
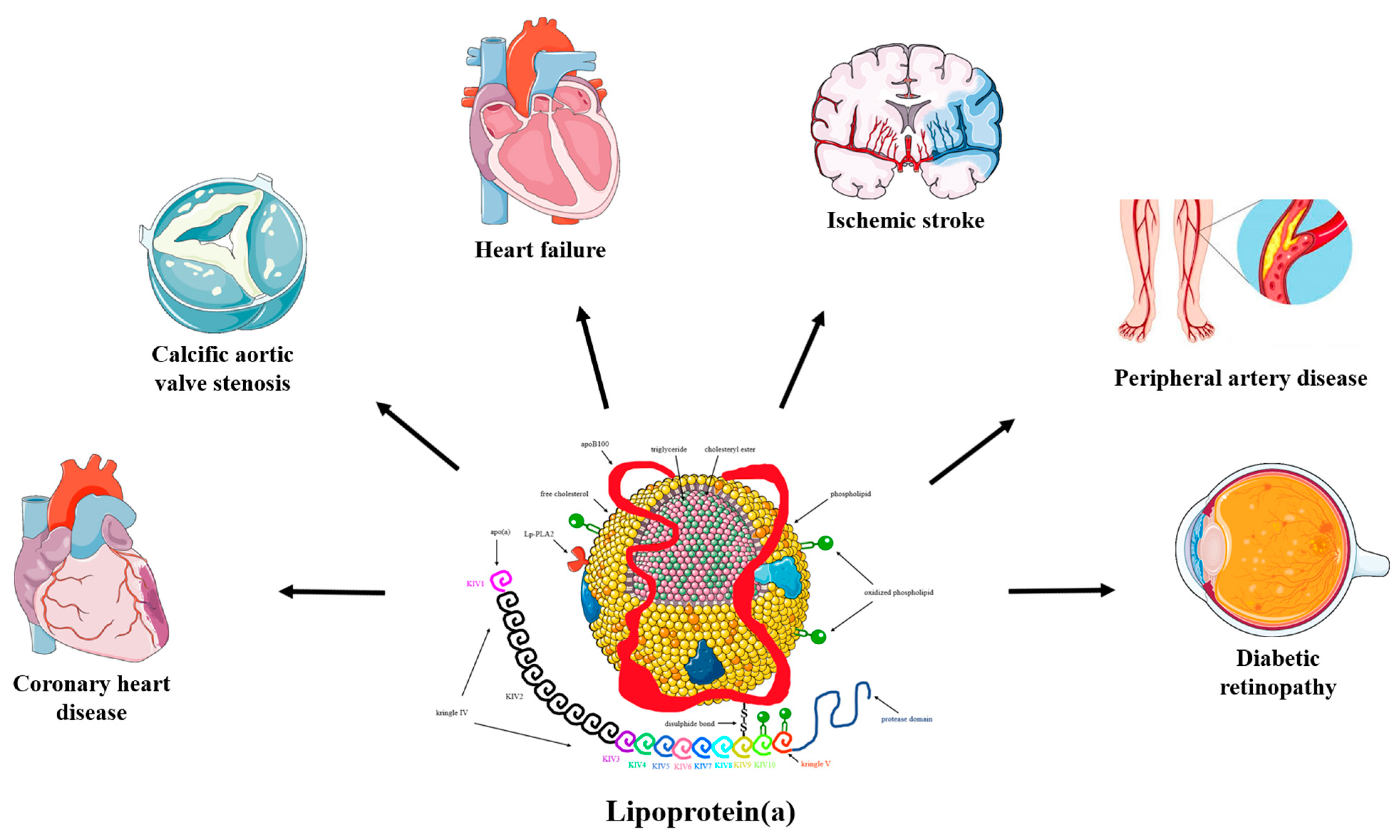
Effects of Lipid-Modifying and Other Drugs on Lipoprotein(a) Levels—Potent Clinical Implications


 ,
,
Abstract
1. Introduction
2. Methods
3. Lp(a)-Lowering Therapies
3.1. Effects of Lipid-Modifying Interventions on Lp(a) Levels
3.1.1. Lipoprotein Apheresis
3.1.2. Statins
3.1.3. Niacin
3.1.4. Ezetimibe
3.1.5. PCSK9 Inhibitors
3.1.6. Fibrates
3.1.7. Lomitapide
3.1.8. Mipomersen
3.1.9. Cholesteryl Transfer Protein (CETP) Inhibitors
3.1.10. Bempedoic Acid
3.1.11. Bile Acid Sequestrants
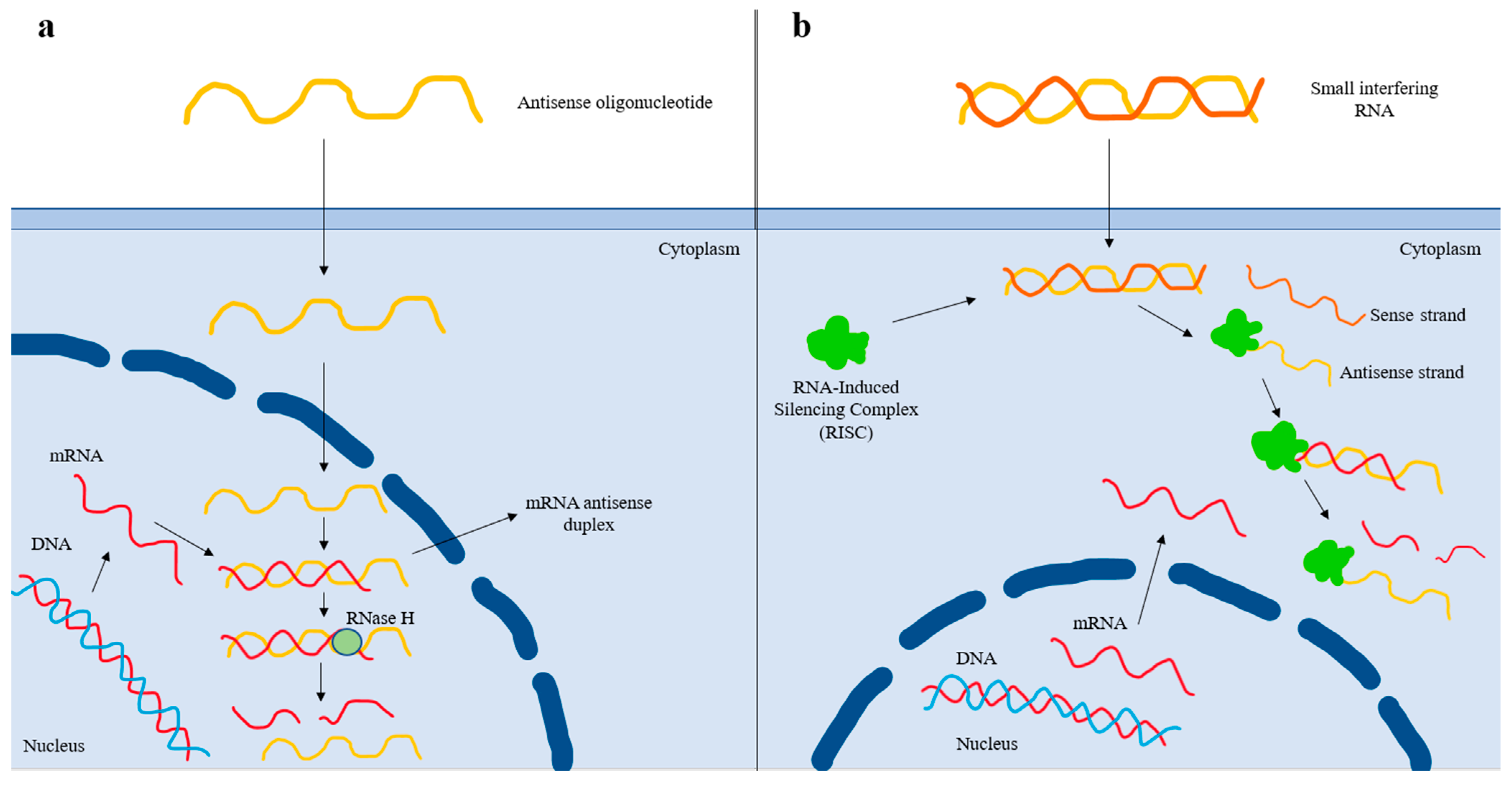
3.1.12. ASO and siRNA Agents
3.2. Effects of Other Drugs on Lp(a) Levels
3.2.1. Sex Hormone Therapies
3.2.2. Thyroid Hormone Therapies
3.2.3. Growth Hormone Replacement Therapy
3.2.4. Aspirin
3.2.5. Anti-Inflammatory Agents
3.3. Effects of Dietary Intervention and Physical Activity on Lp(a) Levels
3.4. Effects of Bariatric Surgery on Lp(a) Levels
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schmidt, K.; Noureen, A.; Kronenberg, F.; Utermann, G. Thematic Review Series: Lipoprotein (a): Coming of Age at Last: Structure, function, and genetics of lipoprotein (a). J. Lipid Res. 2016, 57, 1339. [Google Scholar] [CrossRef]
- de Boer, L.M.; Hof, M.H.; Wiegman, A.; Stroobants, A.K.; Kastelein, J.J.P.; Hutten, B.A. Lipoprotein(a) levels from childhood to adulthood: Data in nearly 3000 children who visited a pediatric lipid clinic. Atherosclerosis 2022, 349, 227–232. [Google Scholar] [CrossRef]
- Derby, C.A.; Crawford, S.L.; Pasternak, R.C.; Sowers, M.; Sternfeld, B.; Matthews, K.A. Lipid Changes During the Menopause Transition in Relation to Age and Weight: The Study of Women’s Health Across the Nation. Am. J. Epidemiol. 2009, 169, 1352. [Google Scholar] [CrossRef]
- Fanshawe, A.E.; Ibrahim, M. The current status of lipoprotein (a) in pregnancy: A literature review. J. Cardiol. 2013, 61, 99–106. [Google Scholar] [CrossRef]
- Patel, A.P.; Wang, M.; Pirruccello, J.P.; Ellinor, P.T.; Ng, K.; Kathiresan, S.; Khera, A.V. Lipoprotein(a) concentrations and incident atherosclerotic cardiovascular disease: New insights from a large national biobank. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 465. [Google Scholar] [CrossRef]
- Erhart, G.; Lamina, C.; Lehtimäki, T.; Marques-Vidal, P.; Kähönen, M.; Vollenweider, P.; Raitakari, O.T.; Waeber, G.; Thorand, B.; Strauch, K.; et al. Genetic factors explain a major fraction of the 50% lower lipoprotein(a) concentrations in finns. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1230–1241. [Google Scholar] [CrossRef]
- Kronenberg, F.; Mora, S.; Stroes, E.S.G.; Ference, B.A.; Arsenault, B.J.; Berglund, L.; Dweck, M.R.; Koschinsky, M.; Lambert, G.; Mach, F.; et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: A European Atherosclerosis Society consensus statement. Eur. Heart J. 2022, 43, 3925–3946. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Langsted, A. Thematic Review Series: Lipoprotein (a): Coming of Age at Last: Lipoprotein (a) as a cause of cardiovascular disease: Insights from epidemiology, genetics, and biology. J. Lipid Res. 2016, 57, 1953. [Google Scholar] [CrossRef]
- Tsimikas, S. A Test in Context: Lipoprotein(a): Diagnosis, Prognosis, Controversies, and Emerging Therapies. J. Am. Coll. Cardiol. 2017, 69, 692–711. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Nordestgaard, B.G. Elevated Lipoprotein(a) Levels, LPA Risk Genotypes, and Increased Risk of Heart Failure in the General Population. JACC Heart Fail. 2016, 4, 78–87. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Elevated Lipoprotein(a) and Risk of Aortic Valve Stenosis in the General Population. J. Am. Coll. Cardiol. 2014, 63, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Langsted, A.; Nordestgaard, B.G.; Kamstrup, P.R. Elevated Lipoprotein(a) and Risk of Ischemic Stroke. J. Am. Coll. Cardiol. 2019, 74, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, P.R.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Genetic evidence that lipoprotein(a) associates with atherosclerotic stenosis rather than venous thrombosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1732–1741. [Google Scholar] [CrossRef]
