

Synthesis, Activity, Toxicity, and In Silico Studies of New Antimycobacterial N-Alkyl Nitrobenzamides

 , , , ,
, , , ,  ,
,  and
and
Abstract

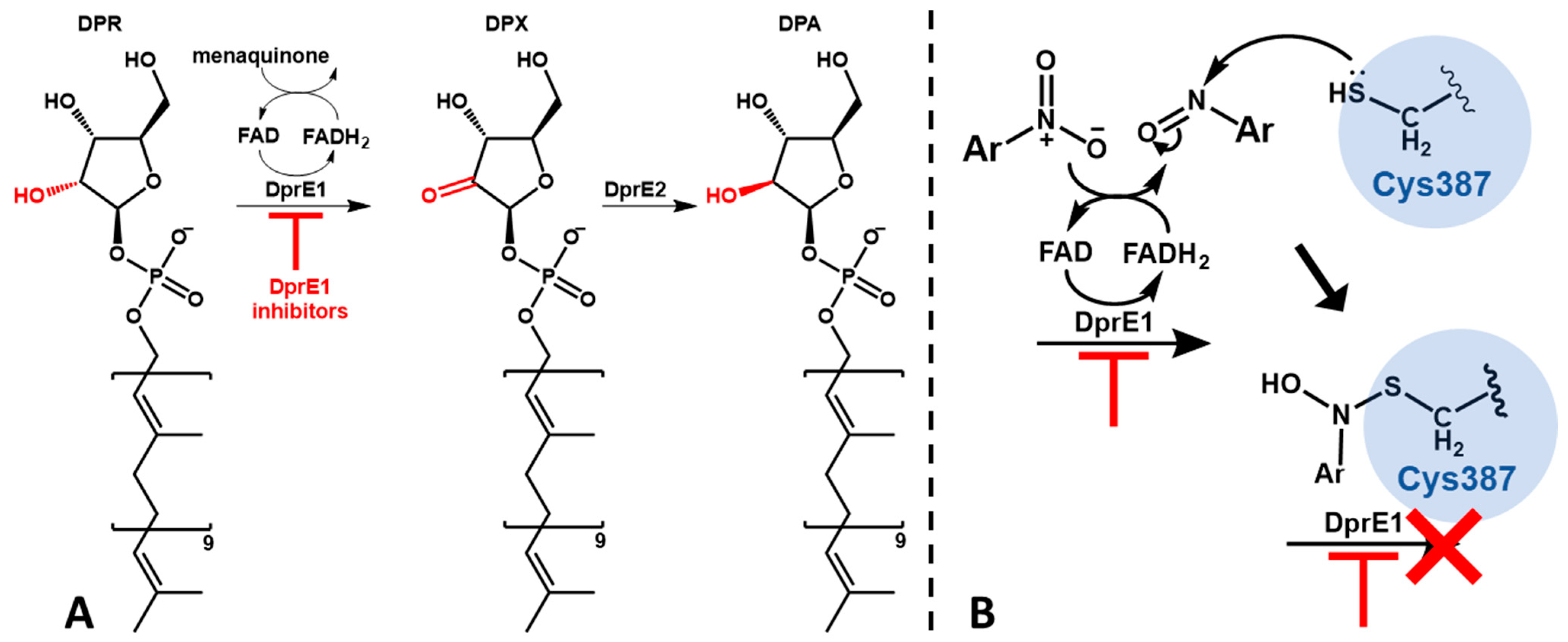
1. Introduction
2. Results
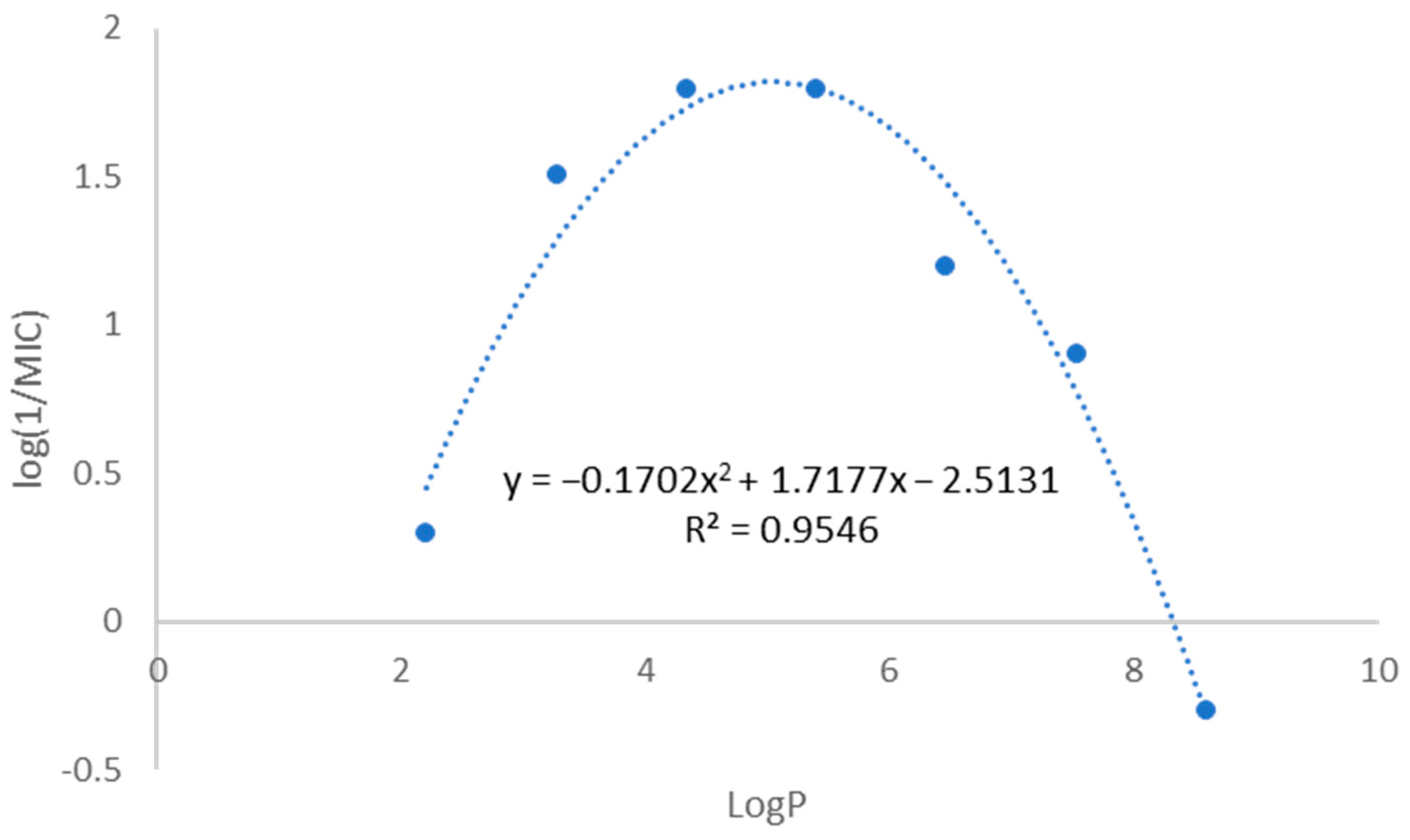
2.1. Compound Library and Antitubercular Activity
2.2. Susceptibility Assessment of Multiple Mycobacterial Species
2.3. Cytotoxicity Evaluation in Human Macrophages
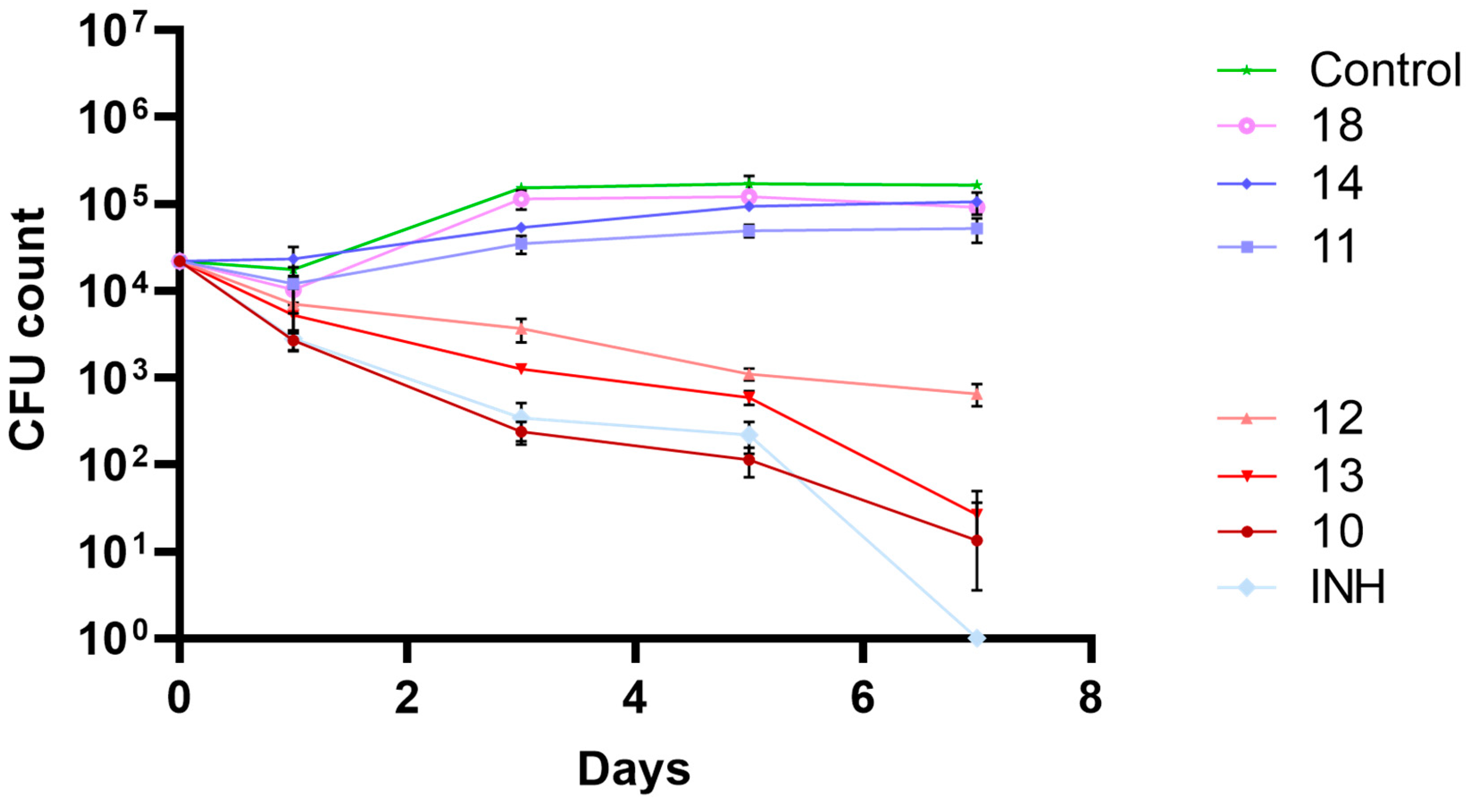
