
Arylamines QSAR-Based Design and Molecular Dynamics of New Phenylthiophene and Benzimidazole Derivatives with Affinity for the C111, Y268, and H73 Sites of SARS-CoV-2 PLpro Enzyme



Abstract

1. Introduction
2. Results and Discussion
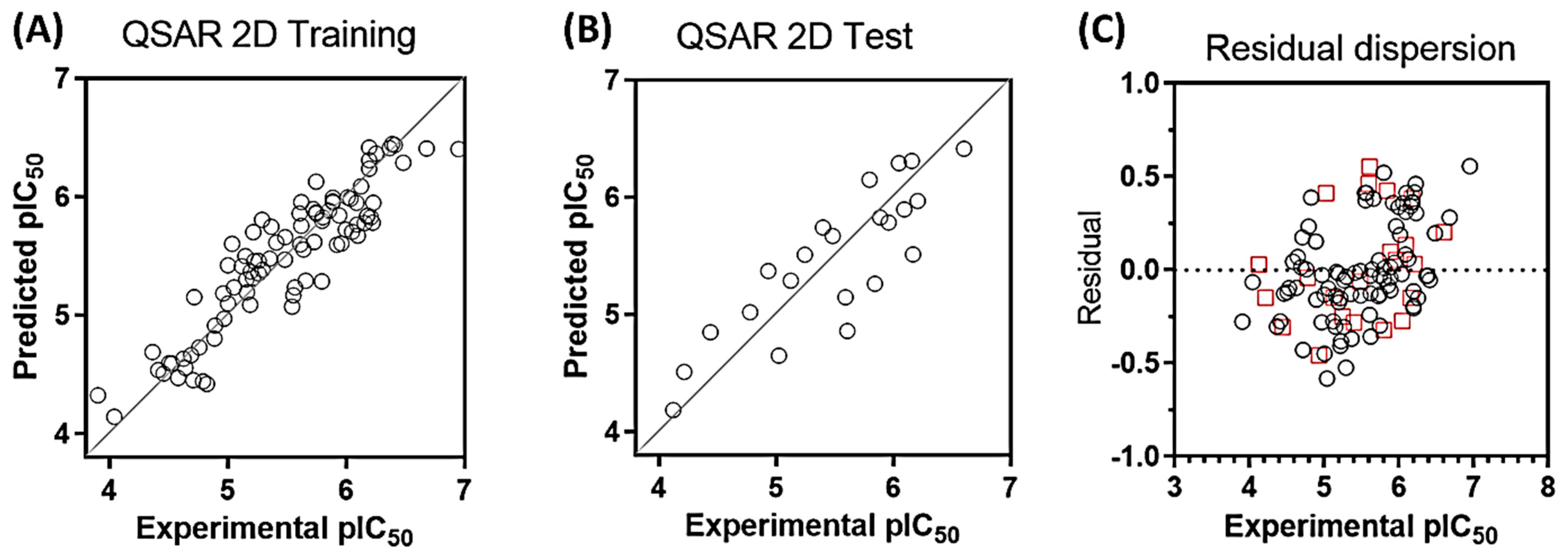
2.1. QSAR
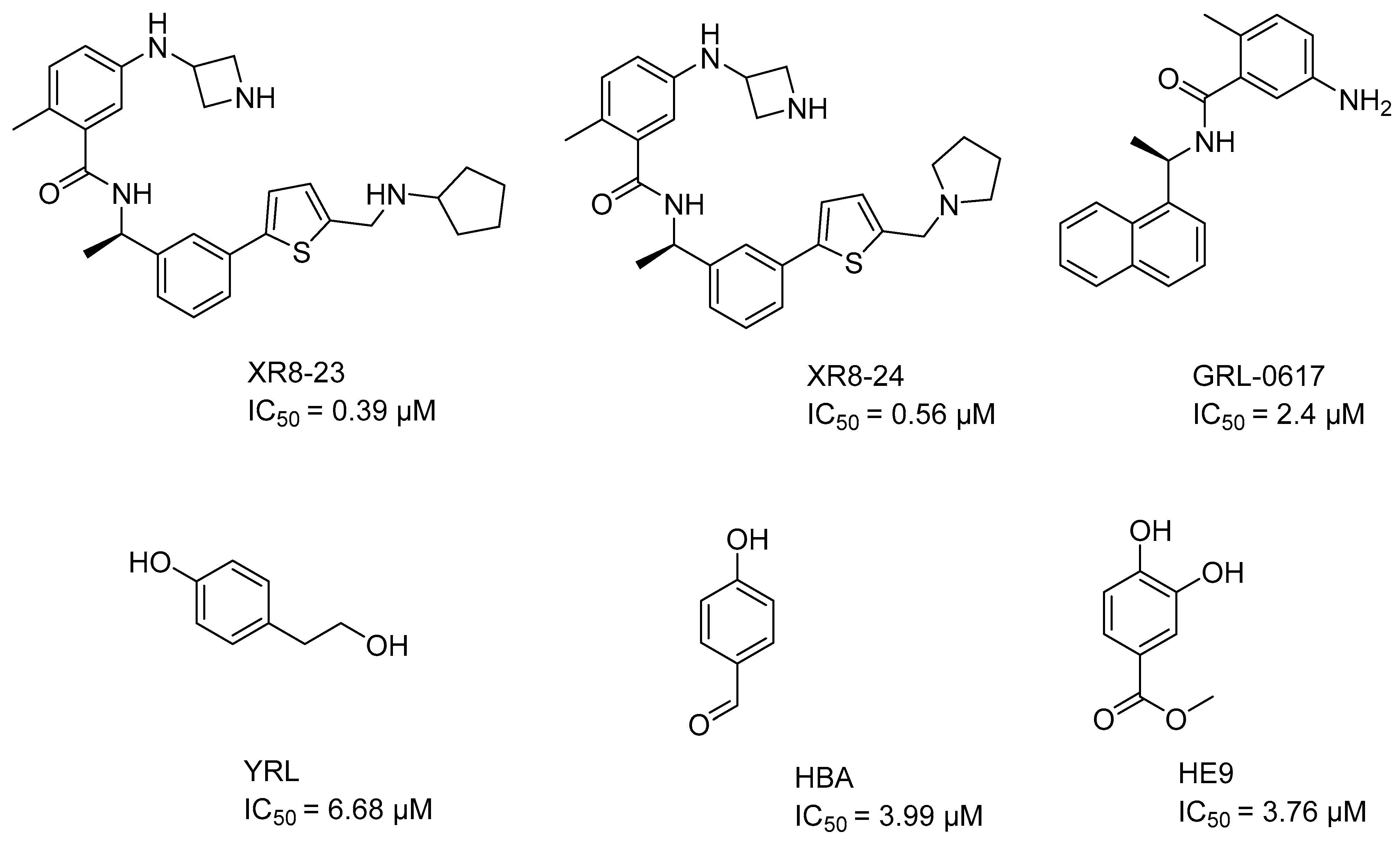
2.2. Evaluation of the Model in Antivirals from the Market
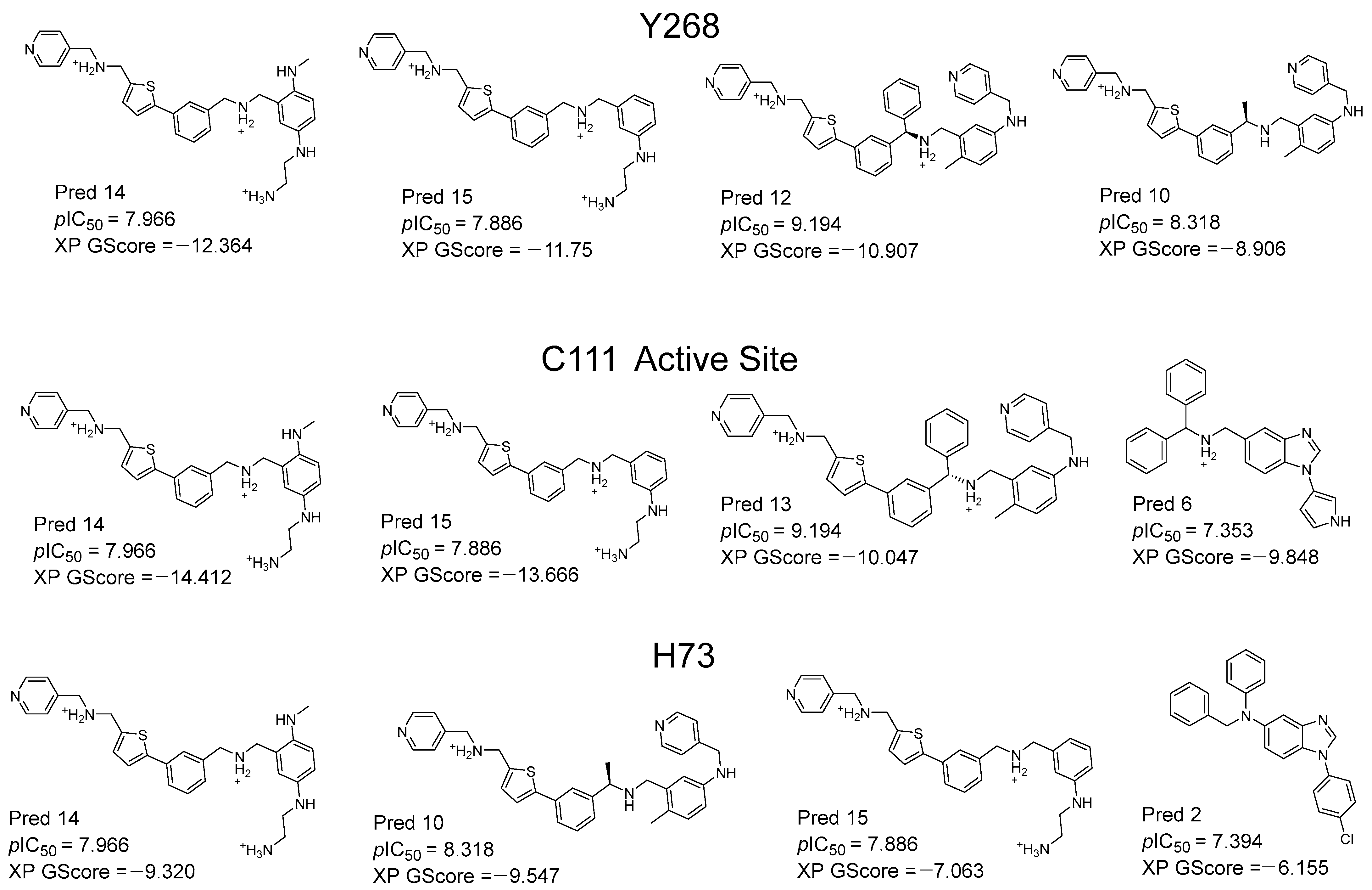
2.3. Design and Proposal of New Compounds
3. Materials and Methods
3.1. Formulations of the QSAR Equation
3.2. Molecular Docking
3.3. Selection QSAR-Docking Criteria
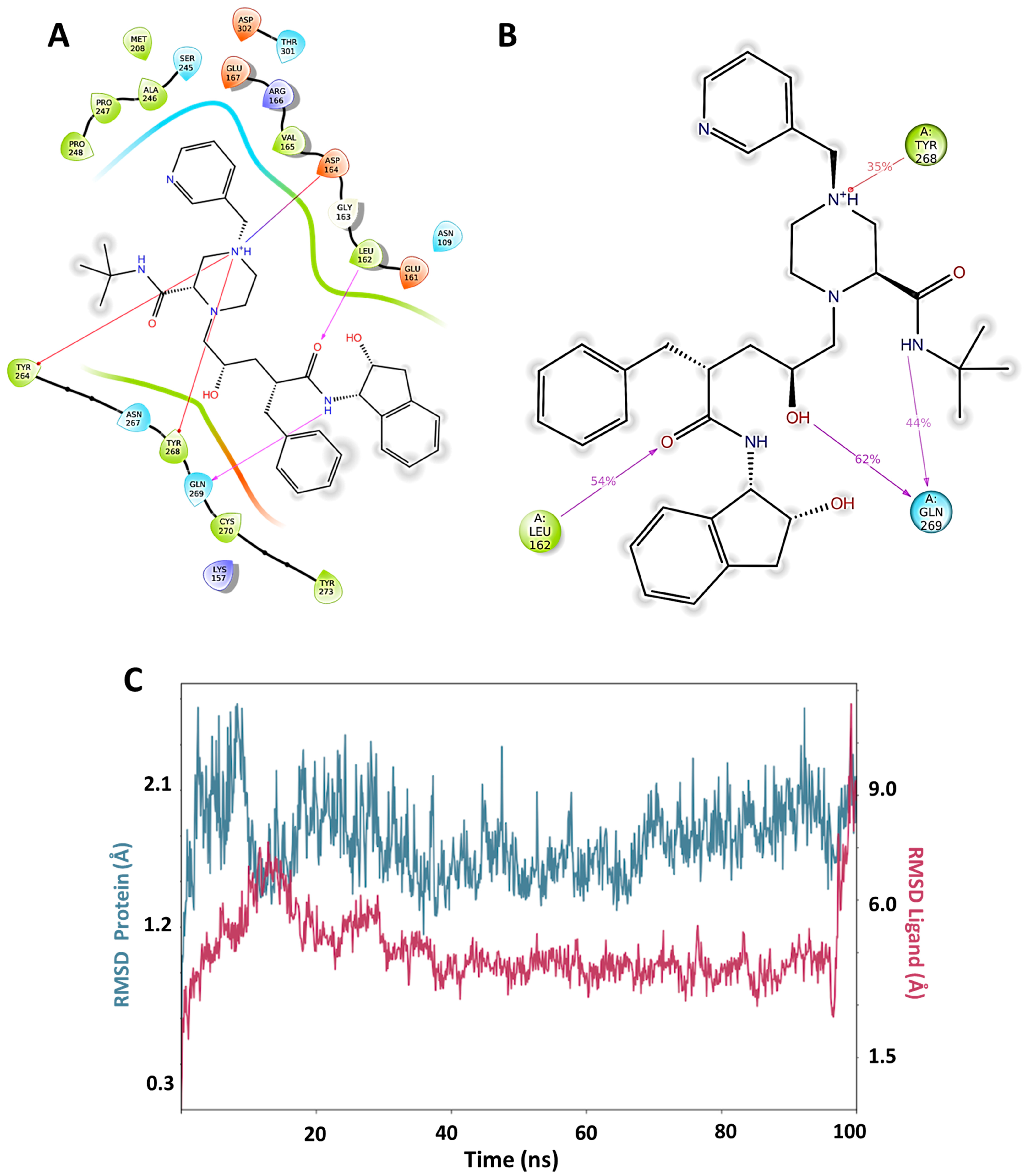
3.4. Molecular Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- COVID—Coronavirus Statistics—Worldometer. Available online: https://www.worldometers.info/coronavirus/ (accessed on 25 March 2024).
- Polack, F.; Thomas, S.; Kitchin, N.; Absalon, J.; Gurtman, A. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Faksova, K.; Walsh, D.; Jiang, Y.; Griffin, J.; Phillips, A.; Gentile, A.; Kwong, J.C.; Macartney, K.; Naus, M.; Grange, Z.; et al. COVID-19 Vaccines and Adverse Events of Special Interest: A Multinational Global Vaccine Data Network (GVDN) Cohort Study of 99 Million Vaccinated Individuals. Vaccine 2024, 42, 2200–2211. [Google Scholar] [CrossRef] [PubMed]
- COVID-19 Vaccine Tracker and Landscape. Available online: https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines (accessed on 25 March 2024).
- Röltgen, K.; Boyd, S.D. Antibody and B Cell Responses to SARS-CoV-2 Infection and Vaccination: The End of the Beginning. Annu. Rev. Pathol.-Mech. Dis. 2024, 19, 69–97. [Google Scholar] [CrossRef] [PubMed]
- Heaney, C.D.; Hempel, H.; DeRosa, K.L.; Pinto, L.A.; Mantis, N.J. Clinical Assessment of SARS-CoV-2 Antibodies in Oral Fluids Following Infection and Vaccination. Clin. Chem. 2024, 70, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Barros-Martins, J.; Hammerschmidt, S.I.; Cossmann, A.; Odak, I.; Stankov, M.V.; Morillas Ramos, G.; Dopfer-Jablonka, A.; Heidemann, A.; Ritter, C.; Friedrichsen, M.; et al. Immune Responses against SARS-CoV-2 Variants after Heterologous and Homologous ChAdOx1 nCoV-19/BNT162b2 Vaccination. Nat. Med. 2021, 27, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Velásquez, P.A.; Hernandez, J.C.; Galeano, E.; Hincapié-García, J.; Rugeles, M.T.; Zapata-Builes, W. Effectiveness of Drug Repurposing and Natural Products Against SARS-CoV-2: A Comprehensive Review. Clin. Pharmacol.-Adv. Appl. 2024, 16, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Moshawih, S.; Jarrar, Q.; Bahrin, A.A.; Lim, A.F.; Ming, L.; Goh, H.P. Evaluating NSAIDs in SARS-CoV-2: Immunomodulatory Mechanisms and Future Therapeutic Strategies. Heliyon 2024, 10, e25734. [Google Scholar] [CrossRef]
