


Synthesis, Physicochemical Characterization, Biological Evaluation, In Silico and Molecular Docking Studies of Pd(II) Complexes with P, S-Donor Ligands

 ,
,
Abstract
1. Introduction
2. Results and Discussion
2.1. FT-IR and Raman Results
 N moiety. The partial double bond character in the C-N bond for dithiocarbamate complexes was confirmed from the appearance of the peak in the range of 1520–1530 cm−1 as the single C-N bond appears at 1251–1361 cm−1 while the C=N shift value is reported at 1640–1690 cm−1 [17]. The presence of a single asymmetric SCS peak in the range of 1092–1097 cm−1 indicates the bidentate bonding mode of the dithiocarbamate ligand with palladium moiety; for monodentate bonding, two peaks are to be observed in this region if the dithiocarbamate [35]. Figure S1 of the Supplementary Materials shows the FTIR spectrum of Complex 2.
N moiety. The partial double bond character in the C-N bond for dithiocarbamate complexes was confirmed from the appearance of the peak in the range of 1520–1530 cm−1 as the single C-N bond appears at 1251–1361 cm−1 while the C=N shift value is reported at 1640–1690 cm−1 [17]. The presence of a single asymmetric SCS peak in the range of 1092–1097 cm−1 indicates the bidentate bonding mode of the dithiocarbamate ligand with palladium moiety; for monodentate bonding, two peaks are to be observed in this region if the dithiocarbamate [35]. Figure S1 of the Supplementary Materials shows the FTIR spectrum of Complex 2.2.2. Multinuclear NMR (1H, 13C and 31P)
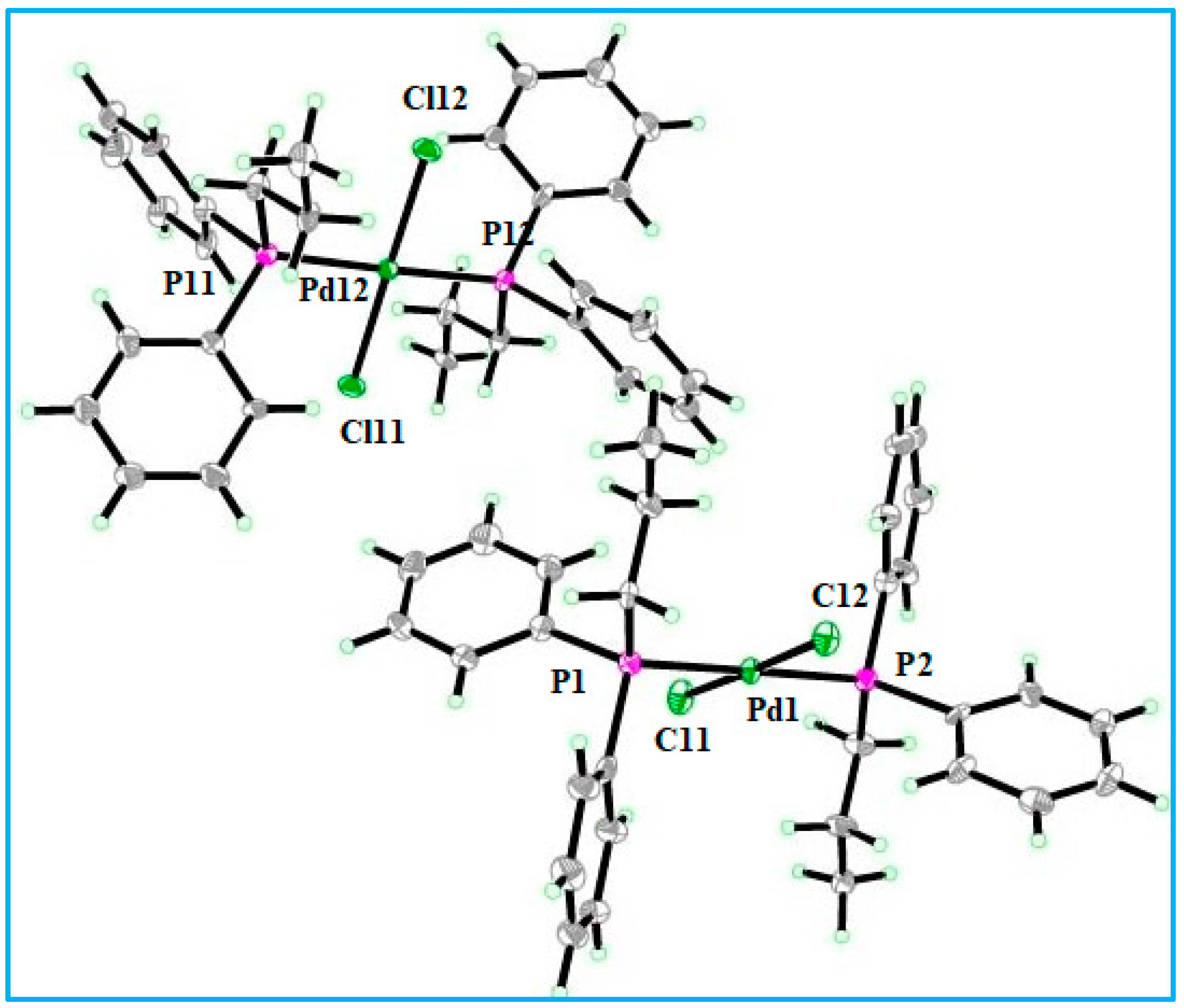
2.3. Structural Study of Complex 1
2.4. Biological Investigation Results
2.4.1. Antitumor Results
2.4.2. Antibacterial Results
2.5. In Silico Assessment
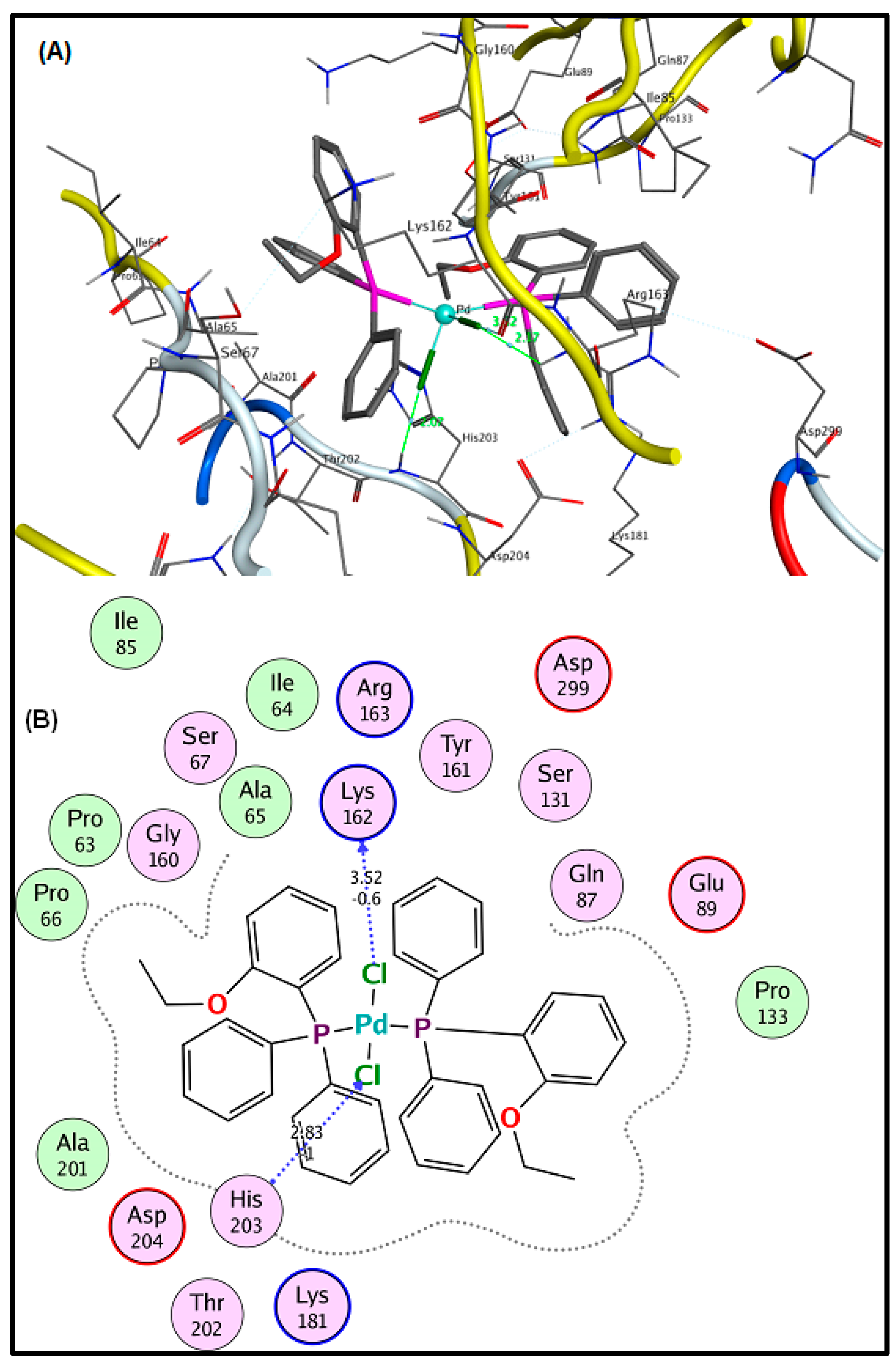
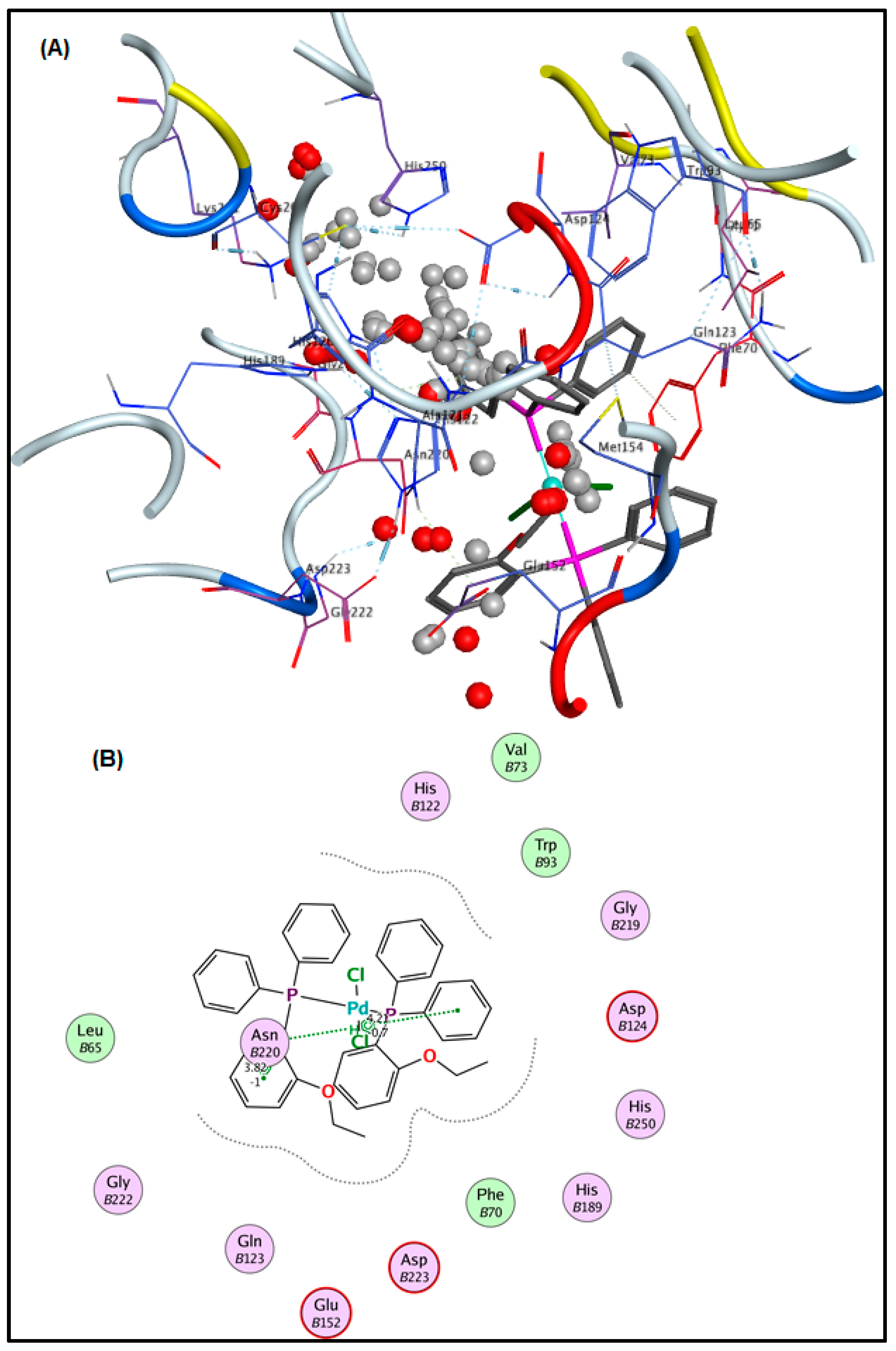
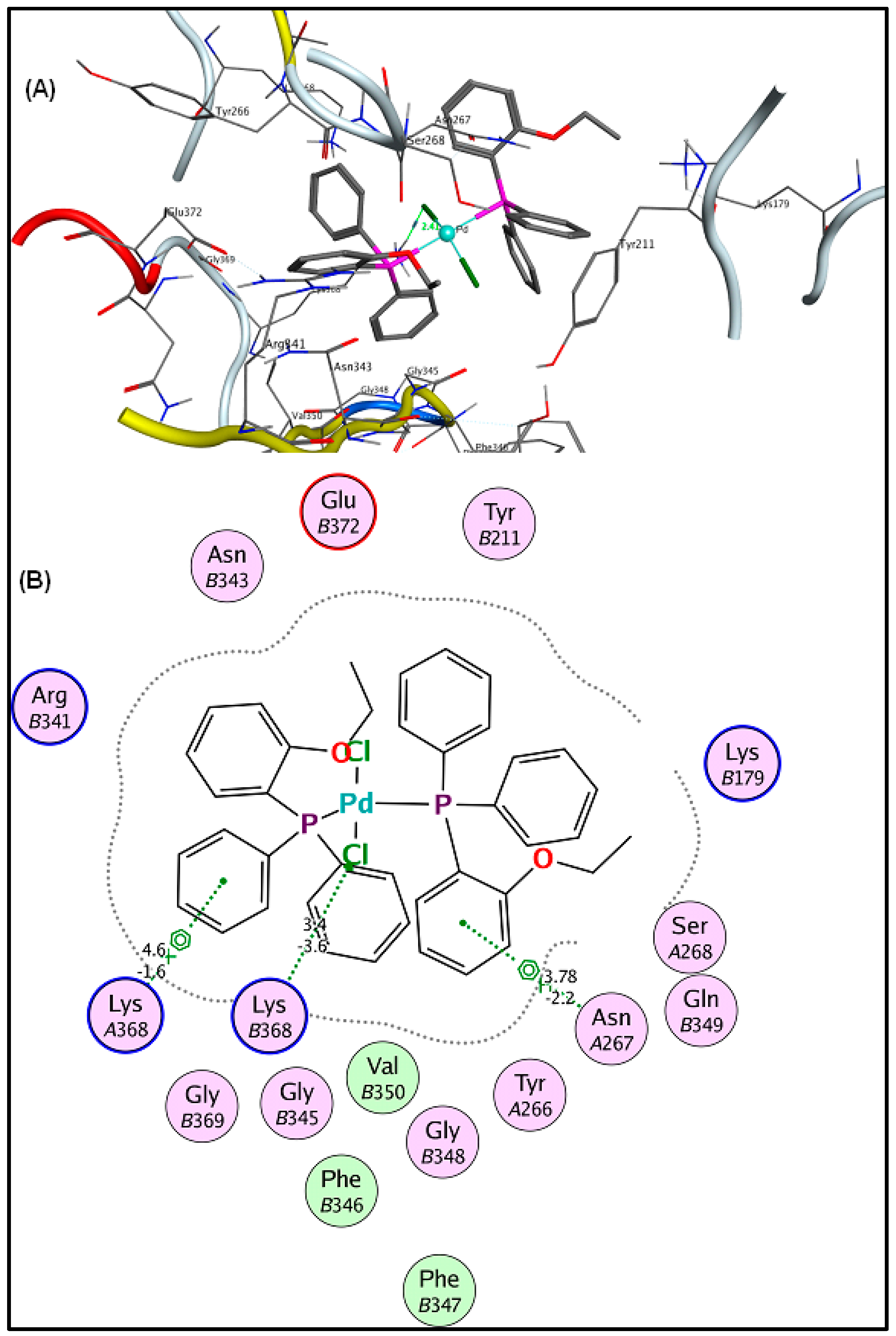
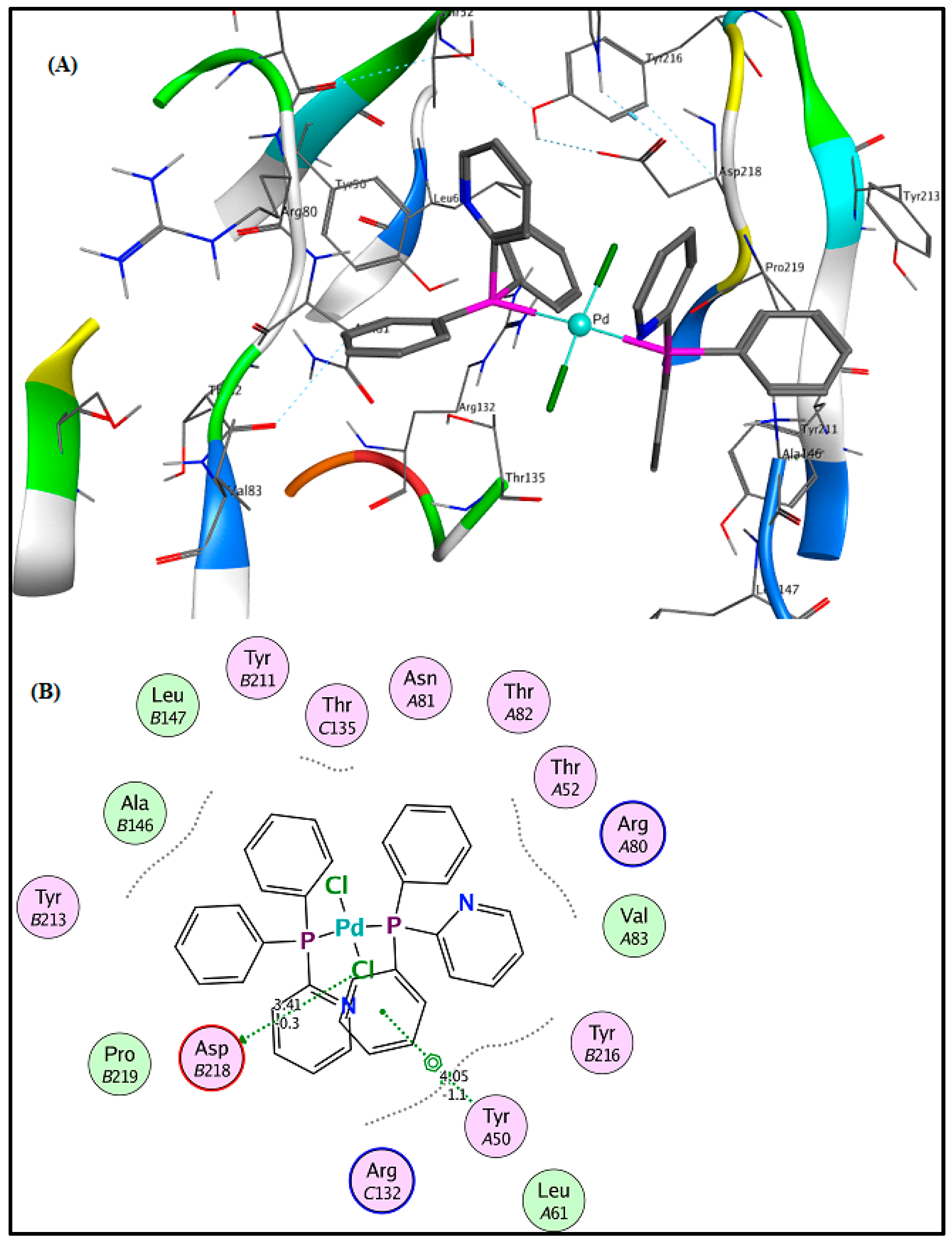
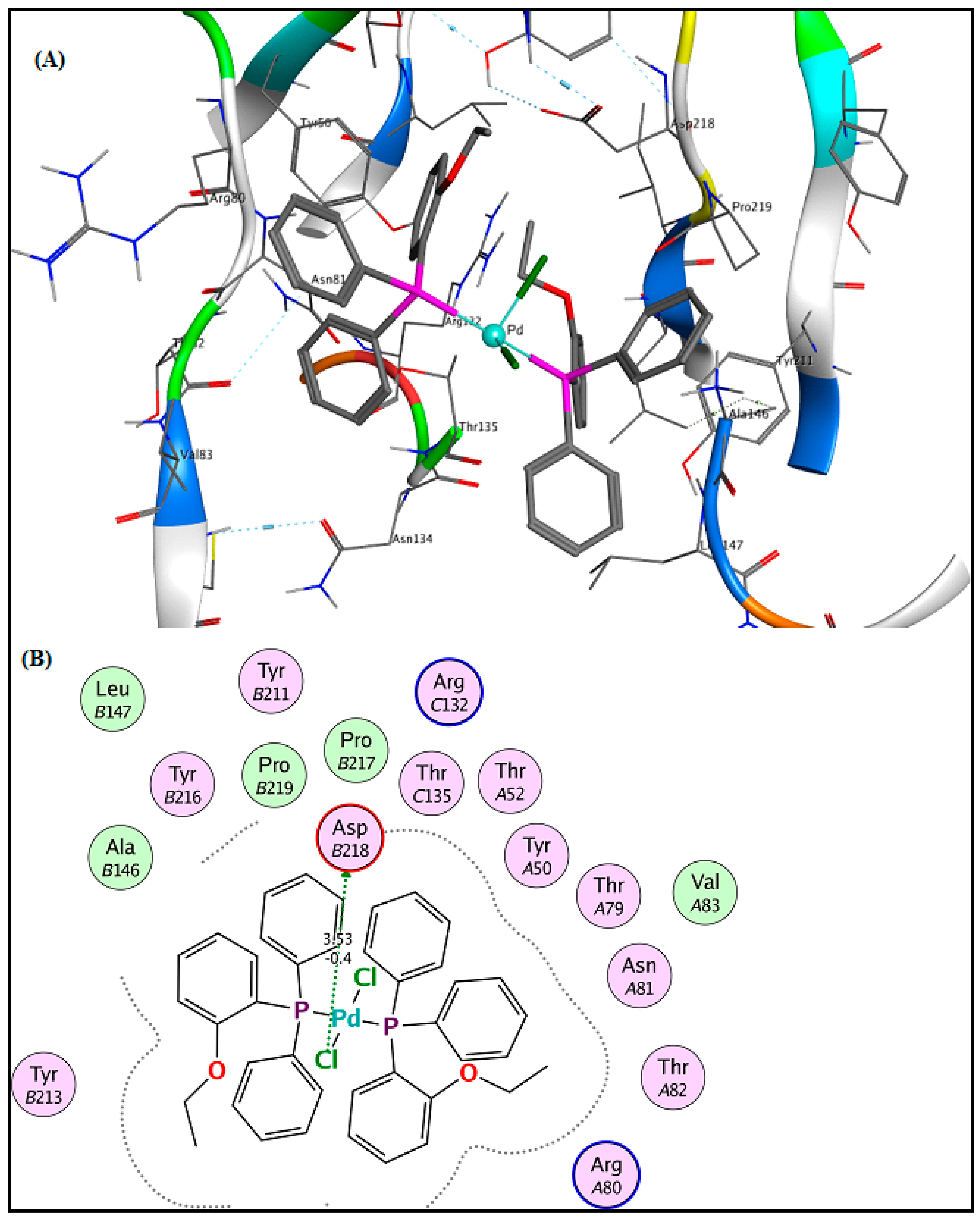
2.6. Molecular Docking Results
3. Experimental
3.1. Materials and Methods
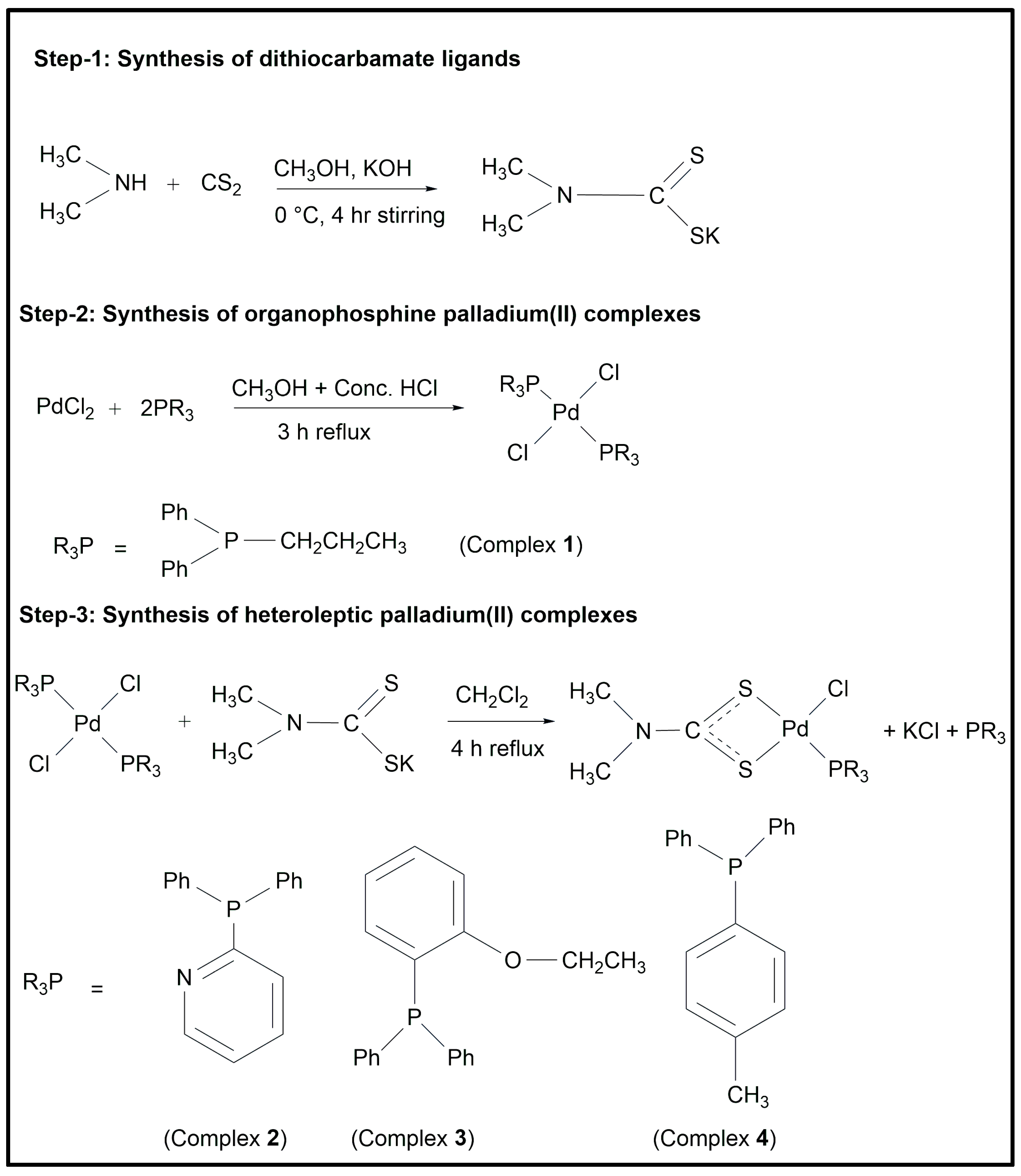
3.2. Synthesis
3.2.1. Dichlorido(diphenyl–n–propylphosphine)palladium(II) (1)
3.2.2. Chlorido–(dimethyldithiocarbamato–κ2S,Sʹ)(diphenyl-2-pyridylphosphine) palladium(II) (2)
N, 2925, 2861, υ(C-H, aliphatic), 3056 υ(C-H, aromatic). Raman (cm−1): 380 υ(Pd–S), 300 υ(Pd–Cl), 218 υ(Pd–P). 1H NMR (ppm): 2.80 (s), 3.58 (s) (CH3), 7.37–8.73 (m) (Ar-H). 13C NMR (ppm): 35.5, 38.6 (CH3), 125.0, 128.9, 130.0, 130.8, 131.3, 134.7, 134.6, 134.9, 149.3, 157.6 (Ar–C) 207.5 (SCS). 31P NMR (ppm): 27.5.3.2.3. Chlorido–(dimethyldithiocarbamato–κ2S,Sʹ)(diphenyl–2–ethoxyphenyl phosphine)palladium(II) (3)
