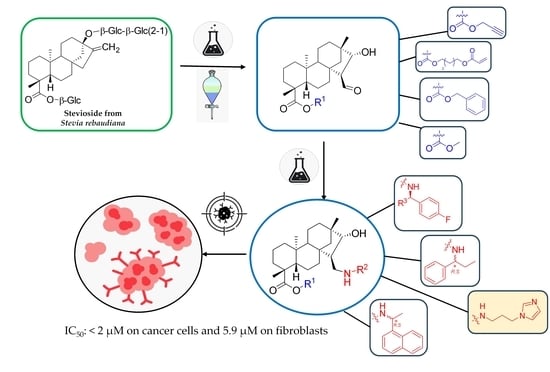
3.1. Chemistry
General methods: commercially available reagents were used as obtained from the suppliers (Molar Chemicals Ltd., Halásztelek, Hungary; Merck Ltd., Budapest, Hungary and VWR International Ltd., Debrecen, Hungary), while solvents were dried according to standard procedures. Optical rotations were measured in MeOH at 20 °C with a Perkin-Elmer 341 polarimeter (PerkinElmer Inc., Shelton, CT, USA). Chromatographic separations and monitoring of the reactions were carried out on Merck Kieselgel 60 (Merck Ltd., Budapest, Hungary). Melting points were determined on a Kofler apparatus (Nagema, Dresden, Germany).
1H- and
13C-NMR spectra were recorded on a Brucker Avance DRX 500 spectrometer (500 MHz (
1H) and 125 MHz (
13C), δ = 0 (TMS)). Chemical shifts were expressed in ppm (δ) relative to the TMS as an internal reference.
J values were presented as Hz. All
1H/
13C NMR, NOESY, 2D-HMBC, and 2D-HSQC spectra are available in the
Supplementary Materials. An HRMS flow injection analysis was performed with a Thermo Scientific Q Exactive Plus hybrid quadrupole-Orbitrap (Thermo Fisher Scientific, Waltham, MA, USA) mass spectrometer coupled to a Waters Acquity I-Class UPLC™ (Waters, Manchester, UK).
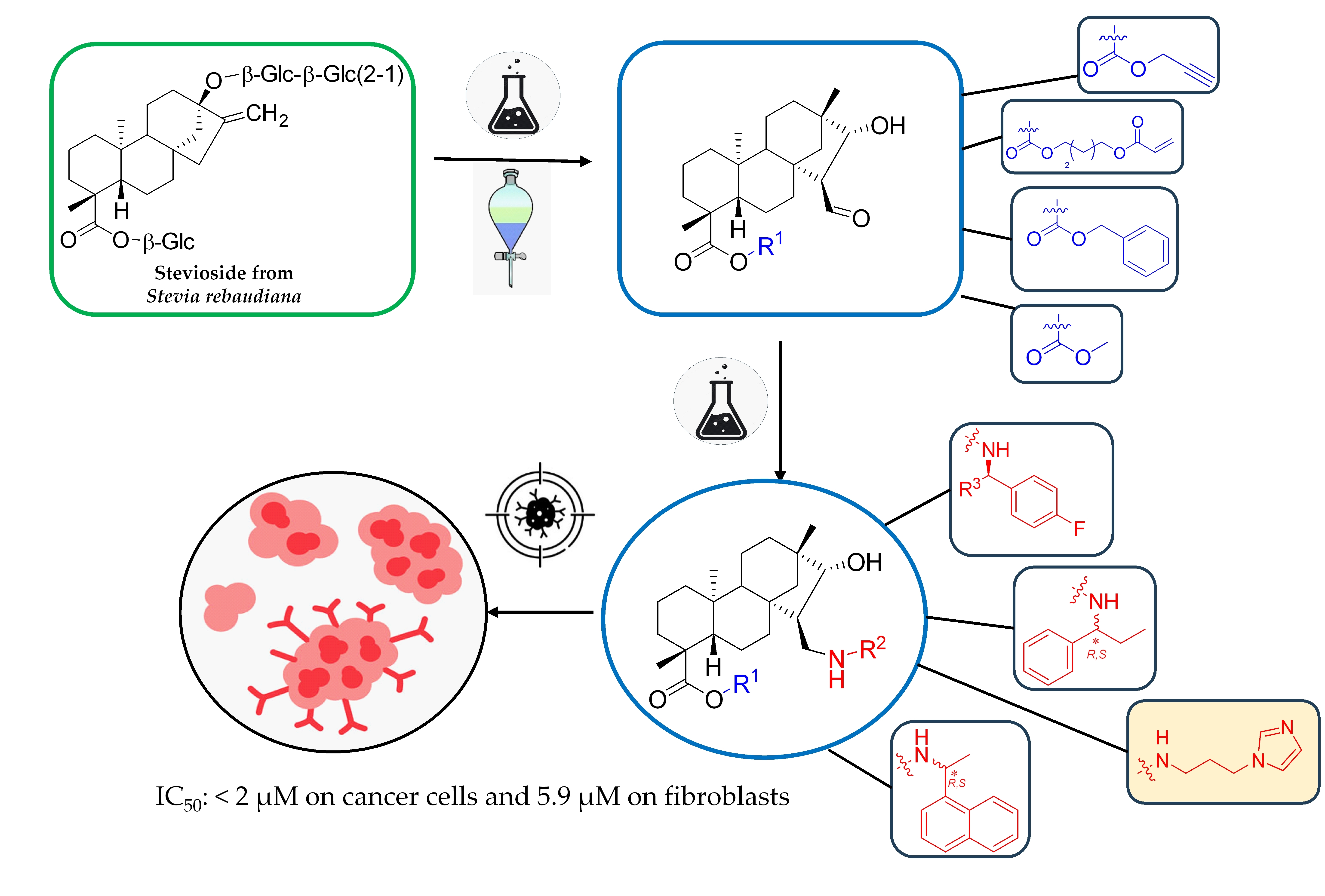
Starting materials: stevioside was obtained from Molar Chemicals Ltd., Halásztelek, Hungary. Isosteviol
1 was prepared from commercially available stevioside or a mixture of steviol glycosides in a one-step synthesis according to the literature, and all spectroscopic data were the same as described in the literature [
29].
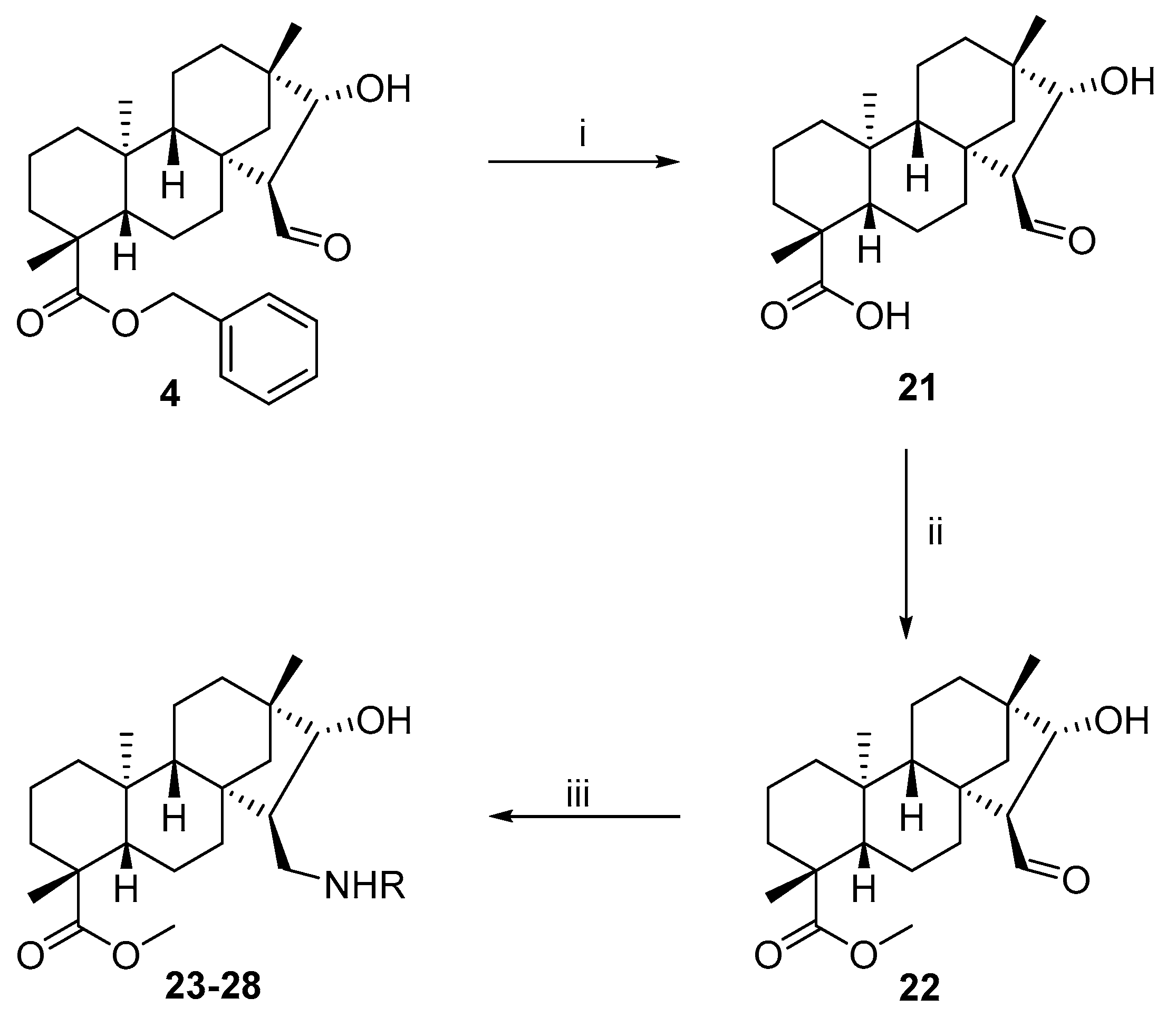
Compounds
2 [
30],
3 [
31],
9 [
32],
21 [
33], and
22 [
26] were prepared by the methods presented in the literature. Their spectroscopic data and physical and chemical properties were similar to those reported in the literature.