- Schultz, D.C.; Johnson, R.M.; Ayyanathan, K.; Miller, J.; Whig, K.; Kamalia, B.; Dittmar, M.; Weston, S.; Hammond, H.L.; Dillen, C.; et al. Pyrimidine Inhibitors Synergize with Nucleoside Analogues to Block SARS-CoV-2. Nature 2022, 604, 134–140. [Google Scholar] [CrossRef]
- Bai, Y.; Du, Z.; Wang, L.; Lau, E.H.Y.; Fung, I.C.-H.; Holme, P.; Cowling, B.J.; Galvani, A.P.; Krug, R.M.; Meyers, L.A. Public Health Impact of Paxlovid as Treatment for COVID-19, United States. Emerg. Infect. Dis. 2024, 30, 262–269. [Google Scholar] [CrossRef]
- Philippidis, A. Rising from the Ashes: FDA Grants EUA to Invivyd COVID-19 Antibody. Genet. Eng. Biotechnol. News 2024, 6, 274–278. [Google Scholar] [CrossRef]
- Cho, C.-C.; Li, S.G.; Lalonde, T.J.; Yang, K.S.; Yu, G.; Qiao, Y.; Xu, S.; Ray Liu, W. Drug Repurposing for the SARS-CoV-2 Papain-Like Protease. ChemMedChem 2022, 17, e202100455. [Google Scholar] [CrossRef]
- Menéndez-Arias, L. Decoding Molnupiravir-Induced Mutagenesis in SARS-CoV-2. J. Biol. Chem. 2021, 297, 100867. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhu, Q.; Xing, X.; Sun, D. A Mini-Review on the Common Antiviral Drug Targets of Coronavirus. Microorganisms 2024, 12, 600. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xie, X.; Luo, H.; Qian, R.; Yang, Y.; Yu, H.; Huang, J.; Shi, P.-Y.; Hu, Q. Resistance Mechanisms of SARS-CoV-2 3CLpro to the Non-Covalent Inhibitor WU-04. Cell Discov. 2024, 10, 40. [Google Scholar] [CrossRef]
- Mody, V.; Ho, J.; Wills, S.; Mawri, A.; Lawson, L.; Ebert, M.C.C.J.C.; Fortin, G.M.; Rayalam, S.; Taval, S. Identification of 3-Chymotrypsin like Protease (3CLPro) Inhibitors as Potential Anti-SARS-CoV-2 Agents. Commun. Biol. 2021, 4, 93. [Google Scholar] [CrossRef]
- Molavi, Z.; Razi, S.; Mirmotalebisohi, S.A.; Adibi, A.; Sameni, M.; Karami, F.; Niazi, V.; Niknam, Z.; Aliashrafi, M.; Taheri, M.; et al. Identification of FDA Approved Drugs against SARS-CoV-2 RNA Dependent RNA Polymerase (RdRp) and 3-Chymotrypsin-like Protease (3CLpro), Drug Repurposing Approach. Biomed. Pharmacother. 2021, 138, 111544. [Google Scholar] [CrossRef]