2.4. Biological Activity in Macrophage Model of Infection
2.5. Stability Studies
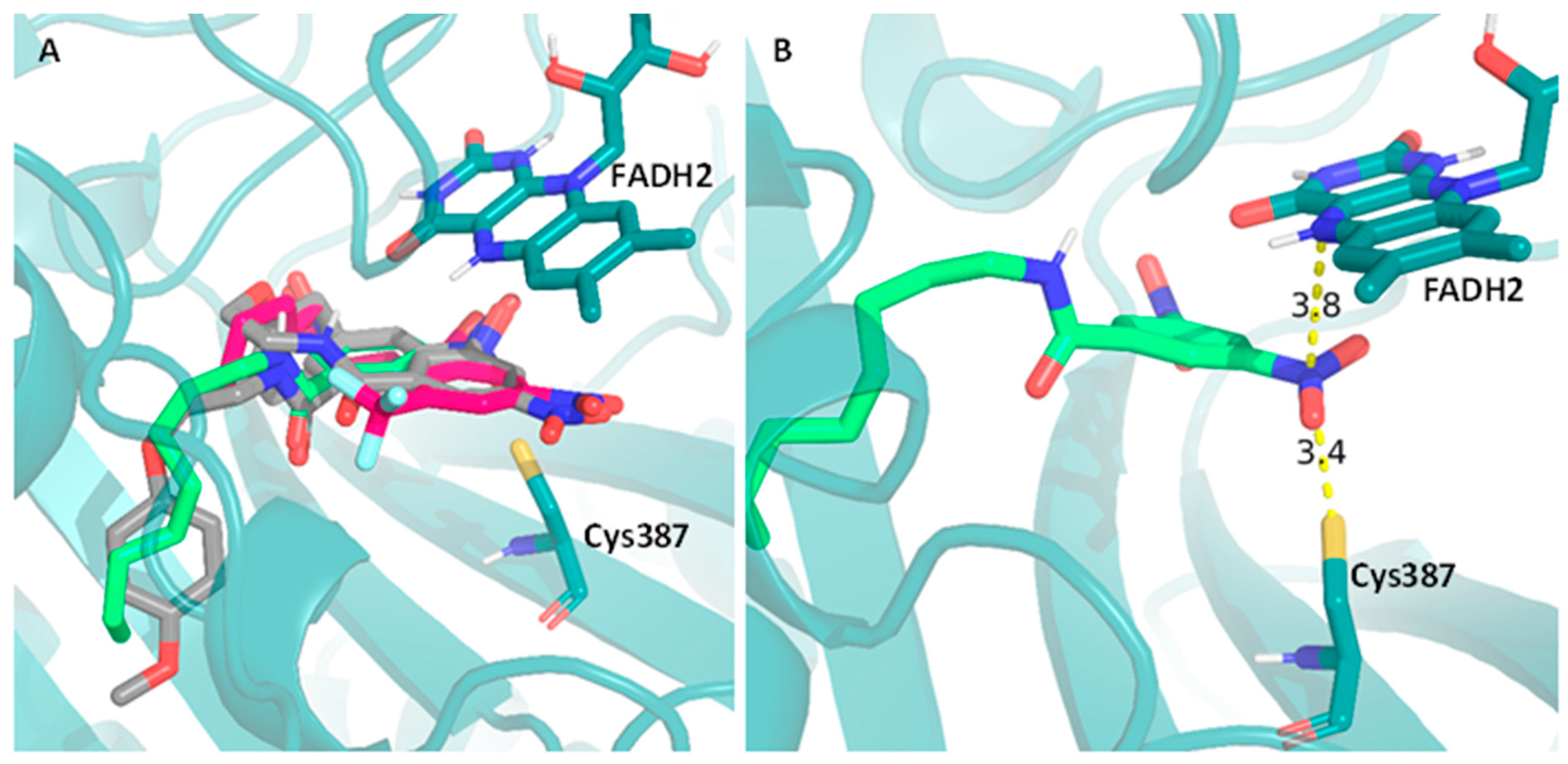
2.6. Computational Studies
3. Discussion
4. Materials and Methods
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cambier, C.J.; Falkow, S.; Ramakrishnan, L. Host Evasion and Exploitation Schemes of Mycobacterium Tuberculosis. Cell 2014, 159, 1497–1509. [Google Scholar] [CrossRef]
- Gill, C.M.; Dolan, L.; Piggott, L.M.; McLaughlin, A.M. New Developments in Tuberculosis Diagnosis and Treatment. Breathe 2022, 18, 210149. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Aarnoutse, R.; Chesov, D.; van Crevel, R.; Gillespie, S.H.; Grobbel, H.P.; Kalsdorf, B.; Kontsevaya, I.; van Laarhoven, A.; Nishiguchi, T.; et al. Perspective for Precision Medicine for Tuberculosis. Front. Immunol. 2020, 11, 566608. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report 2023; World Health Organization: Geneva, Switzerland, 2023; ISBN 978-92-4-008385-1.
