

Synthesis, In Silico Logp Study, and In Vivo Analgesic Activity of Analogs of Tetrapeptide FELL

 , ,
, ,  , and
, and
Abstract
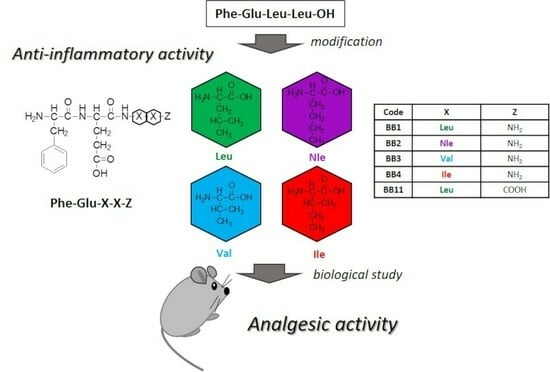
:
1. Introduction
2. Results
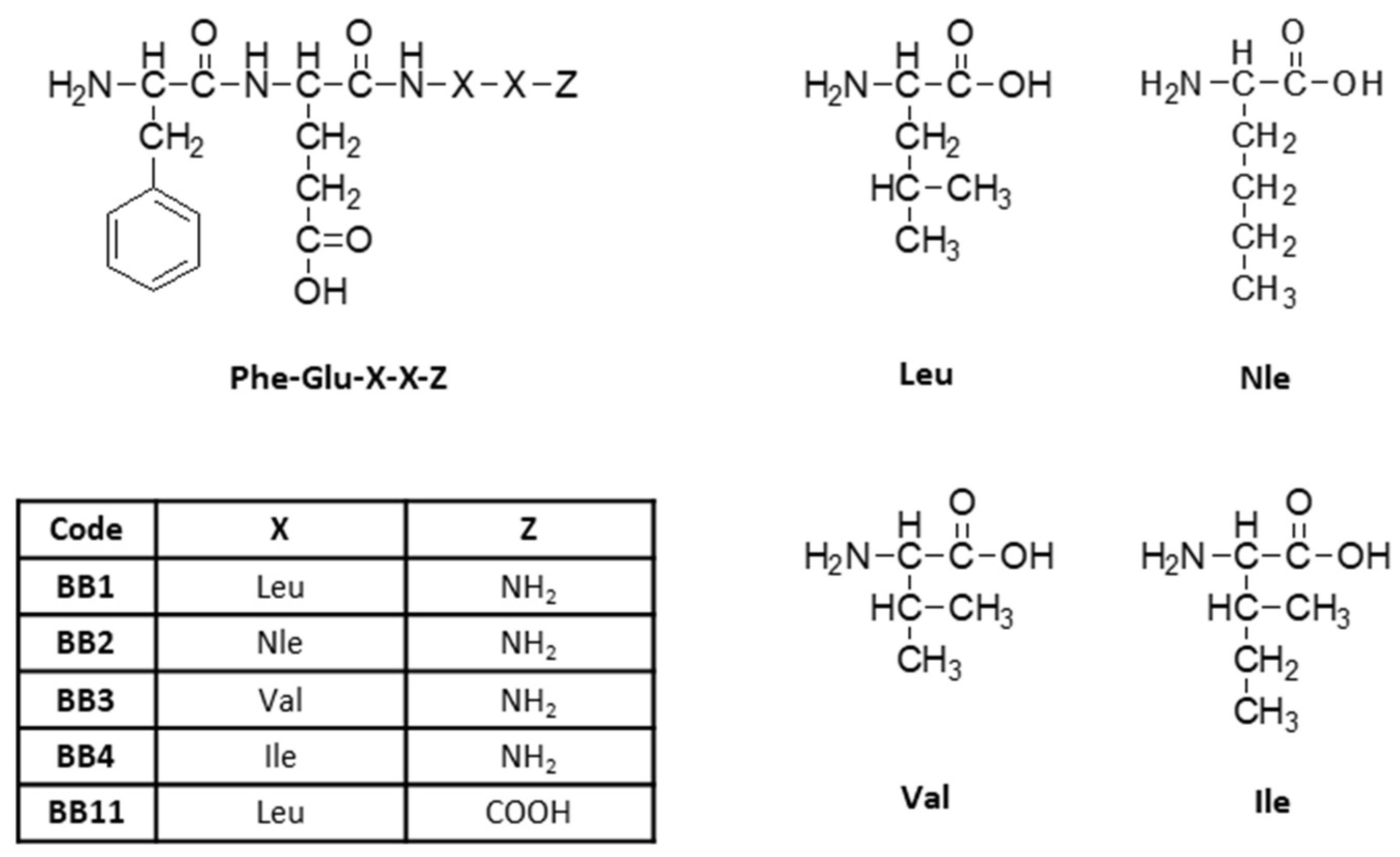
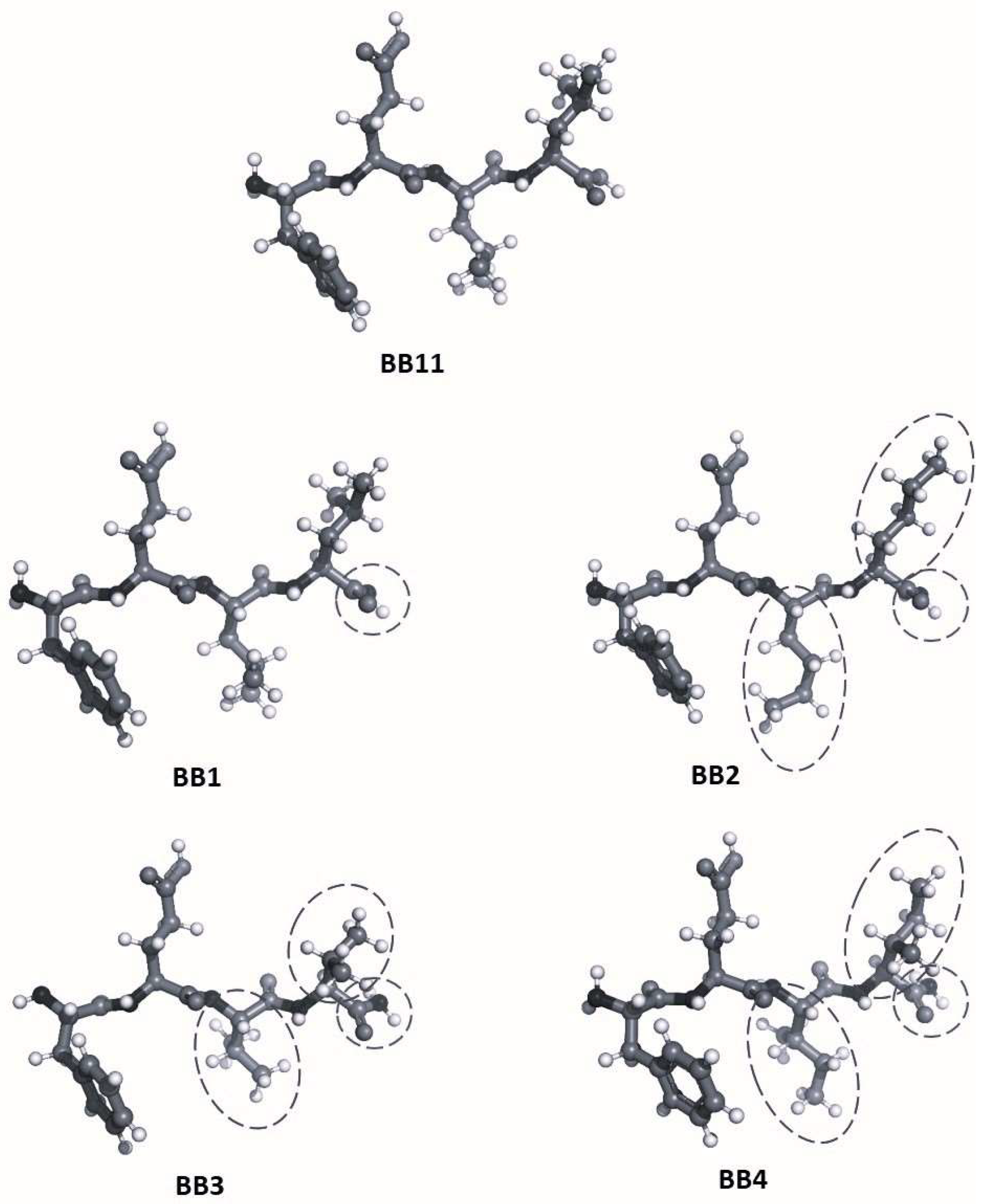
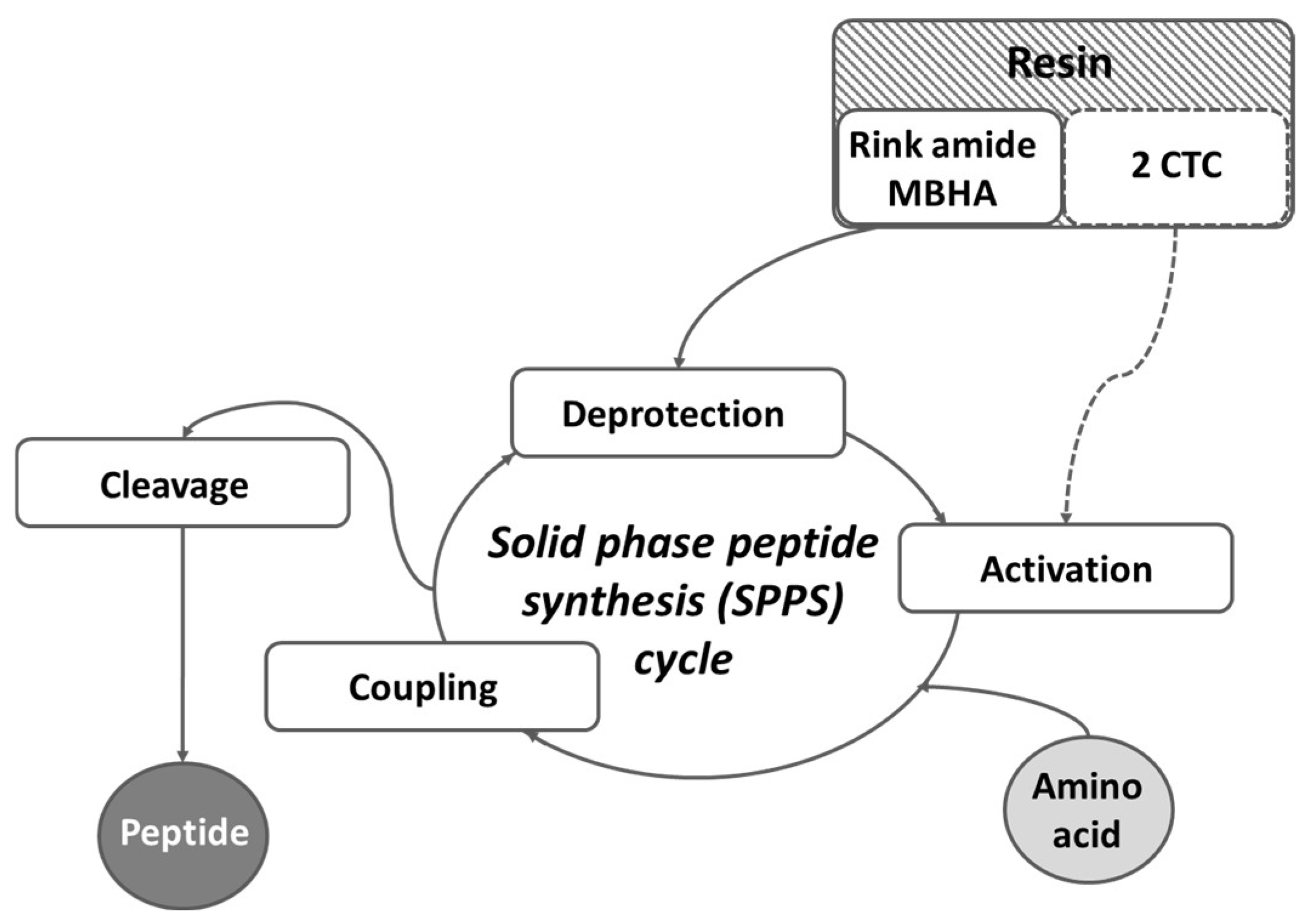
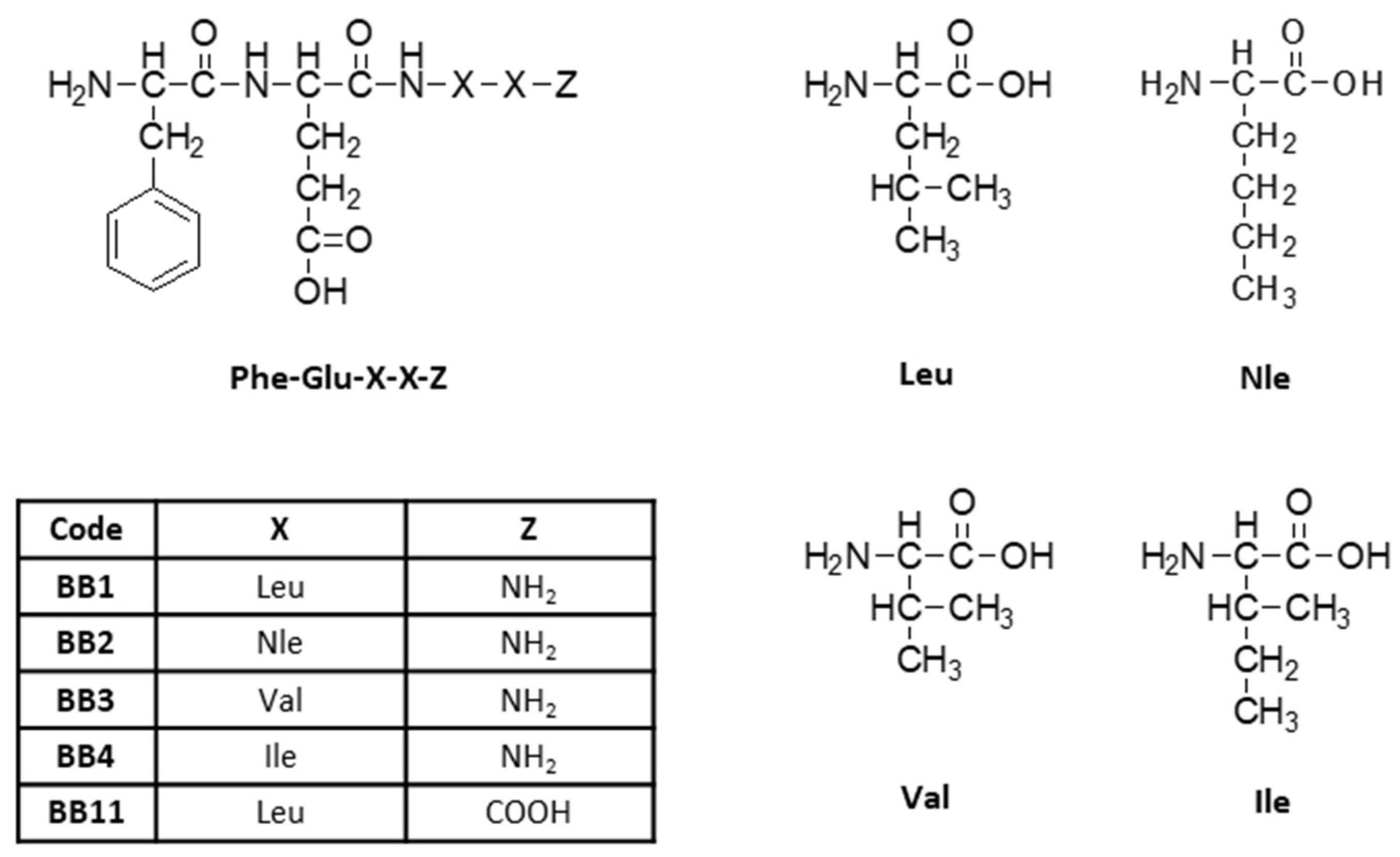
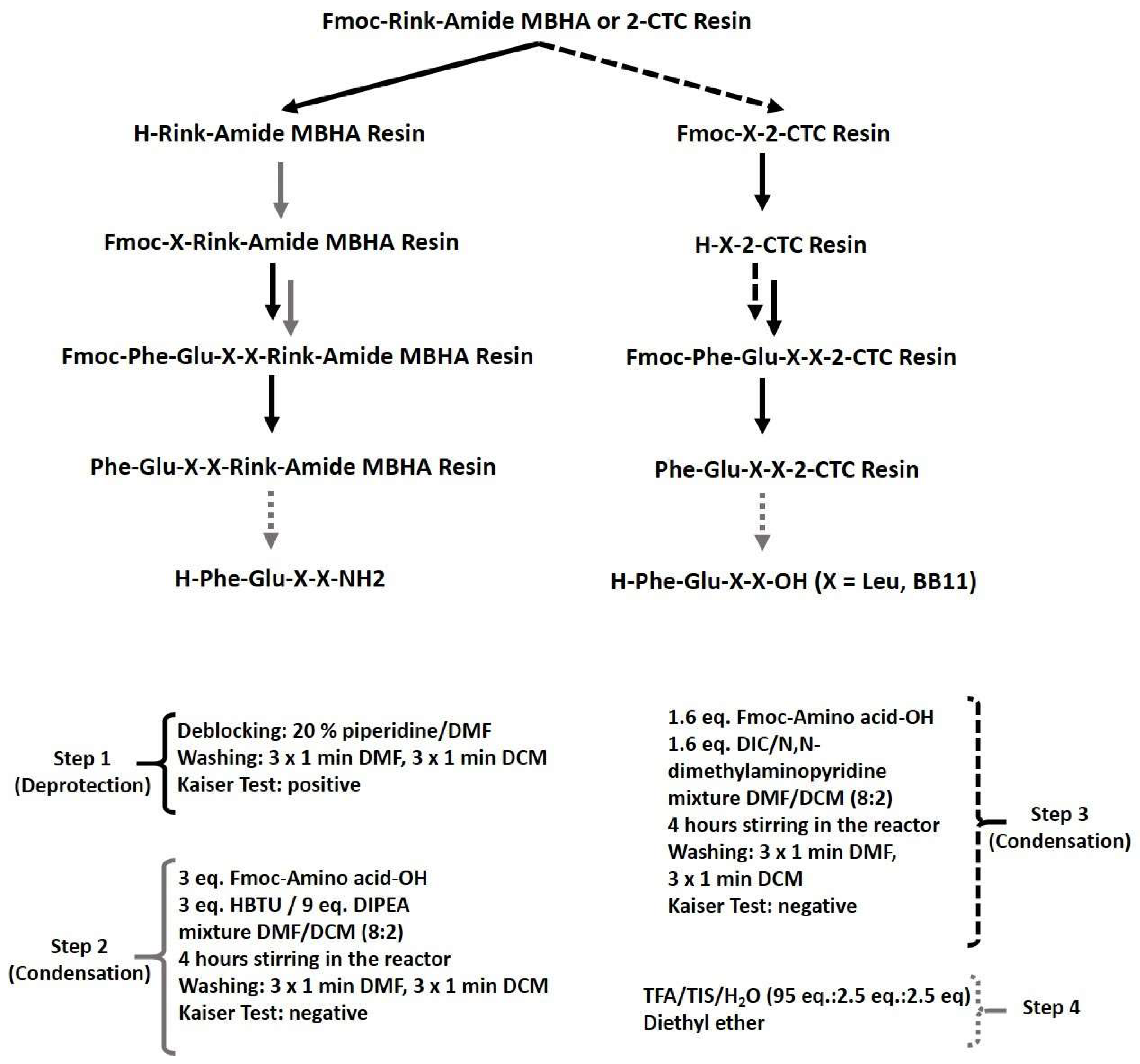