- Yao, Z.; Zhang, L.; Duan, Y.; Tang, X.; Lu, J. Molecular Insights into the Adaptive Evolution of SARS-CoV-2 Spike Protein. J. Infect. 2024, 88, 106121. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.; Xu, W.; Liu, S. Structural and Functional Properties of SARS-CoV-2 Spike Protein: Potential Antivirus Drug Development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Zhou, B.; Chen, D.; Zhang, T.; Song, C.; Zhang, X.; Lin, L.; Huang, J.; Peng, X.; Liu, Y.; Wu, G.; et al. Recent Advancements in the Discovery of Small-Molecule Non-Nucleoside Inhibitors Targeting SARS-CoV-2 RdRp. Biomed. Pharmacother. 2024, 171, 116180. [Google Scholar] [CrossRef]
- Elfiky, A.A. SARS-CoV-2 RNA Dependent RNA Polymerase (RdRp) Targeting: An in Silico Perspective. J. Biomol. Struct. Dyn. 2021, 39, 3204–3212. [Google Scholar] [CrossRef]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-Dependent RNA Polymerase from COVID-19 Virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Yu, R.; Xue, H.; Jin, Z.; Zhang, M.; Bao, Y.; Wang, Z.; Wei, H.; Qiao, X.; Yang, H. Chrysin 7-O-β-D-Glucuronide, a Dual Inhibitor of SARS-CoV-2 3CLpro and PLpro, for the Prevention and Treatment of COVID-19. Int. J. Antimicrob. Agents 2024, 63, 107039. [Google Scholar] [CrossRef]
- Amin, S.A.; Banerjee, S.; Ghosh, K.; Gayen, S.; Jha, T. Protease Targeted COVID-19 Drug Discovery and Its Challenges: Insight into Viral Main Protease (Mpro) and Papain-like Protease (PLpro) Inhibitors. Bioorg. Med. Chem. 2021, 29, 115860. [Google Scholar] [CrossRef] [PubMed]
- Báez-Santos, Y.M.; St John, S.E.; Mesecar, A.D. The SARS-Coronavirus Papain-like Protease: Structure, Function and Inhibition by Designed Antiviral Compounds. Antiviral Res. 2015, 115, 21–38. [Google Scholar] [CrossRef]
- Ibrahim, T.M.; Ismail, M.I.; Bauer, M.R.; Bekhit, A.A.; Boeckler, F.M. Supporting SARS-CoV-2 Papain-Like Protease Drug Discovery: In Silico Methods and Benchmarking. Front. Chem. 2020, 8, 592289. [Google Scholar] [CrossRef]
- van Huizen, M.; Horst, J.R.B.; de Gruyter, H.L.M.; Geurink, P.P.; van der Heden van Noort, G.J.; Knaap, R.C.M.; Nelemans, T.; Ogando, N.S.; Leijs, A.A.; Urakova, N.; et al. Deubiquitinating Activity of SARS-CoV-2 Papain-like Protease Does Not Influence Virus Replication or Innate Immune Responses in Vivo. PLoS Pathog. 2024, 20, e1012100. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K. Papain-like Protease Regulates SARS-CoV-2 Viral Spread and Innate Immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Harcourt, B.H.; Jukneliene, D.; Kanjanahaluethai, A.; Bechill, J.; Severson, K.M.; Smith, C.M.; Rota, P.A.; Baker, S.C. Identification of Severe Acute Respiratory Syndrome Coronavirus Replicase Products and Characterization of Papain-Like Protease Activity. J. Virol. 2004, 78, 13600–13612. [Google Scholar] [CrossRef]
