

Novel Opioids in the Setting of Acute Postoperative Pain: A Narrative Review

 , and
, and
Abstract
:1. Introduction
1.1. Endogenous Opioid Receptors and Ligands
1.1.1. Mu (μ) Receptor
1.1.2. Mu Receptor Endogenous Ligands
1.1.3. Delta (δ) Receptor and Ligands
1.1.4. Kappa (κ) Receptor and Ligands
1.1.5. Nociceptin Opioid Peptide (NOP) Receptor
1.2. Conventional Opioids and Mechanism of Action
1.3. Opioid-Related Adverse Effects and the Opioid Crisis
2. Novel Opioids
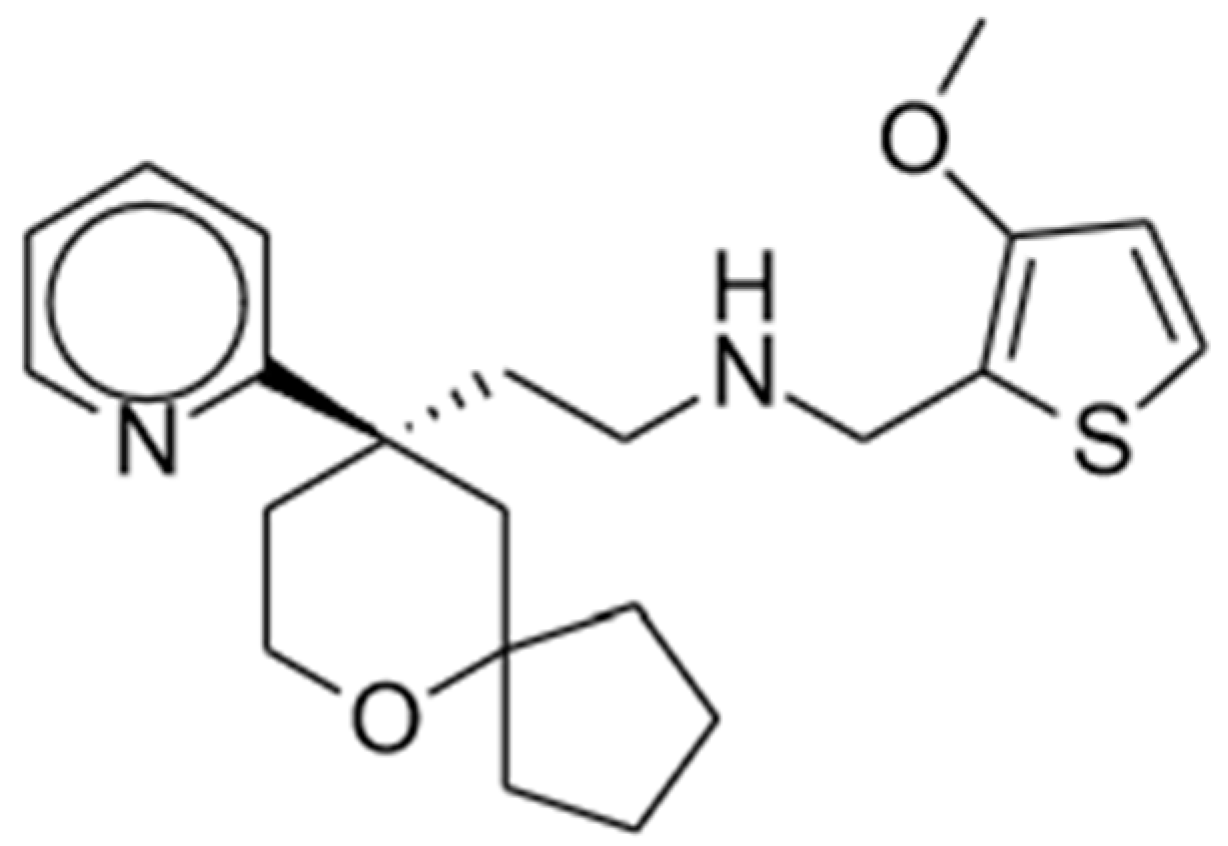
2.1. Oliceridine
2.1.1. Clinical Studies
2.1.2. Potential Advantages
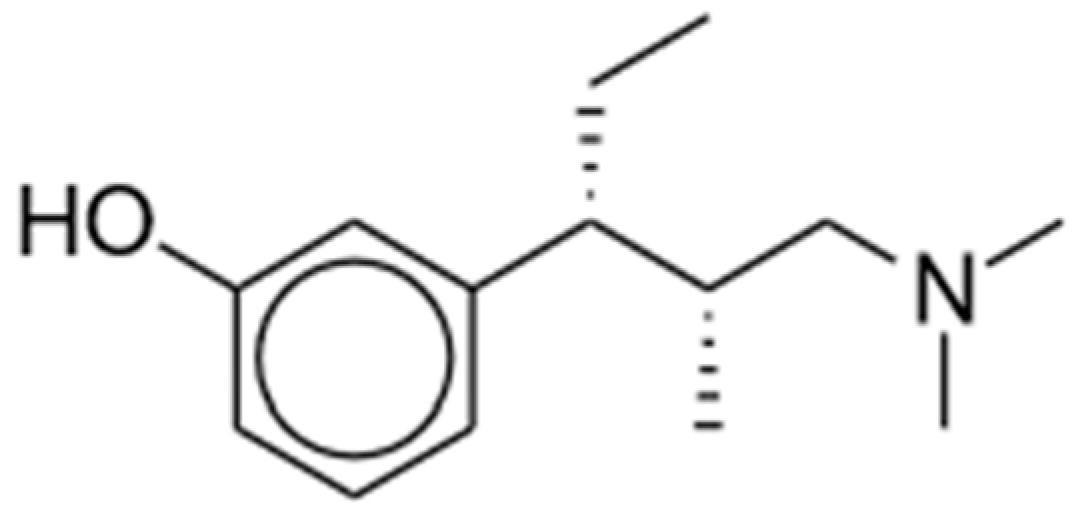
2.2. Tapentadol
2.2.1. Mechanisms of Action and Preclinical Studies
2.2.2. Clinical Studies
2.2.3. Potential Advantages
2.3. Cebranopadol
2.3.1. Preclinical Studies
2.3.2. Clinical Studies and Potential Advantages
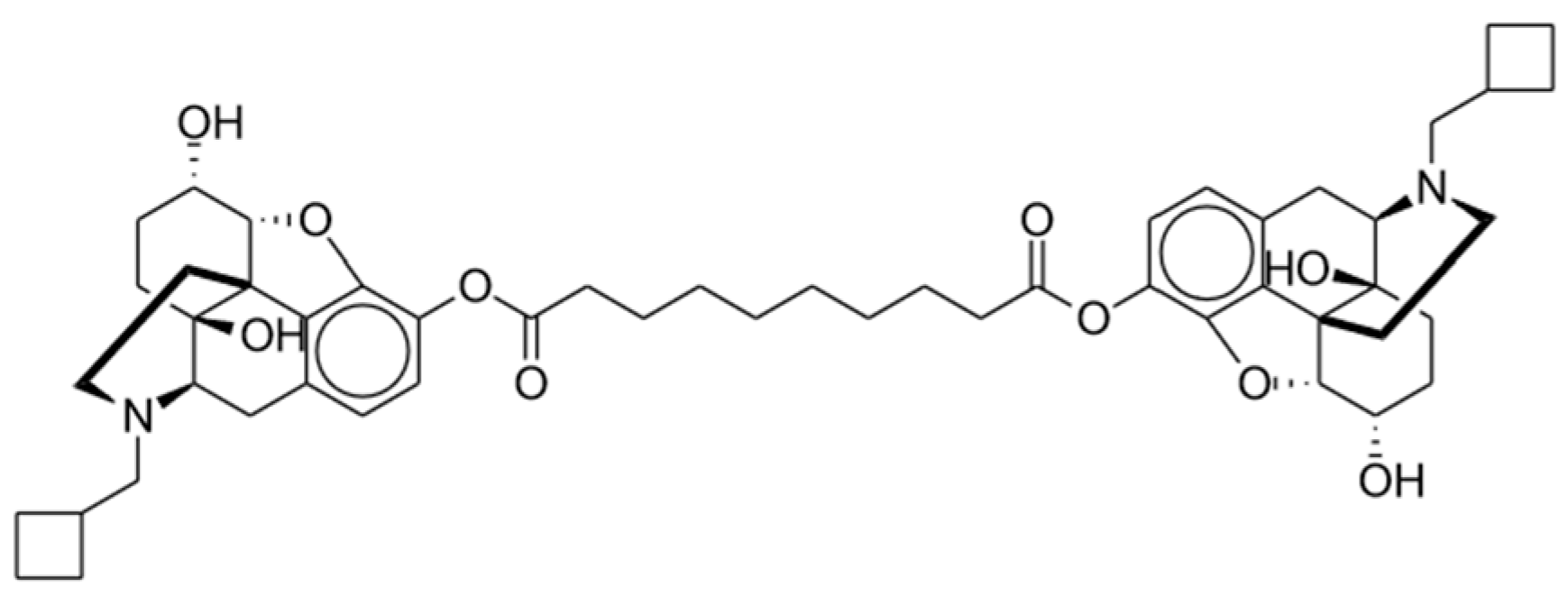
2.4. Dinalbuphine
2.4.1. Clinical Studies
2.4.2. Potential Advantages
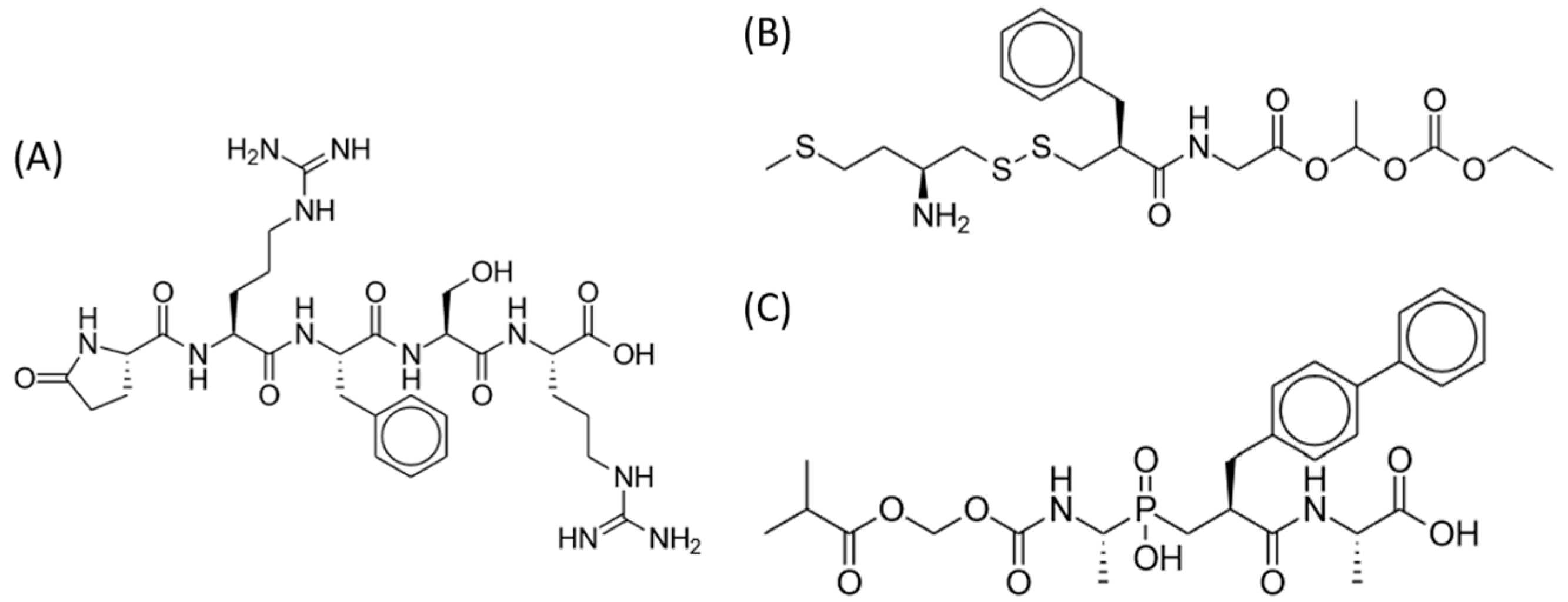
2.5. Dual Enkephalinase Inhibitors (STR-324, PL37, PL265)
2.5.1. Preclinical Studies
2.5.2. Clinical Studies
2.6. Endomorphin-1 Analog (CYT-1010)
Clinical Studies and Potential Advantages
3. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gan, T.J. Poorly controlled postoperative pain: Prevalence, consequences, and prevention. J. Pain Res. 2017, 10, 2287–2298. [Google Scholar] [CrossRef]
- Apfelbaum, J.L.; Chen, C.; Mehta, S.S.; Gan, T.J. Postoperative Pain Experience: Results from a National Survey Suggest Postoperative Pain Continues to Be Undermanaged. Anesth. Analg. 2003, 97, 534–540. [Google Scholar] [CrossRef]
- Garimella, V.; Cellini, C. Postoperative Pain Control. Clin. Colon Rectal Surg. 2013, 26, 191–196. [Google Scholar] [CrossRef]
- Faouzi, A.; Varga, B.R.; Majumdar, S. Biased Opioid Ligands. Molecules 2020, 25, 4257. [Google Scholar] [CrossRef]
- Liu, X.; Xu, X.; Hilger, D.; Aschauer, P.; Tiemann, J.K.; Du, Y.; Liu, H.; Hirata, K.; Sun, X.; Guixà-González, R.; et al. Structural Insights into the Process of GPCR-G Protein Complex Formation. Cell 2019, 177, 1243–1251.e12. [Google Scholar] [CrossRef]
- Deupi, X.; Kobilka, B. Activation of G Protein-Coupled Receptors. Adv. Protein Chem. 2007, 74, 137–166. [Google Scholar]