- Langsted, A.; Kamstrup, P.R.; Nordestgaard, B.G. High lipoprotein(a) and high risk of mortality. Eur. Heart J. 2019, 40, 2760–2770. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, G.; Gagliano, C.; Bucolo, C.; Vacante, M.; Salomone, S.; Malaguarnera, M.; Leonardi, D.G.; Motta, M.; Drago, F.; Avitabile, T. Lipoprotein(a) Serum Levels in Diabetic Patients with Retinopathy. BioMed Res. Int. 2013, 2013, 943505. [Google Scholar] [CrossRef]
- Kaltoft, M.; Sigvardsen, P.E.; Afzal, S.; Langsted, A.; Fuchs, A.; Kühl, J.T.; Køber, L.; Kamstrup, P.R.; Kofoed, K.F.; Nordestgaard, B.G. Elevated lipoprotein(a) in mitral and aortic valve calcification and disease: The Copenhagen General Population Study. Atherosclerosis 2021, 349, 166–174. [Google Scholar] [CrossRef]
- Stürzebecher, P.E.; Schorr, J.J.; Klebs, S.H.G.; Laufs, U. Trends and consequences of lipoprotein(a) testing: Cross-sectional and longitudinal health insurance claims database analyses. Atherosclerosis 2023, 367, 24–33. [Google Scholar] [CrossRef]
- Zierfuss, B.; Höbaus, C.; Feldscher, A.; Hannes, A.; Mrak, D.; Koppensteiner, R.; Stangl, H.; Schernthaner, G.H. Lipoprotein (a) and long-term outcome in patients with peripheral artery disease undergoing revascularization. Atherosclerosis 2022, 363, 94–101. [Google Scholar] [CrossRef]
- Gudbjartsson, D.F.; Thorgeirsson, G.; Sulem, P.; Helgadottir, A.; Gylfason, A.; Saemundsdottir, J.; Bjornsson, E.; Norddahl, G.L.; Jonasdottir, A.; Jonasdottir, A.; et al. Lipoprotein(a) Concentration and Risks of Cardiovascular Disease and Diabetes. J. Am. Coll. Cardiol. 2019, 74, 2982–2994. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular riskThe Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Koutsogianni, A.D.; Adamidis, P.S.; Barkas, F.; Liberopoulos, E.; Su, T.C.; Yamashita, S.; Liamis, G.; Rizzo, M. Familial Hypercholesterolemia and Lipoprotein(a): A Gordian Knot in Cardiovascular Prevention. Metabolites 2022, 12, 1065. [Google Scholar] [CrossRef]
- Amiri, M.; Raeisi-Dehkordi, H.; Verkaar, A.J.C.F.; Wu, Y.; van Westing, A.C.; Berk, K.A.; Bramer, W.M.; Aune, D.; Voortman, T. Circulating lipoprotein (a) and all-cause and cause-specific mortality: A systematic review and dose-response meta-analysis. Eur. J. Epidemiol. 2023, 38, 485–499. [Google Scholar] [CrossRef]
- Jawi, M.M.; Frohlich, J.; Chan, S.Y. Lipoprotein(a) the Insurgent: A New Insight into the Structure, Function, Metabolism, Pathogenicity, and Medications Affecting Lipoprotein(a) Molecule. J. Lipids 2020, 2020, 3491764. [Google Scholar] [CrossRef]
- Koutsogianni, A.-D.; Liberopoulos, E.; Tselepis, A.D. Lipoprotein(a): An update on its role in human health and disease. J. Atheroscler. Prev. Treat. 2021, 12, 92–102. [Google Scholar] [CrossRef]
- Koutsogianni, A.D.; Liberopoulos, E.; Tellis, K.; Tselepis, A.D. Oxidized phospholipids and lipoprotein(a): An update. Eur. J. Clin. Investig. 2022, 52, e13710. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, E.; Parhofer, K.G. Lipoprotein apheresis to treat elevated lipoprotein (a). J. Lipid Res. 2016, 57, 1751–1757. [Google Scholar] [CrossRef]
- Roeseler, E.; Julius, U.; Heigl, F.; Spitthoever, R.; Heutling, D.; Breitenberger, P.; Leebmann, J.; Lehmacher, W.; Kamstrup, P.R.; Nordestgaard, B.G.; et al. Lipoprotein apheresis for lipoprotein(a)-Associated cardiovascular disease: Prospective 5 years of follow-up and apolipoprotein(a) characterization. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2019–2027. [Google Scholar] [CrossRef]