N), 2930, 2850 υ(C–H, aliphatic), 3052 υ(C–H, aromatic). Raman (cm−1): 390 υ(Pd–S), 301 υ(Pd–Cl), 215 υ(Pd–P). 1H NMR (ppm): 3.14 (s), 3.26 (s) (CH3), 3.98 (q, –CH2), 1.33 (s, CH3), 6.90–7.76 (m, Ar–H). 13C NMR (ppm): 14.3 (-CH2CH3), 65.2 (-CH2CH3), 120.1, 126.3, 128.2, 130.4, 131.7, 133.2, 164.0 (Ar-C), 208.2 (SCS). 31P NMR (ppm): 20.3.3.2.4. Chlorido–(dimethyldithiocarbamato–κ2S,Sʹ)(diphenyl–p–tolylphosphine) palladium(II) (4)
N) 2935, 2852, υ(C–H, aliphatic), 3040 υ(C–H, aromatic). Raman (cm−1): 395 υ(Pd–S), 308 υ(Pd–Cl), 210 υ(Pd–P). 1H NMR (ppm): 3.09 (s), 3.27 (s) (CH3), 2.38 (s) (Ph–CH3), 7.43–7.92 (m) (Ar–H). 13C NMR (ppm): 36.4, 39.1 (CH3), 128.3, 129.5, 134.0, 136.5, 136.8, 137.1, 137.6, 141.7 (Ar–C), 22.5 (Ph–CH3), 207.3 (SCS). 31P NMR (ppm): 22.8.3.3. Biological Activity Assays
3.3.1. Antibacterial Activity Assay
3.3.2. Antitumor Activity
3.4. In Silico Studies
3.5. Molecular Docking Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parvez, T. Cancer treatment: What’s ahead? J. Coll. Physicians Surg. Pak. 2005, 15, 738–745. [Google Scholar] [PubMed]
- Huq, F. Molecular modelling analysis of the metabolism of benzene. Int. J. Pure Appl. Chem. 2006, 1, 461–467. [Google Scholar]
- Meyskens, F.L., Jr.; Tully, P. Principles of cancer prevention. Semin. Oncol. Nurs. 2005, 21, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.A.; Khan, H.; Sirajuddin, M.; Salman, S.M. DNA Interaction and Biological Activities of Heteroleptic Palladium (II) Complexes. J. Chem. Soc. Pak. 2021, 43, 227–243. [Google Scholar]
- Verweij, J.; De Jonge, M. Achievements and future of chemotherapy. Eur. J. Cancer 2000, 36, 1479–1487. [Google Scholar] [CrossRef]
- Galanski, M.; Arion, V.; Jakupec, M.; Keppler, B. Recent developments in the field of tumor-inhibiting metal complexes. Curr. Pharm. Des. 2003, 9, 2078–2089. [Google Scholar] [CrossRef]
- Loehrer, P.J.; Einhorn, L.H. Cisplatin. Ann. Intern. Med. 1984, 100, 704–713. [Google Scholar] [CrossRef]
- Goyns, M.H. Cancer and You: How to Stack the Odds in Your Favour; CRC Press: Boca Raton, FL, USA, 1999. [Google Scholar]
- Stern, T.A.; Sekeres, M.A. Facing Cancer: A Complete Guide for People with Cancer, Their Families, and Caregivers; McGraw Hill Professional: New York City, NY, USA, 2004. [Google Scholar]
- Mehdi, I. Second Malignancy-A Rare Phenomenon. J. Pak. Med. Assoc. 1998, 48, 345–346. [Google Scholar]