- Dhaliwal, A.; Gupta, M. Physiology, Opioid Receptor. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Herman, T.F.; Cascella, M.; Muzio, M.R. Mu Receptors. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Nockemann, D.; Rouault, M.; Labuz, D.; Hublitz, P.; McKnelly, K.; Reis, F.C.; Stein, C.; Heppenstall, P.A. The K+ Channel GIRK2 Is Both Necessary and Sufficient for Peripheral Opioid-mediated Analgesia. EMBO Mol. Med. 2013, 5, 1263–1277. [Google Scholar] [CrossRef]
- Cullen, J.M.; Cascella, M. Physiology, Enkephalin. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Schwarzer, C. 30 years of dynorphins—New insights on their functions in neuropsychiatric diseases. Pharmacol. Ther. 2009, 123, 353–370. [Google Scholar] [CrossRef]
- Quirion, B.; Bergeron, F.; Blais, V.; Gendron, L. The Delta-Opioid Receptor; A Target for the Treatment of Pain. Front. Mol. Neurosci. 2020, 13, 52. [Google Scholar] [CrossRef]
- Dalefield, M.L.; Scouller, B.; Bibi, R.; Kivell, B.M. The Kappa Opioid Receptor: A Promising Therapeutic Target for Multiple Pathologies. Front. Pharmacol. 2022, 13, 837671. [Google Scholar] [CrossRef]
- Toll, L.; Bruchas, M.R.; Calo’, G.; Cox, B.M.; Zaveri, N.T. Nociceptin/Orphanin FQ Receptor Structure, Signaling, Ligands, Functions, and Interactions with Opioid Systems. Pharmacol. Rev. 2016, 68, 419–457. [Google Scholar] [CrossRef]
- Ziemichod, W.; Kotlinska, J.; Gibula-Tarlowska, E.; Karkoszka, N.; Kedzierska, E. Cebranopadol as a Novel Promising Agent for the Treatment of Pain. Molecules 2022, 27, 3987. [Google Scholar] [CrossRef]
- Kantonen, T.; Karjalainen, T.; Isojärvi, J.; Nuutila, P.; Tuisku, J.; Rinne, J.; Hietala, J.; Kaasinen, V.; Kalliokoski, K.; Scheinin, H.; et al. Interindividual variability and lateralization of μ-opioid receptors in the human brain. NeuroImage 2020, 217, 116922. [Google Scholar] [CrossRef]
- Stein, C. Opioid Receptors. Annu. Rev. Med. 2016, 67, 433–451. [Google Scholar] [CrossRef]
- Williams, J.T.; Ingram, S.L.; Henderson, G. Regulation of µ-Opioid Receptors: Desensitization, Phosphorylation, Internalization, and Tolerance. Pharmacol. Rev 2013, 65, 223–254. [Google Scholar] [CrossRef]
- Cox, B.M. Recent Developments in the Study of Opioid Receptors. Mol. Pharmacol. 2013, 83, 723–728. [Google Scholar] [CrossRef]