- Pai, M.; Kasaeva, T.; Swaminathan, S. COVID-19’s Devastating Effect on Tuberculosis Care—A Path to Recovery. N. Engl. J. Med. 2022, 386, 1490–1493. [Google Scholar] [CrossRef] [PubMed]
- Blondiaux, N.; Moune, M.; Desroses, M.; Frita, R.; Flipo, M.; Mathys, V.; Soetaert, K.; Kiass, M.; Delorme, V.; Djaout, K.; et al. Reversion of Antibiotic Resistance in Mycobacterium Tuberculosis by Spiroisoxazoline SMARt-420. Science 2017, 355, 1206–1211. [Google Scholar] [CrossRef] [PubMed]
- Shetye, G.S.; Franzblau, S.G.; Cho, S. New Tuberculosis Drug Targets, Their Inhibitors, and Potential Therapeutic Impact. Transl. Res. 2020, 220, 68–97. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, G.; Pasca, M.R.; Chiarelli, L.R.; Manina, G.; Mattevi, A.; Binda, C. The DprE1 Enzyme, One of the Most Vulnerable Targets of Mycobacterium Tuberculosis. Appl. Microbiol. Biotechnol. 2013, 97, 8841–8848. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Alshrari, A.S.; Thabet, H.K.; Abida; Afroz Bakht, M. Synthetic Molecules as DprE1 Inhibitors: A Patent Review. Expert Opin. Ther. Pat. 2021, 31, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Gawad, J.; Bonde, C. Decaprenyl-Phosphoryl-Ribose 2′-Epimerase (DprE1): Challenging Target for Antitubercular Drug Discovery. Chem. Cent. J. 2018, 12, 1–12. [Google Scholar] [CrossRef]
- Amado, P.S.M.; Woodley, C.; Cristiano, M.L.S.; O’Neill, P.M. Recent Advances of DprE1 Inhibitors against Mycobacterium Tuberculosis: Computational Analysis of Physicochemical and ADMET Properties. ACS Omega 2022, 7, 40659–40681. [Google Scholar] [CrossRef]
- Yadav, S.; Soni, A.; Tanwar, O.; Bhadane, R.; Besra, G.S.; Kawathekar, N. DprE1 Inhibitors: Enduring Aspirations for Future Antituberculosis Drug Discovery. ChemMedChem 2023, 18, e202300099. [Google Scholar] [CrossRef]
- Brecik, M.; Centárová, I.; Mukherjee, R.; Kolly, G.S.; Huszár, S.; Bobovská, A.; Kilacsková, E.; Mokošová, V.; Svetlíková, Z.; Šarkan, M.; et al. DprE1 Is a Vulnerable Tuberculosis Drug Target Due to Its Cell Wall Localization. ACS Chem. Biol. 2015, 10, 1631–1636. [Google Scholar] [CrossRef]
- Richter, A.; Rudolph, I.; Möllmann, U.; Voigt, K.; Chung, C.-W.; Singh, O.M.P.; Rees, M.; Mendoza-Losana, A.; Bates, R.; Ballell, L.; et al. Novel Insight into the Reaction of Nitro, Nitroso and Hydroxylamino Benzothiazinones and of Benzoxacinones with Mycobacterium Tuberculosis DprE1. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Neres, J.; Pojer, F.; Molteni, E.; Chiarelli, L.R.; Dhar, N.; Boy-Röttger, S.; Buroni, S.; Fullam, E.; Degiacomi, G.; Lucarelli, A.P.; et al. Structural Basis for Benzothiazinone-Mediated Killing of Mycobacterium Tuberculosis. Sci. Transl. Med. 2012, 4, 150ra121. [Google Scholar] [CrossRef]
- Foo, C.S.Y.; Lechartier, B.; Kolly, G.S.; Boy-Röttger, S.; Neres, J.; Rybniker, J.; Lupien, A.; Sala, C.; Piton, J.; Cole, S.T. Characterization of DprE1-Mediated Benzothiazinone Resistance in Mycobacterium Tuberculosis. Antimicrob. Agents Chemother. 2016, 60, 6451–6459. [Google Scholar] [CrossRef] [PubMed]
- de Jesus Lopes Ribeiro, A.L.; Degiacomi, G.; Ewann, F.; Buroni, S.; Incandela, M.L.; Chiarelli, L.R.; Mori, G.; Kim, J.; Contreras-Dominguez, M.; Park, Y.S.; et al. Analogous Mechanisms of Resistance to Benzothiazinones and Dinitrobenzamides in Mycobacterium Smegmatis. PLoS ONE 2011, 6, e26675. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, H.; Sun, Z. Susceptibility of Mycobacterium Tuberculosis to Weak Acids. J. Antimicrob. Chemother. 2003, 52, 56–60. [Google Scholar] [CrossRef]