- Davis, M.E.; Gack, M.U. Ubiquitination in the Antiviral Immune Response. Virology 2015, 479–480, 52–65. [Google Scholar] [CrossRef]
- Kikkert, M. Innate Immune Evasion by Human Respiratory RNA Viruses. J. Innate Immun. 2019, 12, 4–20. [Google Scholar] [CrossRef]
- Ma-Lauer, Y.; Carbajo-Lozoya, J.; Hein, M.Y.; Müller, M.A.; Deng, W. P53 Down-Regulates SARS Coronavirus Replication and Is Targeted by the SARS-Unique Domain and PLpro via E3 Ubiquitin Ligase RCHY1. Proc. Natl. Acad. Sci. USA 2016, 113, E5192–E5201. [Google Scholar] [CrossRef] [PubMed]
- Leng, R.P.; Lin, Y.; Ma, W.; Wu, H.; Lemmers, B.; Chung, S.; Parant, J.M.; Lozano, G.; Hakem, R.; Benchimol, S. Pirh2, a P53-Induced Ubiquitin-Protein Ligase, Promotes P53 Degradation. Cell 2003, 112, 779–791. [Google Scholar] [CrossRef]
- Li, M.; Brooks, C.L.; Wu-Baer, F.; Chen, D.; Baer, R.; Gu, W. Mono- Versus Polyubiquitination: Differential Control of P53 Fate by Mdm2. Science 2003, 302, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Qin, B.; Chen, P.; Zhu, K.; Hou, P.; Wojdyla, J.A.; Wang, M.; Cui, S. Crystal Structure of SARS-CoV-2 Papain-like Protease. Acta Pharm. Sin. B 2021, 11, 237–245. [Google Scholar] [CrossRef]
- Sanachai, K.; Mahalapbutr, P.; Lee, V.S.; Rungrotmongkol, T.; Hannongbua, S. In Silico Elucidation of Potent Inhibitors and Rational Drug Design against SARS-CoV-2 Papain-like Protease. J. Phys. Chem. B 2021, 125, 13644–13656. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Shaw, N.; Yan, L.; Lou, Z.; Rao, Z. Structural View and Substrate Specificity of Papain-like Protease from Avian Infectious Bronchitis Virus. J. Biol. Chem. 2015, 290, 7160–7168. [Google Scholar] [CrossRef]
- Tan, B.; Zhang, X.; Ansari, A.; Jadhav, P.; Tan, H.; Li, K.; Chopra, A.; Ford, A.; Chi, X.; Ruiz, F.X.; et al. Design of a SARS-CoV-2 Papain-like Protease Inhibitor with Antiviral Efficacy in a Mouse Model. Science 2024, 383, 1434–1440. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, P.; Huang, B.; Osipiuk, J.; Zhang, X.; Tan, H.; Tesar, C.; Endres, M.; Jedrzejczak, R.; Tan, B.; Deng, X.; et al. Structure-Based Design of SARS-CoV-2 Papain-like Protease Inhibitors. Eur. J. Med. Chem. 2024, 264, 116011. [Google Scholar] [CrossRef]