- Zhu, Y.; Ouyang, Z.; Du, H.; Wang, M.; Wang, J.; Sun, H.; Kong, L.; Xu, Q.; Ma, H.; Sun, Y. New opportunities and challenges of natural products research: When target identification meets single-cell multiomics. Acta Pharm. Sin. B 2022, 12, 4011–4039. [Google Scholar] [CrossRef]
- Benyamin, R.; Trescot, A.; Datta, S.; Buenaventura, R.; Adlaka, R.; Sehgal, N.; Glaser, S.; Vallejo, R. Opioid Complications and Side Effects. Pain Physician 2008, 11, S105–S120. [Google Scholar] [CrossRef]
- Dahan, A.; Romberg, R.; Teppema, L.; Sarton, E.; Bijl, H.; Olofsen, E. Simultaneous Measurement and Integrated Analysis of Analgesia and Respiration after an Intravenous Morphine Infusion. Anesthesiology 2004, 101, 1201–1209. [Google Scholar] [CrossRef]
- Khanna, A.K.; Bergese, S.D.; Jungquist, C.R.; Morimatsu, H.; Uezono, S.; Lee, S.; Ti, L.K.; Urman, R.D.; McIntyre, R.; Tornero, C.; et al. Prediction of Opioid-Induced Respiratory Depression on Inpatient Wards Using Continuous Capnography and Oximetry: An International Prospective, Observational Trial. Anesth. Analg. 2020, 131, 1012–1024. [Google Scholar] [CrossRef]
- Webster, L.; Schmidt, W.K. Dilemma of Addiction and Respiratory Depression in the Treatment of Pain: A Prototypical Endomorphin as a New Approach. Pain Med. 2019, 21, 992–1004. [Google Scholar] [CrossRef]
- Shafi, S.; Collinsworth, A.W.; Copeland, L.A.; Ogola, G.O.; Qiu, T.; Kouznetsova, M.; Liao, I.-C.; Mears, N.; Pham, A.T.; Wan, G.J.; et al. Association of Opioid-Related Adverse Drug Events with Clinical and Cost Outcomes Among Surgical Patients in a Large Integrated Health Care Delivery System. JAMA Surg. 2018, 153, 757–763. [Google Scholar] [CrossRef]
- Rudd, R.A.; Seth, P.; David, F.; Scholl, L. Increases in Drug and Opioid-Involved Overdose Deaths—United States, 2010–2015. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 1445–1452. [Google Scholar] [CrossRef]
- Han, B.; Compton, W.M.; Blanco, C.; Crane, E.; Lee, J.; Jones, C.M. Prescription Opioid Use, Misuse, and Use Disorders in U.S. Adults: 2015 National Survey on Drug Use and Health. Ann. Intern. Med. 2017, 167, 293–301. [Google Scholar] [CrossRef]
- Substance Abuse and Mental Health Administration. National Survey on Drug Use and Health: Detailed Tables; Department of Health and Human Services: Rockville, MD, USA, 2019.