- Fichtinger-Schepman, A.; Veer, J.; Lohman, P.; Reedijk, J. A simple method for the inactivation of monofunctionally DNA-bound cis-diamminedichloroplatinum (II). J. Inorg. Biochem. 1984, 21, 103–111. [Google Scholar] [CrossRef]
- Lippard, S.J. New chemistry of an old molecule: Cis-[Pt(NH3)2Cl2]. Science 1982, 218, 1075–1082. [Google Scholar] [CrossRef]
- Imran, M.; ur Rehman, Z.; Hogarth, G.; Tocher, D.A.; Butler, I.S.; Bélanger-Gariepy, F.; Kondratyuk, T. Two new monofunctional platinum (ii) dithiocarbamate complexes: Phenanthriplatin-type axial protection, equatorial-axial conformational isomerism, and anticancer and DNA binding studies. Dalton Trans. 2020, 49, 15385–15396. [Google Scholar] [CrossRef] [PubMed]
- Martinho, N.; Marquês, J.M.; Todoriko, I.; Prieto, M.; de Almeida, R.F.; Silva, L.C. Effect of Cisplatin and Its Cationic Analogues in the Phase Behavior and Permeability of Model Lipid Bilayers. Mol. Pharm. 2023, 20, 918–928. [Google Scholar] [CrossRef]
- Abbotto, A.; Beverina, L.; Bradamante, S.; Facchetti, A.; Klein, C.; Pagani, G.A.; Redi-Abshiro, M.; Wortmann, R. A distinctive example of the cooperative interplay of structure and environment in tuning of intramolecular charge transfer in second-order nonlinear optical chromophores. Chem. A Eur. J. 2003, 9, 1991–2007. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, V.M.; Fuertes, M.A.; Alonso, C.; Perez, J.M. Is cisplatin-induced cell death always produced by apoptosis? Mol. Pharmacol. 2001, 59, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.; Badshah, A.; Murtaz, G.; Said, M.; Neuhausen, C.; Todorova, M.; Jean-Claude, B.J.; Butler, I.S. Synthesis, characterization and anticancer studies of mixed ligand dithiocarbamate palladium (II) complexes. Eur. J. Med. Chem. 2011, 46, 4071–4077. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.; Badshah, A.; Said, M.; Murtaza, G.; Ahmad, J.; Jean-Claude, B.J.; Todorova, M.; Butler, I.S. Anticancer metallopharmaceutical agents based on mixed-ligand palladium (II) complexes with dithiocarbamates and tertiary organophosphine ligands. Appl. Organomet. Chem. 2013, 27, 387–395. [Google Scholar] [CrossRef]
- Khan, H.; Badshah, A.; Said, M.; Murtaza, G.; Shah, A.; Butler, I.S.; Ahmed, S.; Fontaine, F.-G. New dimeric and supramolecular mixed ligand Palladium (II) dithiocarbamates as potent DNA binders. Polyhedron 2012, 39, 1–8. [Google Scholar] [CrossRef]