2.1. Synthesis and Characterization of Target Peptides
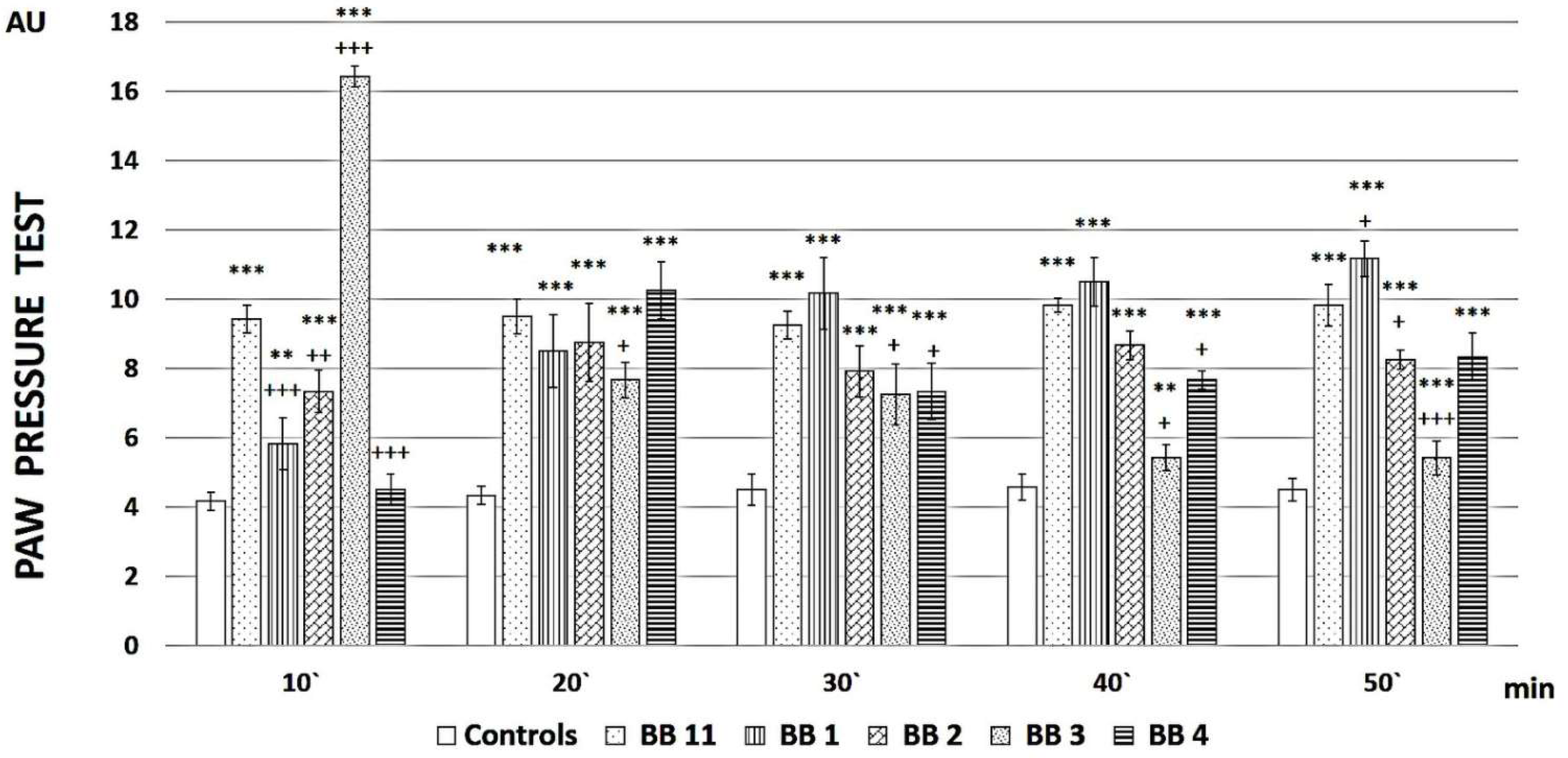
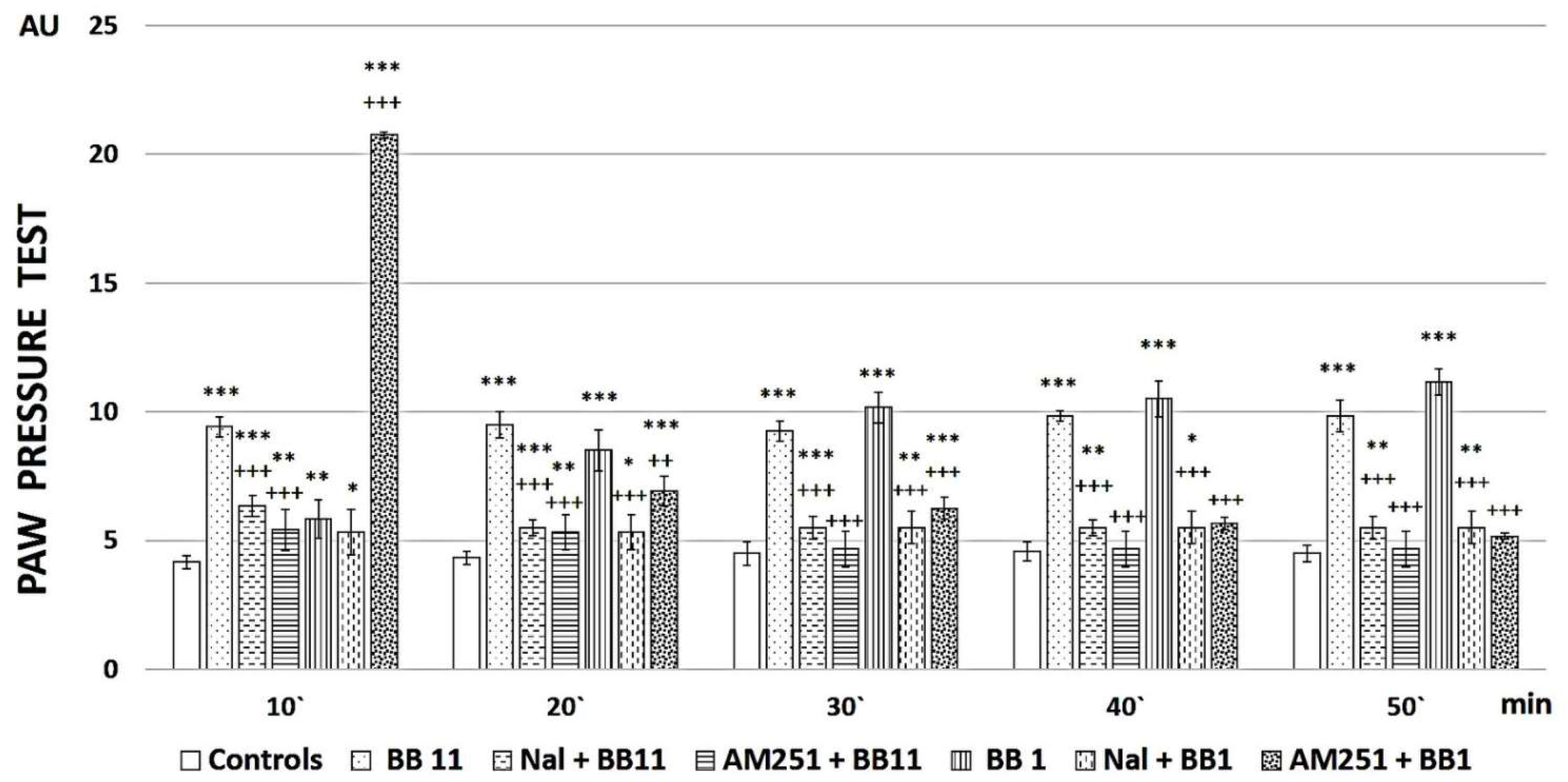
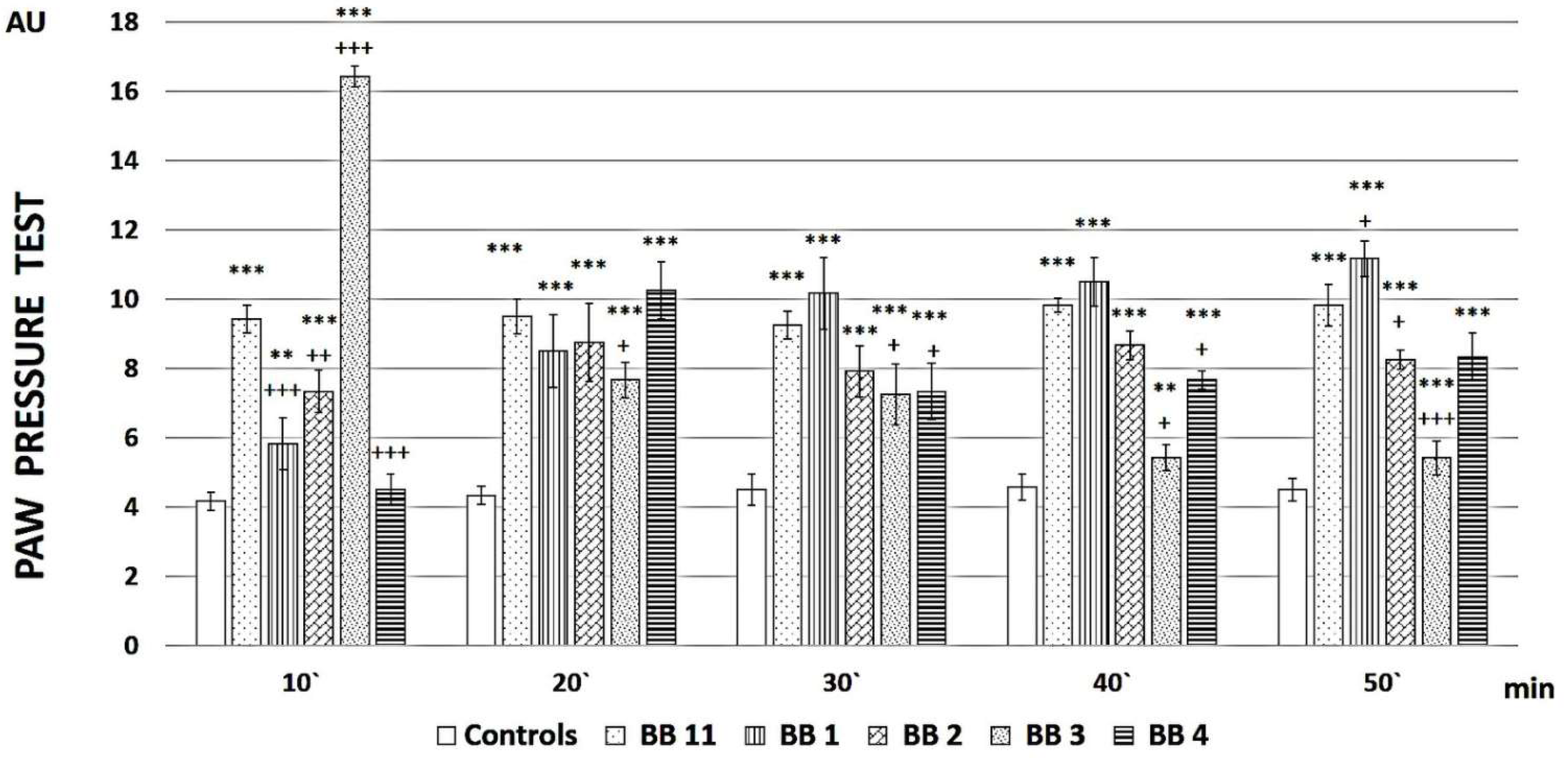
2.2. Evaluation of the Analgesic Properties of the New FELL Analogs
2.3. Prediction of the Logarithm of the N-Octanol Water Partition Coefficient (Logp)
3. Discussion
3.1. Synthesis and Characterization of Target Peptides