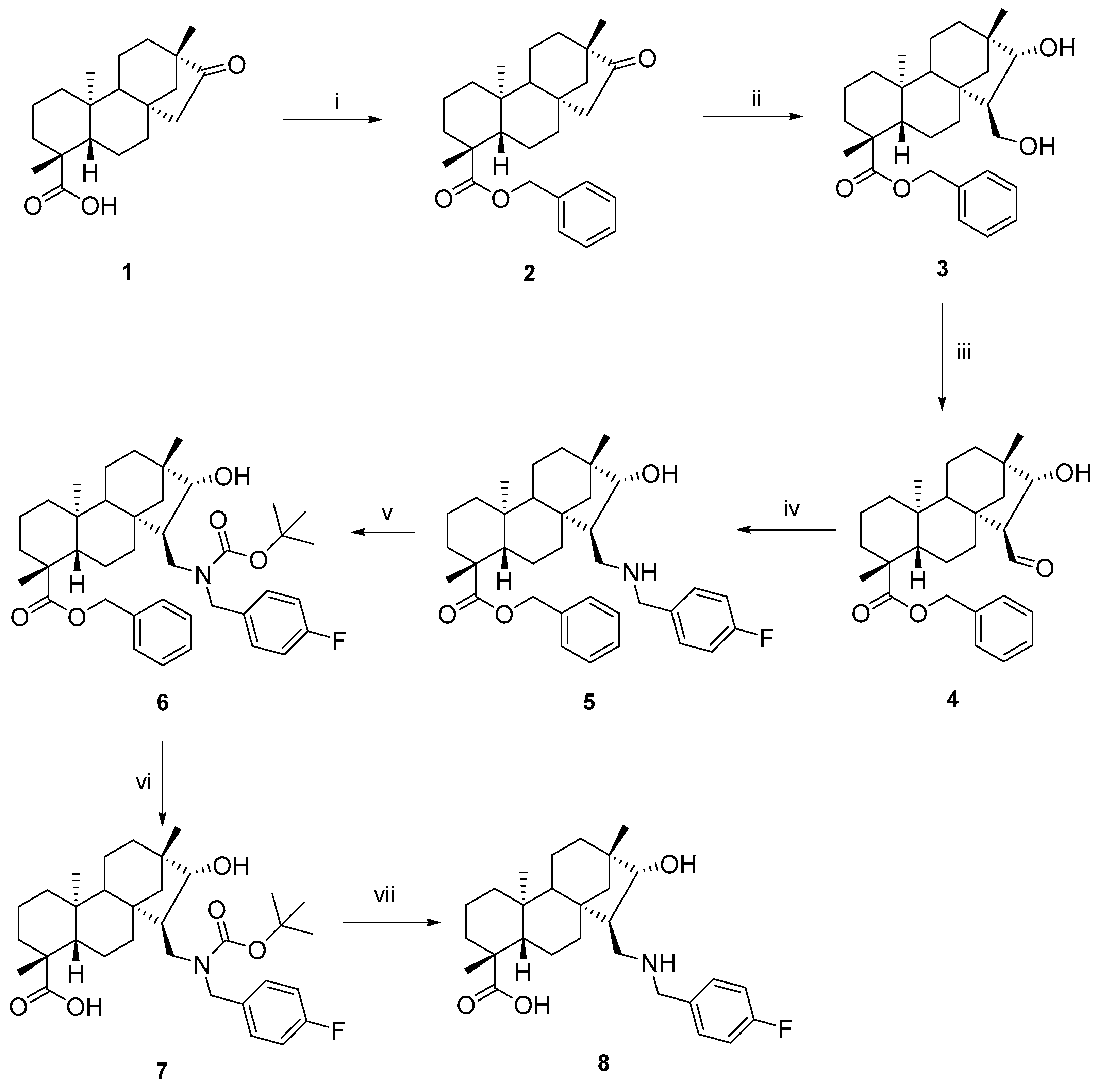
3.1.1. (4R,4aS,6aS,7S,8R,9S,11bS)-Benzyl 7-formyl-8-hydroxy-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (4)
To a solution of 3 (6.81 mmol, 3.00 g) in 1/1 DCM/H2O (300 mL), TEMPO (10 mol%, 106 mg), NBS (13.62 mmol, 2.42 g), and TBAB (6.81 mmol, 2.20 g) were added. After a 12 h reflux, the reaction was found to be completed (indicated by TLC), and the mixture was extracted with DCM (3 × 100 mL). The combined organic phase was extracted with water (1 × 100 mL), dried (Na2SO4), filtered, and concentrated. The purification of the crude product was accomplished by column chromatography on silica gel with an appropriate solvent mixture (n-hexane/EtOAc = 3:1). Yield: 2.48 g (83%); colourless oil; [α]D20 = −69 (c 0.71 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.79 (s, 3H), 0.86–0.91 (m, 1H), 0.95 (s, 3H), 0.99–1.04 (m, 3H), 1.12–1.15 (m, 1H), 1.19 (s, 3H), 1.22–1.23 (m, 1H), 1.33–1.36 (m, 1H), 1.41–1.45 (m, 1H), 1.50–1.4 (m, 1H), 1.64–1.85 (m, 8H), 2.22 (d, 1H, J = 13.2 Hz), 2.85–2.86 (m, 1H), 4.27 (d, 1H, J = 4.9 Hz), 5.03 (d, 1H, J = 12.2 Hz), 5.14 (d, 1H, J = 12.2 Hz), 7.31–7.39 (m, 5H), 9.77 (d, 1H, J = 2.2 Hz); 13C-NMR (125 MHz, CDCl3) δ (ppm) 13.1 (CH3), 18.8 (CH2), 19.7 (CH2), 21.8 (CH2), 24.5 (CH3), 28.8 (CH3), 33.0 (CH2), 36.0 (CH2), 38.1 (Cq), 38.4 (Cq), 39.6 (CH2), 41.1 (Cq), 43.9 (Cq), 46.5 (Cq), 53.9 (CH2), 56.9 (CH), 57.3 (CH), 61.7 (CH), 66.3 (CH2), 78.2 (CH), 128.3 (CH), 128.6 (2 × CH), 128.6 (2 × CH), 135.9 (Cq), 177.1 (C=O), 204.0 (CHO). C28H38O4: 438.5989. HRMS (ESI+): m/z calcd. for C28H39O4 [M + H]+ 439.2848; found 439.28230.
3.1.2. (4R,4aS,6aS,7R,8R,9S,11bS)-Benzyl 7-(((4-fluorobenzyl)amino)methyl)-8-hydroxy-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (5)
To a solution of 4 (1.14 mmol, 500 mg) in dry EtOH (20 mL), 4-fluorobenzylamine (1.14 mmol, 130 µL) was added in one portion, and the solution was stirred at room temperature for 3 h and then evaporated to dryness. The residue was dissolved in dry EtOH (20 mL), stirred for a further 1 h, and evaporated to dryness again. The product was dissolved in dry MeOH (20 mL), and NaBH4 (2.28 mmol, 90 mg) was added in small portions to the mixture under ice cooling. After stirring for 4 h at room temperature, the mixture was evaporated to dryness, and the residue was dissolved in H2O (50 mL) and extracted with DCM (3 × 50 mL). The combined organic layer was dried (Na2SO4), filtered, and evaporated to dryness. The crude product obtained was purified by column chromatography on silica gel (CHCl3/MeOH = 19:1). Yield: 440 mg (70%); colourless oil; [α]D20 = −38 (c 1.6 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.65 (s, 3H), 0.83–0.88 (m, 2H), 0.90 (s, 3H), 0.93–1.07 (m, 5H), 1.13–1.15 (m, 1H), 1.19 (s, 3H), 1.31 (d, 1H, J = 11.5 Hz), 1.40 (d, 1H, J = 14.2 Hz), 1.53–1.84 (m, 9H), 2.20 (d, 1H, J = 13.4 Hz), 2.33 (t, 1H, J = 11.9 Hz), 2.89 (dd, 1H, J = 3.7 Hz, 11.2 Hz), 3.44 (d, 1H, J = 4.8 Hz), 3.65 (d, 1H, J = 13.1 Hz), 3.85 (d, 1H, J = 13.1 Hz), 5.07 (dd, 2H, J = 12.4 Hz, J = 12.4 Hz), 7.01 (t, 2H, J = 8.5 Hz), 7.30–7.36 (m, 7H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.3 (CH3), 18.9 (CH2), 19.5 (CH2), 22.2 (CH2), 25.1 (CH3), 29.0 (CH3), 33.0 (CH2), 35.0 (CH2), 38.0 (CH2), 38.2 (Cq), 39.6 (CH2), 40.7 (Cq), 42.3 (Cq), 43.9 (Cq), 48.0 (CH), 51.6 (CH2), 53.2 (CH2), 54.2 (CH2), 57.2 (CH), 57.7 (CH), 66.0 (CH2), 88.3 (CH), 115.2 (CH), 115.3 (CH), 128.0 (CH), 128.3 (CH), 128.4 (2 × CH), 129.8 (2 × CH), 129.9 (CH), 135.4 (Cq-F), 136.1 (2 × Cq), 136.2 (Cq-F), 161.1 (Cq-F), 163.0 (Cq-F), 177.1 (C=O); 19F-NMR (470 MHz, CDCl3) δ (ppm): −116.2 (Cq-F). C35H46FNO3: 547.7430. HRMS (ESI+): m/z calcd. for C35H47FNO3 [M + H]+ 548.3540; found 548.3533.
3.1.3. (4R,4aS,6aS,7R,8R,9S,11bS)-Benzyl 7-(((tert-butoxycarbonyl)(4-fluorobenzyl)amino)methyl)-8-hydroxy-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (6)
To a solution of 5 (0.91 mmol, 500 mg) in dry DCM (10 mL), tert-butyloxycarbonyl anhydride (0.91 mmol, 200 mg) was added in one portion. The solution was stirred at room temperature for 2 h and then evaporated to dryness, and the residue was dissolved in DCM (10 mL) and extracted with H2O (3 × 10 mL). The organic layer was dried (Na2SO4), filtered, and evaporated to dryness. The crude product obtained was purified by column chromatography on silica gel (n-hexane/EtOAc = 4:1). Yield: 470 mg (79%); colourless oil; [α]D20 = −1 (c 2.80 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.70 (s, 3H), 0.83–0.87 (m, 1H), 0.90 (s, 3H), 0.92–1.06 (m, 5H), 1.12–1.18 (m, 4H), 1.37–1.42 (m, 2H), 1.47 (s, 9H), 1.56–1.84 (m, 8H), 2.14–2.21 (m, 2H), 3.03 (dd, 1H, J = 4.5 Hz, J = 13.8 Hz), 3.55 (t, 1H, J = 13.1 Hz), 3.64 (d, 1H, J = 4.4 Hz), 4.18 (d, 1H, J = 15.5 Hz), 4.66 (d, 1H, J = 15.5 Hz), 4.87 (d, 1H, J = 12.5 Hz), 5.18 (d, 1H, J = 12.5 Hz), 6.99 (t, 2H, J = 8.6 Hz), 7.23–7.31 (m, 7H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.5 (CH3), 19.0 (CH2), 19.5 (CH2), 22.0 (CH2), 25.1 (CH3), 28.5 (3 × CH3), 29.0 (CH3), 33.2 (CH2), 34.9 (CH2), 37.9 (CH2), 38.2 (Cq), 39.5 (CH2), 41.0 (Cq), 42.9 (Cq), 43.8 (Cq), 45.6 (CH), 47.5 (CH2), 49.3 (CH2), 53.9 (CH2), 57.3 (CH), 57.7 (CH), 65.8 (CH2), 80.0 (Cq), 85.6 (CH), 115.3 (CH), 115.5 (CH), 127.9 (CH), 127.9 (2 × CH), 128.4 (2 × CH), 129.4 (CH), 129.5 (CH), 134.3 (2 × Cq), 136.3 (Cq), 156.5 (Cq-F), 161.1 (Cq-F), 163.1 (Cq-F), 176.8 (C=O); 19F-NMR (470 MHz, CDCl3) δ (ppm): −115.0 (Cq-F). C40H54FNO5: 647.3986. HRMS (ESI+): m/z calcd. for C40H55FNO5 [M + H]+ 648.4064; found 648.4074.