- Guerra, W.; de Andrade Azevedo, E.; de Souza Monteiro, A.R.; Bucciarelli-Rodriguez, M.; Chartone-Souza, E.; Nascimento, A.M.A.; Fontes, A.P.S.; Le Moyec, L.; Pereira-Maia, E.C. Synthesis, characterization, and antibacterial activity of three palladium (II) complexes of tetracyclines. J. Inorg. Biochem. 2005, 99, 2348–2354. [Google Scholar] [CrossRef]
- Khan, B.T.; Najmuddin, K.; Shamsuddin, S.; Annapoorna, K.; Bhatt, J. Synthesis, antimicrobial, and antitumor activity of a series of palladium (II) mixed ligand complexes. J. Inorg. Biochem. 1991, 44, 55–63. [Google Scholar] [CrossRef]
- Khan, H.; Badshah, A.; Said, M.; Murtaza, G.; Sirajuddin, M.; Ahmad, J.; Butler, I.S. Synthesis, structural characterization and biological screening of heteroleptic palladium (II) complexes. Inorg. Chim. Acta 2016, 447, 176–182. [Google Scholar] [CrossRef]
- Hogarth, G. Metal-dithiocarbamate complexes: Chemistry and biological activity. Mini Rev. Med. Chem. 2012, 12, 1202–1215. [Google Scholar] [CrossRef]
- Marta Nagy, E.; Ronconi, L.; Nardon, C.; Fregona, D. Noble metal-dithiocarbamates precious allies in the fight against cancer. Mini Rev. Med. Chem. 2012, 12, 1216–1229. [Google Scholar] [CrossRef]
- Adeyemi, J.O.; Onwudiwe, D.C. Organotin (IV) dithiocarbamate complexes: Chemistry and biological activity. Molecules 2018, 23, 2571. [Google Scholar] [CrossRef]
- Sirajuddin, M.; Ali, S.; Tahir, M.N. Pharmacological investigation of mono-, di-and tri-organotin (IV) derivatives of carbodithioates: Design, spectroscopic characterization, interaction with SS-DNA and POM analyses. Inorg. Chim. Acta 2016, 439, 145–158. [Google Scholar] [CrossRef]
- Micklitz, W.; Sheldrick, W.S.; Lippert, B. Mono-and dinuclear palladium (II) complexes of uracil and thymine model nucleobases and the x-ray structure of [(bpy) Pd (1-MeT) 2Pd (bpy)](NO3) 2. cntdot. 5.5 H2O (head-head). Inorg. Chem. 1990, 29, 211–216. [Google Scholar] [CrossRef]
- Barnham, K.J.; Bauer, C.J.; Djuran, M.I.; Mazid, M.A.; Rau, T.; Sadler, P.J. Outer-Sphere Macrochelation in [Pd (en)(5′-GMP-N7) 2]. cntdot. 9H2O and [Pt (en)(5′-GMP-N7) 2]. cntdot. 9H2O: X-ray Crystallography and NMR Spectroscopy in Solution. Inorg. Chem. 1995, 34, 2826–2832. [Google Scholar] [CrossRef]
- Shen, W.-Z.; Gupta, D.; Lippert, B. Cyclic Trimer versus Head—Tail Dimer in Metal—Nucleobase Complexes: Importance of Relative Orientation (Syn, Anti) of the Metal Entities and Relevance as a Metallaazacrown Compound. Inorg. Chem. 2005, 44, 8249–8258. [Google Scholar] [CrossRef]
