


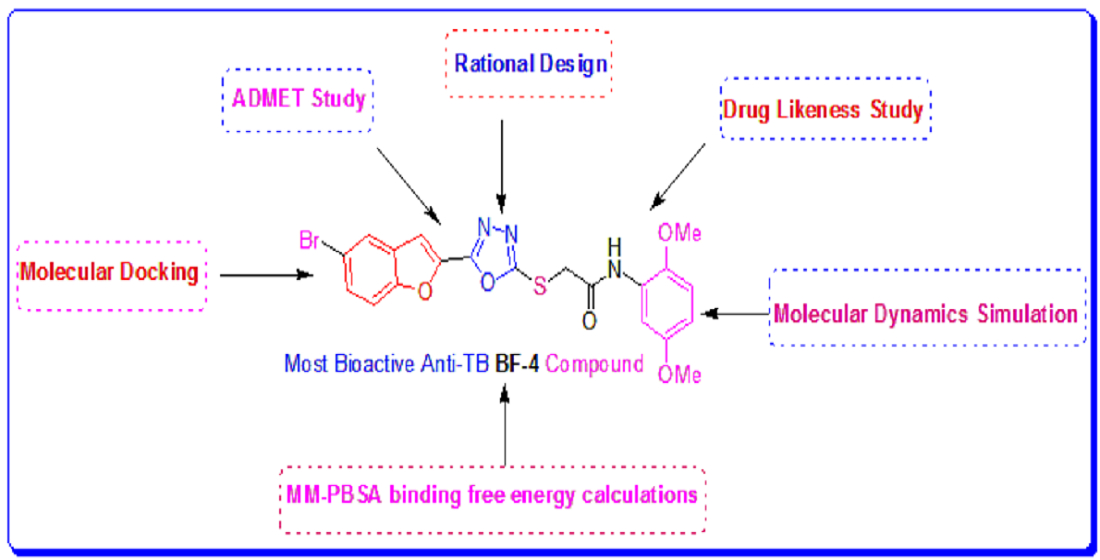
In Silico Development of Novel Benzofuran-1,3,4-Oxadiazoles as Lead Inhibitors of M. tuberculosis Polyketide Synthase 13

 ,
,  ,
,  , , , ,
, , , ,  and
and 
Abstract

1. Introduction
2. Results and Discussion
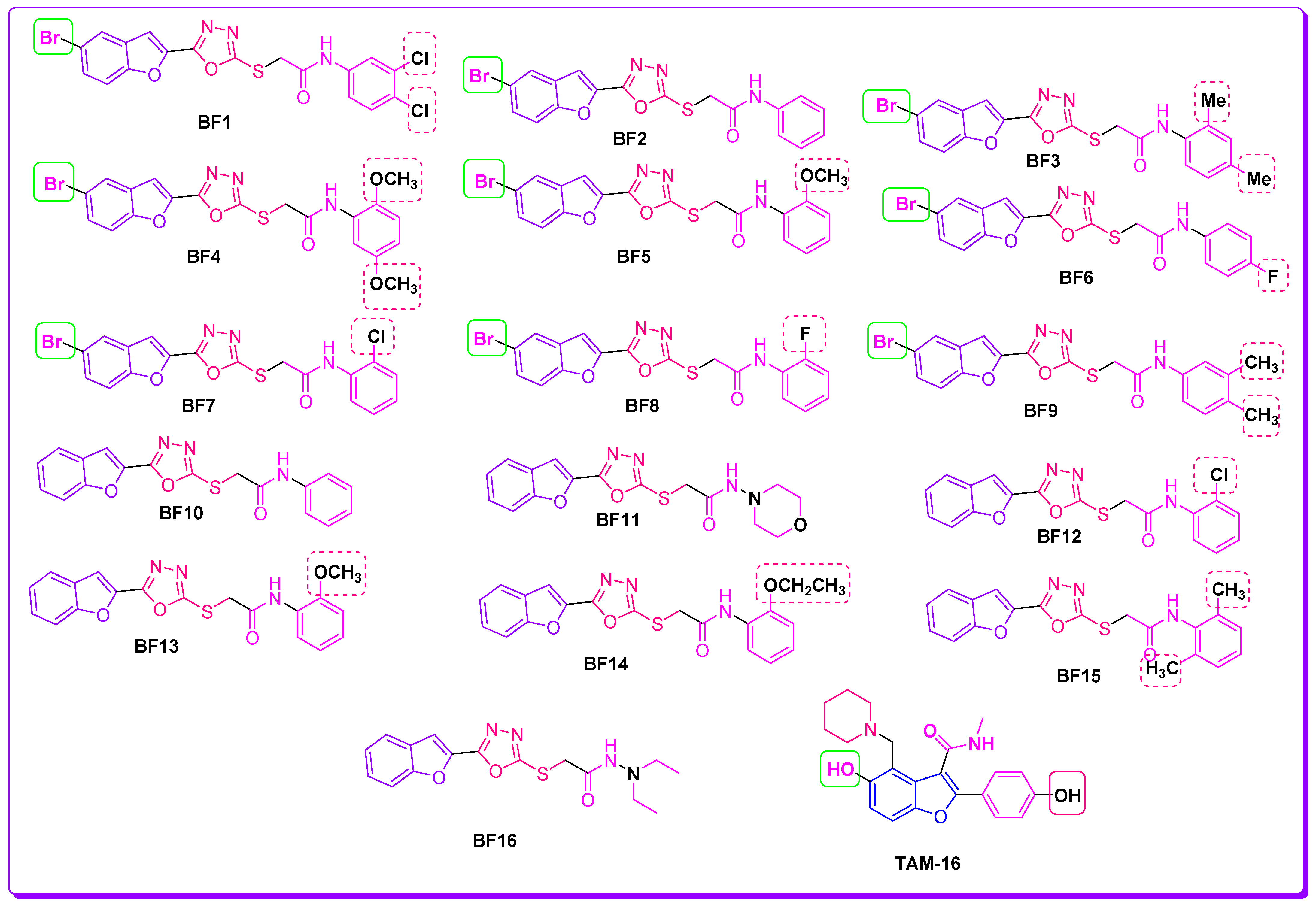
2.1. Evalaution of Anti-Mtb Potenial of Benzofurans-1,3,4-Oxadiazoles Using Computational Approaches
Mycobacterium Tuberculosis
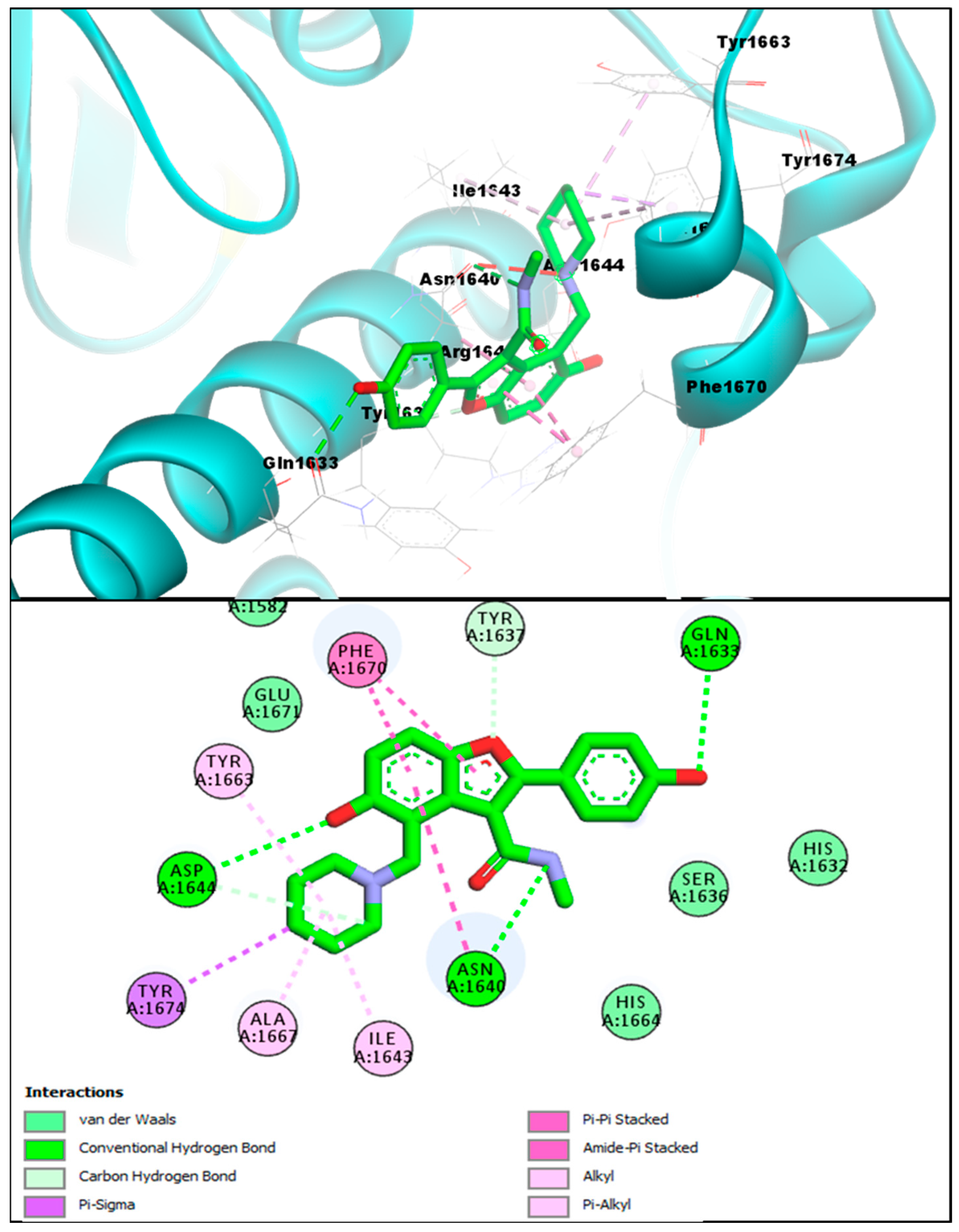
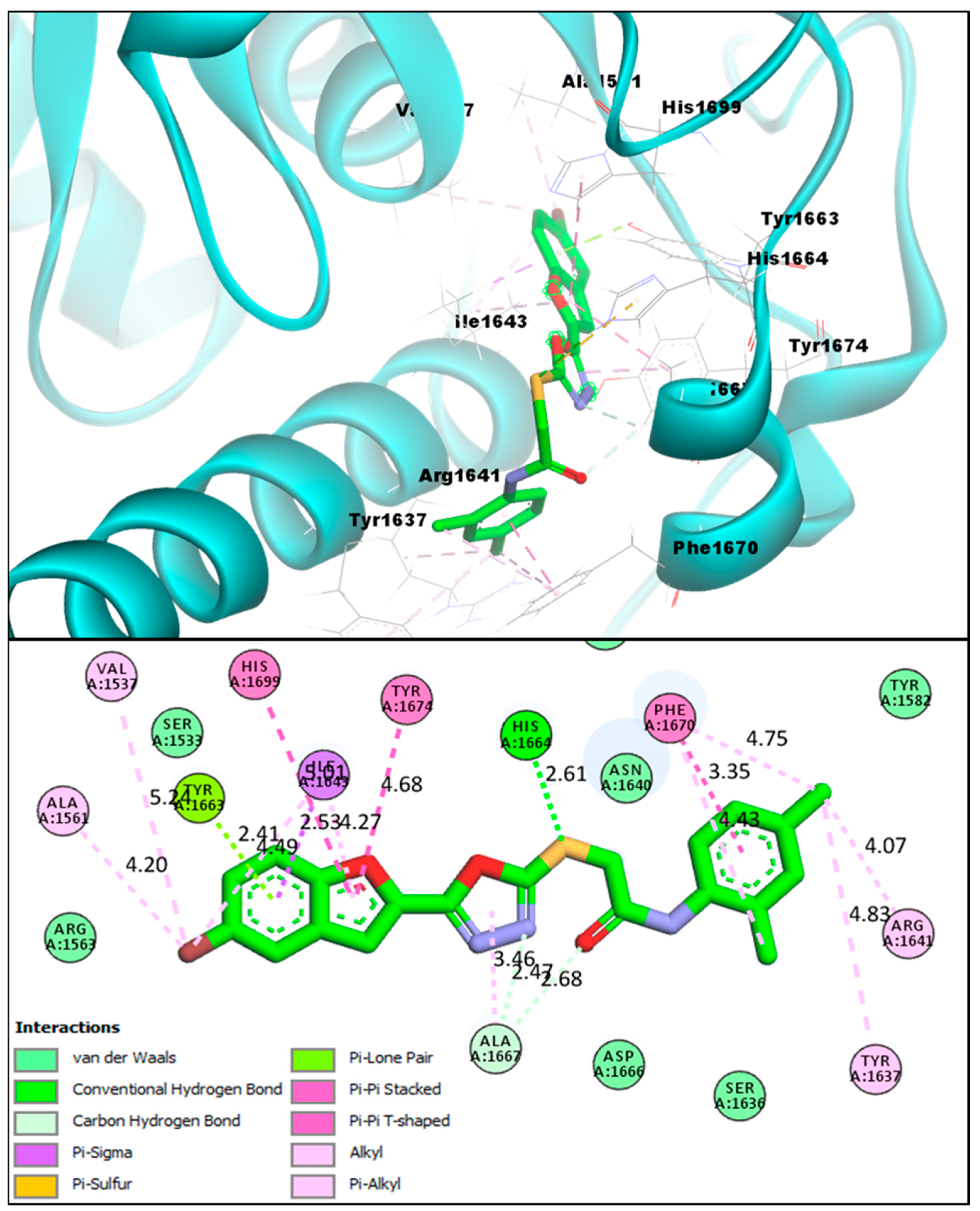
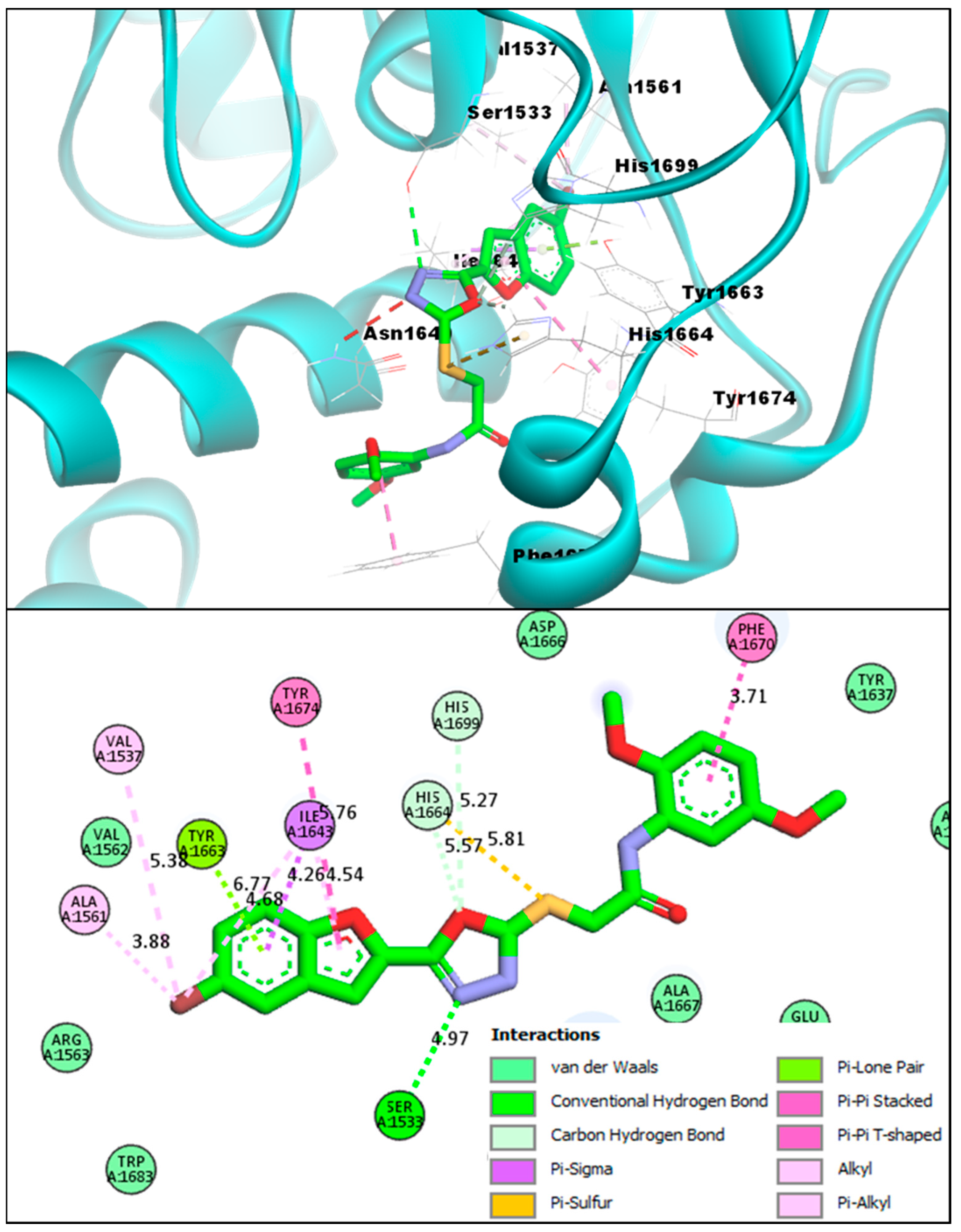
2.2. Molecular Docking Investigations of BF1–BF16 against the Pks13 Enzyme
2.3. Structure-Activity Relationship (SAR) of Bromobenzofuran-1,3,4-Oxadiazoles BF3, BF4, and BF8
2.4. ADMET and Drug-Likeness Studies of Benzofuran-1,3,4-Oxadiazoles BF1–BF16
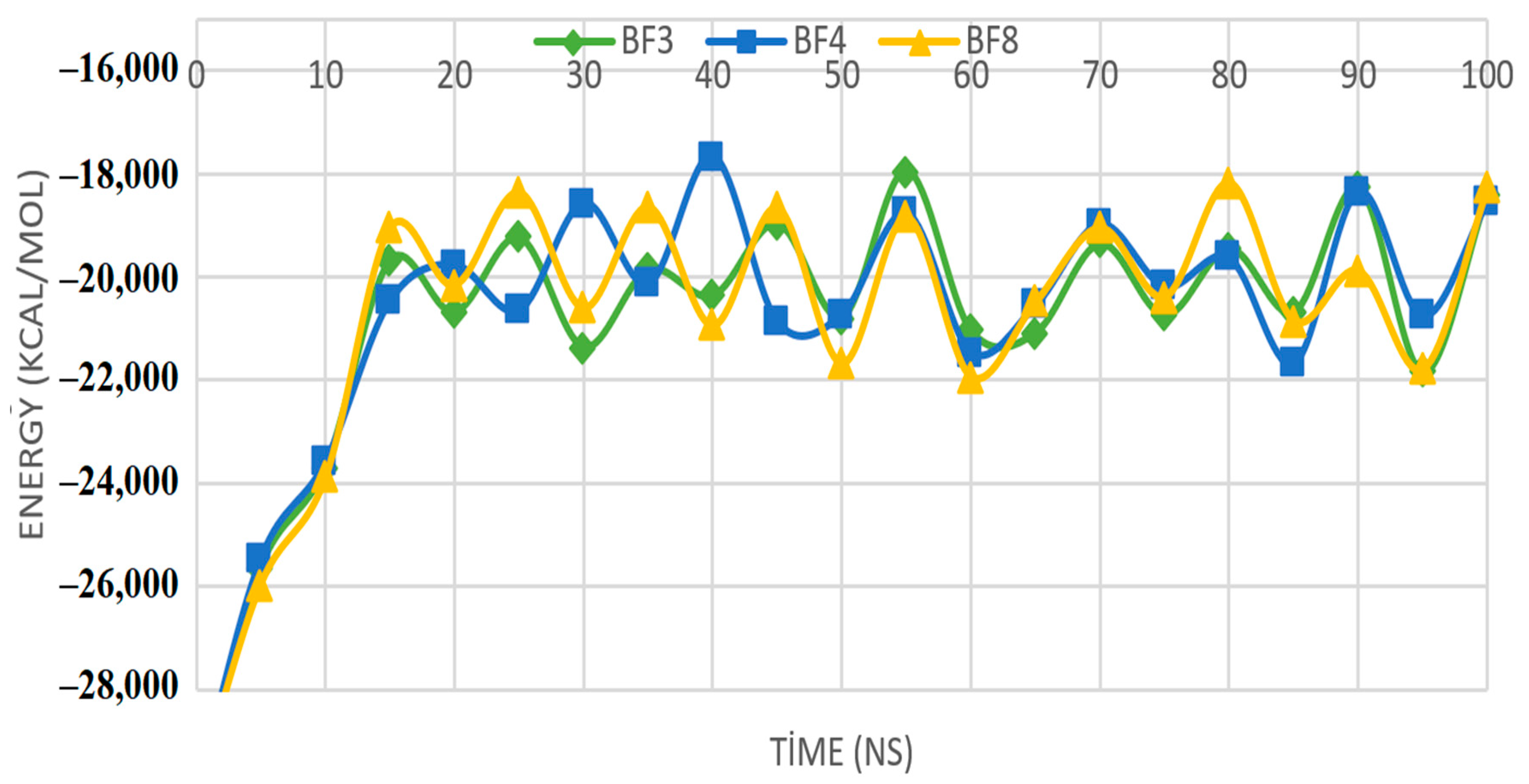
2.5. MD Simulations Study of Benzofuran-1,3,4-oxadiazoles BF3, BF4, and BF8
2.6. MM-PBSA Investigations of the Most In Silico Bioactive Benzofuran-1,3,4-oxadiazoles
3. Materials and Methods
3.1. Chemistry
3.2. Molecular Docking of Benzofuran-1,3,4-oxadiazoles BF1–BF16
3.3. ADMET and Drug-Likeness Investigations of Benzofuran-1,3,4-oxadiazoles
3.4. MD Simulation Study of the Most In Silico Bioactive BF3, BF4, and BF8 Derivatives
3.5. MM-PBSA Binding Free Energy Calculations of the Most In Silico Bioactive BF3, BF4, and BF8 Derivatives
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Global Tuberculosis Report 2018; WHO: Geneva, Switzerland, 2018; pp. 1–277. ISBN 978-92-4-156564-6. Available online: https://apps.who.int/iris/handle/10665/274453 (accessed on 8 May 2023).
- Espinal, M.A. The Global Situation of MDR-TB. Proc. Tuberc. 2003, 83, 44–51. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Tuberculosis Report 2022; WHO: Geneva, Switzerland, 2022; pp. 1–68. ISBN 978-92-4-006173-6. [Google Scholar]
- Prasad, R.; Gupta, N.; Banka, A. Multidrug-resistant tuberculosis/rifampicin-resistant tuberculosis: Principles of management. Lung India 2018, 35, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Sotgiu, G.; Migliori, G.B. Facing Multi-Drug Resistant Tuberculosis. Pulm. Pharmacol. Ther. 2015, 32, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Aspatwar, A.; Kairys, V.; Rala, S.; Parikka, M.; Bozdag, M.; Carta, F.; Supuran, C.T.; Parkkila, S. Mycobacterium Tuberculosis β-Carbonic Anhydrases: Novel Targets for Developing Antituberculosis Drugs. Int. J. Mol. Sci. 2019, 20, 5153. [Google Scholar] [CrossRef]
- Kaul, G.; Kapoor, E.; Dasgupta, A.; Chopra, S. Management of multidrug-resistant tuberculosis in the 21st century. Drugs Today 2019, 55, 215–224. [Google Scholar] [CrossRef]
- Saxena, A.K.; Singh, A. Mycobacterial tuberculosis Enzyme Targets and their Inhibitors. Curr. Top. Med. Chem. 2019, 19, 337–355. [Google Scholar] [CrossRef]
- Harding, E. WHO Global Progress Report on Tuberculosis Elimination. Lancet. Respir. Med. 2020, 8, 19. [Google Scholar] [CrossRef]