- Kralj, S.; Jukič, M.; Bahun, M.; Kranjc, L.; Kolarič, A.; Hodošček, M.; Ulrih, N.P.; Bren, U. Identification of Triazolopyrimidinyl Scaffold SARS-CoV-2 Papain-Like Protease (PLpro) Inhibitor. Pharmaceutics 2024, 16, 169. [Google Scholar] [CrossRef]
- Osipiuk, J.; Azizi, S.-A.; Dvorkin, S.; Endres, M.; Jedrzejczak, R.; Jones, K.A.; Kang, S.; Kathayat, R.S.; Kim, Y.; Lisnyak, V.G.; et al. Structure of Papain-like Protease from SARS-CoV-2 and Its Complexes with Non-Covalent Inhibitors. Nat. Commun. 2021, 12, 743. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, V.; Brognaro, H.; Prabhu, P.R.; de Souza, E.E.; Günther, S.; Reinke, P.Y.A.; Lane, T.J.; Ginn, H.; Han, H.; Ewert, W.; et al. SARS-CoV-2 Papain-like Protease PLpro in Complex with Natural Compounds Reveal Allosteric Sites for Antiviral Drug Design. Commun. Biol. 2022, 5, 805. [Google Scholar] [CrossRef]
- Ratia, K.; Pegan, S.; Takayama, J.; Sleeman, K.; Coughlin, M. A Noncovalent Class of Papain-like Protease/Deubiquitinase Inhibitors Blocks SARS Virus Replication. Proc. Natl. Acad. Sci. USA 2008, 105, 16119–16124. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Ratia, K.; Cooper, L.; Kong, D.; Lee, H. Design of SARS-CoV-2 PLpro Inhibitors for COVID-19 Antiviral Therapy Leveraging Binding Cooperativity. J. Med. Chem. 2022, 65, 2940–2955. [Google Scholar] [CrossRef]
- Freitas, B.T.; Durie, I.A.; Murray, J.; Longo, J.E.; Miller, H.C.; Crich, D.; Hogan, R.J.; Tripp, R.A.; Pegan, S.D. Characterization and Noncovalent Inhibition of the Deubiquitinase and deISGylase Activity of SARS-CoV-2 Papain-Like Protease. ACS Infect. Dis. 2020, 6, 2099–2109. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.A.; Kato-Weinstein, J.; Li, Y.; Deng, Y.; Granet, R. Potential Therapeutic Agents and Associated Bioassay Data for COVID-19 and Related Human Coronavirus Infections. ACS Pharmacol. Transl. Sci. 2020, 3, 813–834. [Google Scholar] [CrossRef]
- Welker, A.; Kersten, C.; Müller, C.; Madhugiri, R.; Zimmer, C.; Müller, P.; Zimmermann, R.; Hammerschmidt, S.; Maus, H.; Ziebuhr, J.; et al. Structure-Activity Relationships of Benzamides and Isoindolines Designed as SARS-CoV Protease Inhibitors Effective against SARS-CoV-2. ChemMedChem 2021, 16, 340–354. [Google Scholar] [CrossRef] [PubMed]
- Di Sarno, V.; Lauro, G.; Musella, S.; Ciaglia, T.; Vestuto, V.; Sala, M.; Scala, M.C.; Smaldone, G.; Di Matteo, F.; Novi, S.; et al. Identification of a Dual Acting SARS-CoV-2 Proteases Inhibitor through in Silico Design and Step-by-Step Biological Characterization. Eur. J. Med. Chem. 2021, 226, 113863. [Google Scholar] [CrossRef]