- Kayikçioʇlu, M.; Kismali, E.; Can, L.; Payzin, S. Long-term follow-up in patients with homozygous familial hypercholesterolemia; 13-year experience of a university hospital lipid clinic. Turk Kardiyol. Dern. Ars. 2014, 42, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Kolovou, G.; Kolovou, V.; Bilianou, H.; Goumas, G.; Foussas, S.; Grapsa, E.; Garoufi, A.; Karavolias, G.; Mavrogieni, S.; Melidonis, A.; et al. Lipoprotein apheresis: A Hellenic consensus on its clinical use. Hell. J. Cardiol. 2021, 62, 460–462. [Google Scholar] [CrossRef]
- Tsimikas, S.; Gordts, P.L.S.M.; Nora, C.; Yeang, C.; Witztum, J.L. Statin therapy increases lipoprotein(a) levels. Eur. Heart J. 2020, 41, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- De Boer, L.M.; Oorthuys, A.O.J.; Wiegman, A.; Langendam, M.W.; Kroon, J.; Spijker, R.; Zwinderman, A.H.; Hutten, B.A. Statin therapy and lipoprotein(a) levels: A systematic review and meta-analysis. Eur. J. Prev. Cardiol. 2022, 29, 779–792. [Google Scholar] [CrossRef]
- Willeit, P.; Ridker, P.M.; Nestel, P.J.; Simes, J.; Tonkin, A.M.; Pedersen, T.R.; Schwartz, G.G.; Olsson, A.G.; Colhoun, H.M.; Kronenberg, F.; et al. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: Individual patient-data meta-analysis of statin outcome trials. Lancet 2018, 392, 1311–1320. [Google Scholar] [CrossRef]
- Khera, A.V.; Everett, B.M.; Caulfield, M.P.; Hantash, F.M.; Wohlgemuth, J.; Ridker, P.M.; Mora, S. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: An analysis from the JUPITER trial (justification for the use of statins in prevention: An intervention trial evaluating rosuvastatin). Circulation 2014, 129, 635–642. [Google Scholar] [CrossRef]
- Villard, E.F.; Thedrez, A.; Blankenstein, J.; Croyal, M.; Tran, T.T.T.; Poirier, B.; Le Bail, J.C.; Illiano, S.; Nobécourt, E.; Krempf, M.; et al. PCSK9 Modulates the Secretion But Not the Cellular Uptake of Lipoprotein(a) Ex Vivo: An Effect Blunted by Alirocumab. JACC Basic Transl. Sci. 2016, 1, 419–427. [Google Scholar] [CrossRef]
- Awan, Z.; Seidah, N.G.; MacFadyen, J.G.; Benjannet, S.; Chasman, D.I.; Ridker, P.M.; Genest, J. Rosuvastatin, Proprotein Convertase Subtilisin/Kexin Type 9 Concentrations, and LDL Cholesterol Response: The JUPITER Trial. Clin. Chem. 2012, 58, 183–189. [Google Scholar] [CrossRef]
- Guo, Y.L.; Liu, J.; Xu, R.X.; Zhu, C.G.; Wu, N.Q.; Jiang, L.X.; Li, J.J. Short-term impact of low-dose atorvastatin on serum proprotein convertase subtilisin/kexin type 9. Clin. Drug Investig. 2013, 33, 877–883. [Google Scholar] [CrossRef]
- Scanu, A.M.; Bamba, R. Niacin and Lipoprotein(a): Facts, Uncertainties, and Clinical Considerations. Am. J. Cardiol. 2008, 101, S44–S47. [Google Scholar] [CrossRef]
- Parish, S.; Hopewell, J.C.; Hill, M.R.; Marcovina, S.; Valdes-Marquez, E.; Haynes, R.; Offer, A.; Pedersen, T.R.; Baigent, C.; Collins, R.; et al. Impact of Apolipoprotein(a) Isoform Size on Lipoprotein(a) Lowering in the HPS2-THRIVE Study. Circ. Genom. Precis. Med. 2018, 11, e001696. [Google Scholar] [CrossRef]
- Chemello, K.; Chan, D.C.; Lambert, G.; Watts, G.F. Recent advances in demystifying the metabolism of lipoprotein(a). Atherosclerosis 2022, 349, 82–91. [Google Scholar] [CrossRef]
- Anderson, T.J.; Boden, W.E.; Desvigne-Nickens, P.; Fleg, J.L.; Kashyap, M.L.; McBride, R.; Probstfield, J.L.; for the AIM-HIGH Investigators. Safety Profile of Extended-Release Niacin in the AIM-HIGH Trial. N. Engl. J. Med. 2014, 371, 288. [Google Scholar] [CrossRef]