- Rau, T.; Van Eldik, R. Mechanistic insight from kinetic studies on the interaction of model palladium (II) complexes with nucleic acid components. Met. Ions Biol. Syst. 1996, 32, 339. [Google Scholar] [PubMed]
- Gao, E.-J.; Wang, L.; Zhu, M.-C.; Liu, L.; Zhang, W.-Z. Synthesis, characterization, interaction with DNA and cytotoxicity in vitro of the complexes [M (dmphen)(CO3)]· H2O [M = Pt (II), Pd (II)]. Eur. J. Med. Chem. 2010, 45, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Budzisz, E.; Małecka, M.; Lorenz, I.-P.; Mayer, P.; Kwiecień, R.A.; Paneth, P.; Krajewska, U.; Rózalski, M. Synthesis, cytotoxic effect, and structure—Activity relationship of Pd (II) complexes with coumarin derivatives. Inorg. Chem. 2006, 45, 9688–9695. [Google Scholar] [CrossRef] [PubMed]
- Ewais, H.A.; Taha, M.; Salm, H.N. Palladium (II) Complexes Containing Dipicolinic Acid (DPA), Iminodiacetic Acid (IDA), and Various Biologically Important Ligands. J. Chem. Eng. Data 2010, 55, 754–758. [Google Scholar] [CrossRef]
- Imran, M.; Kondratyuk, T.; Bélanger-Gariepy, F. New ternary platinum (II) dithiocarbamates: Synthesis, characterization, anticancer, DNA binding and DNA denaturing studies. Inorg. Chem. Commun. 2019, 103, 12–20. [Google Scholar] [CrossRef]
- Shaheen, F.; Badshah, A.; Gielen, M.; Croce, G.; Florke, U.; de Vos, D.; Ali, S. In vitro assessment of cytotoxicity, anti-inflammatory, antifungal properties and crystal structures of metallacyclic palladium (II) complexes. J. Organomet. Chem. 2010, 695, 315–322. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Zhu, W.; Li, Y.; Gao, L. Cisplatin in combination with programmed cell death protein 5 increases antitumor activity in prostate cancer cells by promoting apoptosis. Mol. Med. Rep. 2015, 11, 4561–4566. [Google Scholar] [CrossRef]
- Chohan, Z.H.; Sumrra, S.H.; Youssoufi, M.H.; Hadda, T.B. Metal based biologically active compounds: Design, synthesis, and antibacterial/antifungal/cytotoxic properties of triazole-derived Schiff bases and their oxovanadium (IV) complexes. Eur. J. Med. Chem. 2010, 45, 2739–2747. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Valente, C.; Ferreira, M.J.; Abreu, P.M.; Pedro, M.; Cerqueira, F.; Nascimento, M.S.J. Three new jatrophane-type diterpenes from Euphorbia pubescens. Planta Med. 2003, 69, 361–366. [Google Scholar] [CrossRef]
- Kim, Y.; Bang, S.-C.; Lee, J.-H.; Ahn, B.-Z. Pulsatilla saponin D: The antitumor principle from Pulsatilla koreana. Arch. Pharmacal Res. 2004, 27, 915–918. [Google Scholar] [CrossRef]
- Fleming, N. How artificial intelligence is changing drug discovery. Nature 2018, 557, S55. [Google Scholar] [CrossRef] [PubMed]