- Ma, C.; Sacco, M.D.; Xia, Z.; Lambrinidis, G.; Townsend, J.A.; Hu, Y.; Meng, X.; Szeto, T.; Ba, M.; Zhang, X.; et al. Discovery of SARS-CoV-2 Papain-like Protease Inhibitors through a Combination of High-Throughput Screening and a FlipGFP-Based Reporter Assay. ACS Cent. Sci. 2021, 7, 1245–1260. [Google Scholar] [CrossRef]
- Li, L.; Ma, L.; Hu, Y.; Li, X.; Yu, M.; Shang, H.; Zou, Z. Natural Biflavones Are Potent Inhibitors against SARS-CoV-2 Papain-like Protease. Phytochemistry 2022, 193, 112984. [Google Scholar] [CrossRef]
- Meewan, I.; Kattoula, J.; Kattoula, J.Y.; Skinner, D.; Fajtová, P.; Giardini, M.A.; Woodworth, B.; McKerrow, J.H.; Lage de Siqueira-Neto, J.; O’Donoghue, A.J.; et al. Discovery of Triple Inhibitors of Both SARS-CoV-2 Proteases and Human Cathepsin L. Pharmaceuticals 2022, 15, 744. [Google Scholar] [CrossRef] [PubMed]
- Puhl, A.C.; Gomes, G.F.; Damasceno, S.; Godoy, A.S.; Noske, G.D.; Nakamura, A.M.; Gawriljuk, V.O.; Fernandes, R.S.; Monakhova, N.; Riabova, O.; et al. Pyronaridine Protects Against SARS-CoV-2 in Mouse. ACS Infect. Dis. 2022, 8, 1147–1160. [Google Scholar] [CrossRef]
- Xu, H.; Li, J.; Song, S.; Xiao, Z.; Chen, X.; Huang, B.; Sun, M.; Su, G.; Zhou, D.; Wang, G.; et al. Effective Inhibition of Coronavirus Replication by Polygonum Cuspidatum. Front. Biosci. 2021, 26, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.A.; Ghosh, K.; Gayen, S.; Jha, T. Chemical-Informatics Approach to COVID-19 Drug Discovery: Monte Carlo Based QSAR, Virtual Screening and Molecular Docking Study of Some in-House Molecules as Papain-like Protease (PLpro) Inhibitors. J. Biomol. Struct. Dyn. 2021, 39, 4764–4773. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Du, X.; Duan, Y.; Pan, X.; Sun, Y.; You, T.; Han, L.; Jin, Z.; Shang, W.; Yu, J.; et al. High-Throughput Screening Identifies Established Drugs as SARS-CoV-2 PLpro Inhibitors. Protein Cell 2021, 12, 877–888. [Google Scholar] [CrossRef] [PubMed]

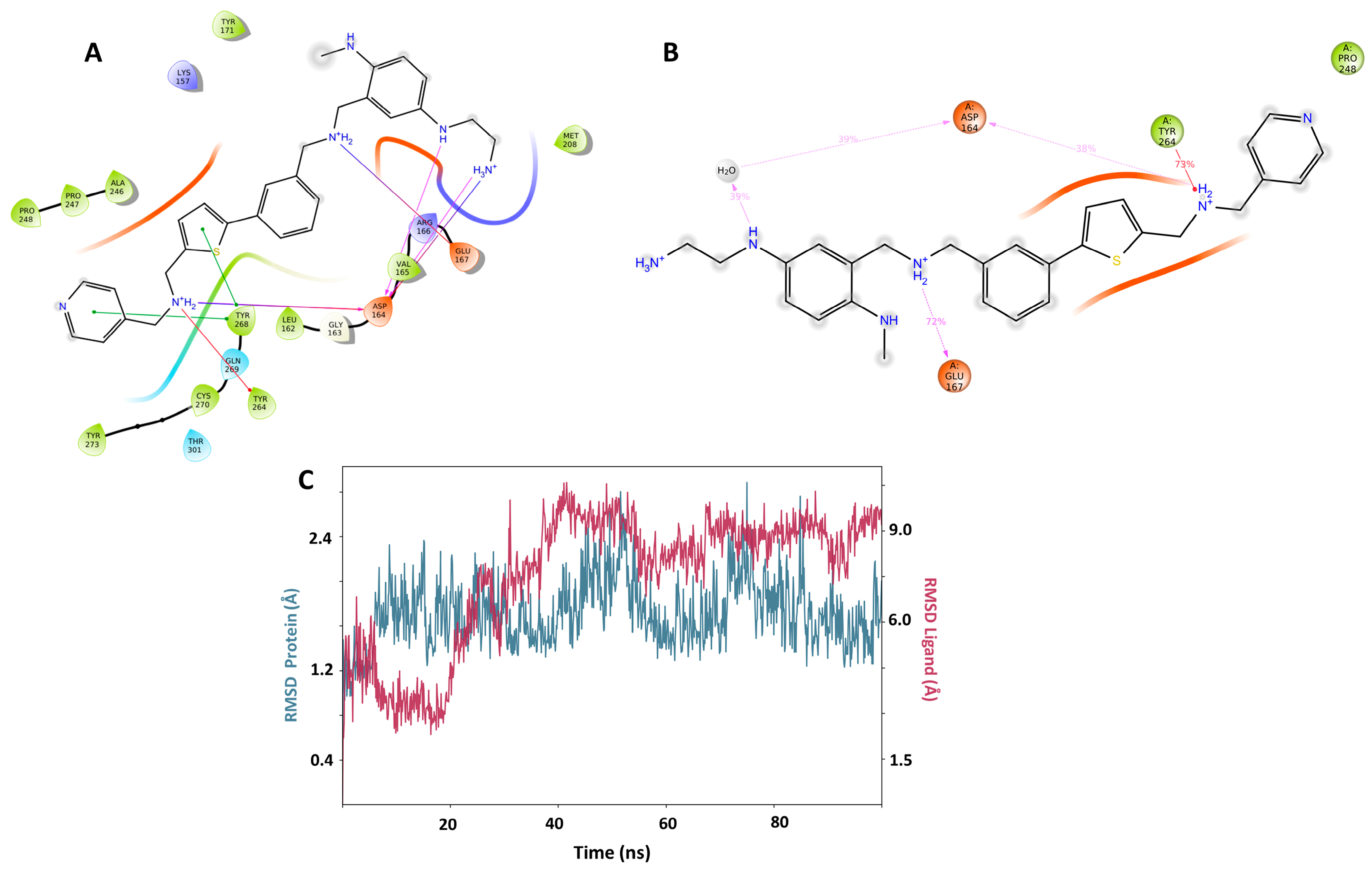
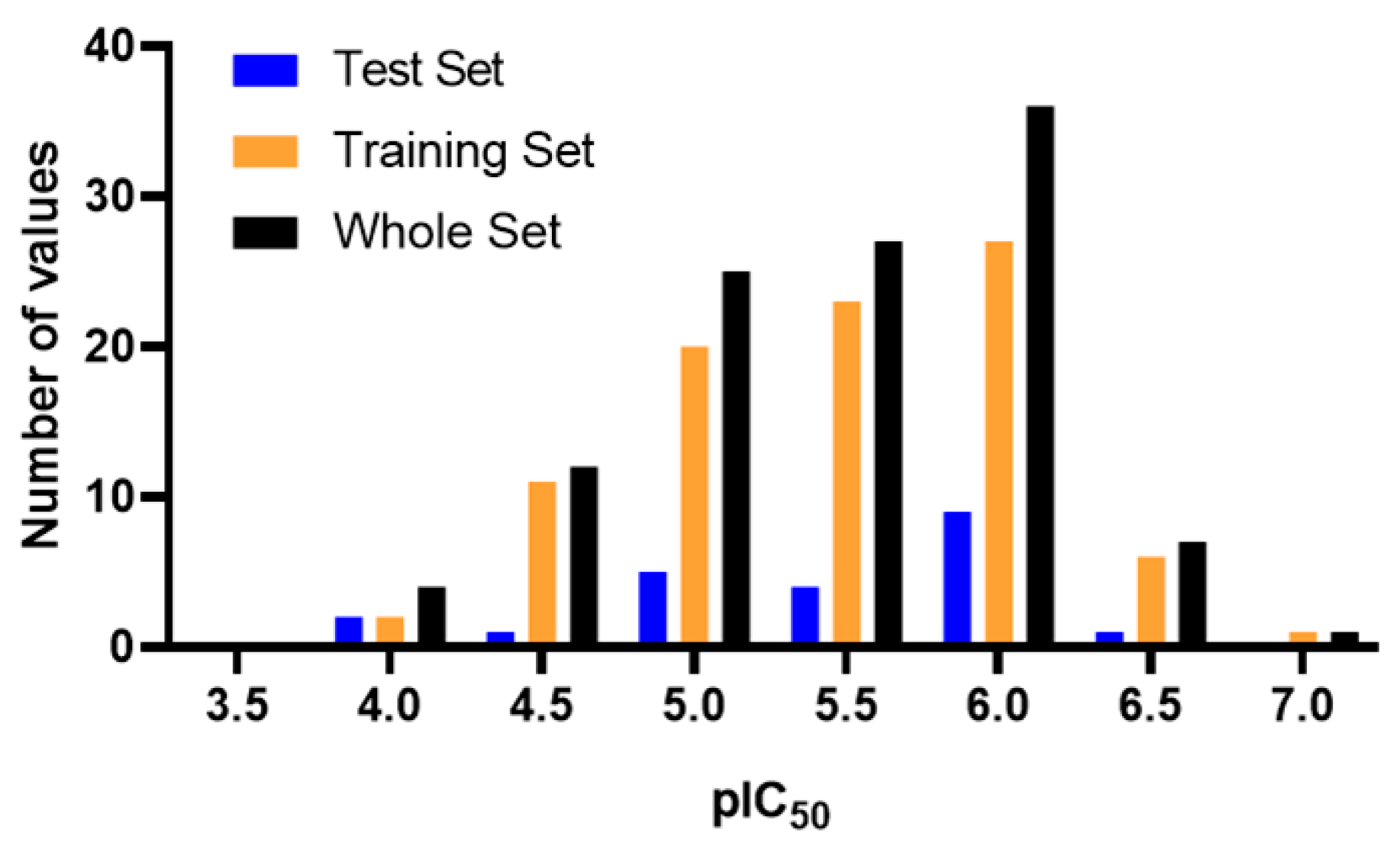

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Parameter | Threshold Value | Result |
|---|---|---|---|
| 1 | r2 | high | 0.8333 |
| 2 | >0.5 | ||
| 3 | >0.7 | ||
| 4 | >0.7 | ||
| 5 | >0.7 | ||
| 6 | Low | ||
| 7 | >0.85 | ||
| 8 | <0.5 | ||
| 9 | <0.6 | ||
| 10 | r2test | >0.6 | 0.721 |