- Landray, M.J.; Haynes, R.; Hope-well, J.C.; Parish, S.; Aung, T.; Tomson, J.; Wallendszus, K.; Craig, M.; Jiang, L.; Collins, R.; et al. Effects of extended-release niacin with laropiprant in high-risk patients. N. Engl. J. Med. 2014, 371, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Warden, B.A.; Minnier, J.; Watts, G.F.; Fazio, S.; Shapiro, M.D. Impact of PCSK9 inhibitors on plasma lipoprotein(a) concentrations with or without a background of niacin therapy. J. Clin. Lipidol. 2019, 13, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Simental-Mendía, L.E.; Pirro, M.; Banach, M.; Watts, G.F.; Sirotri, C.; Al-Rasadi, K.; Atkin, S.L. Impact of ezetimibe on plasma lipoprotein(a) concentrations as monotherapy or in combination with statins: A systematic review and meta-analysis of randomized controlled trials. Sci. Rep. 2018, 8, 17887. [Google Scholar] [CrossRef]
- Gaudet, D.; Kereiakes, D.J.; McKenney, J.M.; Roth, E.M.; Hanotin, C.; Gipe, D.; Du, Y.; Ferrand, A.C.; Ginsberg, H.N.; Stein, E.A. Effect of alirocumab, a monoclonal proprotein convertase subtilisin/kexin 9 antibody, on lipoprotein(a) concentrations (a pooled analysis of 150 mg every two weeks dosing from phase 2 trials). Am. J. Cardiol. 2014, 114, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Giugliano, R.P.; Sabatine, M.S.; Koren, M.J.; Langslet, G.; Bays, H.; Blom, D.; Eriksson, M.; Dent, R.; Wasserman, S.M.; et al. Reduction in Lipoprotein(a) with PCSK9 Monoclonal Antibody Evolocumab (AMG 145): A Pooled Analysis of More Than 1,300 Patients in 4 Phase II Trials. J. Am. Coll. Cardiol. 2014, 63, 1278–1288. [Google Scholar] [CrossRef]
- Stiekema, L.C.A.; Stroes, E.S.G.; Verweij, S.L.; Kassahun, H.; Chen, L.; Wasserman, S.M.; Sabatine, M.S.; Mani, V.; Fayad, Z.A. Persistent arterial wall inflammation in patients with elevated lipoprotein(a) despite strong low-density lipoprotein cholesterol reduction by proprotein convertase subtilisin/kexin type 9 antibody treatment. Eur. Heart J. 2019, 40, 2775. [Google Scholar] [CrossRef]
- Bittner, V.A.; Szarek, M.; Aylward, P.E.; Bhatt, D.L.; Diaz, R.; Edelberg, J.M.; Fras, Z.; Goodman, S.G.; Halvorsen, S.; Hanotin, C.; et al. Effect of Alirocumab on Lipoprotein(a) and Cardiovascular Risk after Acute Coronary Syndrome. J. Am. Coll. Cardiol. 2020, 75, 133–144. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Fazio, S.; Giugliano, R.P.; Stroes, E.S.G.; Kanevsky, E.; Gouni-Berthold, I.; Im, K.A.; Pineda, A.L.; Wasserman, S.M.; Češka, R.; et al. Lipoprotein(a), PCSK9 Inhibition, and Cardiovascular Risk. Circulation 2019, 139, 1483–1492. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Szarek, M.; Bittner, V.A.; Diaz, R.; Goodman, S.G.; Jukema, J.W.; Landmesser, U.; López-Jaramillo, P.; Manvelian, G.; Pordy, R.; et al. Lipoprotein(a) and Benefit of PCSK9 Inhibition in Patients with Nominally Controlled LDL Cholesterol. J. Am. Coll. Cardiol. 2021, 78, 421–433. [Google Scholar] [CrossRef]
- Liberopoulos, E. Lipoprotein(a) reduction with proprotein convertase subtilisin/kexin type 9 inhibitors: An unsolved mystery. Eur. J. Prev. Cardiol. 2021, 28, 813–815. [Google Scholar] [CrossRef]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- Tenenbaum, A.; Fisman, E.Z. Balanced pan-PPAR activator bezafibrate in combination with statin: Comprehensive lipids control and diabetes prevention? Cardiovasc. Diabetol. 2012, 11, 140. [Google Scholar] [CrossRef]
- Fereshetian, A.G.; Davidson, M.; Haber, H.; Black, D.M. Gemfibrozil Treatment in Patients with Elevated Lipoprotein A. Clin. Drug Investig. 2012, 16, 1–7. [Google Scholar] [CrossRef]
- Von Eckardstein, A. Lipoprotein(a). Eur. Heart J. 2017, 38, 1530–1532. [Google Scholar] [CrossRef]
- Cuchel, M.; Meagher, E.A.; Theron, H.D.T.; Blom, D.J.; Marais, A.D.; Hegele, R.A.; Averna, M.R.; Sirtori, C.R.; Shah, P.K.; Gaudet, D.; et al. Efficacy and Safety of a Microsomal Triglyceride Transfer Protein Inhibitor in Homozygous Familial Hypercholesterolemia. Lancet 2013, 381, 40. [Google Scholar] [CrossRef]
- Vuorio, A.; Watts, G.F.; Kovanen, P.T. Depicting new pharmacological strategies for familial hypercholesterolaemia involving lipoprotein (a). Eur. Heart J. 2017, 38, 3555–3559. [Google Scholar] [CrossRef]