- Kinch, M.S.; Griesenauer, R.H. 2017 in review: FDA approvals of new molecular entities. Drug Discov. Today 2018, 23, 1469–1473. [Google Scholar] [CrossRef] [PubMed]
- Sertkaya, A.; Wong, H.-H.; Jessup, A.; Beleche, T. Key cost drivers of pharmaceutical clinical trials in the United States. Clin. Trials 2016, 13, 117–126. [Google Scholar] [CrossRef]
- M Honorio, K.; L Moda, T.; Andricopulo, A.D. Pharmacokinetic properties and in silico ADME modeling in drug discovery. Med. Chem. 2013, 9, 163–176. [Google Scholar] [CrossRef] [PubMed]
- King, D.T.; Worrall, L.J.; Gruninger, R.; Strynadka, N.C. New Delhi metallo-β-lactamase: Structural insights into β-lactam recognition and inhibition. J. Am. Chem. Soc. 2012, 134, 11362–11365. [Google Scholar] [CrossRef] [PubMed]
- Dalal, V.; Kumar, P.; Rakhaminov, G.; Qamar, A.; Fan, X.; Hunter, H.; Tomar, S.; Golemi-Kotra, D.; Kumar, P. Repurposing an ancient protein core structure: Structural studies on FmtA, a novel esterase of Staphylococcus aureus. J. Mol. Biol. 2019, 431, 3107–3123. [Google Scholar] [CrossRef] [PubMed]
- Dalal, V.; Kumar, P.; Rakhaminov, G.; Qamar, A.; Fan, X.; Hunter, H.; Tomar, S.; Kotra, D.; Kumar, P. Repurposing an ancient protein core structure: Structural studies on FmtA, a novel esterase of Staphylococcus aureus. Acta Crystallogr. Sect. A 2021, 77, C706. [Google Scholar] [CrossRef]
- Levy, N.; Bruneau, J.-M.; Le Rouzic, E.; Bonnard, D.; Le Strat, F.; Caravano, A.; Chevreuil, F.; Barbion, J.; Chasset, S.; Ledoussal, B. Structural basis for E. coli penicillin binding protein (PBP) 2 inhibition, a platform for drug design. J. Med. Chem. 2019, 62, 4742–4754. [Google Scholar] [CrossRef]
- Naz, N.; Sirajuddin, M.; Haider, A.; Abbas, S.M.; Ali, S.; Wadood, A.; Ghufran, M.; Rehman, G.; Mirza, B. Synthesis, characterization, biological screenings and molecular docking study of Organotin (IV) derivatives of 2, 4-dichlorophenoxyacetic acid. J. Mol. Struct. 2019, 1179, 662–671. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Data |
|---|---|
| Molecular formula and weigth | C30H34Cl2P2Pd & 633.81 |
| Temperature and λ | 200 K & 1.54178 Å |
| Crystal system and Space group | Monoclinic and P21 |
| dimensions of unit cell | a = 16.3128(4) Å, b = 10.2144(2) Å, c = 18.7283(4)α, γ = 90°, β = 115.793(1)° |
| Volume and Z | 2809.71(11)Å3 & 4 |
| ρ, μ & F(000) | 1.498 g/cm3, 8.280 mm−1 & 1296 |
| θ range | 2.62 to 72.50° |
| Index | −19 ≤ h ≤ 20, −12 ≤ k ≤ 12, −23 ≤ l ≤ 23 |