- Tsai, Y.-P. Effectiveness of Tuberculosis Case Management. J. Microbiol. Immunol. Infect. 2015, 48, S121. [Google Scholar] [CrossRef]
- Zumla, A.; Nahid, P.; Cole, S.T. Advances in the Development of New Tuberculosis Drugs and Treatment Regimens. Nat. Rev. Drug Discov. 2013, 12, 388–404. [Google Scholar] [CrossRef]
- Chimeh, R.A.; Gafar, F.; Pradipta, I.S.; Akkerman, O.W.; Hak, E.; Alffenaar, J.W.C.; van Boven, J.F.M. Clinical and Economic Impact of Medication Non-Adherence in Drug-Susceptible Tuberculosis: A Systematic Review. Int. J. Tuberc. Lung Dis. 2020, 24, 811–819. [Google Scholar] [CrossRef]
- Wang, X.; Dowd, C.S. The Methylerythritol Phosphate Pathway: Promising Drug Targets in the Fight against Tuberculosis. ACS Infect. Dis. 2018, 4, 278–290. [Google Scholar] [CrossRef]
- Hiremathad, A.; Patil, M.R.; Chethana, K.R.; Chand, K.; Santos, M.A.; Keri, R.S. Benzofuran: An Emerging Scaffold for Anti-microbial Agents. RSC Adv. 2015, 5, 96809–96828. [Google Scholar] [CrossRef]
- Xu, Z.; Zhao, S.; Lv, Z.; Feng, L.; Wang, Y.; Zhang, F.; Bai, L.; Deng, J. Benzofuran Derivatives and Their Anti-Tubercular, Antibacterial Activities. Eur. J. Med. Chem. 2019, 162, 266–276. [Google Scholar] [CrossRef]
- Gill, C.; Jadhav, G.; Shaikh, M.; Kale, R.; Ghawalkar, A.; Nagargoje, D.; Shiradkar, M. Clubbed [1,2,3] Triazoles by Fluorine Benzimidazole: A Novel Approach to H37Rv Inhibitors as a Potential Treatment for Tuberculosis. Bioorganic Med. Chem. Lett. 2008, 18, 6244–6247. [Google Scholar] [CrossRef]
- Irfan, A.; Sabeeh, I.; Umer, M.; Naqvi, A.Z.; Fatima, H.; Yousaf, S.; Fatima, Z. A Review On The Therapeutic Potential of Quinoxaline Derivatives. World J. Pharm. Res. 2017, 6, 47–68. [Google Scholar]
- Rubab, L.; Afroz, S.; Ahmad, S.; Hussain, S.; Nawaz, I.; Irfan, A.; Batool, F.; Kotwica-Mojzych, K.; Mojzych, M. An Update on Synthesis of Coumarin Sulfonamides as Enzyme Inhibitors and Anticancer Agents. Molecules 2022, 27, 1604. [Google Scholar] [CrossRef]
- Irfan, A.; Batool, F.; Irum, S.; Ullah, S.; Umer, M.; Shaheen, R.; Chand, A.J. A Therapeutic Journey Of Sulfonamide Derivatives As Potent Anti-Cancer Agents: A Review. WJPR 2018, 7, 257–270. [Google Scholar]
- Aziz, H.; Zahoor, A.F.; Shahzadi, I.; Irfan, A. Recent Synthetic Methodologies Towards the Synthesis of Pyrazoles. Polycycl. Aromat. Compd. 2019, 41, 698–720. [Google Scholar] [CrossRef]
- Irfan, A.; Batool, F.; Ahmad, S.; Ullah, R.; Sultan, A.; Sattar, R.; Nisar, B.; Rubab, L. Recent trends in the synthesis of 1,2,3-thiadiazoles. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 1098–1115. [Google Scholar] [CrossRef]
- Irfan, A.; Faisal, S.; Ahmad, S.; Al-Hussain, S.A.; Javed, S.; Zahoor, A.F.; Parveen, B.; Zaki, M.E.A. Structure-Based Virtual Screening of Furan-1,3,4-Oxadiazole Tethered N-phenylacetamide Derivatives as Novel Class of hTYR and hTYRP1 Inhibitors. Pharmaceuticals 2023, 16, 344. [Google Scholar] [CrossRef]