- Gu, P.; Constantino, L.; Zhang, Y. Enhancement of the Antituberculosis Activity of Weak Acids by Inhibitors of Energy Metabolism but Not by Anaerobiosis Suggests That Weak Acids Act Differently from the Front-Line Tuberculosis Drug Pyrazinamide. J. Med. Microbiol. 2008, 57, 1129–1134. [Google Scholar] [CrossRef]
- Valente, E.; Simões, M.F.; Testa, B.; Constantino, L. Development of a Method to Investigate the Hydrolysis of Xenobiotic Esters by a Mycobacterium Smegmatis Homogenate. J. Microbiol. Methods 2011, 85, 98–102. [Google Scholar] [CrossRef]
- Valente, E.; Testa, B.; Constantino, L. Activation of Benzoate Model Prodrugs by Mycobacteria. Comparison with Mammalian Plasma and Liver Hydrolysis. Eur. J. Pharm. Sci. 2021, 162, 105831. [Google Scholar] [CrossRef]
- Simões, M.F.; Valente, E.; Gómez, M.J.R.; Anes, E.; Constantino, L. Lipophilic Pyrazinoic Acid Amide and Ester Prodrugs Stability, Activation and Activity against M. Tuberculosis. Eur. J. Pharm. Sci. 2009, 37, 257–263. [Google Scholar] [CrossRef]
- Pires, D.; Valente, E.; Simões, M.F.; Carmo, N.; Testa, B.; Constantino, L.; Anes, E. Esters of Pyrazinoic Acid Are Active against Pyrazinamide-Resistant Strains of Mycobacterium Tuberculosis and Other Naturally Resistant Mycobacteria in Vitro and Ex Vivo within Macrophages. Antimicrob. Agents Chemother. 2015, 59, 7693–7699. [Google Scholar] [CrossRef]
- Pais, J.P.; Magalhães, M.; Antoniuk, O.; Barbosa, I.; Freire, R.; Pires, D.; Valente, E.; Testa, B.; Anes, E.; Constantino, L. Benzoic Acid Derivatives as Prodrugs for the Treatment of Tuberculosis. Pharmaceuticals 2022, 15, 1118. [Google Scholar] [CrossRef]
- Pais, J.P.; Antoniuk, O.; Freire, R.; Pires, D.; Valente, E.; Anes, E.; Constantino, L. Nitrobenzoates and Nitrothiobenzoates with Activity against M. Tuberculosis. Microorganisms 2023, 11, 969. [Google Scholar] [CrossRef]
- Munagala, G.; Yempalla, K.R.; Aithagani, S.K.; Kalia, N.P.; Ali, F.; Ali, I.; Rajput, V.S.; Rani, C.; Chib, R.; Mehra, R.; et al. Synthesis and Biological Evaluation of Substituted N-Alkylphenyl-3,5- Dinitrobenzamide Analogs as Anti-TB Agents. Medchemcomm 2014, 5, 521–527. [Google Scholar] [CrossRef]
- Nepali, K.; Lee, H.Y.; Liou, J.P. Nitro-Group-Containing Drugs. J. Med. Chem. 2019, 62, 2851–2893. [Google Scholar] [CrossRef]
- Tetko, I.V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.A.; Radchenko, E.V.; Zefirov, N.S.; Makarenko, A.S.; et al. Virtual Computational Chemistry Laboratory–Design and Description. J. Comput. Aided. Mol. Des. 2005, 19, 453–463. [Google Scholar] [CrossRef]
- Christophe, T.; Jackson, M.; Hee, K.J.; Fenistein, D.; Contreras-Dominguez, M.; Kim, J.; Genovesio, A.; Carralot, J.P.; Ewann, F.; Kim, E.H.; et al. High Content Screening Identifies Decaprenyl-Phosphoribose 2′ Epimerase as a Target for Intracellular Antimycobacterial Inhibitors. PLoS Pathog. 2009, 5, e1000645. [Google Scholar] [CrossRef]
- Li, L.; Lv, K.; Yang, Y.; Sun, J.; Tao, Z.; Wang, A.; Wang, B.; Wang, H.; Geng, Y.; Liu, M.; et al. Identification of N-Benzyl 3,5-Dinitrobenzamides Derived from PBTZ169 as Antitubercular Agents. ACS Med. Chem. Lett. 2018, 9, 741–745. [Google Scholar] [CrossRef]
- Trefzer, C.; Rengifo-Gonzalez, M.; Hinner, M.J.; Schneider, P.; Makarov, V.; Cole, S.T.; Johnsson, K. Benzothiazinones: Prodrugs That Covalently Modify the Decaprenylphosphoryl-β-D-Ribose 2′-Epimerase DprE1 of Mycobacterium tuberculosis. J. Am. Chem. Soc. 2010, 132, 13663–13665. [Google Scholar] [CrossRef]