3.1.4. (4R,4aS,6aS,7R,8R,9S,11bS)-7-(((tert-butoxycarbonyl)(4-fluorobenzyl)amino)methyl)-8-hydroxy-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylic Acid (7)
To a suspension of palladium on carbon (5% Pd/C, 120 mg) in n-hexane/EtOAc (1:1, 20 mL) we added 6 (0.77 mmol, 500 mg) in n-hexane/EtOAc (1:1, 20 mL), and the mixture was stirred in an H2 atmosphere (1 atm) at room temperature. After the completion of the reaction (monitored by TLC for 24 h), the mixture was filtered through a Celite pad, and the solution was evaporated to dryness. The crude product was purified by column chromatography on silica gel (CHCl3/MeOH = 9:1). Yield: 350 mg (82%), colourless oil; [α]D20 = −9 (c 0.67 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.85–0.89 (m, 1H), 0.90 (s, 3H), 0.92 (s, 3H), 0.94–1.05 (m, 4H), 1.09–1.11 (m, 1H), 1.13–1.18 (m, 1H), 1.20 (s, 3H), 1.37–1.42 (m, 1H), 1.49 (s, 9H), 1.58–1.81 (m, 9H), 2.16–2.19 (m, 2H), 2.99–3.02 (m, 1H), 3.56 (t, 1H, J = 12.5 Hz), 3.76 (d, 1H, J = 4.4 Hz), 3.95 (d, 1H, J = 15.3 Hz), 4.86–4.88 (m, 1H), 7.00 (t, 2H, J = 8.6 Hz), 7.24–7.26 (m, 2H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.6 (CH3), 18.8 (CH2), 19.6 (CH2), 21.9 (CH2), 25.2 (CH3), 28.5 (3 × CH3), 29.0 (CH3), 33.2 (CH2), 34.8 (CH2), 37.8 (CH2), 38.4 (Cq), 39.6 (CH2), 41.0 (Cq), 42.9 (Cq), 43.7 (Cq), 45.9 (CH), 46.8 (CH2), 48.6 (CH2), 53.9 (CH2), 57.2 (CH), 57.7 (CH), 80.1 (Cq), 115.4 (CH), 115.5 (CH), 129.8 (2 × CH), 156.4 (Cq-F), 161.2 (Cq-F), 163.2 (Cq-F), 171.3 (Cq), 183.4 (C=O); 19F-NMR (470 MHz, CDCl3) δ (ppm): −116.2 (Cq-F). C33H48FNO5: 557.7363. HRMS (ESI+): m/z calcd. for C33H49FNO5 [M + H]+ 558.3595; found 558.3607.
3.1.5. (4R,4aS,6aS,7R,8R,9S,11bS)-7-(((4-Fluorobenzyl)amino)methyl)-8-hydroxy-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylic Acid (8)
To a solution of 7 (0.54 mmol, 300 mg) in dry DCM (15 mL) at 0 °C we added TFA (6.53 mmol, 0.50 mL). The ice bath was removed and the reaction mixture was stirred at room temperature for 3 h. The solvent and TFA were removed in vacuo and the residue was diluted with dry DCM. The mixture was cooled to 0 °C and we added TEA (1.23 mmol, 171.10 µL). The resulting homogeneous mixture was allowed to warm to room temperature and stirred for 1 h. The solution was evaporated to dryness and the crude product was purified by column chromatography on silica gel (CHCl3/MeOH = 9:1). Yield: 190 mg (77%); white crystals; m.p. 149–150 °C; [α]D20 = −39 (c 0.28 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.77 (s, 3H), 0.80–0.84 (m, 2H), 0.90 (s, 3H), 0.94–1.03 (m, 3H), 1.06–1.09 (m, 4H), 1.13–1.18 (m, 1H), 1.29 (d, 1H, J = 11.4 Hz), 1.35 (d, 1H, J = 14.1 Hz), 1.52–1.85 (m, 8H), 2.09 (d, 1H, J = 13.1 Hz), 2.21 (d, 1H, J = 12.0 Hz), 2.52 (d, 1H, J = 11.6 Hz), 3.10 (d, 1H, J = 8.4 Hz), 3.42 (s, 1H), 3.86 (d, 1H, J = 12.4 Hz), 3.97 (d, 1H, J = 12.4 Hz), 7.04 (t, 2H, J = 8.4 Hz), 7.39 (t, 2H, J = 6.9 Hz); 13C-NMR (125 MHz, CDCl3) δ (ppm): 14.6 (CH3), 18.9 (CH2), 19.4 (CH2), 22.0 (CH2), 25.1 (CH3), 29.2 (CH3), 33.0 (CH2), 34.8 (CH2), 37.8 (CH2), 38.1 (Cq), 39.6 (CH2), 41.5 (Cq), 43.3 (Cq), 43.9 (Cq), 46.0 (CH), 51.1 (CH2), 52.3 (CH2), 56.7 (CH), 56.9 (CH), 86.1 (CH), 115.6 (CH), 115.8 (CH), 130.1 (Cq), 131.4 (CH), 131.5 (CH), 161.8 (Cq-F), 163.8 (Cq-F), 182.5 (C=O). C28H40FNO3: 457.6205. HRMS (ESI+): m/z calcd. for C28H41FNO3 [M + H]+ 458.3070; found 458.3060.
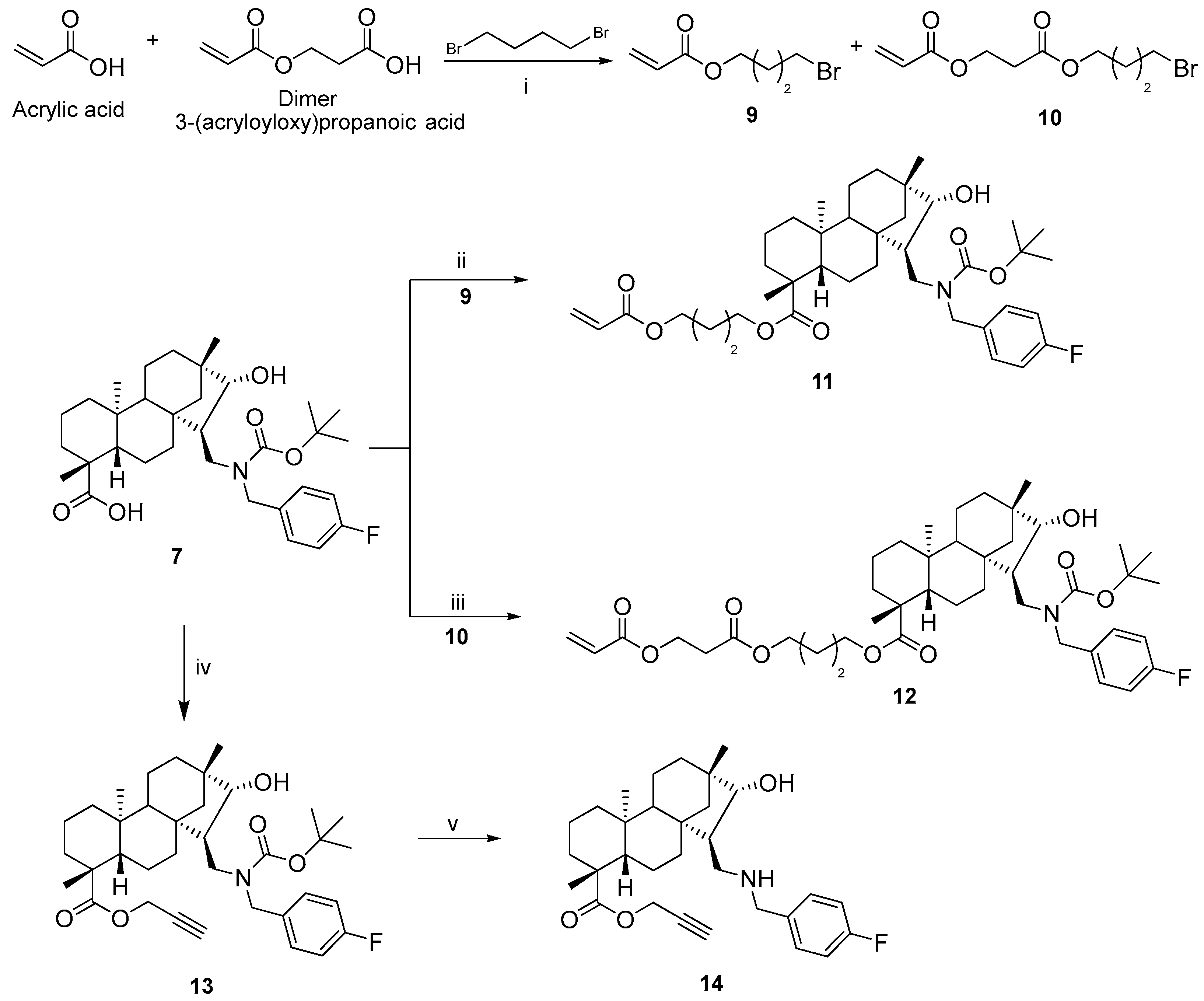
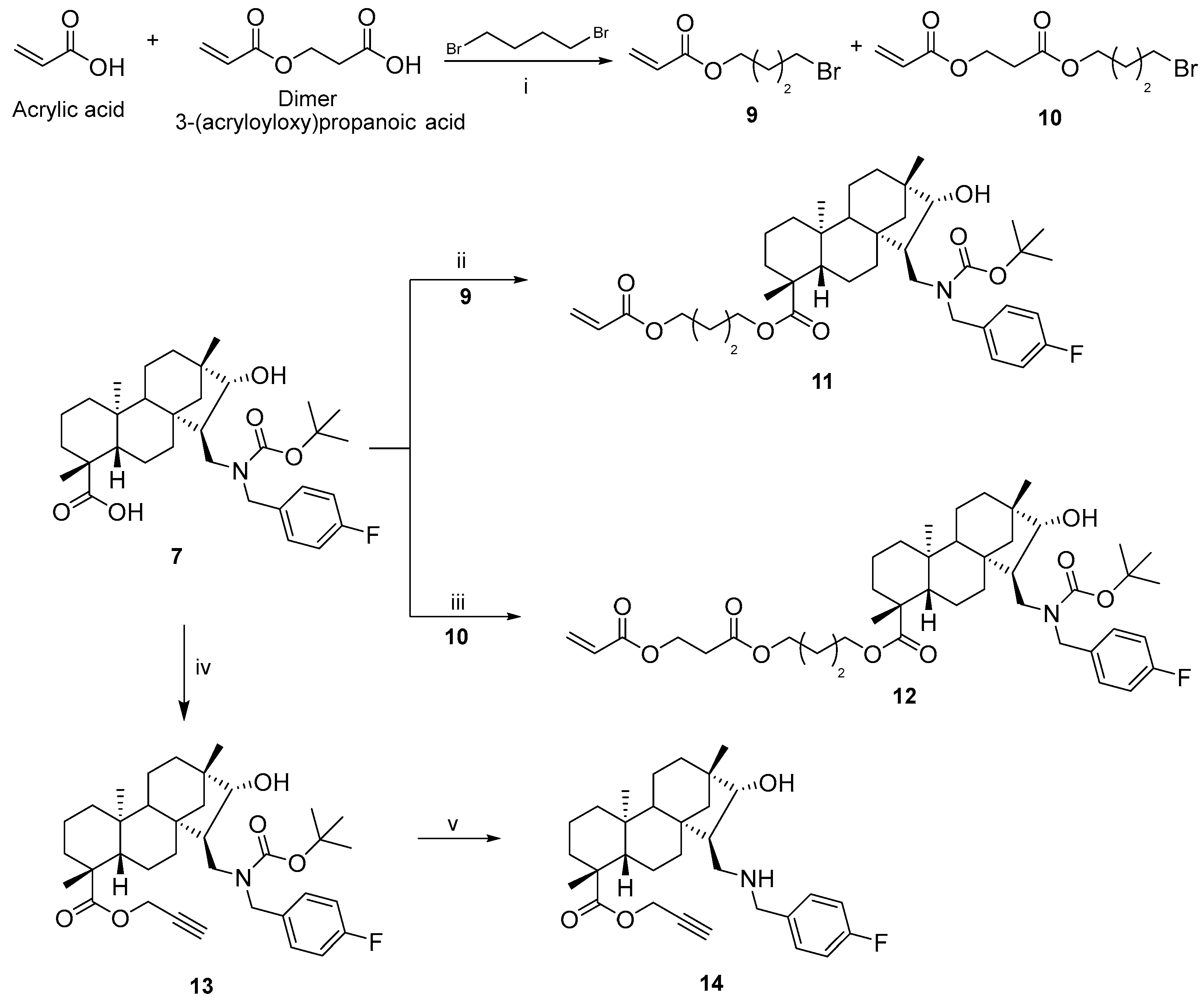
3.1.6. 4-Bromobutyl Acrylate (9) and 3-(4-Bromobutoxy)-3-oxopropyl Acrylate (10)
To a suspension of potassium carbonate (2.78 mmol, 380 mg) in dry acetone (50 mL), acrylic acid containing approx. 20% dimer (2.78 mmol, 200 mg) and 1,4-dibromobutane (2.78 mmol, 332 µL) was added and the mixture was stirred at room temperature for 1 day. After the completion of the reaction (monitored by TLC for 24 h), the mixture was filtered through filter paper and the solution was evaporated to dryness. The crude product was purified by column chromatography on silica gel (n-hexane/EtOAc = 3:1). Yield: 351 mg (product 9, 61%), 202 mg (product 10, 26%); colourless oil; 1H-NMR (500 MHz, CDCl3) δ (ppm): 1.18–1.84 (m, 2H), 1.91–1.96 (m, 2H), 2.69 (t, 2H, J = 6.3 Hz), 3.42 (t, 2H, J = 6.6 Hz), 4.16 (t, 2H, J = 6.2 Hz), 4.44 (t, 2H, J = 6.4 Hz), 5.83 (d, 1H J = 10.5 Hz), 6.11 (dd, 1H, J = 10.5 Hz, J = 17.0 Hz), 6.40 (d, 1H, J = 17,1 Hz); 13C-NMR (125 MHz, CDCl3) δ (ppm): 27.3 (CH2), 29.2 (CH2), 32.7 (CH2), 34.0 (CH2), 59.9 (CH2), 63.8 (CH2), 128.1 (CH), 131.0 (CH2), 165.8 (Cq), 170.5 (Cq); C10H15BrO4: 279.1277. HRMS (ESI+): m/z calcd. for C10H16BrO4 [M + H]+ 280.1356; found 280.1368.