- Santos, R.D.; Raal, F.J.; Catapano, A.L.; Witztum, J.L.; Steinhagen-Thiessen, E.; Tsimikas, S. Mipomersen, an Antisense Oligonucleotide to Apolipoprotein B-100, Reduces Lipoprotein(a) in Various Populations with Hypercholesterolemia: Results of 4 Phase III Trials. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 689–699. [Google Scholar] [CrossRef]
- Fogacci, F.; Ferri, N.; Toth, P.P.; Ruscica, M.; Corsini, A.; Cicero, A.F.G. Efficacy and Safety of Mipomersen: A Systematic Review and Meta-Analysis of Randomized Clinical Trials. Drugs 2019, 79, 751–766. [Google Scholar] [CrossRef]
- Merki, E.; Graham, M.J.; Mullick, A.E.; Miller, E.R.; Crooke, R.M.; Pitas, R.E.; Witztum, J.L.; Tsimikas, S. Antisense Oligonucleotide Directed to Human Apolipoprotein B-100 Reduces Lipoprotein(a) Levels and Oxidized Phospholipids on Human Apolipoprotein B-100 Particles in Lipoprotein(a) Transgenic Mice. Circulation 2008, 118, 743–753. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Ruotolo, G.; Brewer, H.B.; Wang, M.D.; Liu, L.; Willey, M.B.; Deeg, M.A.; Krueger, K.A.; Nissen, S.E. Evacetrapib alone or in combination with statins lowers lipoprotein(a) and total and small LDL particle concentrations in mildly hypercholesterolemic patients. J. Clin. Lipidol. 2016, 10, 519–527.e4. [Google Scholar] [CrossRef]
- Bowman, L.; Chen, F.; Sammons, E.; Hopewell, J.C.; Wallendszus, K.; Stevens, W.; Valdes-Marquez, E.; Wiviott, S.; Cannon, C.P.; Braunwald, E.; et al. Randomized Evaluation of the Effects of Anacetrapib through Lipid-modification (REVEAL)—A large-scale, randomized, placebo-controlled trial of the clinical effects of anacetrapib among people with established vascular disease: Trial design, recruitment, and baseline characteristics. Am. Heart J. 2017, 187, 182. [Google Scholar] [CrossRef] [PubMed]
- Arsenault, B.J.; Petrides, F.; Tabet, F.; Bao, W.; Hovingh, G.K.; Boekholdt, S.M.; Ramin-Mangata, S.; Meilhac, O.; DeMicco, D.; Rye, K.A.; et al. Effect of atorvastatin, cholesterol ester transfer protein inhibition, and diabetes mellitus on circulating proprotein subtilisin kexin type 9 and lipoprotein(a) levels in patients at high cardiovascular risk. J. Clin. Lipidol. 2018, 12, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Ballantyne, C.M.; Barter, P.J.; Kallend, D.; Leiter, L.A.; Leitersdorf, E.; McMurray, J.J.V.; Nicholls, S.J.; Olsson, A.G.; Shah, P.K.; et al. Association of Lipoprotein(a) with Risk of Recurrent Ischemic Events Following Acute Coronary Syndrome: Analysis of the dal-Outcomes Randomized Clinical Trial. JAMA Cardiol. 2018, 3, 164. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.J.; Sniderman, A.D.; Ditmarsch, M.; Dicklin, M.R.; Nicholls, S.J.; Davidson, M.H.; Kastelein, J.J.P. Cholesteryl Ester Transfer Protein Inhibition Reduces Major Adverse Cardiovascular Events by Lowering Apolipoprotein B Levels. Int. J. Mol. Sci. 2022, 23, 9417. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Ditmarsch, M.; Kastelein, J.J.; Rigby, S.P.; Kling, D.; Curcio, D.L.; Alp, N.J.; Davidson, M.H. Lipid lowering effects of the CETP inhibitor obicetrapib in combination with high-intensity statins: A randomized phase 2 trial. Nat. Med. 2022, 28, 1672–1678. [Google Scholar] [CrossRef]
- Bempedoic Acid for LDL-C Lowering: What Do We Know?—American College of Cardiology. Available online: https://www.acc.org/latest-in-cardiology/articles/2020/08/10/08/21/bempedoic-acid-for-ldl-c-lowering (accessed on 4 November 2022).
- Thompson, P.D.; Rubino, J.; Janik, M.J.; Macdougall, D.E.; McBride, S.J.; Margulies, J.R.; Newton, R.S. Use of ETC-1002 to treat hypercholesterolemia in patients with statin intolerance. J. Clin. Lipidol. 2015, 9, 295–304. [Google Scholar] [CrossRef]