3.2. Evaluation of the Analgesic Properties of the New Analogs
3.3. Hydrolytic Stability
3.4. Lipophilicity
4. Materials and Methods
4.1. Synthesis and Analysis of Targeted Peptides
4.1.1. Materials
4.1.2. Peptide Synthesis and Analyses
4.1.3. Model Systems for Hydrolytic Stability Study
- (i)
- A buffer with pH 2.0–6.57 g KCl is dissolved in water (CO2 free), and 119.0 mL of 0.1 mol/L HCl is added. A 0.5-gram aliquot of pepsin was added to the solution in order to obtain a 0.5 mg/mL final concentration. The obtained solution is diluted to 1000.0 mL with dH2O.
- (ii)
- Buffer with pH 7.4–2.38 g Na2HPO4, 0.19 g KH2PO4, and 8.0 g NaCl are dissolved in dH2O. A 0.1-gram aliquot of trypsin was added to the solution in order to obtain a final concentration of 0.1 mg/mL. The obtained solution is diluted to 1000.0 mL with dH2O.
- (iii)
- A buffer with pH 9.0–1000.0 mL of solution I is mixed with 420.0 mL of solution II. Solution I: 6.18 g H3BO3 is dissolved in 0.1 mol/L KCl, and it is completed to 1000.0 mL with the same solvent; Solution II: 0.1 mol/L NaOH. A 0.1-gram aliquot of trypsin was added to the solution in order to obtain a 0.1 mg/mL final concentration.
4.1.4. Lipophilicity Calculations
4.2. In-Vivo Analysis
4.2.1. Animals
4.2.2. Nociceptive Test
Paw-Pressure Test (Randall-Selitto Test)
4.2.3. Pretreatments
4.2.4. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Explanation of Inflammation by National Institute of Environmental Health Sciences. Available online: https://www.niehs.nih.gov/health/topics/conditions/inflammation/index.cfm (accessed on 9 August 2023).