3.1.7. (4R,4aS,6aS,7R,8R,9S,11bS)-4-(Acryloyloxy)butyl 7-(((tert-butoxycarbonyl)(4-fluorobenzyl)amino)methyl)-8-hydroxy-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (11)
To a suspension of potassium carbonate (0.44 mmol, 30 mg) in dry acetone (15 mL) we added 7 (0.22 mmol, 100 mg) and 4-bromobutyl acrylate (0.22 mmol, 35 µL), and the mixture was stirred at room temperature for one day. After the completion of the reaction (monitored by TLC, 24 h), the mixture was filtered through filter paper, and the solution was evaporated to dryness. The crude product was purified by column chromatography on silica gel (n-hexane/EtOAc = 3:1). Yield: 120 mg (83%); colourless oil; [α]D20 = −17 (c 0.05 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.72 (s, 3H), 0.84–0.87 (m, 1H), 0.90 (s, 3H), 0.95–1.06 (m, 5H), 1.13 (s, 3H), 1.15–1.20 (m, 1H), 1.39–1.43 (m, 3H), 1.47 (s, 9H), 1.56–1.83 (m, 11H), 2.13–2.20 (m, 2H), 3.02 (dd, 1H, J = 4.6 Hz, J = 13.8 Hz), 3.58–3.65 (m, 2H), 3.81–3.86 (m, 1H), 4.08–4.17 (m, 4H), 4.78 (d, 1H, J = 15.6 Hz), 5.81 (d, 1H, J = 10.5 Hz), 6.11 (dd, 1H, J = 10.5 Hz, J = 17.6 Hz), 6.39 (d, 1H, J = 17.6 Hz), 7.01 (t, 2H, J = 8.7 Hz), 7.22–7.25 (m, 2H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.4 (CH3), 19.0 (CH2), 19.5 (CH2), 22.1 (CH2), 25.1 (CH3), 25.3 (CH2), 25.6 (CH2), 28.5 (3 × CH3), 29.0 (CH3), 33.3 (CH2), 34.9 (CH2), 38.0 (CH2), 38.2 (Cq), 39.5 (CH2), 41.0 (Cq), 42.8 (Cq), 43.8 (Cq), 45.5 (CH), 47.1 (CH2), 48.8 (CH2), 54.0 (CH2), 57.2 (CH), 57.8 (CH), 63.4 (CH2), 64.0 (CH2), 80.1 (Cq), 85.7 (CH), 115.4 (CH), 115.6 (CH), 128.5 (CH), 129.3 (2 × CH), 130.6 (CH2), 134.2 (Cq), 134.3 (Cq), 156.5 (Cq-F), 161.2 (Cq-F), 163.1 (Cq-F), 166.2 (C=O), 177.1 (C=O); 19F-NMR (470 MHz, CDCl3) δ (ppm): −115.0 (Cq-F). C40H58FNO7: 683.8894. HRMS (ESI+): m/z calcd. for C40H59FNO7 [M + H]+ 684.4064; found 684.4283.
3.1.8. (4R,4aS,6aS,7R,8R,9S,11bS)-4-((3-(Acryloyloxy)propanoyl)oxy)butyl 7-(((tert-butoxycarbonyl)(4-fluorobenzyl)amino)methyl)-8-hydroxy-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (12)
To a suspension of potassium carbonate (0.44 mmol, 30 mg) in dry acetone (15 mL) we added 7 (0.22 mmol, 100 mg) and 3-(4-bromobutoxy)-3-oxopropyl acrylate-product 10 (0.22 mmol, 60 mg), and the mixture was stirred at room temperature for one day. After the completion of the reaction (monitored by TLC for 24 h), the mixture was filtered through filter paper, and the solution was evaporated to dryness. The crude product was purified by column chromatography on silica gel (n-hexane/EtOAc = 3:1). Yield: 130 mg (77%); colourless oil; [α]D20 = +8.7 (c 0.11 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.72 (s, 3H), 0.84–0.88 (m, 1H), 0.90 (s, 3H), 0.95–1.06 (m, 5H), 1.12 (s, 3H), 1.15–1.20 (m, 1H), 1.39–1.43 (m, 3H), 1.47 (s, 9H), 1.57–1.82 (m, 11H), 2.12–2.20 (m, 2H), 2.68 (t, 2H, J = 6.3 Hz), 3.02 (dd, 1H, J = 4.6 Hz, J = 13.8 Hz), 3.57–3.65 (m, 2H), 3.80–3.84 (m, 1H), 4.07–4.16 (m, 4H), 4.43 (t, 2H, J = 6.5 Hz), 4.79 (d, 1H, J = 15.6 Hz), 5.81 (d, 1H, J = 10.5 Hz), 6.10 (dd, 1H, J = 10.5 Hz, J = 17.6 Hz), 6.39 (d, 1H, J = 17.6 Hz), 7.01 (t, 2H, J = 8.9 Hz), 7.22–7.25 (m, 2H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.4 (CH3), 19.0 (CH2), 19.5 (CH2), 22.1 (CH2), 25.1 (CH3), 25.2 (CH2), 25.5 (CH2), 28.5 (3 × CH3), 29.1 (CH3), 33.3 (CH2), 34.0 (CH2), 34.9 (CH2), 38.0 (CH2), 38.2 (Cq), 39.5 (CH2), 41.0 (Cq), 42.8 (Cq), 43.8 (Cq), 45.5 (CH), 47.1 (CH2), 48.8 (CH2), 54.0 (CH2), 57.2 (CH), 57.8 (CH), 59.9 (CH2), 63.3 (CH2), 64.3 (CH2), 80.1 (Cq), 85.6 (CH), 115.4 (CH), 115.5 (CH), 128.2 (CH), 129.3 (2 × CH), 131.0 (CH2), 134.2 (Cq), 134.3 (Cq), 156.5 (Cq-F), 161.2 (Cq-F), 163.1 (Cq-F), 165.8 (C=O), 170.5 (C=O), 177.1 (C=O); 19F-NMR (470 MHz, CDCl3) δ (ppm): −115.0 (Cq-F). 19F-NMR (470 MHz, CDCl3) δ (ppm): −115.3 (Cq-F). C43H62FNO9: 755.9521. HRMS (ESI+): m/z calcd. for C43H63FNO9 [M + H]+ 756.9600; found 756.4495.