- Rubino, J.; MacDougall, D.E.; Sterling, L.R.; Kelly, S.E.; McKenney, J.M.; Lalwani, N.D. Lipid lowering with bempedoic acid added to a proprotein convertase subtilisin/kexin type 9 inhibitor therapy: A randomized, controlled trial. J. Clin. Lipidol. 2021, 15, 593–601. [Google Scholar] [CrossRef]
- CLEAR Outcomes: Bempedoic Acid Reduces Cardiovascular Events. Available online: https://www.practicalcardiology.com/view/clear-outcomes-bempedoic-acid-reduces-cardiovascular-events (accessed on 20 February 2023).
- Eraikhuemen, N.; Lazaridis, D.; Dutton, M.T. Emerging Pharmacotherapy to Reduce Elevated Lipoprotein(a) Plasma Levels. Am. J. Cardiovasc. Drugs 2020, 21, 255–265. [Google Scholar] [CrossRef]
- Greco, M.F.; Sirtori, C.R.; Corsini, A.; Ezhov, M.; Sampietro, T.; Ruscica, M. Lipoprotein(a) Lowering-From Lipoprotein Apheresis to Antisense Oligonucleotide Approach. J. Clin. Med. 2020, 9, 2103. [Google Scholar] [CrossRef]
- Viney, N.J.; van Capelleveen, J.C.; Geary, R.S.; Xia, S.; Tami, J.A.; Yu, R.Z.; Marcovina, S.M.; Hughes, S.G.; Graham, M.J.; Crooke, R.M.; et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): Two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016, 388, 2239–2253. [Google Scholar] [CrossRef]
- Tsimikas, S.; Karwatowska-Prokopczuk, E.; Gouni-Berthold, I.; Tardif, J.-C.; Baum, S.J.; Steinhagen-Thiessen, E.; Shapiro, M.D.; Stroes, E.S.; Moriarty, P.M.; Nordestgaard, B.G.; et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N. Engl. J. Med. 2020, 382, 244–255. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; López, J.A.G.; Knusel, B.; Gencer, B.; Wang, H.; Wu, Y.; Kassahun, H.; Sabatine, M.S. Study design and rationale for the Olpasiran trials of Cardiovascular Events And lipoproteiN(a) reduction-DOSE finding study (OCEAN(a)-DOSE). Am. Heart J. 2022, 251, 61–69. [Google Scholar] [CrossRef]
- Koren, M.J.; Moriarty, P.M.; Neutel, J.; Baum, S.J.; Hernandez-Illas, M.; Weintraub, H.S.; Hellawell, J.; Varrieur, T.; Sohn, W.; Wang, H.; et al. Abstract 13951: Safety, Tolerability and Efficacy of Single-dose Amg 890, a Novel Sirna Targeting Lp(a), in Healthy Subjects and Subjects with Elevated Lp(a). Circulation 2020, 142, A13951. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Rosenson, R.S.; Gencer, B.; López, J.A.G.; Lepor, N.E.; Baum, S.J.; Stout, E.; Gaudet, D.; Knusel, B.; Kuder, J.F.; et al. Small Interfering RNA to Reduce Lipoprotein(a) in Cardiovascular Disease. N. Engl. J. Med. 2022, 387, 1855–1864. [Google Scholar] [CrossRef]
- Rider, D.A.; Eisermann, M.; Löffler, K.; Aleku, M.; Swerdlow, D.I.; Dames, S.; Hauptmann, J.; Morrison, E.; Lindholm, M.W.; Schubert, S.; et al. Pre-clinical assessment of SLN360, a novel siRNA targeting LPA, developed to address elevated lipoprotein (a) in cardiovascular disease. Atherosclerosis 2022, 349, 240–247. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K.; Balog, C.; Swerdlow, D.I.; Scrimgeour, A.C.; Rambaran, C.; Wilson, R.J.; Boyce, M.; Ray, K.K.; Cho, L.; et al. Single Ascending Dose Study of a Short Interfering RNA Targeting Lipoprotein(a) Production in Individuals with Elevated Plasma Lipoprotein(a) Levels. JAMA 2022, 327, 1679–1687. [Google Scholar] [CrossRef]
- Boffelli, D.; Zajchowski, D.A.; Yang, Z.; Lawn, R.M. Estrogen Modulation of Apolipoprotein(a) Expression. J. Biol. Chem. 1999, 274, 15569–15574. [Google Scholar] [CrossRef]
- Anagnostis, P.; Galanis, P.; Chatzistergiou, V.; Stevenson, J.C.; Godsland, I.F.; Lambrinoudaki, I.; Theodorou, M.; Goulis, D.G. The effect of hormone replacement therapy and tibolone on lipoprotein (a) concentrations in postmenopausal women: A systematic review and meta-analysis. Maturitas 2017, 99, 27–36. [Google Scholar] [CrossRef]
- Salpeter, S.R.; Walsh, J.M.E.; Ormiston, T.M.; Greyber, E.; Buckley, N.S.; Salpeter, E.E. Meta-analysis: Effect of hormone-replacement therapy on components of the metabolic syndrome in postmenopausal women. Diabetes Obes. Metab. 2006, 8, 538–554. [Google Scholar] [CrossRef]