| Collec. Ref. | 37,079 |
| Indep. Ref. | 5866 [Rint = 0.045] |
| Max. and min. transmission | 0.5601 and 0.3968 |
| Data/restraints/parameters | 5866/1/636 |
| S on F2 | 1.144 |
| Final R indices [I > 2sigma(I)] | R1 = 0.0584, wR2 = 0.1387 |
| R indices (all data) | R1 = 0.0586, wR2 = 0.1389 |
| Largest diff. peak and hole | 5.871 and −1.371 e/Å3 |
| CCDC# | 2,218,216 |
| Selected Bond Length/Å | |||
|---|---|---|---|
| Atom–Atom | Length | Atom–Atom | Length |
| Pd1–Cl1 | 2.301(2) | Pd12–Cl11 | 2.301(2) |
| Pd1–Cl2 | 2.302(2) | Pd12–Cl12 | 2.300(2) |
| Pd1–P1 | 2.328(3) | Pd12–P11 | 2.318(3) |
| Pd1–P2 | 2.326(3) | Pd12–P12 | 2.328(3) |
| P1–C11 | 1.853(13) | P11–C111 | 1.833(11) |
| P1–C21 | 1.822(11) | P11–C121 | 1.846(13) |
| P1–C31 | 1.829(10) | P11–C131 | 1.841(11) |
| P2–C41 | 1.858(12) | P12–C141 | 1.807(12) |
| P2–C51 | 1.814(10) | P12–C151 | 1.824(11) |
| P2–C61 | 1.825(12) | P12–C161 | 1.841(12) |
| Selected Bond Length/° | |||
| Atom–Atom–Atom | Angle | Atom–Atom–Atom | Angle |
| Cl1–Pd1–Cl2 | 178.40(11) | Cl11–Pd12–P11 | 90.56(10) |
| Cl1–d1–P1 | 90.46(10) | Cl11–Pd12–P12 | 90.25(10) |
| Cl1–Pd1–P2 | 89.09(10) | Cl12–Pd12–Cl11 | 178.86(11) |
| Cl2–Pd1–P1 | 89.68(10) | Cl12–Pd12–P11 | 89.45(10) |
| Cl2–Pd1–P2 | 90.77(10) | Cl12–Pd12–P12 | 89.77(10) |
| P2–Pd1–P1 | 179.39(13) | P11–Pd12–P12 | 178.11(11) |
| C11–P1–Pd1 | 106.6(4) | C111–P11–Pd12 | 119.3(4) |
| C21–P1–Pd1 | 117.5(4) | C121–P11–Pd12 | 105.9(4) |
| C31–P1–Pd1 | 116.4(3) | C131–P11–Pd12 | 117.9(4) |
| C41–P2–Pd1 | 108.0(4) | C141–P12–Pd12 | 117.2(4) |
| C51–P2–Pd1 | 118.7(4) | C151–P12–Pd12 | 109.9(4) |
| C61–P2–Pd1 | 115.8(4) | C161–P12–Pd12 | 114.6(4) |
| Compound # | IC50 (µM) |
|---|---|
| 1 | 6.94 ± 0.58 |
| 2 | 3.67 ± 0.17 |
| 3 | 4.57 ± 0.57 |
| 4 | 21.7 ± 1.15 |
| cisplatin | >200 [38] |
| Sample | E. coli | K. pneumoniae | S. aureus | B. subtilis | S. epidermidis |
|---|---|---|---|---|---|
| 1 | 19 ± 0.33 | 17 ± 0.31 | 21 ± 0.41 | 17 ± 0.31 | 16 ± 0.15 |
| 2 | 18 ± 0.30 | 14 ± 0.24 | 17 ± 0.31 | 18 ± 0.30 | 16 ± 0.15 |
| 3 | 19 ± 0.33 | 2 ± 0.11 | 18 ± 0.30 | 18 ± 0.30 | 20 ± 0.31 |
| 4 | 20 ± 0.34 | 16 ± 0.15 | 17 ± 0.31 | 16 ± 0.15 | 19 ± 0.33 |
| SD | 26 ± 0.21 | 26 ± 0.23 | 35 ± 0.11 | 35 ± 0.11 | 35 ± 0.11 |
| DMSO | 0 | 0 | 0 | 0 | 0 |
| Comp. # | Physicochemical Properties | Lipophilicity | Water Solubility (mg/mL) | Pharmacokinetics | Drug Likeness | Medicinal Chemistry | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M. W. | RT | LogPo/w(iLOGP) | LogPo/w(XLOGP) | S | Class | BBB P | GI A | Lipinsky | Veber | PAINS | Brenk | |