3.1.9. (4R,4aS,6aS,7R,8R,9S,11bS)-Prop-2-yn-1-yl 7-(((tert-butoxycarbonyl)(4-fluorobenzyl)amino)methyl)-8-hydroxy-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (13)
To a suspension of potassium carbonate (0.72 mmol, 100 mg) in dry acetone (15 mL) we added 7 (0.36 mmol, 200 mg) and propargyl bromide (0.36 mmol, 28 µL), and the mixture was stirred at room temperature for one day. After the completion of the reaction (monitored by TLC for 24 h), the mixture was filtered through filter paper, and the solution was evaporated to dryness. The crude product was purified by column chromatography on silica gel (n-hexane/EtOAc = 4:1). Yield: 190 mg (88%); colourless oil; [α]D20 = −17 (c 0.23 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.75 (s, 3H), 0.84–0.88 (m, 1H), 0.93 (s, 3H), 0.95–1.08 (m, 5H), 1.13–1.16 (m, 4H), 1.57–1.80 (m, 7H), 2.15–2.19 (m, 2H), 2.26 (s, 1H), 2.26–2.30 (m, 2H), 3.06 (dd, 1H, J = 4.2 Hz, J = 13.5 Hz), 3.60 (t, 1H, J = 12.9 Hz), 3.67 (m, 1H), 4.16 (d, 1H, J = 15.5 Hz), 4.51 (d, 1H, J = 15.6 Hz), 4.66 (d, 1H, J = 15.5 Hz), 4.74 (d, 1H, J = 15.7 Hz), 7.03 (t, 2H, J = 9.1 Hz), 7.25–7.28 (m, 2H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.6 (CH3), 18.9 (CH2), 19.5 (CH2), 21.9 (CH2), 25.1 (CH3), 28.4 (3 × CH3), 28.8 (CH3), 33.2 (CH2), 34.9 (CH2), 37.9 (CH2), 38.2 (Cq), 39.5 (CH2), 41.0 (Cq), 42.9 (Cq), 43.8 (Cq), 45.7 (CH), 47.5 (CH2), 49.1 (CH2), 51.3 (CH2), 53.9 (CH2), 57.2 (CH), 57.7 (CH), 74.4 (Cq), 80.0 (Cq), 85.6 (CH), 115.4 (CH), 115.5 (CH), 129.3 (CH), 129.4 (CH), 134.3 (2 × Cq), 156.5 (Cq), 161.1 (Cq-F), 163.1 (Cq-F), 176.2 (C=O). C36H50FNO5: 595.3673. HRMS (ESI+): m/z calcd. for C36H51FNO3 [M + H]+ 596.3751; found 596.3759.
3.1.10. (4R,4aS,6aS,7R,8R,9S,11bS)-Prop-2-yn-1-yl 7-(((4-fluorobenzyl)amino)methyl)-8-hydroxy-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (14)
To a solution of 13 (0.34 mmol, 200 mg) in dry DCM (15 mL) at 0 °C we added TFA (6.53 mmol, 0.50 mL). The ice bath was removed and the reaction mixture was stirred at RT for 3 h. The solvent and TFA were removed in vacuo and the residue was diluted with dry DCM. The mixture was cooled to °C and TEA (0.68 mmol, 95 µL) was added. The resulting homogeneous mixture was allowed to warm to RT and stirred for 1 h, and the solution was evaporated to dryness. The crude product was purified by column chromatography on silica gel (CHCl3/MeOH = 19:1). Yield: 130 mg (79%); white crystals; m.p. 104–105 °C; [α]D20 = −52 (c 1.06 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.73 (s, 3H), 0.83–0.87 (m, 1H), 0.90 (s, 3H), 0.95–1.08 (m, 5H), 1.14–1.20 (m, 4H), 1.31 (d, 1H, J = 10.7 Hz), 1.42 (m, 1H, J = 14.2 Hz), 1.51–1.63 (m, 4H), 1.69–1.84 (m, 4H), 2.06 (d, 1H, J = 12.0 Hz), 2.17 (d, 1H, J = 13.3 Hz), 2.52–2.56 (m, 2H), 3.03 (dd, 1H, J = 3.4 Hz, J = 11.4 Hz), 3.53 (d, 1H, J = 4.7 Hz), 3.84 (d, 1H, J = 13.1 Hz), 4.01 (d, 1H, J = 13.1 Hz), 4.61–4.69 (m, 2H), 7.05 (t, 2H, J = 8.6 Hz), 7.42–7.45 (m, 2H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.5 (CH3), 18.8 (CH2), 19.5 (CH2), 21.9 (CH2), 24.9 (CH3), 28.8 (CH3), 33.0 (CH2), 34.9 (CH2), 37.8 (CH2), 38.1 (Cq), 39.4 (CH2), 41.0 (Cq), 42.5 (Cq), 43.8 (Cq), 46.2 (CH), 50.7 (CH2), 51.4 (CH2), 51.9 (CH2), 53.8 (CH2), 57.0 (CH), 57.6 (CH), 74.8 (Cq), 77.9 (Cq), 87.1 (CH), 115.5 (CH), 115.7 (CH), 130.9 (2 × CH), 131.8 (Cq), 161.5 (Cq-F), 163.5 (Cq-F), 176.3 (C=O); 19F-NMR (470 MHz, CDCl3) δ (ppm): −113.9 (Cq-F). C31H42FNO3: 495.6685. HRMS (ESI+): m/z calcd. for C31H43FNO3 [M + H]+ 496.3227; found 496.3233.
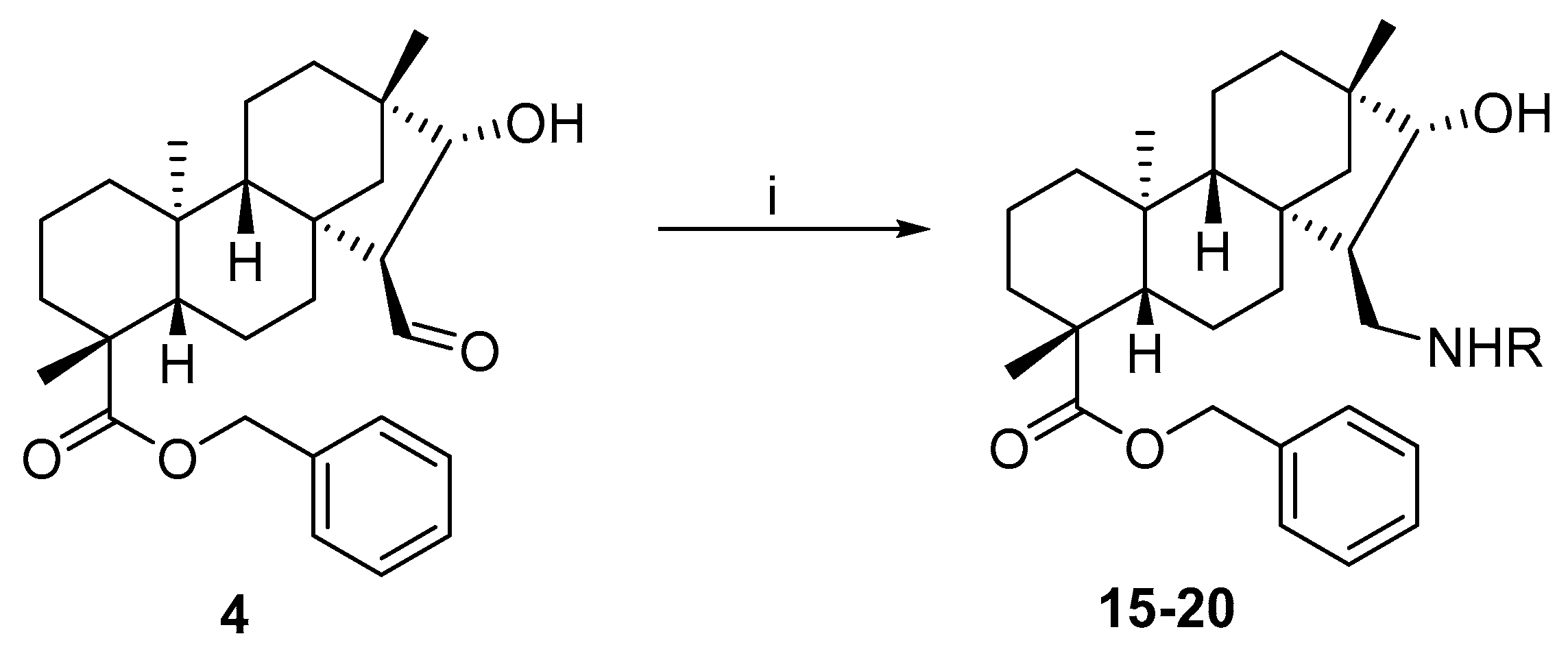
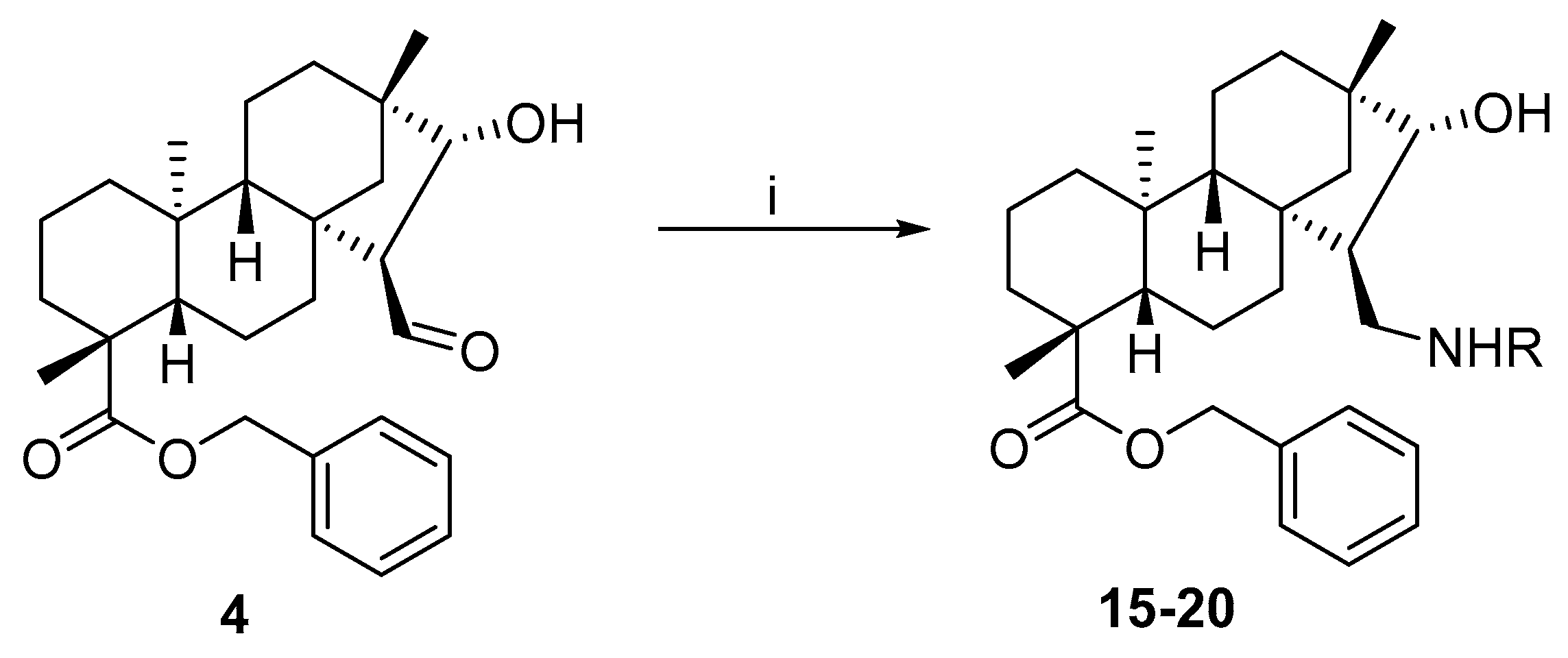
3.1.11. General Procedure for the Preparation of Aminoalcohol with Primary Amines and Benzyl Ester Aldehydes
To a solution of 4 (0.23 mmol, 100 mg) in dry EtOH (10 mL), the appropriate primary amine (0.23 mmol) was added in one portion, and the solution was stirred at room temperature for 3 h and then evaporated to dryness. The residue was dissolved in dry EtOH (10 mL), stirred for a further 1 h, and evaporated to dryness again. The product was dissolved in dry MeOH (10 mL), and NaBH4 (0.46 mmol, 0.02 g) was added in small portions to the mixture under ice cooling. After stirring for 4 h at room temperature, the mixture was evaporated to dryness, and the residue was dissolved in H2O (20 mL) and extracted with DCM (3 × 20 mL). The combined organic layer was dried (Na2SO4), filtered, and evaporated to dryness. The crude product obtained was purified by column chromatography on silica gel (CHCl3/MeOH = 19:1).