- Han, B.; Volkow, N.D.; Compton, W.M.; McCance-Katz, E.F. Reported Heroin Use, Use Disorder, and Injection among Adults in the United States, 2002–2018. J. Am. Med. Assoc. 2020, 323, 568–571. [Google Scholar] [CrossRef]
- Soergel, D.G.; Subach, R.A.; Burnham, N.; Lark, M.W.; James, I.E.; Sadler, B.M.; Skobieranda, F.; Violin, J.D.; Webster, L.R. Biased agonism of the μ-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: A randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain 2014, 155, 1829–1835. [Google Scholar] [CrossRef]
- Viscusi, E.R.; Webster, L.; Kuss, M.; Daniels, S.; Bolognese, J.A.; Zuckerman, S.; Soergel, D.G.; Subach, R.A.; Cook, E.; Skobieranda, F. A randomized, phase 2 study investigating TRV130, a biased ligand of the μ-opioid receptor, for the intravenous treatment of acute pain. Pain 2016, 157, 264–272. [Google Scholar] [CrossRef]
- Viscusi, E.R.; Skobieranda, F.; Soergel, D.G.; Cook, E.; Burt, D.A.; Singla, N. APOLLO-1: A randomized placebo and active-controlled phase III study investigating oliceridine (TRV130), a G protein-biased ligand at the µ-opioid receptor, for management of moderate-to-severe acute pain following bunionectomy. J. Pain Res. 2019, 12, 927–943. [Google Scholar] [CrossRef]
- Singla, N.K.; Skobieranda, F.; Soergel, D.G.; Salamea, M.; Burt, D.A.; Demitrack, M.A.; Viscusi, E.R. APOLLO-2: A Randomized, Placebo and Active-Controlled Phase III Study Investigating Oliceridine (TRV130), a G Protein–Biased Ligand at the μ-Opioid Receptor, for Management of Moderate to Severe Acute Pain Following Abdominoplasty. Pain Pract. 2019, 19, 715–731. [Google Scholar] [CrossRef]
- Bergese, S.D.; Brzezinski, M.; Hammer, G.B.; Beard, T.L.; Pan, P.H.; Mace, S.E.; Berkowitz, R.D.; Cochrane, K.; Wase, L.; Minkowitz, H.S.; et al. ATHENA: A Phase 3, Open-Label Study of the Safety And Effectiveness of Oliceridine (TRV130), A G-Protein Selective Agonist at the μ-Opioid Receptor, In Patients with Moderate to Severe Acute Pain Requiring Parenteral Opioid Therapy. J. Pain Res. 2019, 12, 3113–3126. [Google Scholar] [CrossRef]
- Wang, X.B.; Narayan, S.W.; Penm, J.; Patanwala, A.E. Efficacy and Safety of Tapentadol Immediate Release for Acute Pain. Clin. J. Pain 2020, 36, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Boom, M.; Sarton, E.; Hay, J.; Groeneveld, G.J.; Neukirchen, M.; Bothmer, J.; Aarts, L.; Olofsen, E. Respiratory Effects of the Nociceptin/Orphanin FQ Peptide and Opioid Receptor Agonist, Cebranopadol, in Healthy Human Volunteers. Anesthesiology 2017, 126, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Göhler, K.; Sokolowska, M.; Schoedel, K.A.; Nemeth, R.; Kleideiter, E.; Szeto, I.; Eerdekens, M.-H. Assessment of the Abuse Potential of Cebranopadol in Nondependent Recreational Opioid Users: A Phase 1 Randomized Controlled Study. J. Clin. Psychopharmacol. 2019, 39, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Scholz, A.; Bothmer, J.; Kok, M.; Hoschen, K.; Daniels, S. Cebranopadol: A Novel, First-in-Class, Strong Analgesic: Results from a Randomized Phase IIa Clinical Trial in Postoperative Acute Pain. Pain Physician 2018, 21, E193–E206. [Google Scholar] [CrossRef] [PubMed]
- Christoph, A.; Eerdekens, M.-H.; Kok, M.; Volkers, G.; Freynhagen, R. Cebranopadol, a novel first-in-class analgesic drug candidate: First experience in patients with chronic low back pain in a randomized clinical trial. Pain 2017, 158, 1813–1824. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.Y.; Jao, S.W.; Chen, J.S.; Fan, C.W.; Chen, H.H.; Hsieh, P.S.; Wu, C.C.; Lee, C.C.; Kuo, Y.H.; Hsieh, M.C.; et al. Sebacoyl Dinalbuphine Ester Extended-release Injection for Long-acting Analgesia: A Multicenter, Randomized, Double-Blind, And Placebo-controlled Study in Hemorrhoidectomy Patients. Clin. J. Pain 2017, 33, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.E.; Wang, S.Y.; Chen, J.H.; Chen, C.Y.; Shiue, Y.L.; Soong, T.C.; Lam, C.F. Efficacy and Safety of Parenteral Injection of an Extended Release κ-receptor Opioid Sebacoyl Dinalbuphine Ester for Acute and Chronic Pain After Laparoscopic Bariatric Surgery: A Randomized, Placebo-Controlled, Double-Blind Trial. Obes. Surg. 2023, 33, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.-K.; Huang, C.-W.; Su, W.-C.; Tsai, H.-L.; Ma, C.-J.; Yeh, Y.-S.; Chen, Y.-C.; Li, C.-C.; Cheng, K.-I.; Su, M.-P.; et al. Extended-Release Dinalbuphine Sebacate Versus Intravenous Patient-Controlled Analgesia with Fentanyl for Postoperative Moderate-to-Severe Pain: A Randomized Controlled Trial. Pain Ther. 2020, 9, 671–681. [Google Scholar] [CrossRef]
- Moss, L.M.; Berends, C.L.; van Brummelen, E.M.J.; Kamerling, I.M.C.; Klaassen, E.S.; Bergmann, K.; Ville, V.; Juarez-Perez, V.; Benichou, A.; Groeneveld, G.J. First-in-human trial to assess the safety, tolerability, pharmacokinetics and pharmacodynamics of STR-324, a dual enkephalinase inhibitor for pain management. Br. J. Clin. Pharmacol. 2022, 88, 103–114. [Google Scholar] [CrossRef]
- Daksla, N.; Wang, A.; Jin, Z.; Gupta, A.; Bergese, S.D. Oliceridine for the Management of Moderate to Severe Acute Postoperative Pain: A Narrative Review. Drug Des. Dev. Ther. 2023, 17, 875–886. [Google Scholar] [CrossRef]
- Food and Drug Administration (FDA). Highlights of Prescribing Information–Olinvyk (Oliceridine). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/210730s001lbl.pdf (accessed on 7 August 2023).