- Makarov, V.; Manina, G.; Mikusova, K.; Möllmann, U.; Ryabova, O.; Saint-Joanis, B.; Dhar, N.; Pasca, M.R.; Buroni, S.; Lucarelli, A.P.; et al. Benzothiazinones Kill Mycobacterium Tuberculosis by Blocking Arabinan Synthesis. Science 2009, 324, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Favrot, L.; Ronning, D.R. Targeting the Mycobacterial Envelope for Tuberculosis Drug Development. Expert Rev. Anti. Infect. Ther. 2012, 10, 1023. [Google Scholar] [CrossRef] [PubMed]
- Manina, G.; Bellinzoni, M.; Pasca, M.R.; Neres, J.; Milano, A.; De Jesus Lopes Ribeiro, A.L.; Buroni, S.; Škovierová, H.; Dianišková, P.; Mikušová, K.; et al. Biological and Structural Characterization of the Mycobacterium Smegmatis Nitroreductase NfnB, and Its Role in Benzothiazinone Resistance. Mol. Microbiol. 2010, 77, 1172–1185. [Google Scholar] [CrossRef] [PubMed]
- Neres, J.; Hartkoorn, R.C.; Chiarelli, L.R.; Gadupudi, R.; Pasca, M.R.; Mori, G.; Venturelli, A.; Savina, S.; Makarov, V.; Kolly, G.S.; et al. 2-Carboxyquinoxalines Kill Mycobacterium Tuberculosis through Noncovalent Inhibition of DprE1. ACS Chem. Biol. 2015, 10, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Nakamura, R.; Kadokami, K.; Ogawa, H.I. Relationship between Mutagenicity and Reactivity or Biodegradability for Nitroaromatic Compounds. Environ. Toxicol. Chem. 2007, 26, 237–241. [Google Scholar] [CrossRef]
- Purohit, V.; Basu, A.K. Mutagenicity of Nitroaromatic Compounds. Chem. Res. Toxicol. 2000, 13, 673–692. [Google Scholar] [CrossRef] [PubMed]
- Polaquini, C.; Torrezan, G.; Santos, V.; Nazaré, A.; Campos, D.; Almeida, L.; Silva, I.; Ferreira, H.; Pavan, F.; Duque, C.; et al. Antibacterial and Antitubercular Activities of Cinnamylideneacetophenones. Molecules 2017, 22, 1685. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.M.; Zhang, Y. Effects of Weak Acids, UV and Proton Motive Force Inhibitors on Pyrazinamide Activity against Mycobacterium Tuberculosis in Vitro. J. Antimicrob. Chemother. 2006, 58, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Karabanovich, G.; Dušek, J.; Savková, K.; Pavliš, O.; Pávková, I.; Korábečný, J.; Kučera, T.; Kočová Vlčková, H.; Huszár, S.; Konyariková, Z.; et al. Development of 3,5-Dinitrophenyl-Containing 1,2,4-Triazoles and Their Trifluoromethyl Analogues as Highly Efficient Antitubercular Agents Inhibiting Decaprenylphosphoryl-β- d -Ribofuranose 2′-Oxidase. J. Med. Chem. 2019, 62, 8115–8139. [Google Scholar] [CrossRef]
- Chikhale, R.V.; Barmade, M.A.; Murumkar, P.R.; Yadav, M.R. Overview of the Development of DprE1 Inhibitors for Combating the Menace of Tuberculosis. J. Med. Chem. 2018, 61, 8563–8593. [Google Scholar] [CrossRef]
- Egorova, A.; Salina, E.G.; Makarov, V. Targeting Non-Replicating Mycobacterium Tuberculosis and Latent Infection: Alternatives and Perspectives (Mini-Review). Int. J. Mol. Sci. 2021, 22, 13317. [Google Scholar] [CrossRef] [PubMed]
- de Santos, N.C.S.; Scodro, R.B.d.L.; Sampiron, E.G.; Ieque, A.L.; Carvalho, H.C.d.; Santos, T.d.S.; Ghiraldi Lopes, L.D.; Campanerut-Sá, P.A.Z.; Siqueira, V.L.D.; Caleffi-Ferracioli, K.R.; et al. Minimum Bactericidal Concentration Techniques in Mycobacterium Tuberculosis: A Systematic Review. Microb. Drug Resist. 2020, 26, 752–765. [Google Scholar] [CrossRef] [PubMed]
- Landrum, G. RDKit: Open-Source Cheminformatics. Release 2023.03.3. Available online: https://zenodo.org/records/8254217 (accessed on 3 May 2024).