(4R,4aS,6aS,7R,8R,9S,11bS)-Benzyl 7-((((R)-1-(4-fluorophenyl)ethyl)amino)methyl)-8-hydroxy-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (15)
The reaction was performed starting from 4 with (R)-4-fluoro-α-methylbenzylamine (0.23 mmol, 38 µL) according to the general procedure. Yield: 30 mg (21%); white crystals; m.p. 135–136 °C; [α]D20 = −33 (c 0.97 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.58 (s, 3H), 0.81–0.83 (m, 1H), 0.87 (s, 3H), 0.93–1.06 (m, 5H), 1.15 (s, 3H), 1.30–1.44 (m, 6H), 1.55–1.57 (m, 2H), 1.66 (d, 3H, J = 6.7 Hz), 1.72–1.80 (m, 3H), 2.13–2.19 (m, 2H), 2.82 (t, 1H, J = 12.5 Hz), 3.00 (d, 1H, J = 9.4 Hz), 3.70 (d, 1H, J = 4.5 Hz), 4.32–4.33 (m, 1H), 5.02 (d, 1H, J = 12.7 Hz), 5.20 (d, 1H, J = 12.7 Hz), 7.07 (t, 2H, J = 8.4 Hz), 7.31–7.39 (m, 5H), 7.56–7.58 (m, 2H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.2 (CH3), 18.9 (CH2), 19.2 (CH3), 19.4 (CH2), 21.7 (CH2), 24.7 (CH3), 29.0 (CH3), 33.0 (CH2), 34.8 (CH2), 37.8 (CH2), 38.0 (Cq), 39.5 (CH2), 41.4 (Cq), 42.9 (Cq), 43.7 (Cq), 44.4 (CH), 48.4 (CH2), 53.4 (CH2), 57.0 (CH), 57.5 (CH), 57.7 (Cq), 65.8 (CH2), 85.3 (CH), 116.2 (CH), 116.4 (CH), 127.8 (2 × CH), 128.0 (CH), 128.6 (2 × CH), 129.9 (CH), 129.9 (CH), 132.9 (Cq) 136.4 (Cq), 162.1 (Cq-F), 164.0 (Cq-F), 176.6 (C=O). C36H48FNO3: 561.7696. HRMS (ESI+): m/z calcd. for C36H49FNO3 [M + H]+ 562.3696; found 562.3702.
(4R,4aS,6aS,7R,8R,9S,11bS)-Benzyl 8-hydroxy-4,9,11b-trimethyl-7-((((R)-1-phenylpropyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (16)
The reaction was performed starting from 4 with (R)-(+)-α-ethylbenzylamine (0.23 mmol, 37 µL) according to the general procedure. Yield: 60 mg (52%); white crystals; m.p. 154–155 °C; [α]D20 = −24 (c 0.26 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.56 (s, 3H), 0.79 (t, 3H, J = 7.4 Hz), 0.82–0.90 (m, 3H), 0.92 (s, 3H), 0.95–1.03 (m, 3H), 1.13 (s, 3H), 1.15–1.17 (m, 1H), 1.33–1.39 (m, 2H), 1.52–1.81 (m, 11H), 2.16 (d, 1H, J = 13.2 Hz), 2.32 (t, 1H, J = 11.6 Hz), 2.82 (dd, 1H, J = 4.1 Hz, J = 11.0 Hz), 3.42–3.48 (m, 2H), 4.94 (d, 1H, J = 12.7 Hz), 5.12 (d, 1H, J = 12.7 Hz), 7.18–7.22 (m, 1H), 7.25–7.33 (m, 9H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 10.5 (CH3), 13.0 (CH3), 18.9 (CH2), 19.5 (CH2), 22.2 (CH2), 25.1 (CH3), 28.9 (CH3), 30.2 (CH2), 33.0 (CH2), 35.0 (CH2), 38.1 (CH2), 38.1 (Cq), 39.6 (CH2), 40.6 (Cq), 42.3 (Cq), 43.8 (Cq), 49.0 (CH), 50.2 (CH2), 54.4 (CH2), 57.2 (CH), 57.9 (CH), 65.7 (CH), 65.7 (CH2), 88.6 (CH), 127.0 (CH), 127.1 (2 × CH), 127.9 (CH), 128.1 (2 × CH), 128.4 (2 × CH), 128.4 (2 × CH), 136.3 (Cq), 144.4 (Cq), 177.0 (C=O). C37H51NO3: 557.8057. HRMS (ESI+): m/z calcd. for C37H52NO3 [M + H]+ 558.8137; found 558.3951.
(4R,4aS,6aS,7R,8R,9S,11bS)-Benzyl 8-hydroxy-4,9,11b-trimethyl-7-((((S)-1-phenylpropyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (17)
The reaction was performed starting from 4 with (S)-(+)-α-ethylbenzylamine (0.23 mmol, 37 µL) according to the general procedure. Yield: 80 mg (68%); white crystals; m.p. 62–63 °C; [α]D20 = −32 (c 0.19 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.67 (s, 3H), 0.78–0.82 (m, 4H), 0.84 (s, 3H), 0.84 (m, 1H), 0.90–1.04 (m, 4H), 1.15 (m, 1H), 1.16 (s, 3H), 1.39–1.43 (m, 2H), 1.56–1.79 (m, 11H), 2.02 (t, 1H, J = 11.9 Hz), 2.19 (d, 1H, J = 13.6 Hz), 2.75 (dd, 1H, J = 3.7 Hz, J = 10.8 Hz), 3.30 (d, 1H, J = 4.9 Hz), 3.48 (t, 1H, J = 6.8 Hz), 5.06 (d, 1H, J = 12.5 Hz), 5.10 (d, 1H, J = 12.5 Hz), 7.23–7.38 (m, 10H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 10.7 (CH3), 13.3 (CH3), 19.0 (CH2), 19.6 (CH2), 22.2 (CH2), 25.1 (CH3), 29.0 (CH3), 31.7 (CH2), 33.0 (CH2), 35.0 (CH2), 38.0 (CH2), 38.2 (Cq), 39.6 (CH2), 40.4 (Cq), 42.1 (Cq), 43.9 (Cq), 48.7 (CH), 49.8 (CH2), 54.2 (CH2), 57.0 (CH), 57.8 (CH), 65.3 (CH), 66.0 (CH2), 88.5 (CH), 127.0 (CH), 127.6 (2 × CH), 128.0 (CH), 128.2 (2 × CH), 128.3 (2 × CH), 128.4 (2 × CH), 136.2 (Cq), 143.8 (Cq), 177.2 (C=O). C37H51NO3: 557.8057. HRMS (ESI+): m/z calcd. for C37H52NO3 [M + H]+ 558.8137; found 558.3947.
(4R,4aS,6aS,7R,8R,9S,11bS)-Benzyl 8-hydroxy-4,9,11b-trimethyl-7-((((R)-1-(naphthalen-1-yl)ethyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (18)
The reaction was performed starting from 4 with (R)-(+)-1-(1-naphthyl)ethylamine (0.23 mmol, 37 µL) according to the general procedure. Yield: 60 mg (40%); white crystals; m.p. 118–119 °C; [α]D20 = −36 (c 0.43 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.64 (s, 3H), 0.82–0.90 (m, 2H), 0.93 (s, 3H), 0.95–1.04 (m, 4H), 1.13 (s, 3H), 1.16–1.20 (m, 1H), 1.37 (t, 3H, J = 15.7 Hz), 1.49 (d, 3H, J = 6.5 Hz), 1.56–1.79 (m, 9H), 2.16 (d, 1H, J = 13.1 Hz), 2.41 (t, 1H, J = 11.5 Hz), 2.94 (dd, 1H, J = 3.1 Hz, J = 10.6 Hz), 3.52 (d, 1H, J = 4.4 Hz), 3.58–3.60 (m, 1H), 4.89 (d, 1H, J = 12.2 Hz), 5.09 (d, 1H, J = 12.2 Hz), 7.13–7.15 (m, 1H), 7.21–7.26 (m, 4H), 7.43–7.53 (m, 3H), 7.62 (d, 1H, J = 6.8 Hz), 7.72 (d, 1H, J = 7.9 Hz), 7.85 (d, 1H, J = 7.9 Hz), 8.18 (d, 1H, J = 8.4 Hz); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.1 (CH3), 18.9 (CH2), 19.5 (CH2), 22.3 (CH2), 23.1 (CH3), 25.2 (CH3), 28.9 (CH3), 33.0 (CH2), 35.1 (CH2), 38.1 (CH), 38.2 (Cq), 39.6 (CH2), 40.6 (Cq), 42.3 (Cq), 43.8 (Cq), 49.2 (CH), 50.3 (CH2), 54.3 (CH), 54.4 (CH2), 57.1 (CH), 57.8 (CH), 65.8 (CH2), 88.8 (CH), 122.5 (CH), 123.1 (CH), 125.4 (CH), 125.7 (CH), 125.8 (CH), 127.3 (CH), 127.9 (CH), 128.2 (2 × CH), 128.4 (2 × CH), 129.0 (CH), 131.0 (Cq), 134.0 (Cq), 136.2 (Cq), 141.7 (Cq), 177.1 (C=O); C40H51NO3: 593.8378. HRMS (ESI+): m/z calcd. for C40H52NO3 [M + H]+ 594.3947; found 594.3953.