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory Responses and Inflammation-Associated Diseases in Organs. Oncotarget 2018, 9, 7204. [Google Scholar] [CrossRef] [PubMed]
- Ferrero-Miliani, L.; Nielsen, O.H.; Andersen, P.S.; Girardin, S.E. Chronic Inflammation: Importance of NOD2 and NALP3 in Interleukin-1β Generation. Clin. Exp. Immunol. 2007, 147, 227–235. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Raja, S.N.; Carr, D.B.; Cohen, M.; Finnerup, N.B.; Flor, H.; Gibson, S.; Keefe, F.J.; Mogil, J.S.; Ringkamp, M.; Sluka, K.A.; et al. The Revised International Association for the Study of Pain Definition of Pain: Concepts, Challenges, and Compromises. Pain 2020, 161, 1976–1982. [Google Scholar] [CrossRef] [PubMed]
- Rainville, P.; Duncan, G.H.; Price, D.D.; Carrier, B.; Bushnell, M.C. Pain Affect Encoded in Human Anterior Cingulate But Not Somatosensory Cortex. Science 1997, 277, 968–971. [Google Scholar] [CrossRef]
- Price, D.D. Psychological and Neural Mechanisms of the Affective Dimension of Pain. Science 2000, 288, 1769–1772. [Google Scholar] [CrossRef]
- Danalev, D.L.; Vladimirova, S.P.; Borisov, B.P.; Nocheva, H.H.; Bocheva, A.I.; Marinkova, D.A.; Naydenova, E.D.; Lozanov, V.S. Synthesis and Analgesic Activity of New Analogues of Tyr-MIF Including Pyrrole Moiety. Int. J. Pept. Res. Ther. 2016, 22, 243–248. [Google Scholar] [CrossRef]
- McNicol, E.; Horowicz-Mehler, N.; Fisk, R.A.; Bennett, K.; Gialeli-Goudas, M.; Chew, P.W.; Lau, J.; Carr, D. Management of Opioid Side Effects in Cancer-Related and Chronic Noncancer Pain: A Systematic Review. J. Pain 2003, 4, 231–256. [Google Scholar] [CrossRef] [PubMed]
- Hyman, S.E.; Malenka, R.C.; Nestler, E.J. Neural mechanisms of addiction: The Role of Reward-Related Learning and Memory. Annu. Rev. Neurosci. 2006, 29, 565–598. [Google Scholar] [CrossRef]
- Le Merrer, J.; Becker, J.A.J.; Befort, K.; Kieffer, B.L. Reward Processing by the Opioid System in the Brain. Physiol. Rev. 2009, 89, 1379–1412. [Google Scholar] [CrossRef]
- Al-Hasani, R.; Bruchas, M.R. Molecular Mechanisms of Opioid Receptor-Dependent Signaling and Behavior. Anesthesiology 2011, 115, 1363–1381. [Google Scholar] [CrossRef] [PubMed]
- Castro, D.C.; Berridge, K.C. Opioid Hedonic Hotspot in Nucleus Accumbens Shell: Mu, Delta, and Kappa Maps for Enhancement of Sweetness “Liking” and “Wanting”. J. Neurosci. 2014, 34, 4239–4250. [Google Scholar] [CrossRef]
- Wang, Y.; Zhuang, Y.; DiBerto, J.F.; Zhou, X.E.; Schmitz, G.P.; Yuan, Q.; Jain, M.K.; Liu, W.; Melcher, K.; Jiang, Y.; et al. Structures of the Entire Human Opioid Receptor Family. Cell 2023, 186, 413–427.e17. [Google Scholar] [CrossRef]
- Cuitavi, J.; Torres-Pérez, J.V.; Lorente, J.D.; Campos-Jurado, Y.; Andrés-Herrera, P.; Polache, A.; Agustín-Pavón, C.; Hipólito, L. Crosstalk between Mu-Opioid Receptors and Neuroinflammation: Consequences for Drug Addiction and Pain. Neurosci. Biobehav. Rev. 2023, 145, 105011. [Google Scholar] [CrossRef]
- Bodnar, R.J. Endogenous Opiates and Behavior: 2020. Peptides 2022, 151, 170752. [Google Scholar] [CrossRef] [PubMed]
- Hasler, W.L.; Wilson, L.A.; Nguyen, L.A.; Snape, W.J.; Abell, T.L.; Koch, K.L.; McCallum, R.W.; Pasricha, P.J.; Sarosiek, I.; Farrugia, G.; et al. Opioid Use and Potency Are Associated with Clinical Features, Quality of Life, and Use of Resources in Patients with Gastroparesis. Clin. Gastroenterol. Hepatol. 2019, 17, 1285–1294.e1. [Google Scholar] [CrossRef]
- Rudd, R.A.; Seth, P.; David, F.; Scholl, L. Increases in Drug and Opioid-Involved Overdose Deaths—United States, 2010–2015. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 1445–1452. [Google Scholar] [CrossRef]
- Pertwee, R.G. Cannabinoid Receptors and Pain. Prog. Neurobiol. 2001, 63, 569–611. [Google Scholar] [CrossRef]
- Liu, Q.Q.; Yao, X.X.; Gao, S.H.; Li, R.; Li, B.J.; Yang, W.; Cui, R.J. Role of 5-HT Receptors in Neuropathic Pain: Potential Therapeutic Implications. Pharmacol. Res. 2020, 159, 104949. [Google Scholar] [CrossRef]
- Zheng, H.; Lim, J.Y.; Kim, Y.; Jung, S.T.; Hwang, S.W. The Role of Oxytocin, Vasopressin, and Their Receptors at Nociceptors in Peripheral Pain Modulation. Front. Neuroendocrinol. 2021, 63, 100942. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.-M.; Peyton, L.; Essa, H.; Choi, D.-S. Adenosine Receptors: Emerging Non-Opioids Targets for Pain Medications. Neurobiol. Pain 2022, 11, 100087. [Google Scholar] [CrossRef]
- Luo, Y.; Kusay, A.S.; Jiang, T.; Chebib, M.; Balle, T. Delta-Containing GABAA Receptors in Pain Management: Promising Targets for Novel Analgesics. Neuropharmacology 2021, 195, 108675. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J. Synthesis of Best-Seller Drugs. By Ruben Vardanyan and Victor Hruby. Angew. Chemie Int. Ed. 2017, 56, 2541. [Google Scholar] [CrossRef]
- Ramamoorthy, S.; Cidlowski, J.A. Corticosteroids. Rheum. Dis. Clin. N. Am. 2016, 42, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Zöllner, C.; Stein, C. Opioids. In Analgesia; Springer: Berlin/Heidelberg, Germany; pp. 31–63.