- Fogacci, F.; Borghi, C.; Davinelli, S.; Scapagnini, G.; Cicero, A.F.G. Impact of anti-oestrogen therapy on lipoprotein(a) in postmenopausal women: A systematic review and meta-analysis of double-blind placebo-controlled clinical studies. Endocrine 2022, 80, 292–302. [Google Scholar] [CrossRef]
- Hulley, S.; Grady, D.; Bush, T.; Furberg, C.; Herrington, D.; Riggs, B.; Vittinghoff, E. Randomized Trial of Estrogen Plus Progestin for Secondary Prevention of Coronary Heart Disease in Postmenopausal Women. JAMA 1998, 280, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Shlipak, M.G.; Simon, J.A.; Vittinghoff, E.; Lin, F.; Barrett-Connor, E.; Knopp, R.H.; Levy, R.I.; Hulley, S.B. Estrogen and Progestin, Lipoprotein(a), and the Risk of Recurrent Coronary Heart Disease Events after Menopause. JAMA 2000, 283, 1845–1852. [Google Scholar] [CrossRef] [PubMed]
- Marcovina, S.M.; Lippi, G.; Bagatell, C.J.; Bremner, W.J. Testosterone-induced suppression of lipoprotein(a) in normal men; relation to basal lipoprotein(a) level. Atherosclerosis 1996, 122, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Giannoulis, M.G.; Jackson, N.; Shojaee-Moradie, F.; Sonksen, P.H.; Martin, F.C.; Umpleby, A.M. Effects of growth hormone and/or testosterone on very low density lipoprotein apolipoprotein B100 kinetics and plasma lipids in healthy elderly men: A randomised controlled trial. Growth Horm. IGF Res. 2006, 16, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Hartgens, F.; Rietjens, G.; Keizer, H.A.; Kuipers, H.; Wolffenbuttel, B.H.R. Effects of androgenic-anabolic steroids on apolipoproteins and lipoprotein (a). Br. J. Sports Med. 2004, 38, 253–259. [Google Scholar] [CrossRef]
- Shewmon, D.A.; Stock, J.L.; Abusamra, L.C.; Kristan, M.A.; Baker, S.; Heiniluoma, K.M. Tamoxifen decreases lipoprotein(a) in patients with breast cancer. Metab.-Clin. Exp. 1994, 43, 531–532. [Google Scholar] [CrossRef]
- Liberopoulos, E.; Karabina, S.A.; Tselepis, A.; Bairaktari, E.; Nicolaides, C.; Pavlidis, N.; Elisaf, M. Are the Effects of Tamoxifen on the Serum Lipid Profile Modified by Apolipoprotein E Phenotypes? Oncology 2002, 62, 115–120. [Google Scholar] [CrossRef]
- Kotwal, A.; Cortes, T.; Genere, N.; Hamidi, O.; Jasim, S.; Newman, C.B.; Prokop, L.J.; Hassan Murad, M.; Alahdab, F. Treatment of Thyroid Dysfunction and Serum Lipids: A Systematic Review and Meta-analysis. J. Clin. Endocrinol. Metab. 2020, 105, 3683–3694. [Google Scholar] [CrossRef]
- Angelin, B.; Kristensen, J.D.; Eriksson, M.; Carlsson, B.; Klein, I.; Olsson, A.G.; Chester Ridgway, E.; Ladenson, P.W. Reductions in serum levels of LDL cholesterol, apolipoprotein B, triglycerides and lipoprotein(a) in hypercholesterolaemic patients treated with the liver-selective thyroid hormone receptor agonist eprotirome. J. Intern. Med. 2015, 277, 331–342. [Google Scholar] [CrossRef]
- Harrison, S.A.; Bashir, M.R.; Guy, C.D.; Zhou, R.; Moylan, C.A.; Frias, J.P.; Alkhouri, N.; Bansal, M.B.; Baum, S.; Neuschwander-Tetri, B.A.; et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2019, 394, 2012–2024. [Google Scholar] [CrossRef]
- Zhao, M.; Xie, H.; Shan, H.; Zheng, Z.; Li, G.; Li, M.; Hong, L. Development of Thyroid Hormones and Synthetic Thyromimetics in Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 1102. [Google Scholar] [CrossRef]
- Eden, S.; Wiklund, O.; Oscarsson, J.; Rosen, T.; Bengtsson, B.A. Growth hormone treatment of growth hormone-deficient adults results in a marked increase in Lp(a) and HDL cholesterol concentrations. Arterioscler. Thromb. J. Vasc. Biol. 1993, 13, 296–301. [Google Scholar] [CrossRef]