- Fossler, M.J.; Sadler, B.M.; Farrell, C.; Burt, D.A.; Pitsiu, M.; Skobieranda, F.; Soergel, D.G. Oliceridine (TRV130), a Novel G Protein–Biased Ligand at the μ-Opioid Receptor, Demonstrates a Predictable Relationship between Plasma Concentrations and Pain Relief. I: Development of a Pharmacokinetic/Pharmacodynamic Model. J. Clin. Pharmacol. 2018, 58, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Nafziger, A.N.; Arscott, K.A.; Cochrane, K.; Skobieranda, F.; Burt, D.A.; Fossler, M.J. The Influence of Renal or Hepatic Impairment on the Pharmacokinetics, Safety, and Tolerability of Oliceridine. Clin. Pharmacol. Drug Dev. 2019, 9, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Singla, N.; Minkowitz, H.S.; Soergel, D.G.; Burt, D.; Subach, R.A.; Salamea, M.Y.; Fossler, M.J.; Skobieranda, F. A randomized, Phase IIb study investigating oliceridine (TRV130), a novel µ-receptor G-protein pathway selective (µ-GPS) modulator, for the management of moderate to severe acute pain following abdominoplasty. J. Pain Res. 2017, 10, 2413–2424. [Google Scholar] [CrossRef] [PubMed]
- Beard, T.L.; Michalsky, C.; Candiotti, K.A.; Rider, P.; Wase, L.; Habib, A.S.; Demitrack, M.A.; Fossler, M.J.; Viscusi, E.R. Oliceridine is Associated with Reduced Risk of Vomiting and Need for Rescue Antiemetics Compared to Morphine: Exploratory Analysis from Two Phase 3 Randomized Placebo and Active Controlled Trials. Pain Ther. 2020, 10, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Ayad, S.; Demitrack, M.A.; Burt, D.A.; Michalsky, C.; Wase, L.; Fossler, M.J.; Khanna, A.K. Evaluating the Incidence of Opioid-Induced Respiratory Depression Associated with Oliceridine and Morphine as Measured by the Frequency and Average Cumulative Duration of Dosing Interruption in Patients Treated for Acute Postoperative Pain. Clin. Drug Investig. 2020, 40, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Bergese, S.; Berkowitz, R.; Rider, P.; Ladouceur, M.; Griffith, S.; Vasi, A.S.; Cochrane, K.; Wase, L.; Demitrack, M.A.; Habib, A.S. Low Incidence of Postoperative Respiratory Depression with Oliceridine Compared to Morphine: A Retrospective Chart Analysis. Pain Res. Manag. 2020, 2020, 7492865. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration (FDA). Highlights of Prescribing Information–Nucynta IR (Tapentadol) Immediate-Release Oral Tablets. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/022304s003lbl.pdf (accessed on 7 August 2023).
- Food and Drug Administration (FDA). Highlights of Prescribing Information–Nucynta ER (Tapentadol) Extended-Release Oral Tablets. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/200533s020lbl.pdf (accessed on 7 August 2023).
- Alshehri, F.S. Tapentadol: A Review of Experimental Pharmacology Studies, Clinical Trials, and Recent Findings. Drug Des. Dev. Ther. 2023, 17, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Manandhar, P.; Connor, M.; Santiago, M. Tapentadol shows lower intrinsic efficacy at µ receptor than morphine and oxycodone. Pharmacol. Res. Perspect. 2022, 10, e00921. [Google Scholar] [CrossRef]
- Tzschentke, T.M.; Christoph, T.; Kögel, B.; Schiene, K.; Hennies, H.-H.; Englberger, W.; Haurand, M.; Jahnel, U.; Cremers, T.I.F.H.; Friderichs, E.; et al. (–)-(1R,2R)-3-(3-Dimethylamino-1-ethyl-2-methyl-propyl)-phenol Hydrochloride (Tapentadol HCl): A Novel μ-Opioid Receptor Agonist/Norepinephrine Reuptake Inhibitor with Broad-Spectrum Analgesic Properties. J. Pharmacol. Exp. Ther. 2007, 323, 265–276. [Google Scholar] [CrossRef]
- Viscusi, E.R.; Allard, R.; Sohns, M.; Eerdekens, M. Tapentadol immediate release for moderate to severe acute post-surgery pain. J. Opioid Manag. 2019, 15, 51–67. [Google Scholar] [CrossRef]
- Kleinert, R.; Lange, C.; Steup, A.; Black, P.; Goldberg, J.; Desjardins, P. Single Dose Analgesic Efficacy of Tapentadol in Postsurgical Dental Pain: The Results of a Randomized, Double-Blind, Placebo-Controlled Study. Anesth. Analg. 2008, 107, 2048–2055. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.; Mohan, G.; Ramakrishnan, S.; Theodore, S. Comparison of tapentadol with tramadol for analgesia after cardiac surgery. Ann. Card. Anaesth. 2015, 18, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Moorthy, S.; Codi, R.S.; Surendher, R.; Manimekalai, K. Comparison of the Efficacy and Safety of Tramadol versus Tapentadol in Acute Osteoarthritic Knee Pain: A Randomized, Controlled Trial. Asian J. Pharm. Clin. Res. 2016, 9, 253–256. [Google Scholar]
- Romualdi, P.; Grilli, M.; Canonico, P.L.; Collino, M.; Dickenson, A.H. Pharmacological rationale for tapentadol therapy: A review of new evidence. J. Pain Res. 2019, 12, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Freynhagen, R.; Elling, C.; Radic, T.; Sohns, M.; Liedgens, H.; James, D.; McCool, R.; Edwards, M. Safety of tapentadol compared with other opioids in chronic pain treatment: Network meta-analysis of randomized controlled and withdrawal trials. Curr. Med. Res. Opin. 2021, 37, 89–100. [Google Scholar] [CrossRef] [PubMed]
- van der Schrier, R.; Jonkman, K.; van Velzen, M.; Olofsen, E.; Drewes, A.M.; Dahan, A.; Niesters, M. An experimental study comparing the respiratory effects of tapentadol and oxycodone in healthy volunteers. Br. J. Anaesth. 2017, 119, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- Roulet, L.; Rollason, V.; Desmeules, J.; Piguet, V. Tapentadol Versus Tramadol: A Narrative and Comparative Review of Their Pharmacological, Efficacy and Safety Profiles in Adult Patients. Drugs 2021, 81, 1257–1272. [Google Scholar] [CrossRef] [PubMed]
- Medicines and Healthcare Products Regulatory Agency (UK). Tapentadol (Palexia): Risk of Seizures and Reports of Serotonin Syndrome When Co-Administered with Other Medicines. 2019. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/770006/PDF-Jan-2019-publication.pdf (accessed on 7 August 2023).