| 1 | 633.4 | 10 | 0.00 | 9.49 | 1.61 × 10−7 | PS | No | Low | No; 2 vio: MW > 500, MLOGP > 4.15 | Yes | 0 alert | 1 alert: phosphor |
| 2 | 526.4 | 5 | 0.00 | 6.46 | 2.42 × 10−5 | PS | No | High | Yes; 1 vio: MW > 500 | Yes | 0 alert | 1 alert: phosphor |
| 3 | 568.4 | 7 | 0.00 | 7.54 | 4.55 × 10−6 | PS | No | High | No; 2 vio: MW > 500, MLOGP > 4.15 | Yes | 0 alert | 2 alerts: phosphor, thiocarbonyl group |
| 4 | 539.4 | 5 | 0.00 | 7.56 | 4.36 × 10−6 | PS | No | High | No; 2 vio: MW > 500, | Yes | 0 alert | 1 alert: phosphor |
| Receptor | Ligand | Binding Affinity (kcal/mol) |
|---|---|---|
| E. coli (PDB_ID: 6G9S) | 1 | −5.3894 |
| 2 | −6.1962 | |
| 3 | −8.6569 | |
| 4 | −6.8761 | |
| K. pneumonia (PDB_ID: 4EXS) | 1 | −5.5038 |
| 2 | −5.9045 | |
| 3 | −6.5716 | |
| 4 | −6.4517 | |
| S. aureus (PDB_ID: 5ZH8) | 1 | −6.1450 |
| 2 | −7.5425 | |
| 3 | −7.6966 | |
| 4 | −7.4745 | |
| DR5 (PDB_ID: 1DU3) | 1 | −6.4104 |
| 2 | −7. 5148 | |
| 3 | −7. 0343 | |
| 4 | −6.6559 | |
| Cisplatin | −3.5666 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, H.; Sirajuddin, M.; Badshah, A.; Ahmad, S.; Bilal, M.; Salman, S.M.; Butler, I.S.; Wani, T.A.; Zargar, S. Synthesis, Physicochemical Characterization, Biological Evaluation, In Silico and Molecular Docking Studies of Pd(II) Complexes with P, S-Donor Ligands. Pharmaceuticals 2023, 16, 806. https://doi.org/10.3390/ph16060806
Khan H, Sirajuddin M, Badshah A, Ahmad S, Bilal M, Salman SM, Butler IS, Wani TA, Zargar S. Synthesis, Physicochemical Characterization, Biological Evaluation, In Silico and Molecular Docking Studies of Pd(II) Complexes with P, S-Donor Ligands. Pharmaceuticals. 2023; 16(6):806. https://doi.org/10.3390/ph16060806
Chicago/Turabian StyleKhan, Hizbullah, Muhammad Sirajuddin, Amin Badshah, Sajjad Ahmad, Muhammad Bilal, Syed Muhammad Salman, Ian S. Butler, Tanveer A. Wani, and Seema Zargar. 2023. "Synthesis, Physicochemical Characterization, Biological Evaluation, In Silico and Molecular Docking Studies of Pd(II) Complexes with P, S-Donor Ligands" Pharmaceuticals 16, no. 6: 806. https://doi.org/10.3390/ph16060806
APA StyleKhan, H., Sirajuddin, M., Badshah, A., Ahmad, S., Bilal, M., Salman, S. M., Butler, I. S., Wani, T. A., & Zargar, S. (2023). Synthesis, Physicochemical Characterization, Biological Evaluation, In Silico and Molecular Docking Studies of Pd(II) Complexes with P, S-Donor Ligands. Pharmaceuticals, 16(6), 806. https://doi.org/10.3390/ph16060806