| pIC50 | pIC50 | pIC50 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mol | Exp | Pred | Res | Mol | Exp | Pred | Res | Mol | Exp | Pred | Res |
| 1 | 5.7932 | 5.2703 | 0.52 | 39 | 6.1938 | 6.3979 | 0.20 | 77 | 3.9031 | 4.1783 | 0.28 |
| 2 | 5.9586 | 5.7228 | 0.24 | 40 | 6.3872 | 6.4158 | 0.03 | 78 * | 4.2157 | 4.3637 | 0.15 |
| 3 | 5.2596 | 5.5638 | 0.30 | 41 | 6.6778 | 6.3946 | 0.28 | 79 * | 5.6073 | 5.0538 | 0.55 |
| 4 | 5.2218 | 5.6027 | 0.38 | 42 | 6.3665 | 6.3969 | 0.03 | 80 | 5.2924 | 5.3276 | 0.04 |
| 5 | 5.9957 | 5.6569 | 0.34 | 43 | 6.9469 | 6.3889 | 0.56 | 81 | 5.1938 | 5.3670 | 0.17 |
| 6 | 6.2218 | 5.7595 | 0.46 | 44 * | 6.6021 | 6.3989 | 0.20 | 82 | 5.1549 | 5.2915 | 0.14 |
| 7 | 5.9208 | 5.5601 | 0.36 | 45 * | 6.0915 | 5.9576 | 0.13 | 83 * | 4.7773 | 4.8182 | 0.04 |
| 8 | 6.0969 | 5.6826 | 0.41 | 46 | 5.7447 | 5.8732 | 0.13 | 84 | 4.7077 | 4.5312 | 0.18 |
| 9 | 6.1549 | 5.8123 | 0.34 | 47 * | 5.9586 | 5.9038 | 0.05 | 85 | 4.7905 | 4.5555 | 0.23 |
| 10 | 5.7959 | 5.8425 | 0.05 | 48 | 5.6383 | 5.6343 | 0.00 | 86 | 4.8928 | 5.0501 | 0.16 |
| 11 | 5.3665 | 5.7334 | 0.37 | 49 | 5.8539 | 5.9360 | 0.08 | 87 | 4.6253 | 4.7200 | 0.09 |
| 12 | 5.6198 | 5.7446 | 0.12 | 50 | 5.5436 | 5.1288 | 0.41 | 88 | 4.7595 | 4.7559 | 0.00 |
| 13 | 5.4089 | 5.4229 | 0.01 | 51 | 5.1612 | 5.1707 | 0.01 | 89 | 4.6882 | 4.6716 | 0.02 |
| 14 | 5.8861 | 5.8705 | 0.02 | 52 | 4.8239 | 4.4353 | 0.39 | 90 | 4.4123 | 4.6861 | 0.27 |
| 15 * | 5.4815 | 5.5415 | 0.06 | 53 * | 5.1249 | 5.2728 | 0.15 | 91 | 4.7160 | 5.1436 | 0.43 |
| 16 | 5.2147 | 5.6203 | 0.41 | 54 | 5.0000 | 5.4479 | 0.45 | 92 | 5.1487 | 5.4517 | 0.30 |
| 17 | 4.9706 | 4.9969 | 0.03 | 55 | 5.0000 | 5.1307 | 0.13 | 93 * | 4.9314 | 5.3882 | 0.46 |
| 18 | 5.4815 | 5.4849 | 0.00 | 56 | 5.1192 | 5.3934 | 0.27 | 94 | 5.2899 | 5.8119 | 0.52 |
| 19 | 5.6198 | 5.9756 | 0.36 | 57 * | 5.2441 | 5.4936 | 0.25 | 95 | 6.2076 | 5.7904 | 0.42 |
| 20 | 4.9626 | 5.2422 | 0.28 | 58 | 4.8861 | 4.7325 | 0.15 | 96 | 5.8013 | 5.7898 | 0.01 |
| 21 * | 5.7959 | 6.1161 | 0.32 | 59 | 4.3645 | 4.6669 | 0.30 | 97 * | 5.8861 | 5.7878 | 0.10 |
| 22 | 5.7212 | 5.8610 | 0.14 | 60 | 4.5258 | 4.6212 | 0.10 | 98 | 5.6073 | 5.8467 | 0.24 |
| 23 | 5.7447 | 6.0421 | 0.30 | 61 | 4.5058 | 4.6240 | 0.12 | 99 | 6.1805 | 5.8153 | 0.37 |
| 24 | 5.5528 | 5.1787 | 0.37 | 62 | 4.4584 | 4.5846 | 0.13 | 100 | 6.1739 | 5.8121 | 0.36 |
| 25 | 6.2291 | 5.9227 | 0.31 | 63 * | 4.4389 | 4.7455 | 0.31 | 101 | 5.9393 | 5.8987 | 0.04 |
