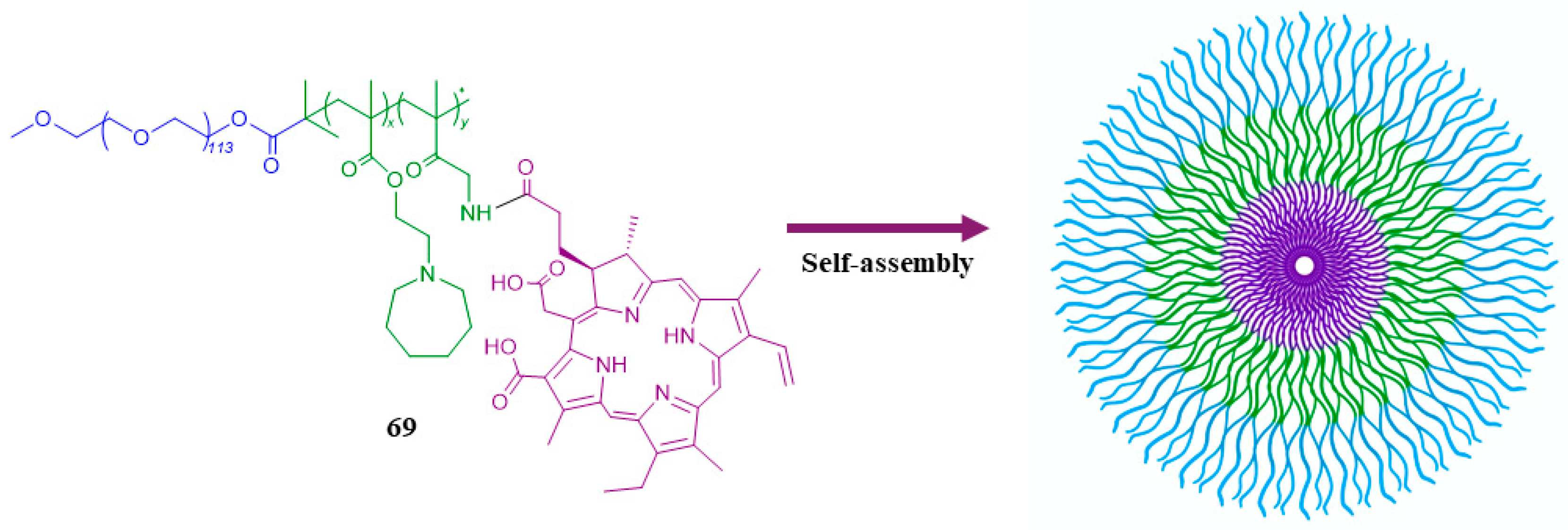
| 69 | 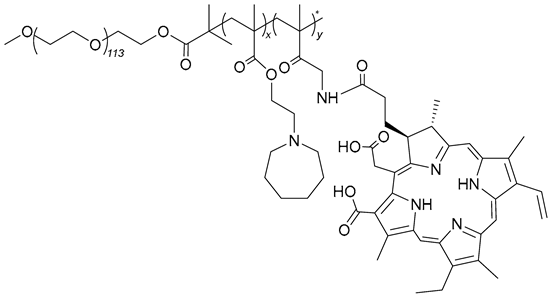 | - | X | X | [50] |
| 70 | 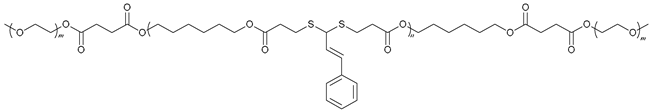 | - | X | X | [52] |
| 71. Diacerin |  | - | X | X | [53] |
| 72. Rhein | 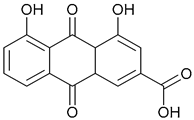 | ||||
| 73. Bet6-IL | 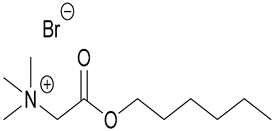 | ||||
| 74. Carn6-IL | 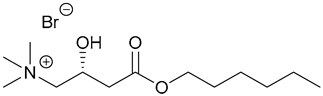 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maria, C.; de Matos, A.M.; Rauter, A.P. Antibacterial Prodrugs to Overcome Bacterial Antimicrobial Resistance. Pharmaceuticals 2024, 17, 718. https://doi.org/10.3390/ph17060718
Maria C, de Matos AM, Rauter AP. Antibacterial Prodrugs to Overcome Bacterial Antimicrobial Resistance. Pharmaceuticals. 2024; 17(6):718. https://doi.org/10.3390/ph17060718
Chicago/Turabian StyleMaria, Catarina, Ana M. de Matos, and Amélia P. Rauter. 2024. "Antibacterial Prodrugs to Overcome Bacterial Antimicrobial Resistance" Pharmaceuticals 17, no. 6: 718. https://doi.org/10.3390/ph17060718
APA StyleMaria, C., de Matos, A. M., & Rauter, A. P. (2024). Antibacterial Prodrugs to Overcome Bacterial Antimicrobial Resistance. Pharmaceuticals, 17(6), 718. https://doi.org/10.3390/ph17060718