- Gressler, L.E.; Hammond, D.A.; Painter, J.T. Serotonin Syndrome in Tapentadol Literature: Systematic Review of Original Research. J. Pain Palliat. Care Pharmacother. 2017, 31, 228–236. [Google Scholar] [CrossRef]
- Vosburg, S.K.; Severtson, S.G.; Dart, R.C.; Cicero, T.J.; Kurtz, S.P.; Parrino, M.W.; Green, J.L. Assessment of Tapentadol API Abuse Liability with the Researched Abuse, Diversion and Addiction-Related Surveillance System. J. Pain 2018, 19, 439–453. [Google Scholar] [CrossRef]
- Butler, S.F.; McNaughton, E.C.; Black, R.A. Tapentadol Abuse Potential: A Postmarketing Evaluation Using a Sample of Individuals Evaluated for Substance Abuse Treatment. Pain Med. 2015, 16, 119–130. [Google Scholar] [CrossRef]
- Rizzi, A.; Cerlesi, M.C.; Ruzza, C.; Malfacini, D.; Ferrari, F.; Bianco, S.; Costa, T.; Guerrini, R.; Trapella, C.; Calo’, G. Pharmacological characterization of cebranopadol a novel analgesic acting as mixed nociceptin/orphanin FQ and opioid receptor agonist. Pharmacol. Res. Perspect. 2016, 4, e00247. [Google Scholar] [CrossRef] [PubMed]
- Tzschentke, T.M.; Linz, K.; Frosch, S.; Christoph, T. Antihyperalgesic, Antiallodynic, and Antinociceptive Effects of Cebranopadol, a Novel Potent Nociceptin/Orphanin FQ and Opioid Receptor Agonist, after Peripheral and Central Administration in Rodent Models of Neuropathic Pain. Pain Pract. 2017, 17, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Christoph, T.; Raffa, R.; De Vry, J.; Schröder, W. Synergistic Interaction between the Agonism of Cebranopadol at Nociceptin/Orphanin FQ and Classical Opioid Receptors in the Rat Spinal Nerve Ligation Model. Pharmacol. Res. Perspect. 2018, 6, e00444. [Google Scholar] [CrossRef] [PubMed]
- Kleideiter, E.; Piana, C.; Wang, S.; Nemeth, R.; Gautrois, M. Clinical Pharmacokinetic Characteristics of Cebranopadol, a Novel First-in-Class Analgesic. Clin. Pharmacokinet. 2018, 57, 31–50. [Google Scholar] [CrossRef] [PubMed]
- Schiene, K.; Schröder, W.; Linz, K.; Frosch, S.; Tzschentke, T.M.; Jansen, U.; Christoph, T. Nociceptin/orphanin FQ opioid peptide (NOP) receptor and µ-opioid peptide (MOP) receptors both contribute to the anti-hypersensitive effect of cebranopadol in a rat model of arthritic pain. Eur. J. Pharmacol. 2018, 832, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Schiene, K.; Schröder, W.; Linz, K.; Frosch, S.; Tzschentke, T.M.; Christoph, T.; Xie, J.Y.; Porreca, F. Inhibition of experimental visceral pain in rodents by cebranopadol. Behav. Pharmacol. 2019, 30, 320–326. [Google Scholar] [CrossRef]
- Rizzi, A.; Ruzza, C.; Bianco, S.; Trapella, C.; Calo’, G. Antinociceptive action of NOP and opioid receptor agonists in the mouse orofacial formalin test. Peptides 2017, 94, 71–77. [Google Scholar] [CrossRef]
- Ding, H.; Trapella, C.; Kiguchi, N.; Hsu, F.-C.; Caló, G.; Ko, M.-C. Functional Profile of Systemic and Intrathecal Cebranopadol in Nonhuman Primates. Anesthesiology 2021, 135, 482–493. [Google Scholar] [CrossRef]
- Linz, K.; Schröder, W.; Frosch, S.; Christoph, T. Opioid-type Respiratory Depressant Side Effects of Cebranopadol in Rats Are Limited by Its Nociceptin/Orphanin FQ Peptide Receptor Agonist Activity. Anesthesiology 2017, 126, 708–715. [Google Scholar] [CrossRef]
- Tzschentke, T.M.; Kögel, B.Y.; Frosch, S.; Linz, K. Limited Potential of Cebranopadol to Produce Opioid-Type Physical Dependence in Rodents: Weak Cebranopadol Dependence. Addict. Biol. 2018, 23, 1010–1019. [Google Scholar] [CrossRef]
- Ruzza, C.; Holanda, V.A.; Gavioli, E.C.; Trapella, C.; Calo, G. NOP agonist action of cebranopadol counteracts its liability to promote physical dependence. Peptides 2019, 112, 101–105. [Google Scholar] [CrossRef]
- Wei, H.; Zhang, T.; Zhan, C.-G.; Zheng, F. Cebranopadol reduces cocaine self-administration in male rats: Dose, treatment and safety consideration. Neuropharmacology 2020, 172, 108128. [Google Scholar] [CrossRef]
- Shen, Q.; Deng, Y.; Ciccocioppo, R.; Cannella, N. Cebranopadol, a Mixed Opioid Agonist, Reduces Cocaine Self-administration through Nociceptin Opioid and Mu Opioid Receptors. Front. Psychiatry 2017, 8, 234. [Google Scholar] [CrossRef]
- de Guglielmo, G.; Matzeu, A.; Kononoff, J.; Mattioni, J.; Martin-Fardon, R.; George, O. Cebranopadol Blocks the Escalation of Cocaine Intake and Conditioned Reinstatement of Cocaine Seeking in Rats. J. Pharmacol. Exp. Ther. 2017, 362, 378–384. [Google Scholar] [CrossRef]
- Wei, H.; Shang, L.; Zhan, C.-G.; Zheng, F. Effects of Cebranopadol on Cocaine-induced Hyperactivity and Cocaine Pharmacokinetics in Rats. Sci. Rep. 2020, 10, 9254. [Google Scholar] [CrossRef]
- Eerdekens, M.-H.; Kapanadze, S.; Koch, E.D.; Kralidis, G.; Volkers, G.; Ahmedzai, S.H.; Meissner, W. Cancer-Related Chronic Pain: Investigation of the Novel Analgesic Drug Candidate Cebranopadol in a Randomized, Double-Blind, Noninferiority Trial. Eur. J. Pain 2019, 23, 577–588. [Google Scholar] [CrossRef]
- Koch, E.D.; Kapanadze, S.; Eerdekens, M.-H.; Kralidis, G.; Létal, J.; Sabatschus, I.; Ahmedzai, S.H. Cebranopadol, a Novel First-in-Class Analgesic Drug Candidate: First Experience with Cancer-Related Pain for up to 26 Weeks. J. Pain Symptom Manag. 2019, 58, 390–399. [Google Scholar] [CrossRef]
- CTG Labs–NCBI. Clinicaltrials.gov. Available online: http://clinicaltrials.gov (accessed on 7 August 2023).
- Tien, Y.E.; Huang, W.; Kuo, H.; Tai, L.; Uang, Y.; Chern, W.H.; Huang, J. Pharmacokinetics of dinalbuphine sebacate and nalbuphine in human after intramuscular injection of dinalbuphine sebacate in an extended-release formulation. Biopharm. Drug Dispos. 2017, 38, 494–497. [Google Scholar] [CrossRef]
- Drug Enforcement Administration (DEA). Diversion Control Division. Nalbuphine Hydrochloride (Brand Name: Nubain). Available online: https://www.deadiversion.usdoj.gov/drug_chem_info/nalbuphine.pdf (accessed on 10 July 2023).