- Irfan, A.; Faiz, S.; Rasul, A.; Zafar, R.; Zahoor, A.F.; Kotwica-Mojzych, K.; Mojzych, M. Exploring the Synergistic Anticancer Potential of Benzofuran–Oxadiazoles and Triazoles: Improved Ultrasound-and Microwave-Assisted Synthesis, Molecular Docking, Hemolytic, Thrombolytic and Anticancer Evaluation of Furan-Based Molecules. Molecules 2022, 27, 1023. [Google Scholar] [CrossRef] [PubMed]
- Faiz, S.; Zahoor, A.F.; Ajmal, M.; Kamal, S.; Ahmad, S.; Abdelgawad, A.M.; Elnaggar, M.E. Design, Synthesis, Antimicrobial Evaluation, and Laccase Catalysis Effect of Novel Benzofuran–Oxadiazole and Benzofuran–Triazole Hybrids. J. Heterocycl. Chem. 2019, 56, 2839–2852. [Google Scholar] [CrossRef]
- Bhargava, S.; Rathore, D. Synthetic Routes and Biological Activities of Benzofuran and Its Derivatives: A Review. Lett. Org. Chem. 2017, 14, 381–402. [Google Scholar] [CrossRef]
- Nevagi, R.J.; Dighe, S.N.; Dighe, S.N. Biological and Medicinal Significance of Benzofuran. Eur. J. Med. Chem. 2015, 97, 561–581. [Google Scholar] [CrossRef] [PubMed]
- Hiremathad, A.; Chand, K.; Tolayan, L.; Rajeshwari; Keri, R.S.; Esteves, A.R.; Cardoso, S.M.; Chaves, S.; Santos, M.A. Hydroxypyridinone-Benzofuran Hybrids with Potential Protective Roles for Alzheimer’s Disease Therapy. J. Inorg. Biochem. 2018, 179, 82–96. [Google Scholar] [CrossRef]
- Goyal, D.; Kaur, A.; Goyal, B. Benzofuran and Indole: Promising Scaffolds for Drug Development in Alzheimer’s Disease. ChemMedChem 2018, 13, 1275–1299. [Google Scholar] [CrossRef]
- Zeni, G.; Lüdtke, D.S.; Nogueira, C.W.; Panatieri, R.B.; Braga, A.L.; Silveira, C.C.; Stefani, H.A.; Rocha, J.B.T. New Acetylenic Furan Derivatives: Synthesis and Anti-Inflammatory Activity. Tetrahedron Lett. 2001, 42, 8927–8930. [Google Scholar] [CrossRef]
- Thévenin, M.; Thoret, S.; Grellier, P.; Dubois, J. Synthesis of Polysubstituted Benzofuran Derivatives as Novel Inhibitors of Parasitic Growth. Bioorganic Med. Chem. 2013, 21, 4885–4892. [Google Scholar] [CrossRef]
- Zhong, M.; Peng, E.; Huang, N.; Huang, Q.; Huq, A.; Lau, M.; Colonno, R.; Li, L. Discovery of Novel Potent HCV NS5B Polymerase Non-Nucleoside Inhibitors Bearing a Fused Benzofuran Scaffold. Bioorganic Med. Chem. Lett. 2018, 28, 963–968. [Google Scholar] [CrossRef]
- Xie, Y.S.; Kumar, D.; Bodduri, V.D.V.; Tarani, P.S.; Zhao, B.X.; Miao, J.Y.; Jang, K.; Shin, D.S. Microwave-Assisted Parallel Synthesis of Benzofuran-2-Carboxamide Derivatives Bearing Anti-Inflammatory, Analgesic and Antipyretic Agents. Tetrahedron Lett. 2014, 55, 2796–2800. [Google Scholar] [CrossRef]
- Rangaswamy, J.; Kumar, H.V.; Harini, S.T.; Naik, N. Functionalized 3-(Benzofuran-2-Yl)-5-(4-Methoxyphenyl)-4,5-Dihydro-1H-Pyrazole Scaffolds: A New Class of Antimicrobials and Antioxidants. Arab. J. Chem. 2017, 10, S2685–S2696. [Google Scholar] [CrossRef]
- Singh, F.V.; Chaurasia, S.; Joshi, M.D.; Srivastava, A.K.; Goel, A. Synthesis and in Vivo Antihyperglycemic Activity of Nature-Mimicking Furanyl-2-Pyranones in STZ-S Model. Bioorganic Med. Chem. Lett. 2007, 17, 2425–2429. [Google Scholar] [CrossRef]