(4R,4aS,6aS,7R,8R,9S,11bS)-Benzyl 8-hydroxy-4,9,11b-trimethyl-7-((((S)-1-(naphthalen-1-yl)ethyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (19)
The reaction was performed starting from 4 with (S)-(+)-1-(1-naphthyl)ethylamine (0.23 mmol, 37 µL) according to the general procedure. Yield: 60 mg (46%); white crystals; m.p. 126–127 °C; [α]D20 = −31 (c 0.12 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.04 (s, 3H), 0.69–0.73 (m, 1H), 0.85–0.90 (m, 5H), 0.91 (s, 3H), 1.05 (s, 3H), 1.09–1.17 (m, 2H), 1.24–1.30 (m, 3H), 1.53–1.64 (m, 5H), 1.82 (d, 1H, J = 12.3 Hz), 2.05–2.07 (m, 4H), 2.26 (d, 1H, J = 12.8 Hz), 2.89 (t, 1H, J = 12.5 Hz), 3.15–3.17 (m, 1H), 3.87–3.88 (m, 1H), 4.86 (d, 1H, J = 12.5 Hz), 5.05 (m, 1H, J = 12.5 Hz), 5.60 (s, 1H), 7.36–7.63 (m, 8H), 7.84 (d, 2H, J = 8.2 Hz), 8.00 (d, 1H, J = 8.5 Hz), 8.24 (d, 1H, J = 6.9 Hz); 13C-NMR (125 MHz, CDCl3) δ (ppm): 12.3 (CH3), 18.8 (CH2), 19.3 (CH2), 21.7 (CH2), 22.1 (CH3), 24.6 (CH3), 28.9 (CH3), 33.1 (CH2), 34.6 (CH2), 37.7 (CH), 37.8 (Cq), 39.4 (CH2), 41.4 (Cq), 42.8 (Cq), 43.0 (CH), 43.5 (Cq), 48.7 (CH2), 53.0 (CH), 53.4 (CH2), 57.1 (CH), 58.0 (CH), 65.8 (CH2), 84.6 (CH), 121.9 (CH), 124.5 (2 × CH), 126.1 (2 × CH), 127.1 (CH), 128.2 (2 × CH), 128.5 (2 × CH), 129.1 (CH), 129.3 (CH), 130.7 (Cq), 133.0 (Cq) 134.0 (Cq), 136.6 (Cq), 178.0 (C=O); C40H51NO3: 593.8378. HRMS (ESI+): m/z calcd. for C40H52NO3 [M + H]+ 594.3947; found 594.3945.
(4R,4aS,7R,8R,11bS)-Benzyl 7-(((3-(1H-imidazol-1-yl)propyl)amino)methyl)-8-hydroxy-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (20)
The reaction was performed starting from 4 with 1-(3-aminopropyl)imidazole (0.23 mmol, 27 µL) according to the general procedure. Yield: 30 mg (33%); colourless oil; [α]D20 = −11 (c 0.25 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.67 (s, 3H), 0.84–0.89 (m, 1H), 0.91 (s, 3H), 0.93–1.08 (m, 5H), 1.16–1.20 (m, 4H), 1.32–1.35 (m, 1H), 1.39–1.43 (m, 1H), 1.56–1.85 (m, 9H), 1.91–1.95 (m, 2H), 2.19–2.25 (m, 2H), 2.47–2.52 (m, 1H), 2.66–2.71 (m, 1H), 2.83 (dd, 1H, J = 4.0 Hz, J = 10.7 Hz), 3.40 (d, 1H, J = 5.0 Hz), 3.97–4.07 (m, 2H), 5.03 (d, 1H, J = 12.5 Hz), 5.10 (d, 1H, J = 12.5 Hz), 6.91 (s, 1H), 7.05 (s, 1H), 7.27–7.35 (m, 5H), 7.48 (s, 1H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.3 (CH3), 19.0 (CH2), 19.5 (CH2), 22.2 (CH2), 25.1 (CH3), 29.0 (CH3), 31.5 (CH2), 33.0 (CH2), 35.0 (CH2), 38.0 (CH2) 38.2 (Cq), 39.6 (CH2), 40.7 (Cq), 42.4 (Cq), 43.9 (Cq), 44.9 (CH2), 46.9 (CH2), 48.4 (CH), 52.4 (CH2), 54.3 (CH2), 57.2 (CH), 57.8 (CH), 66.0 (CH2), 88.6 (CH), 118.8 (CH), 127.9 (CH), 128.2 (2 × CH), 128.4 (2 × CH), 129.5 (Cq), 136.2 (Cq), 137.2 (CH), 177.1 (C=O). C34H49N3O3: 547.7712. HRMS (ESI+): m/z calcd. For C34H50N3O3 [M + H]+ 548.7791; found 548.3862.
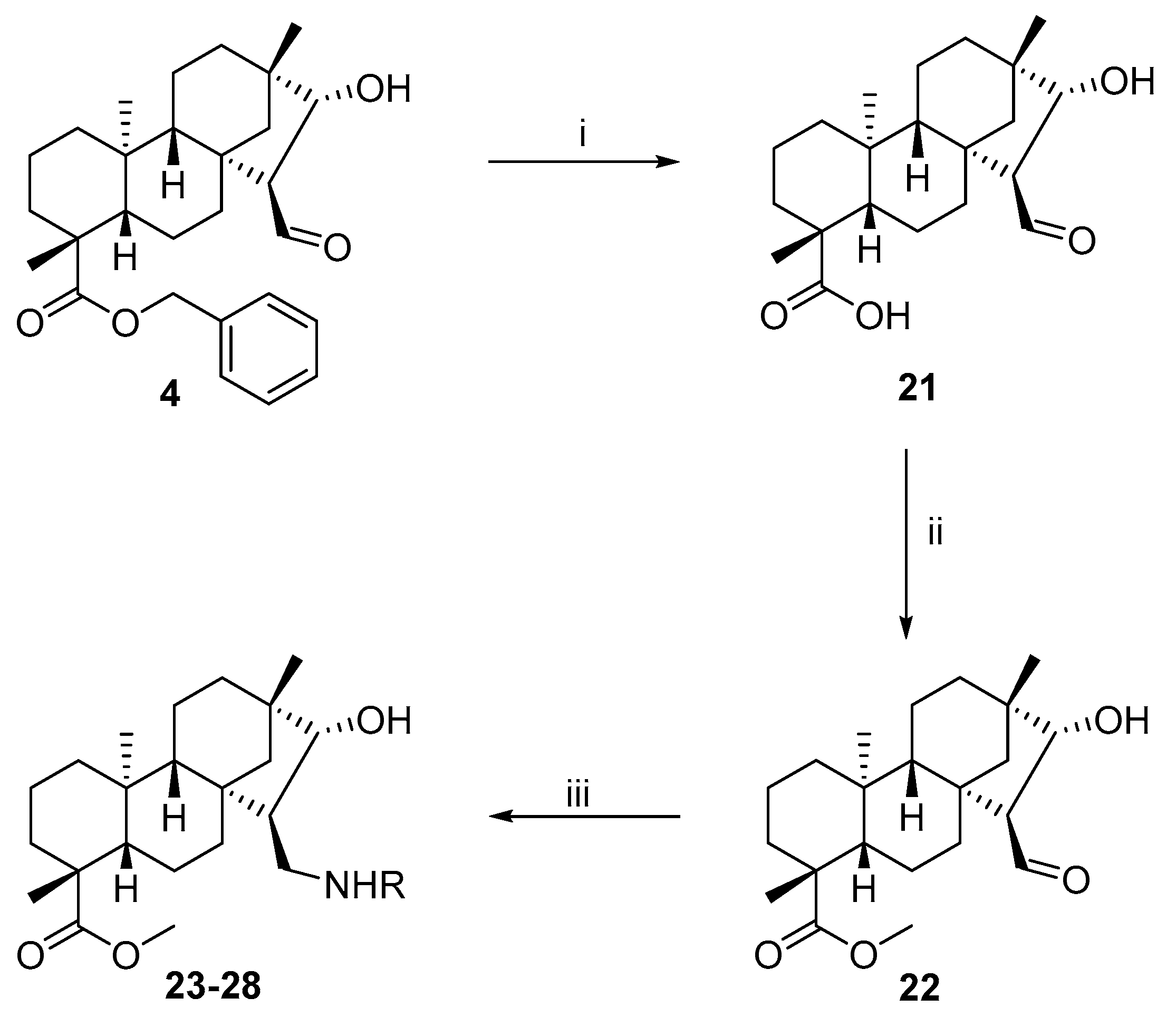
3.1.12. General Procedure for the Preparation of Aminoalcohols by the Reaction of Aldehyde 22 with Primary Amines
To a solution of 22 (100 mg, 0.28 mmol) in dry EtOH (10 mL), the appropriate primary amine (0.28 mmol) was added in one portion, and the solution was stirred at room temperature for 3 h and then evaporated to dryness. The residue was dissolved in dry EtOH (10 mL), stirred for a further 1 h, and evaporated to dryness. The product was dissolved in dry MeOH (10 mL), and NaBH4 (0.56 mmol, 20 mg) was added in small portions to the mixture under ice cooling. After stirring for 4 h at room temperature, the mixture was evaporated to dryness, and the residue was dissolved in H2O (20 mL) and extracted with DCM (3 × 20 mL). The combined organic layer was dried (Na2SO4), filtered, and evaporated to dryness. The crude product obtained was purified by column chromatography on silica gel (CHCl3/MeOH = 19:1).
(4R,4aS,6aS,7R,8R,9S,11bS)-Methyl 7-((((R)-1-(4-fluorophenyl)ethyl)amino)methyl)-8-hydroxy-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (23)
The reaction was performed starting from 22 with (R)-4-fluoro-α-methylbenzylamine (0.28 mmol, 38 µL) according to the general procedure. Yield: 60 mg (50%); white crystals; m.p. 129–130 °C; [α]D20 = −56 (c 0.17 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.58 (s, 3H), 0.81–0.91 (m, 3H), 0.93 (s, 3H), 0.95–1.01 (m, 3H), 1.11 (s, 3H), 1.14–1.19 (m, 1H), 1.33 (d, 3H, J = 6.5 Hz), 1.35 (m, 1H), 1.38–1.47 (m, 2H), 1.51–1.59 (m, 3H), 1.64–1.69 (m, 2H), 1.75–1.79 (m, 3H), 2.13 (d, 1H, J = 13.5 Hz), 2.34 (t, 1H, J = 11.6 Hz), 2.78 (dd, 1H, J = 4.3 Hz, J = 11.0 Hz), 3.47 (d, 1H, J = 5.0 Hz), 3.56 (s, 3H), 3.77 (m, 1H), 7.00 (t, 2H, J = 8.8 Hz), 7.28–7.31 (m, 2Hz); 13C-NMR (125 MHz, CDCl3) δ (ppm): 12.8 (CH3), 18.9 (CH2), 19.5 (CH2), 22.1 (CH2), 24.3 (CH3), 25.1 (CH3), 28.8 (CH3), 33.0 (CH2), 34.9 (CH2), 38.0 (CH2), 38.0 (Cq), 39.6 (CH2), 40.7 (Cq), 42.2 (Cq), 43.7 (Cq), 48.8 (CH), 50.0 (CH2), 51.0 (CH3), 54.3 (CH2), 56.7 (CH), 57.7 (CH), 58.1 (CH), 88.5 (CH), 115.1 (CH), 115.3 (CH), 127.9 (CH), 128.0 (CH), 141.5 (Cq), 160.8 (Cq-F), 162.8 (Cq-F), 177.9 (C=O); 19F-NMR (470 MHz, CDCl3) δ (ppm): −116.3 (Cq-F). C30H44FNO3: 485.6737. HRMS (ESI+): m/z calcd. for C30H45FNO3 [M + H]+ 485.3305; found 485.3321.