- Zeng, Z.; Lu, J.; Shu, C.; Chen, Y.; Guo, T.; Wu, Q.P.; Yao, S.L.; Yin, P. A comparison of nalbuphine with morphine for analgesic effects and safety: Meta-analysis of randomized controlled trials. Sci. Rep. 2015, 5, 10927. [Google Scholar] [CrossRef]
- Chang, S.-H.; Chang, T.-C.; Chen, M.-Y.; Chen, W.-C.; Chou, H.-H. Comparison of the Efficacy and Safety of Dinalbuphine Sebacate, Patient-Controlled Analgesia, and Conventional Analgesia After Laparotomy for Gynecologic Cancers: A Retrospective Study. J. Pain Res. 2021, 14, 1763–1771. [Google Scholar] [CrossRef]
- Zheng, Z.-H.; Yeh, T.-T.; Yeh, C.-C.; Lin, P.-A.; Wong, C.-S.; Lee, P.-Y.; Lu, C.-H. Multimodal Analgesia with Extended-Release Dinalbuphine Sebacate for Perioperative Pain Management in Upper Extremity Trauma Surgery: A Retrospective Comparative Study. Pain Ther. 2022, 11, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-O.; Huang, L.-P.; Wong, C.-S. Preoperative Administration of Extended-Release Dinalbuphine Sebacate Compares with Morphine for Post-Laparoscopic Cholecystectomy Pain Management: A Randomized Study. J. Pain Res. 2020, 13, 2247–2253. [Google Scholar] [CrossRef] [PubMed]
- Menéndez, L.; Hidalgo, A.; Meana, Á.; Poras, H.; Fournié-Zaluski, M.-C.; Roques, B.P.; Baamonde, A. Inhibition of osteosarcoma-induced thermal hyperalgesia in mice by the orally active dual enkephalinase inhibitor PL37. Potentiation by gabapentin. Eur. J. Pharmacol. 2008, 596, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Raffa, R.B.; Pergolizzi, J.V., Jr.; Taylor, R., Jr.; Ossipov, M.H.; The NEMA Research Group. Indirect-acting strategy of opioid action instead of direct receptor activation: Dual-acting enkephalinase inhibitors (DENKIs). J. Clin. Pharm. Ther. 2018, 43, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Hu, Y.; Kapadia, S.; Ouimet, T.; Poras, H.; Dussor, G. Efficacy of dual enkephalinase inhibition in a preclinical migraine model is mediated by activation of peripheral delta opioid receptors. Headache 2023, 63, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Thibault, K.; Elisabeth, B.; Sophie, D.; Claude, F.-Z.M.; Bernard, R.; Bernard, C. Antinociceptive and anti-allodynic effects of oral PL37, a complete inhibitor of enkephalin-catabolizing enzymes, in a rat model of peripheral neuropathic pain induced by vincristine. Eur. J. Pharmacol. 2008, 600, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Reaux-Le Goazigo, A.; Poras, H.; Ben-Dhaou, C.; Ouimet, T.; Baudouin, C.; Wurm, M.; Melik Parsadaniantz, S. Dual Enkephalinase Inhibitor PL265: A Novel Topical Treatment to Alleviate Corneal Pain and Inflammation. Pain 2019, 160, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Bonnard, E.; Poras, H.; Fournié-Zaluski, M.-C.; Roques, B.P. Preventive and alleviative effects of the dual enkephalinase inhibitor (Denki) PL265 in a murine model of neuropathic pain. Eur. J. Pharmacol. 2016, 788, 176–182. [Google Scholar] [CrossRef]
- Bonnard, E.; Poras, H.; Nadal, X.; Maldonado, R.; Fournié-Zaluski, M.; Roques, B.P. Long-lasting oral analgesic effects of N-protected aminophosphinic dual ENKephalinase inhibitors (DENKIs) in peripherally controlled pain. Pharmacol. Res. Perspect. 2015, 3, e00116. [Google Scholar] [CrossRef]
- Van Elstraete, A.; Sitbon, P.; Hamdi, L.; Juarez-Perez, V.; Mazoit, J.-X.; Benhamou, D.; Rougeot, C. The Opiorphin Analog STR-324 Decreases Sensory Hypersensitivity in a Rat Model of Neuropathic Pain. Anesth. Analg. 2018, 126, 2102–2111. [Google Scholar] [CrossRef]
- Sitbon, P.; Van Elstraete, A.; Hamdi, L.; Juarez-Perez, V.; Mazoit, J.X.; Benhamou, D.; Rougeot, C. STR-324, a Stable Analog of Opiorphin, Causes Analgesia in Postoperative Pain by Activating Endogenous Opioid Receptor-dependent Pathways. Surv. Anesthesiol. 2016, 125, 1017–1029. [Google Scholar] [CrossRef] [PubMed]
- Pharmaleads. PL37. Available online: http://www.pharmaleads.com/pharmaleads-pipeline/pl37/ (accessed on 15 August 2023).
- Pharmaleads. PL265. Available online: http://www.pharmaleads.com/pharmaleads-pipeline/pl265/ (accessed on 15 August 2023).
- U.S. National Library of Medicine. First-In-Human PainCart Study for STR-324. Identifier NCT03430232. 2021. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT03430232 (accessed on 10 August 2023).
- Varamini, P.; Blanchfield, J.T.; Toth, I. Endomorphin derivatives with improved pharmacological properties. Curr. Med. Chem. 2013, 20, 2741–2758. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.-H.; Wang, B.; Kou, Z.-Z.; Bai, Y.; Chen, T.; Dong, Y.-L.; Li, H.; Li, Y.-Q. Endomorphins: Promising Endogenous Opioid Peptides for the Development of Novel Analgesics. Neurosignals 2017, 25, 98–116. [Google Scholar] [CrossRef] [PubMed]
- Cytogel Pharma. CYT-1010 Novel Mechanism of Action. Available online: https://cytogelpharma.com/novel-mechanism-of-action/ (accessed on 15 August 2023).