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
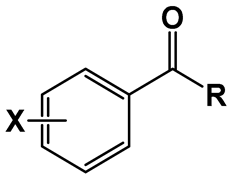 | |||||
|---|---|---|---|---|---|
| Compound | X | R | LogP 1 | MIC (µg/mL) | MBC (µg/mL) |
| 1 | H | NH(CH2)3CH3 | 2.45 | 256 | 256 |
| 2 | NH(CH2)5CH3 | 3.52 | 64 | 64 | |
| 3 | NH(CH2)7CH3 | 4.58 | 32 | 32 | |
| 4 | NH(CH2)11CH3 | 6.7 | >256 | >256 | |
| 5 | 4-NO2 | NH(CH2)3CH3 | 2.62 | 128 | 256 |
| 6 | NH(CH2)5CH3 | 3.68 | 32 | 64 | |
| 7 | NH(CH2)7CH3 | 4.74 | 128 | 256 | |
| 8 | NH(CH2)11CH3 | 6.87 | 512 | >1024 | |
| 9 | 3,5-NO2 | NH(CH2)3CH3 | 2.21 | 0.5 | 0.5 |
| 10 | NH(CH2)5CH3 | 3.28 | 0.031 | 0.031 | |
| 11 | NH(CH2)7CH3 | 4.34 | 0.016 | 0.031 | |
| 12 | NH(CH2)9CH3 | 5.4 | 0.016 | 0.016 | |
| 13 | NH(CH2)11CH3 | 6.46 | 0.063 | 0.063 | |
| 14 | NH(CH2)13CH3 | 7.53 | 0.125 | 0.25 | |
| 15 | NH(CH2)15CH3 | 8.59 | 2 | 2 | |
| 16 | 3-NO2-5-CF3 | NH(CH2)3CH3 | 3.52 | 2 | 2 |
| 17 | NH(CH2)5CH3 | 4.58 | 0.5 | 0.5 | |
| 18 | NH(CH2)7CH3 | 5.65 | 0.016 | 0.031 | |
| 19 | NH(CH2)9CH3 | 6.71 | 0.5 | 0.5 | |
| 20 | NH(CH2)11CH3 | 7.77 | 0.5 | 0.5 | |
| Compounds | M. tuberculosis | M. bovis BCG | M. smegmatis | M. avium | ||||
|---|---|---|---|---|---|---|---|---|
| MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | |
| DNB1 | 0.031 | 0.031 | 0.063 | 0.5 | 1 | 32 | >128 | |
| DNB2 | 0.031 | 0.063 | 0.125 | 0.5 | 1 | 64 | >128 | |
| PAS | ≤0.063 | ≤0.063 | ≤0.008 | ≤0.008 | 1024 | >1024 | 1024 | 1024 |
| INH | 0.05 | 0.05 | 0.025 | 0.025 | 8 | >25.6 | >25.6 | >25.6 |
| 3 | 32 | 32 | 32 | 32 | 128 | 128 | 128 | 256 |
| 7 | 128 | 256 | 128 | 512 | 1024 | 1024 | 1024 | 1024 |
| 9 | 0.5 | 0.5 | 0.5 | 1 | 4 | 4 | 64 | 512 |
| 10 | 0.063 | 0.063 | 0.25 | 0.25 | 1 | 1 | ||
| 11 | 0.016 | 0.031 | 0.016 | 0.016 | 0.25 | 4 | 512 | >512 |
| 12 | 0.016 | 0.016 | 0.063 | 0.063 | 1 | 1 | >512 | >512 |
| 13 | 0.031 | 0.063 | 0.083 | 0.166 | 0.667 | >2.664 | >512 | >512 |
| 14 | 0.125 | 0.25 | 0.25 | 0.25 | 1 | 4 | ||
| 15 | 2 | 2 | 2 | 4 | 32 | >64 | ||
| O9 | 32 | 128 | 16 | 16 | 32 | 64 | 128 | 256 |
| O10 | 32 | 4 | 8 | 8 | 8 | 8 | ||
| O11 | 8 | 8 | 2 | 4 | 4 | 4 | >1024 | >1024 |
| O12 | 2 | 4 | 8 | 8 | 16 | 32 | >512 | >512 |
| O13 | 8 | 16 | 8 | 8 | >512 | >512 | >512 | >512 |
| O14 | 16 | >1024 | 8 | 8 | >1024 | >1024 | >512 | >512 |
| O15 | >1024 | >1024 | 128 | 256 | >1024 | >1024 | >512 | >512 |
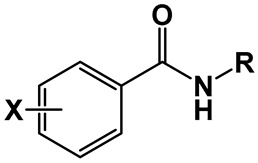 | |||||
|---|---|---|---|---|---|
| Compounds | X | R | MIC | LC50 | LC50/MIC |
| (μg/mL) | (μg/mL) | ||||
| 3 | H | C8H17 | 32 | 188 | 6 |
| 7 | 4-NO2 | C8H17 | 128 | 308 | 2 |
| 9 | 3,5-dNO2 | C4H9 | 0.5 | 26 | 52 |
| 10 | C6H13 | 0.031 | 361 | 11,645 | |
| 11 | C8H17 | 0.016 | 97 | 6063 | |
| 12 | C10H21 | 0.016 | 53 | 3313 | |
| 13 | C12H25 | 0.063 | 72 | 1143 | |
| 14 | C14H29 | 0.125 | 330 | 2640 | |
| 15 | C16H32 | 2 | n/d | - | |
| 16 | 3-NO2-5-CF3 | C4H9 | 2 | 41 | 21 |
| 17 | C6H13 | 0.5 | 23 | 46 | |
| 18 | C8H17 | 0.016 | 14 | 875 | |
| 19 | C10H21 | 0.5 | 53 | 106 | |
| 20 | C12H25 | 0.5 | 113 | 226 | |
| Puromycin | - | 0.35 | - | ||
| Compound | Human Plasma | Mycobacterium Homogenate | ||
|---|---|---|---|---|
| kobs × 100 (h−1) | % (72 h) | kobs × 100 (h−1) | % (48 h) | |
| 1 | ND | 0 | ND | 3 |
| 2 | - | - | ND | 2 |
| 3 | ND | 0 | ND | 2 |
| 4 | ND | 0 | ND | 0 |
| 5 | ND | 32 | 0.91 ± 0.07 | 35 |