(4R,4aS,6aS,7R,8R,9S,11bS)-Methyl 8-hydroxy-4,9,11b-trimethyl-7-((((R)-1-phenylpropyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (24)
The reaction was performed starting from 22 with (R)-(+)-α-ethylbenzylamine (0.28 mmol, 36 µL) according to the general procedure. Yield: 120 mg (88%); white crystals; m.p. 144–145 °C; [α]D20 = −47 (c 0.26 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.58 (s, 3H), 0.81 (t, 3H, J = 7.4 Hz), 0.81–0.91 (m, 3H), 0.93 (s, 3H), 0.95–1.02 (m, 3H), 1.10 (s, 3H), 1.14–1.20 (m, 1H), 1.32–1.79 (m, 14H), 2.12 (d, 1H, J = 13.3 Hz), 2.30 (t, 1H, J = 11.7 Hz), 2.82 (dd, 1H, J = 4.2 Hz, J = 11.0 Hz), 3.47–3.50 (m, 2H), 3.54 (s, 3H), 7.21–7.25 (m, 1H), 7.26–7.33 (m, 4H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 10.7 (CH3), 12.8 (CH3), 18.8 (CH2), 19.5 (CH2), 22.1 (CH2), 25.1 (CH3), 28.8 (CH3), 30.7 (CH2), 33.0 (CH2), 34.9 (CH2), 38.0 (Cq), 38.1 (CH2), 39.6 (CH2), 40.6 (Cq), 42.2 (Cq), 43.7 (Cq), 48.9 (CH), 50.2 (CH2), 51.0 (CH3), 54.4 (CH2), 57.0 (CH), 57.8 (CH), 65.7 (CH), 88.8 (CH), 126.9 (2 × CH), 127.1 (2 × CH), 128.3 (CH), 144.6 (Cq), 178.0 (C=O); C31H47NO3: 481.7098. HRMS (ESI+): m/z calcd. for C31H48NO3 [M + H]+ 482.3634; found 482.3644.
(4R,4aS,6aS,7R,8R,9S,11bS)-Methyl 8-hydroxy-4,9,11b-trimethyl-7-((((S)-1-phenylpropyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (25)
The reaction was performed starting from 22 with (S)-(+)-α-ethylbenzylamine (0.28 mmol, 36 µL) according to the general procedure. Yield: 60 mg (48%); white crystals; m.p. 115–116 °C; [α]D20 = −23 (c 0.27 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.71 (s, 3H), 0.78–0.83 (m, 4H), 0.85–0.88 (m, 4H), 0.91–1.02 (m, 4H), 1.13 (s, 3H), 1.15–1.21 (m, 2H), 1.39–1.83 (m, 14H), 2.08–2.17 (m, 2H), 2.85 (dd, 1H, J = 3.9 Hz, J = 11.1 Hz), 3.34 (d, 1H, J = 4.9 Hz), 3.53 (t, 1H, J = 7.1 Hz), 3.62 (s, 3H), 7.23–7.24 (m, 1H), 7.28–7.34 (m, 4H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 10.7 (CH3), 13.1 (CH3), 19.0 (CH2), 19.6 (CH2), 22.1 (CH2), 25.1 (CH3), 29.0 (CH3), 31.6 (CH2), 33.0 (CH2), 34.9 (CH2), 37.9 (CH), 38.0 (Cq), 39.6 (CH2), 40.4 (Cq), 42.1 (Cq), 43.7 (Cq), 48.5 (CH), 49.6 (CH2), 51.3 (CH3), 54.3 (CH2), 57.1 (CH), 57.9 (CH), 64.9 (CH), 88.5 (CH), 126.9 (CH), 127.5 (2 × CH), 128.3 (2 × CH), 144.0 (Cq), 177.9 (C=O); C31H47NO3: 481.7098. HRMS (ESI+): m/z calcd. for C31H48NO3 [M + H]+ 482.3634; found 482.3631.
(4R,4aS,6aS,7R,8R,9S,11bS)-Methyl 8-hydroxy-4,9,11b-trimethyl-7-((((S)-1-(naphthalen-2-yl)ethyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (26)
The reaction was performed starting from 22 with (S)-(+)-1-(1-naphthyl)ethylamine (0.28 mmol, 45 µL) according to the general procedure. Yield: 60 mg (42%); white crystals; m.p. 194–195 °C; [α]D20 = −65 (c 0.47 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.66 (s, 3H), 0.81–0.87 (m, 2H), 0.89 (s, 3H), 0.91–1.01 (m, 4H), 1.10 (s, 3H), 1.14–1.23 (m, 2H), 1.37–1.40 (m, 1H), 1.43–1.46 (m, 1H), 1.48 (d, 3H, J = 6.6 Hz), 1.59–1.82 (m, 6H), 1.87–1.89 (m, 1H), 2.12 (d, 1H, J = 13.4 Hz), 2.23 (t, 1H, J = 12.0 Hz), 2.31 (s, 1H), 2.95 (dd, 1H, J = 3.9 Hz, J = 11.0 Hz), 3.43 (s, 3H), 3.50 (d, 1H, J = 4.8 Hz), 4.67–4.71 (m, 1H), 7.45–7.52 (m, 3H), 7.69 (d, 1H, J = 7.0 Hz), 7.74 (d, 1H, J = 8.2 Hz), 7.86 (d, 1H, J = 7.8 Hz), 8.20 (d, 1H, J = 8.4 Hz); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.1 (CH3), 18.9 (CH2), 19.6 (CH2), 22.1 (CH2), 24.5 (CH3), 25.1 (CH3), 28.9 (CH3), 33.0 (CH2), 34.9 (CH2), 37.9 (CH), 38.1 (Cq), 39.6 (CH2), 40.5 (Cq), 42.2 (Cq), 43.7 (Cq), 48.6 (CH2), 50.2 (CH2), 51.0 (CH3), 53.7 (CH), 54.3 (CH2), 57.1 (CH), 57.8 (CH), 88.6 (CH), 122.6 (CH), 122.9 (CH), 125.3 (CH), 125.7 (2 × CH), 127.1 (CH), 128.9 (CH), 131.5 (Cq), 134.0 (Cq), 141.2 (Cq), 177.9 (C=O); C34H47NO3: 517.7419. HRMS (ESI+): m/z calcd. for C34H48NO3 [M + H]+ 518.3634; found 518.3629.
(4R,4aS,6aS,7R,8R,9S,11bS)-Methyl 8-hydroxy-4,9,11b-trimethyl-7-((((R)-1-(naphthalen-2-yl)ethyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (27)
The reaction was performed starting from 22 with (R)-(+)-1-(1-naphthyl)ethylamine (0.28 mmol, 45 µL) according to the general procedure. Yield: 60 mg (38%); white crystals; m.p. 153–154 °C; [α]D20 = −40 (c 0.75 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.66 (s, 3H), 0.84–0.92 (m, 2H), 0.93 (s, 3H), 0.95–1.02 (m, 3H), 1.09 (s, 3H), 1.15–1.22 (m, 1H), 1.37–1.47 (m, 3H), 1.51 (d, 3H, J = 6.7 Hz), 1.57–1.86 (m, 9H), 2.12 (d, 1H, J = 13.3 Hz), 2.42 (t, 1H, J = 11.3 Hz), 2.96 (dd, 1H, J = 3.9 Hz, J = 10.9 Hz), 3.50 (s, 3H), 3.52 (d, 1H, J = 5.0 Hz), 4.63 (m, 1H), 7.44–7.54 (m, 3H), 7.64 (d, 1H, J = 7.2 Hz), 7.74 (d, 1H, J = 8.3 Hz), 7.87 (d, 1H, J = 7.6 Hz), 8.19 (d, 1H, J = 8.5 Hz); 13C-NMR (125 MHz, CDCl3) δ (ppm): 12.8 (CH3), 18.9 (CH2), 19.5 (CH2), 22.2 (CH2), 23.5 (CH3), 25.1 (CH3), 28.8 (CH3), 33.0 (CH2), 34.9 (CH2), 38.1 (CH), 38.1 (Cq), 39.6 (CH2), 40.7 (Cq), 42.3 (Cq), 43.7 (Cq), 49.1 (CH2), 50.4 (CH2), 51.0 (CH3), 54.3 (CH2), 54.4 (Cq), 57.0 (CH), 57.8 (CH), 88.8 (CH), 122.4 (CH), 123.0 (CH), 125.4 (CH), 125.7 (CH), 125.8 (CH), 127.2 (CH), 129.0 (CH), 131.2 (Cq), 134.1 (Cq), 141.6 (Cq), 178.0 (C=O); C34H47NO3: 517.7419. HRMS (ESI+): m/z calcd. for C34H48NO3 [M + H]+ 518.3634; found 518.3632.
(4R,4aS,6aS,7R,8R,9S,11bS)-Methyl 7-(((3-(1H-imidazol-1-yl)propyl)amino)methyl)-8-hydroxy-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (28)
The reaction was performed starting from 22 with 1-(3-aminopropyl)imidazole (0.28 mmol, 33 µL) according to the general procedure. Yield: 60 mg (42%); white crystals; m.p. 107–108 °C; [α]D20 = −39 (c 0.20 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.71 (s, 3H), 0.84–0.90 (m, 1H), 0.92 (s, 3H), 0.95–1.08 (m, 5H), 1.16 (s, 3H), 1.18–1.22 (m, 1H), 1.34–1.42 (m, 2H), 1.64–1.81 (m, 9H), 1.94–1.99 (m, 2H), 2.16 (d, 1H, J = 13.3 Hz), 2.31 (t, 1H, J = 11.8 Hz), 2.55–2.60 (m, 1H), 2.71–2.76 (m, 1H), 2.95 (dd, 1H, J = 4.0 Hz, J = 11.0 Hz), 3.43 (d, 1H, J = 4.8 Hz), 3.62 (s, 3H), 4.00–4.09 (m, 2H), 6.92 (s, 1H), 7.05 (s, 3H), 7.48 (s, 1H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.2 (CH3), 19.0 (CH2), 19.6 (CH2), 22.2 (CH2), 25.1 (CH3), 29.0 (CH3), 31.4 (CH2), 33.1 (CH2), 35.1 (CH2), 38.0 (CH2), 38.2 (Cq), 39.7 (CH2), 40.8 (Cq), 42.4 (Cq), 43.8 (Cq), 44.9 (CH2), 46.9 (CH2), 48.3 (CH), 51.2 (CH3), 52.5 (CH2), 54.3 (CH2), 57.3 (CH), 57.8 (CH), 88.7 (CH), 118.9 (CH), 129.6 (CH), 137.3 (CH), 177.9 (C=O); C28H45N3O3: 471.6752. HRMS (ESI+): m/z calcd. For C28H46N3O3 [M + H]+ 472.3539; found 472.3551.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}