- Zadina, J.E.; Nilges, M.R.; Morgenweck, J.; Zhang, X.; Hackler, L.; Fasold, M.B. Endomorphin analog analgesics with reduced abuse liability, respiratory depression, motor impairment, tolerance, and glial activation relative to morphine. Neuropharmacology 2016, 105, 215–227. [Google Scholar] [CrossRef]
- Fowler, S.; Morcos, P.N.; Cleary, Y.; Martin-Facklam, M.; Parrott, N.; Gertz, M.; Yu, L. Progress in Prediction and Interpretation of Clinically Relevant Metabolic Drug-Drug Interactions: A Minireview Illustrating Recent Developments and Current Opportunities. Curr. Pharmacol. Rep. 2017, 3, 36–49. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
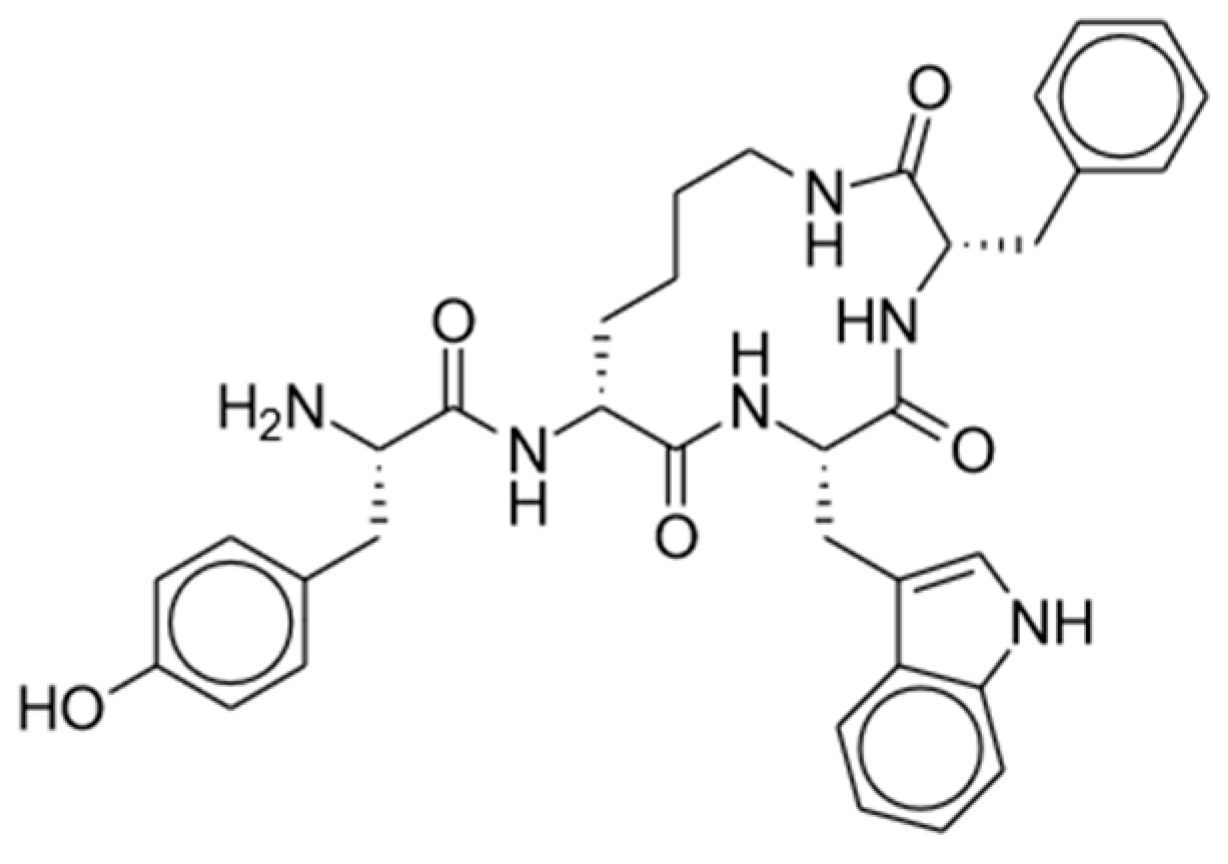
| Novel Compound and Structure | Mechanism of Action | Selected Clinical Trials and Studies |
|---|---|---|
Oliceridine | Mu-opioid agonist | Soergel (2014), Phase I [30]: Oliceridine (3 mg) produced greater analgesia compared to morphine with lower incidences of reduced respiratory drive and severe nausea. |
| Viscusi (2016), Phase II [31]: Oliceridine (2 and 3 mg) after bunionectomy significantly lowered pain score compared to placebo, with the 3 mg dose having a significant improvement over morphine while showing similar tolerability. | ||
| APOLLO-1 & 2 (2019), Phase III [32,33]: Oliceridine is effective as PCA, providing significant relief from moderate/severe postoperative pain compared to placebo with better safety and tolerability vs. morphine. | ||
| ATHENA (2019), Phase III [34]: Oliceridine is safe and well tolerated in patients (both postoperative surgical and non-surgical) with moderate/severe acute pain. | ||
Tapentadol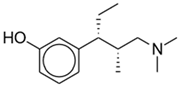 | Mu-opioid agonist | Wang (2020), Systematic Review and Meta-Analysis including 8 RCTs [35]: High-dose tapentadol IR (75–100 mg) is as effective as other opioids for acute pain and is associated with fewer GI adverse effects. |
| Norepinephrine reuptake inhibitor | ||
Cebranopadol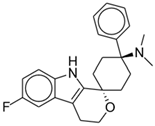 | Mu-opioid agonist | Dahan (2017), Phase I [36]: Cebranopadol (600 mcg) produced respiratory depression but not apnea (as seen in full mu-opioid receptor agonists). |
| Nociceptin opioid peptide receptor agonist | Göhler (2019), Phase I [37]: Cebranopadol has lower abuse potential than hydromorphone IR. | |
| Some delta- and kappa-opioid agonist activity | Scholz (2018), Phase IIa [38]: Single-dose cebranopadol (400 and 600 mcg) produced more effective analgesia after bunionectomy compared to morphine with better tolerability and patient satisfaction rating. | |
| Christoph (2017), Phase II [39]: Cebranopadol demonstrated statistically significant and clinically relevant analgesia for chronic lower back pain compared to placebo with acceptable tolerability. | ||
Dinalbuphine sebacate (DNS)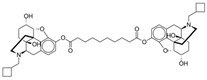 | Partial mu-opioid antagonist | Yeh (2017), Phase III [40]: Single-dose DNS (150 mg) before hemorrhoidectomy produced significant reduction in cumulative pain intensity postoperatively. |
| Kappa-opioid agonist | Lee (2023), Phase II/III [41]: In addition to standard perioperative multimodal analgesia, single-dose DNS (150 mg) allows significantly better pain relief 48 h after laparoscopic bariatric surgery. | |
| Chang (2020), Phase IV [42]: Single-dose DNS (150 mg) resulted in lower pain intensity compared to IV PCA with fentanyl. | ||
STR 324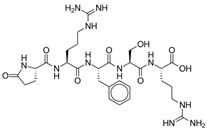 | Dual enkephalinase inhibitor | Moss (2022), Phase I [43]: STR-324 showed favorable safety and tolerability profiles at doses up to 11.475 mg/h in healthy male subjects. |
CYT-1010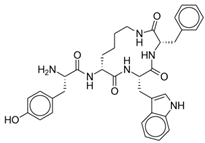 | Endomorphin-1 analog with selective mu-opioid agonism | Webster (2020), Phase I [24]: CYT-1010 demonstrated significant analgesia and no respiratory depression or decrease in plasma oxygen saturation at dose levels tested. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, A.; Murphy, J.; Shteynman, L.; Daksla, N.; Gupta, A.; Bergese, S. Novel Opioids in the Setting of Acute Postoperative Pain: A Narrative Review. Pharmaceuticals 2024, 17, 29. https://doi.org/10.3390/ph17010029
Wang A, Murphy J, Shteynman L, Daksla N, Gupta A, Bergese S. Novel Opioids in the Setting of Acute Postoperative Pain: A Narrative Review. Pharmaceuticals. 2024; 17(1):29. https://doi.org/10.3390/ph17010029
Chicago/Turabian StyleWang, Ashley, Jasper Murphy, Lana Shteynman, Neil Daksla, Abhishek Gupta, and Sergio Bergese. 2024. "Novel Opioids in the Setting of Acute Postoperative Pain: A Narrative Review" Pharmaceuticals 17, no. 1: 29. https://doi.org/10.3390/ph17010029
APA StyleWang, A., Murphy, J., Shteynman, L., Daksla, N., Gupta, A., & Bergese, S. (2024). Novel Opioids in the Setting of Acute Postoperative Pain: A Narrative Review. Pharmaceuticals, 17(1), 29. https://doi.org/10.3390/ph17010029