| 6 | - | - | 0.80 ± 0.05 | 32 |
| 7 | ND | 1 | ND | 2 |
| 8 | ND | 1 | ND | 3 |
| 9 | ND | 0 | 3.03 ± 0.20 | 77 |
| 10 | - | - | 0.84 ± 0.01 | 33 |
| 11 | ND | 0 | ND | 5 |
| 12 | - | - | ND | 2 |
| 13 | ND | 0 | ND | 2 |
| 14 | - | - | ND | 2 |
| 15 | - | - | ND | 0 |
| 16 | ND | 0 | 1.09 ± 0.07 | 41 |
| 17 | - | - | ND | 14 |
| 18 | ND | 0 | ND | 11 |
| 19 | ND | 0 | ND | 3 |
| 20 | ND | 0 | ND | 2 |
| Compound | MIC (µg/mL) | NDCys387(Å) | NDFAD(Å) | ΣNDist | CDCys387(Å) | CDFAD(Å) | ΣCDist | ScoreAutodock |
|---|---|---|---|---|---|---|---|---|
| DNB1 | 0.016 | 3.562 | 3.908 | 7.470 | 5.575 | 5.069 | 10.645 | −8.487 |
| DNB2 | 0.031 | 3.531 | 4.008 | 7.539 | 4.989 | 3.352 | 8.341 | −8.639 |
| 1 | 256 | - | - | - | 9.444 | 13.128 | 22.572 | −5.495 |
| 2 | 64 | - | - | - | 11.572 | 11.664 | 23.235 | −6.021 |
| 3 | 32 | - | - | - | 9.523 | 13.200 | 22.723 | −6.672 |
| 4 | >256 | - | - | - | 12.150 | 11.659 | 23.809 | −7.507 |
| 5 | 128 | 12.765 | 12.345 | 25.110 | 12.079 | 12.044 | 24.124 | −6.431 |
| 6 | 32 | 12.784 | 12.322 | 25.106 | 11.841 | 11.938 | 23.779 | −7.091 |
| 7 | 128 | 12.702 | 12.286 | 24.989 | 11.683 | 11.841 | 23.525 | −7.419 |
| 8 | 512 | 5.079 | 3.862 | 8.941 | 12.218 | 12.185 | 24.403 | −8.261 |
| 9 | 0.5 | 3.371 | 3.814 | 7.185 | 5.015 | 4.935 | 9.950 | −7.313 |
| 10 | 0.031 | 3.440 | 3.745 | 7.185 | 4.949 | 5.085 | 10.034 | −7.688 |
| 11 | 0.016 | 3.421 | 3.927 | 7.348 | 5.461 | 5.221 | 10.682 | −8.305 |
| 12 | 0.016 | 3.370 | 3.760 | 7.131 | 4.806 | 4.813 | 9.619 | −8.584 |
| 13 | 0.063 | 3.484 | 3.667 | 7.151 | 4.944 | 4.989 | 9.932 | −9.064 |
| 14 | 0.125 | 3.471 | 3.698 | 7.169 | 4.928 | 5.064 | 9.992 | −9.356 |
| 15 | 2 | 3.439 | 3.731 | 7.170 | 5.438 | 5.011 | 10.448 | −9.706 |
| 16 | 2 | 6.048 | 7.087 | 13.135 | 5.417 | 4.958 | 10.374 | −6.672 |
| 17 | 0.5 | 5.432 | 4.410 | 9.843 | 5.076 | 3.471 | 8.547 | −7.161 |
| 18 | 0.016 | 5.944 | 4.447 | 10.392 | 5.108 | 3.535 | 8.643 | −7.600 |
| 19 | 0.5 | 5.907 | 4.482 | 10.390 | 5.370 | 4.941 | 10.312 | −8.057 |
| 20 | 0.5 | 5.225 | 3.915 | 9.140 | 4.754 | 3.387 | 8.141 | −8.522 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pais, J.P.; Antoniuk, O.; Pires, D.; Delgado, T.; Fortuna, A.; Costa, P.J.; Anes, E.; Constantino, L. Synthesis, Activity, Toxicity, and In Silico Studies of New Antimycobacterial N-Alkyl Nitrobenzamides. Pharmaceuticals 2024, 17, 608. https://doi.org/10.3390/ph17050608
Pais JP, Antoniuk O, Pires D, Delgado T, Fortuna A, Costa PJ, Anes E, Constantino L. Synthesis, Activity, Toxicity, and In Silico Studies of New Antimycobacterial N-Alkyl Nitrobenzamides. Pharmaceuticals. 2024; 17(5):608. https://doi.org/10.3390/ph17050608
Chicago/Turabian StylePais, João P., Olha Antoniuk, David Pires, Tiago Delgado, Andreia Fortuna, Paulo J. Costa, Elsa Anes, and Luis Constantino. 2024. "Synthesis, Activity, Toxicity, and In Silico Studies of New Antimycobacterial N-Alkyl Nitrobenzamides" Pharmaceuticals 17, no. 5: 608. https://doi.org/10.3390/ph17050608
APA StylePais, J. P., Antoniuk, O., Pires, D., Delgado, T., Fortuna, A., Costa, P. J., Anes, E., & Constantino, L. (2024). Synthesis, Activity, Toxicity, and In Silico Studies of New Antimycobacterial N-Alkyl Nitrobenzamides. Pharmaceuticals, 17(5), 608. https://doi.org/10.3390/ph17050608