- Simonetti, S.O.; Larghi, E.L.; Bracca, A.B.J.; Kaufman, T.S. Angular Tricyclic Benzofurans and Related Natural Products of Fungal Origin. Isolation, Biological Activity and Synthesis. Nat. Prod. Rep. 2013, 30, 941–969. [Google Scholar] [CrossRef]
- Mei, W.; Ji, S.; Xiao, W.; Wang, X.; Jiang, C.; Ma, W.; Zhang, H.; Gong, J.; Guo, Y. Synthesis and Biological Evaluation of Benzothiazol-Based 1,3,4-Oxadiazole Derivatives as Amyloid β-Targeted Compounds against Alzheimer’s Disease. Mon. fur Chem. 2017, 148, 1807–1815. [Google Scholar] [CrossRef]
- Nieddu, V.; Pinna, G.; Marchesi, I.; Sanna, L.; Asproni, B.; Pinna, G.A.; Bagella, L.; Murineddu, G. Synthesis and Antineoplastic Evaluation of Novel Unsymmetrical 1,3,4-Oxadiazoles. J. Med. Chem. 2016, 59, 10451–10469. [Google Scholar] [CrossRef]
- Calcagno, A.; D’Avolio, A.; Bonora, S. Pharmacokinetic and Pharmacodynamic Evaluation of Raltegravir and Experience from Clinical Trials in HIV-Positive Patients. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1167–1176. [Google Scholar] [CrossRef]
- Bondock, S.; Adel, S.; Etman, H.A.; Badria, F.A. Synthesis and Antitumor Evaluation of Some New 1,3,4-Oxadiazole-Based Heterocycles. Eur. J. Med. Chem. 2012, 48, 192–199. [Google Scholar] [CrossRef]
- Sun, J.; Ren, S.Z.; Lu, X.Y.; Li, J.J.; Shen, F.Q.; Xu, C.; Zhu, H.L. Discovery of a Series of 1,3,4-Oxadiazole-2(3H)-Thione Derivatives Containing Piperazine Skeleton as Potential FAK Inhibitors. Bioorganic Med. Chem. 2017, 25, 2593–2600. [Google Scholar] [CrossRef]
- Prakash, O.; Kumar, M.; Kumar, R.; Sharma, C.; Aneja, K.R. Hypervalent Iodine(III) Mediated Synthesis of Novel Unsymmetrical 2,5-Disubstituted 1,3,4-Oxadiazoles as Antibacterial and Antifungal Agents. Eur. J. Med. Chem. 2010, 45, 4252–4257. [Google Scholar] [CrossRef]
- Akhter, M.; Husain, A.; Azad, B.; Ajmal, M. Aroylpropionic Acid Based 2,5-Disubstituted-1,3,4-Oxadiazoles: Synthesis and Their Anti-Inflammatory and Analgesic Activities. Eur. J. Med. Chem. 2009, 44, 2372–2378. [Google Scholar] [CrossRef]
- Janardhanan, J.; Chang, M.; Mobashery, S. The Oxadiazole Antibacterials. Curr. Opin. Microbiol. 2016, 33, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, M.J.; Samy, J.G.; Khalilullah, H.; Nomani, M.S.; Saraswat, P.; Gaur, R.; Singh, A. Molecular Properties Prediction and Synthesis of Novel 1,3,4-Oxadiazole Analogues as Potent Antimicrobial and Antitubercular Agents. Bioorg. Med. Chem. Lett. 2011, 21, 7246–7250. [Google Scholar] [CrossRef] [PubMed]
- Irfan, A.; Zahoor, A.F.; Rasul, A.; Al-Hussain, S.A.; Faisal, S.; Ahmad, S.; Noor, R.; Muhammed, M.T.; Zaki, M.E.A. BTEAC Catalyzed Ultrasonic-Assisted Synthesis of Bromobenzofuran-Oxadiazoles: Unravelling Anti-HepG-2 Cancer Therapeutic Potential through In Vitro and In Silico Studies. Int. J. Mol. Sci. 2023, 24, 3008. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Ray, P.; Zuccotto, F.; Hernandez, J.; Aggarwal, A.; Mackenzie, C.; Caldwell, N.; Taylor, M.; Huggett, M.; Mathieson, M.; et al. Optimization of TAM16, a Benzofuran That Inhibits the Thioesterase Activity of Pks13; Evaluation toward a Preclinical Candidate for a Novel Antituberculosis Clinical Target. J. Med. Chem. 2022, 65, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.K.; Verma, R.; Verma, S.; Vaishnav, Y.; Tiwari, S.P.; Rakesh, K.P. Anti-Tuberculosis Activity and Its Structure-Activity Relationship (SAR) Studies of Oxadiazole Derivatives: A Key Review. Eur. J. Med. Chem. 2021, 209, 112886. [Google Scholar] [CrossRef]
- Makane, V.B.; Krishna, V.S.; Krishna, E.V.; Shukla, M.; Mahizhaveni, B.; Misra, S.; Chopra, S.; Sriram, D.; Azger Dusthackeer, V.N.; Rode, H.B. Novel 1,3,4-Oxadiazoles as Antitubercular Agents with Limited Activity against Drug-Resistant Tuberculosis. Future Med. Chem. 2019, 11, 499–510. [Google Scholar] [CrossRef]
- Desai, N.C.; Somani, H.; Trivedi, A.; Bhatt, K.; Nawale, L.; Khedkar, V.M.; Jha, P.C.; Sarkar, D. Synthesis, Biological Evaluation and Molecular Docking Study of Some Novel Indole and Pyridine Based 1,3,4-Oxadiazole Derivatives as Potential Antitubercular Agents. Bioorgan. Med. Chem. Lett. 2016, 26, 1776–1783. [Google Scholar] [CrossRef]
- Chobe, S.S.; Kamble, R.D.; Patil, S.D.; Acharya, A.P.; Hese, S.V.; Yemul, O.S.; Dawane, B.S. Green Approach towards Synthesis of Substituted Pyrazole-1,4-Dihydro,9-Oxa, 1,2,6,8-Tetrazacyclopentano[b]Naphthalene-5-One Derivatives as Antimycobacterial Agents. Med. Chem. Res. 2013, 22, 5197–5203. [Google Scholar] [CrossRef]
- Dhumal, S.T.; Deshmukh, A.R.; Bhosle, M.R.; Khedkar, V.M.; Nawale, L.U.; Sarkar, D.; Mane, R.A. Synthesis and Antitubercular Activity of New 1,3,4-Oxadiazoles Bearing Pyridyl and Thiazolyl Scaffolds. Bioorganic Med. Chem. Lett. 2016, 26, 3646–3651. [Google Scholar] [CrossRef]
- Irfan, A.; Zahoor, A.F.; Kamal, S.; Hassan, M.; Kloczkowski, A. Ultrasonic-Assisted Synthesis of Benzofuran Appended Oxadiazole Molecules as Tyrosinase Inhibitors: Mechanistic Approach through Enzyme Inhibition, Molecular Docking, Chemoinformatics, ADMET and Drug-Likeness Studies. Int. J. Mol. Sci. 2022, 23, 10979. [Google Scholar] [CrossRef]
- Marimani, M.; Ahmad, A.; Duse, A. The Role of Epigenetics, Bacterial and Host Factors in Progression of Mycobacterium Tuberculosis Infection. Tuberculosis 2018, 113, 200–214. [Google Scholar] [CrossRef]
- Ryndak, M.B.; Laal, S. Mycobacterium tuberculosis Primary Infection and Dissemination: A Critical Role for Alveolar Epithelial Cells. Front. Cell. Infect. Microbiol. 2019, 9, 299. [Google Scholar] [CrossRef]
- Abdoli, A.; Falahi, S.; Kenarkoohi, A. COVID-19-Associated Opportunistic Infections: A Snapshot on the Current Reports. Clin. Exp. Med. 2022, 22, 327–346. [Google Scholar] [CrossRef]
- Oh, S.; Park, Y.; Engelhart, C.A.; Wallach, J.B.; Schnappinger, D.; Arora, K.; Manikkam, M.; Gac, B.; Wang, H.; Murgolo, N.; et al. Discovery and Structure-Activity-Relationship Study of N-Alkyl-5-Hydroxypyrimidinone Carboxamides as Novel Antitubercular Agents Targeting Decaprenylphosphoryl-β-D-Ribose 2′-Oxidase. J. Med. Chem. 2018, 61, 9952–9965. [Google Scholar] [CrossRef]