- Thayer, M. Improving peptides. Chem. Eng. News 2011, 89, 13–20. [Google Scholar] [CrossRef]
- St. Laurent, C.D.; St. Laurent, K.E.; Mathison, R.D.; Befus, A.D. Calcium-Binding Protein, Spermatid-Specific 1 Is Expressed in Human Salivary Glands and Contains an Anti-Inflammatory Motif. Am. J. Physiol. Integr. Comp. Physiol. 2015, 308, R569–R575. [Google Scholar] [CrossRef]
- Morris, K.; Kuo, B.; Wilkinson, M.D.; Davison, J.S.; Befus, A.D.; Mathison, R.D. Vcsa1 Gene Peptides for the Treatment of Inflammatory and Allergic Reactions. Recent Pat. Inflamm. Allergy Drug Discov. 2007, 1, 124–132. [Google Scholar] [CrossRef]
- Dadar, M.; Shahali, Y.; Chakraborty, S.; Prasad, M.; Tahoori, F.; Tiwari, R.; Dhama, K. Antiinflammatory Peptides: Current Knowledge and Promising Prospects. Inflamm. Res. 2019, 68, 125–145. [Google Scholar] [CrossRef]
- Mathison, R. Anti-Inflammatory Peptides. U.S. Patent 7,153,835 B2, 26 December 2006. [Google Scholar]
- Metwally, E.; Pires, J.M.; Moore, G.J.; Befus, D.A.; Davison, J.S.; Mathison, R. Submandibular Gland Tripeptide FEG (Phe-Glu-Gly) and Analogues: Keys to Structure Determination. Peptides 2002, 23, 193–199. [Google Scholar] [CrossRef]
- Omoigui, S. The Biochemical Origin of Pain: The Origin of All Pain Is Inflammation and the Inflammatory Response. Part 2 of 3—Inflammatory Profile of Pain Syndromes. Med. Hypotheses 2007, 69, 1169–1178. [Google Scholar] [CrossRef]
- Mandalari, G.; Adel-Patient, K.; Barkholt, V.; Baro, C.; Bennett, L.; Bublin, M.; Gaier, S.; Graser, G.; Ladics, G.S.; Mierzejewska, D.; et al. In Vitro Digestibility of β-Casein and β-Lactoglobulin under Simulated Human Gastric and Duodenal Conditions: A Multi-Laboratory Evaluation. Regul. Toxicol. Pharmacol. 2009, 55, 372–381. [Google Scholar] [CrossRef]
- Froimowitz, M. HyperChem: A Software Package for Computational Chemistry and Molecular Modeling. Biotechniques 1993, 14, 1010–1013. [Google Scholar] [PubMed]
- Randall, L.O.; Selitto, J.J. A Method for Measurement of Analgesic Activity on Inflamed Tissue. Arch. Int. Pharmacodyn. Ther. 1957, 111, 409–419. [Google Scholar]
- Kanemasa, T.; Koike, K.; Takase, K.; Arai, T.; Nakamura, A.; Morioka, Y.; Hasegawa, M. Pharmacological Profile of Naldemedine, a Peripherally Acting μ -Opioid Receptor Antagonist: Comparison with Naloxone and Naloxegol. J. Pharmacol. Exp. Ther. 2020, 373, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Katsuyama, S.; Mizoguchi, H.; Komatsu, T.; Nagaoka, K.; Sakurada, S.; Sakurada, T. The Cannabinoid 1 Receptor Antagonist AM251 Produces Nocifensive Behavior via Activation of ERK Signaling Pathway. Neuropharmacology 2010, 59, 534–541. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Structure | Molecular Formula | Mm exact, g/mol | [M + nH]+ observed | [M + Na]+observed | M.p., jjj °C | tR Min | αD20, O | Yield, % | Chromatographic Purity, % |
|---|---|---|---|---|---|---|---|---|---|---|
| BB1 | H-Phe-Glu-Leu-Leu-NH2 | C26H41N5O6 | 519.31 | 520.40 | 542.40 | 219 ± 221 | 5.317 | −28 | 100 | 99 |
| BB2 | H-Phe-Glu-Nle-Nle-NH2 | C26H41N5O6 | 519.31 | 520.45 | 542.40 | 221 ± 223 | 5.467 | 10 | 99 | 96 |
| BB3 | H-Phe-Glu-Val-Val-NH2 | C24H37N5O6 | 491.27 | 492.40 | 514.35 | 223 ± 224 | 4.100 | −10 | 83 | 96 |
| BB4 | H-Phe-Glu-Ile-Ile-NH2 | C26H40N4O7 | 520.29 | 520.45 | - | 218 ± 211 | 4.950 | −8 | 76 | 97 |
| BB11 | H-Phe-Glu-Leu-Leu-OH * | C26H40N4O7 | 520.29 | 521.39 | 543.40 | 219 ± 220 | 4.817 | −8 | 46 | 98 |
| Code | Logp |
|---|---|
| BB1 | 0.67 |
| BB2 | 0.80 |
| BB3 | 0.02 |
| BB4 | 0.82 |
| BB11 | 1.54 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borisova, B.; Nocheva, H.; Gérard, S.; Laronze-Cochard, M.; Dobrev, S.; Angelova, S.; Petrin, S.; Danalev, D. Synthesis, In Silico Logp Study, and In Vivo Analgesic Activity of Analogs of Tetrapeptide FELL. Pharmaceuticals 2023, 16, 1183. https://doi.org/10.3390/ph16081183
Borisova B, Nocheva H, Gérard S, Laronze-Cochard M, Dobrev S, Angelova S, Petrin S, Danalev D. Synthesis, In Silico Logp Study, and In Vivo Analgesic Activity of Analogs of Tetrapeptide FELL. Pharmaceuticals. 2023; 16(8):1183. https://doi.org/10.3390/ph16081183
Chicago/Turabian StyleBorisova, Boryana, Hristina Nocheva, Stéphane Gérard, Marie Laronze-Cochard, Stefan Dobrev, Silvia Angelova, Stoyko Petrin, and Dancho Danalev. 2023. "Synthesis, In Silico Logp Study, and In Vivo Analgesic Activity of Analogs of Tetrapeptide FELL" Pharmaceuticals 16, no. 8: 1183. https://doi.org/10.3390/ph16081183
APA StyleBorisova, B., Nocheva, H., Gérard, S., Laronze-Cochard, M., Dobrev, S., Angelova, S., Petrin, S., & Danalev, D. (2023). Synthesis, In Silico Logp Study, and In Vivo Analgesic Activity of Analogs of Tetrapeptide FELL. Pharmaceuticals, 16(8), 1183. https://doi.org/10.3390/ph16081183