| 26 | 5.8861 | 5.9265 | 0.04 | 64 * | 5.0223 | 4.6103 | 0.41 | 102 * | 5.3990 | 5.6804 | 0.28 |
| 27 | 5.7447 | 5.7750 | 0.03 | 65 | 4.5800 | 4.5345 | 0.05 | 103 | 5.8861 | 5.9937 | 0.11 |
| 28 * | 6.0458 | 6.3180 | 0.27 | 66 | 4.6421 | 4.5707 | 0.07 | 104 | 5.1878 | 5.2057 | 0.02 |
| 29 | 6.4089 | 6.4614 | 0.05 | 67 * | 4.1209 | 4.0921 | 0.03 | 105 | 5.0511 | 5.1481 | 0.10 |
| 30 | 6.2518 | 6.4008 | 0.15 | 68 | 4.0400 | 4.1036 | 0.06 | 106 | 5.5654 | 5.1510 | 0.41 |
| 31 | 6.1249 | 6.0665 | 0.06 | 69 | 6.1938 | 6.3859 | 0.19 | 107 | 5.2596 | 5.3131 | 0.05 |
| 32 | 6.0132 | 5.8246 | 0.19 | 70 * | 6.1675 | 5.7847 | 0.38 | 108 | 5.2132 | 5.3635 | 0.15 |
| 33 | 6.0915 | 5.7794 | 0.31 | 71 * | 6.2076 | 6.1755 | 0.03 | 109 | 6.0506 | 5.6907 | 0.36 |
| 34 | 6.0362 | 6.0576 | 0.02 | 72 | 5.7305 | 5.6786 | 0.05 | 110 | 5.3546 | 5.4823 | 0.13 |
| 35 | 6.0862 | 6.0020 | 0.08 | 73 | 5.4841 | 5.6207 | 0.14 | 111 * | 5.8416 | 5.4159 | 0.43 |
| 36 | 6.1938 | 6.3055 | 0.11 | 74 | 5.6126 | 5.6438 | 0.03 | 112 | 5.0353 | 5.6163 | 0.58 |
| 37 * | 6.1549 | 6.3037 | 0.15 | 75 * | 5.5918 | 5.1286 | 0.46 | 113 * | 5.0888 | 5.1672 | 0.08 |
| 38 | 6.4815 | 6.2844 | 0.20 | 76 | 5.6576 | 5.2757 | 0.38 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabadini, G.; Mellado, M.; Morales, C.; Mella, J. Arylamines QSAR-Based Design and Molecular Dynamics of New Phenylthiophene and Benzimidazole Derivatives with Affinity for the C111, Y268, and H73 Sites of SARS-CoV-2 PLpro Enzyme. Pharmaceuticals 2024, 17, 606. https://doi.org/10.3390/ph17050606
Sabadini G, Mellado M, Morales C, Mella J. Arylamines QSAR-Based Design and Molecular Dynamics of New Phenylthiophene and Benzimidazole Derivatives with Affinity for the C111, Y268, and H73 Sites of SARS-CoV-2 PLpro Enzyme. Pharmaceuticals. 2024; 17(5):606. https://doi.org/10.3390/ph17050606
Chicago/Turabian StyleSabadini, Gianfranco, Marco Mellado, César Morales, and Jaime Mella. 2024. "Arylamines QSAR-Based Design and Molecular Dynamics of New Phenylthiophene and Benzimidazole Derivatives with Affinity for the C111, Y268, and H73 Sites of SARS-CoV-2 PLpro Enzyme" Pharmaceuticals 17, no. 5: 606. https://doi.org/10.3390/ph17050606
APA StyleSabadini, G., Mellado, M., Morales, C., & Mella, J. (2024). Arylamines QSAR-Based Design and Molecular Dynamics of New Phenylthiophene and Benzimidazole Derivatives with Affinity for the C111, Y268, and H73 Sites of SARS-CoV-2 PLpro Enzyme. Pharmaceuticals, 17(5), 606. https://doi.org/10.3390/ph17050606