- Mohanty, D.; Sankaranarayanan, R.; Gokhale, R.S. Fatty Acyl-AMP Ligases and Polyketide Synthases Are Unique Enzymes of Lipid Biosynthetic Machinery in Mycobacterium tuberculosis. Tuberculosis 2011, 91, 448–455. [Google Scholar] [CrossRef]
- Singh, S.; Singh, D.; Hameed, S.; Fatima, Z. An Overview of Mycolic Acids. Structure–function–classification, biosynthesis, and beyond. In Biology of Mycobacterial Lipids; Academic Press: Cambridge, MA, USA, 2022; pp. 1–25. [Google Scholar] [CrossRef]
- Maitra, A.; Munshi, T.; Healy, J.; Martin, L.T.; Vollmer, W.; Keep, N.H.; Bhakta, S. Cell Wall Peptidoglycan in Mycobacterium tuberculosis: An Achilles’ Heel for the TB-Causing Pathogen. FEMS Microbiol. Rev. 2019, 43, 548–575. [Google Scholar] [CrossRef]
- Howard, N.C.; Marin, N.D.; Ahmed, M.; Rosa, B.A.; Martin, J.; Bambouskova, M.; Sergushichev, A.; Loginicheva, E.; Kurepina, N.; Rangel-Moreno, J.; et al. Mycobacterium tuberculosis Carrying a Rifampicin Drug Resistance Mutation Reprograms Macrophage Metabolism through Cell Wall Lipid Changes. Nat. Microbiol. 2018, 3, 1099–1108. [Google Scholar] [CrossRef]
- Wellington, S.; Hung, D.T. The Expanding Diversity of Mycobacterium tuberculosis Drug Targets. ACS Infect. Dis. 2018, 4, 696–714. [Google Scholar] [CrossRef]
- Aggarwal, A.; Parai, M.K.; Shetty, N.; Wallis, D.; Woolhiser, L.; Hastings, C.; Dutta, N.K.; Galaviz, S.; Dhakal, R.C.; Shrestha, R.; et al. Development of a Novel Lead That Targets M. Tuberculosis Polyketide Synthase 13. Cell 2017, 170, 249–259.e25. [Google Scholar] [CrossRef]
- Çınaroğlu, S.S.; Timuçin, E. Insights into an Alternative Benzofuran Binding Mode and Novel Scaffolds of Polyketide Synthase 13 Inhibitors. J. Mol. Model. 2019, 25, 130. [Google Scholar] [CrossRef]
- He, Y.; Xu, J.; Yu, Z.H.; Gunawan, A.M.; Wu, L.; Wang, L.; Zhang, Z.Y. Discovery and Evaluation of Novel Inhibitors of Mycobacterium Protein Tyrosine Phosphatase B from the 6-Hydroxy-Benzofuran-5-Carboxylic Acid Scaffold. J. Med. Chem. 2013, 56, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Lokhande, K.B.; Shrivastava, A.; Singh, A. In silico discovery of potent inhibitors against monkeypox’s major structural proteins. J. Biomol. Struct. Dyn. 2023, 25, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Nagare, S.; Lokhande, K.B.; Swamy, K.V. Docking and simulation studies on cyclin D/CDK4 complex for targeting cell cycle arrest in cancer using flavanone and its congener. J. Mol. Model. 2023, 29, 90. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.K.; Vemula, S.; Donde, R.; Gouda, G.; Behera, L.; Vadde, R. In-Silico Approaches to Detect Inhibitors of the Human Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Ion Channel. J. Biomol. Struct. Dyn. 2021, 39, 2617–2627. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S.; et al. RCSB Protein Data Bank: Biological Macromolecular Structures Enabling Research and Education in Fundamental Biology, Biomedicine, Biotechnology and Energy. Nucleic Acids Res. 2019, 47, D464–D474. [Google Scholar] [CrossRef]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal Chemistry and the Molecular Operating Environment (MOE): Application of QSAR and Molecular Docking to Drug Discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Faisal, S.; Lal Badshah, S.; Kubra, B.; Sharaf, M.; Emwas, A.H.; Jaremko, M.; Abdalla, M. Computational Study of SARS-CoV-2 Rna Dependent Rna Polymerase Allosteric Site Inhibition. Molecules 2022, 27, 223. [Google Scholar] [CrossRef]
- Accelrys Software Inc. Discovery Studio Modeling Environment, Release 3.5; Accelrys Software Inc.: San Diego, CA, USA, 2012. [Google Scholar]
- Mills, N. ChemDraw Ultra 10.0 CambridgeSoft, 100 CambridgePark Drive, Cambridge, MA 02140. Www.Cambridgesoft.Com. Commercial Price: $1910 for Download, $2150 for CD-ROM; Academic Price: $710 for Download, $800 for CD-ROM. J. Am. Chem. Soc. 2006, 128, 13649–13650. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An Integrated Online Platform for Accurate and Comprehensive Predictions of ADMET Properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- GFYang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. AdmetSAR 2.0: Web-Service for Prediction and Optimization of Chemical ADMET Properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. AdmetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUIInput Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Aktaş, A.; Tüzün, B.; Aslan, R.; Sayin, K.; Ataseven, H. New Anti-Viral Drugs for the Treatment of COVID-19 Instead of Favipiravir. J. Biomol. Struct. Dyn. 2021, 39, 7263–7273. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Binding Affinities | Interacting Residues of Pks13 | Interaction Types |
|---|---|---|---|
| BF1 | −12.93 kcal/mol | ILE1643, TYR1663, HIS1632, HIS1699, ALA1667, TYR1674, | Carbon-Hydrogen Bond, Van der Waals, Pi-Pi T-Shaped, Pi-Alkyl, and Alkyl. |
| BF2 | −12.71 kcal/mol | ASN1640, ILE1643, TYR1637, TYR1663, HIS1632, ALA1667, TYR1674, PHE1670 | C-Hydrogen Bond, Van der Waals, Pi-Pi T-Shaped, Pi-Pi Stacked, and Alkyl. |
| BF3 | −14.23 kcal/mol | VAL1537, ALA1561, PHE1637, ARG1641, ILE1643, TYR1663, HIS1664, ALA1667, PHE1670, TYR1674, HIS1699 | Conventional H-bond, C-Hydrogen Bond, Van der Waals, Pi-Pi T-Shaped, Pi-Alkyl, Pi-Lone pair, Pi-Sulfur, Pi-Sigma, and Pi-Pi Stacked |
| BF4 | −14.82 kcal/mol | VAL1537, SER1533, ALA1561, VAL1537, TYR1674, ILE1643, PHE1670, ALA1667 | Conventional H-bond, C-Hydrogen Bond, Van der Waals, Pi-Pi T-Shaped, Pi-Alkyl, Pi-Lone pair, Pi-Sulfur, Pi-Sigma, and Pi-Pi Stacked |
| BF5 | −12.31 kcal/mol | ALA1561, TYR1663, ILE1643, HIS1664, TYR1674, ALA1667 | C-Hydrogen Bond, Van der Waals, Pi-Pi T-Shaped, Pi-Alkyl, Pi-Lone pair, and Alkyl. |
| BF6 | −11.89 kcal/mol | SER1533, ALA1667, ALA1561, TYR1663, ILE1643, HIS1664, GLN1633, TYR1674 | Conventional H-bond, C-Hydrogen Bond, Van der Waals, Pi-Pi T-Shaped, Pi-Alkyl, Pi-Lone pair, Halogen, and Alkyl. |
| BF7 | −12.23 kcal/mol | HIS1632, TYR1637, ILE1643, TYR1663, ALA1667, PHE1670, TYR1674 | Carbon-Hydrogen Bond, Van der Waals, Pi-Pi Stacked, Pi-Alkyl, and Alkyl. |
| BF8 | −14.11 kcal/mol | VAL1537, ALA1561, TYR1663, ASN1640, ILE1643, PHE1670, ARG1641, ASP1644, HIS1664 | Conventional H-bond, C-Hydrogen Bond, Van der Waals, Pi-Pi T-Shaped, Pi-Alkyl, Pi-Lone pair, Amide-Pi Stacked |
| BF9 | −13.44 kcal/mol | ILE1643, ALA1667, PHE1670, VAL1562, HIS1699, TYR1674, TYR1637 | Conventional H-bond, C-Hydrogen Bond, Van der Waals, Pi-Pi T-Shaped, Pi-Pi Stacked, Pi-Alkyl, and Alkyl |
| TAM-16 (Standard) | −14.61 kcal/mol | SER1533, GLN1633, ASN1640, ASP1644, ILE1643, TYR1663, ALA1667, PHE1670, TYR1674 | Conventional H-bond, C-Hydrogen Bond, Van der Waals, Pi-Pi Stacked, Pi-Alkyl, Amide Pi-Stacked, Pi-Sigma, and Alkyl |
| Nanoseconds | Pks13+BF3 | Pks13+BF4 | Pks13+BF8 |
|---|---|---|---|
| 10 | −59.4 ± 149.6 | −956.2 ± 586.2 | −87.8 ± 235.6 |
| 20 | −135.2 ± 235.5 | −105.3 ± 387.4 | −508.9 ± 245.1 |
| 30 | −95.8 ± 269.3 | −912.3 ± 189.3 | −354.2 ± 245.3 |
| 40 | −570.3 ± 684.2 | −245.3 ± 245.6 | −150.8 ± 250.4 |
| 50 | −856.2 ± 345.6 | −856.3 ± 409.8 | −750.4 ± 150.6 |
| 60 | −135.2 ± 248.6 | −301.7 ± 204.8 | −723.3 ± 523.6 |
| 70 | −486.3 ± 367.3 | −501.1 ± 193.5 | −685.8 ± 351.2 |
| 80 | −648.8 ± 385.2 | −1101.3 ± 497.6 | −289.7 ± 487.5 |
| 90 | −329.2 ± 301.2 | −687.5 ± 260.1 | −350.4 ± 293.7 |
| 100 | −300.8 ± 283.2 | −423.4 ± 305.3 | −145.8 ± 354.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irfan, A.; Faisal, S.; Zahoor, A.F.; Noreen, R.; Al-Hussain, S.A.; Tuzun, B.; Javaid, R.; Elhenawy, A.A.; Zaki, M.E.A.; Ahmad, S.; et al. In Silico Development of Novel Benzofuran-1,3,4-Oxadiazoles as Lead Inhibitors of M. tuberculosis Polyketide Synthase 13. Pharmaceuticals 2023, 16, 829. https://doi.org/10.3390/ph16060829
Irfan A, Faisal S, Zahoor AF, Noreen R, Al-Hussain SA, Tuzun B, Javaid R, Elhenawy AA, Zaki MEA, Ahmad S, et al. In Silico Development of Novel Benzofuran-1,3,4-Oxadiazoles as Lead Inhibitors of M. tuberculosis Polyketide Synthase 13. Pharmaceuticals. 2023; 16(6):829. https://doi.org/10.3390/ph16060829
Chicago/Turabian StyleIrfan, Ali, Shah Faisal, Ameer Fawad Zahoor, Razia Noreen, Sami A. Al-Hussain, Burak Tuzun, Rakshanda Javaid, Ahmed A. Elhenawy, Magdi E. A. Zaki, Sajjad Ahmad, and et al. 2023. "In Silico Development of Novel Benzofuran-1,3,4-Oxadiazoles as Lead Inhibitors of M. tuberculosis Polyketide Synthase 13" Pharmaceuticals 16, no. 6: 829. https://doi.org/10.3390/ph16060829
APA StyleIrfan, A., Faisal, S., Zahoor, A. F., Noreen, R., Al-Hussain, S. A., Tuzun, B., Javaid, R., Elhenawy, A. A., Zaki, M. E. A., Ahmad, S., & Abdellattif, M. H. (2023). In Silico Development of Novel Benzofuran-1,3,4-Oxadiazoles as Lead Inhibitors of M. tuberculosis Polyketide Synthase 13. Pharmaceuticals, 16(6), 829. https://doi.org/10.3390/ph16060829