- Glynn, N.; Halsall, D.J.; Boran, G.; Cook, P.; McDermott, J.H.; Smith, D.; Tormey, W.; Thompson, C.J.; O’Gorman, D.; McKenna, M.J.; et al. Growth hormone replacement may influence the biological action of thyroid hormone on liver and bone tissue. Growth Horm. IGF Res. 2021, 57–58, 101393. [Google Scholar] [CrossRef]
- Chasman, D.I.; Shiffman, D.; Zee, R.Y.L.; Louie, J.Z.; Luke, M.M.; Rowland, C.M.; Catanese, J.J.; Buring, J.E.; Devlin, J.J.; Ridker, P.M. Polymorphism in the apolipoprotein(a) gene, plasma lipoprotein(a), cardiovascular disease, and low-dose aspirin therapy. Atherosclerosis 2009, 203, 371–376. [Google Scholar] [CrossRef]
- Lacaze, P.; Bakshi, A.; Riaz, M.; Polekhina, G.; Owen, A.; Bhatia, H.S.; Natarajan, P.; Wolfe, R.; Beilin, L.; Nicholls, S.J.; et al. Aspirin for Primary Prevention of Cardiovascular Events in Relation to Lipoprotein(a) Genotypes. J. Am. Coll. Cardiol. 2022, 80, 1287–1298. [Google Scholar] [CrossRef]
- Schultz, O.; Oberhauser, F.; Saech, J.; Rubbert-Roth, A.; Hahn, M.; Krone, W.; Laudes, M. Effects of Inhibition of Interleukin-6 Signalling on Insulin Sensitivity and Lipoprotein (A) Levels in Human Subjects with Rheumatoid Diseases. PLoS ONE 2010, 5, e14328. [Google Scholar] [CrossRef]
- Wade, D.P.; Clarke, J.G.; Lindahl, G.E.; Liu, A.C.; Zysow, B.R.; Meer, K.; Schwartz, K.; Lawn, R.M. 5′ control regions of the apolipoprotein(a) gene and members of the related plasminogen gene family. Proc. Natl. Acad. Sci. USA 1993, 90, 1369. [Google Scholar] [CrossRef]
- Enkhmaa, B.; Anuurad, E.; Zhang, W.; Li, C.S.; Kaplan, R.; Lazar, J.; Merenstein, D.; Karim, R.; Aouizerat, B.; Cohen, M.; et al. Effect of antiretroviral therapy on allele-associated Lp(a) level in women with HIV in the Women’s Interagency HIV Study. J. Lipid Res. 2018, 59, 1967. [Google Scholar] [CrossRef]
- Santos, H.O.; Kones, R.; Rumana, U.; Earnest, C.P.; Izidoro, L.F.M.; Macedo, R.C.O. Lipoprotein(a): Current Evidence for a Physiologic Role and the Effects of Nutraceutical Strategies. Clin. Ther. 2019, 41, 1780–1797. [Google Scholar] [CrossRef]
- Enkhmaa, B.; Petersen, K.S.; Kris-Etherton, P.M.; Berglund, L. Diet and Lp(a): Does Dietary Change Modify Residual Cardiovascular Risk Conferred by Lp(a)? Nutrients 2020, 12, 2024. [Google Scholar] [CrossRef]
- Clevidence, B.A.; Judd, J.T.; Schaefer, E.J.; Jenner, J.L.; Lichtenstein, A.H.; Muesing, R.A.; Wittes, J.; Sunkin, M.E. Plasma Lipoprotein (a) Levels in Men and Women Consuming Diets Enriched in Saturated, Cis-, or Trans-Monounsaturated Fatty Acids. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1657–1661. [Google Scholar] [CrossRef] [PubMed]
- Silaste, M.L.; Rantala, M.; Alfthan, G.; Aro, A.; Witztum, J.L.; Kesäniemi, Y.A.; Hörkkö, S. Changes in Dietary Fat Intake Alter Plasma Levels of Oxidized, Low-Density Lipoprotein and Lipoprotein(a). Arterioscler. Thromb. Vasc. Biol. 2004, 24, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Ebbeling, C.B.; Knapp, A.; Johnson, A.; Wong, J.M.W.; Greco, K.F.; Ma, C.; Mora, S.; Ludwig, D.S. Effects of a low-carbohydrate diet on insulin-resistant dyslipoproteinemia—Arandomized controlled feeding trial. Am. J. Clin. Nutr. 2022, 115, 154. [Google Scholar] [CrossRef] [PubMed]
- Florentin, M.; Elisaf, M.S.; Rizos, C.V.; Nikolaou, V.; Bilianou, E.; Pitsavos, C.; Liberopoulos, E.N. l-Carnitine/Simvastatin Reduces Lipoprotein (a) Levels Compared with Simvastatin Monotherapy: A Randomized Double-Blind Placebo-Controlled Study. Lipids 2017, 52, 1–9. [Google Scholar] [CrossRef]
- Jenner, J.L.; Jacques, P.F.; Seman, L.J.; Schaefer, E.J. Ascorbic acid supplementation does not lower plasma lipoprotein(a) concentrations. Atherosclerosis 2000, 151, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Bostom, A.G.; Hume, A.L.; Eaton, C.B.; Laurino, J.P.; Yanek, L.R.; Regan, M.S.; McQuade, W.H.; Craig, W.Y.; Perrone, G.; Jacques, P.F. The Effect of High-Dose Ascorbate Supplementation on Plasma Lipoprotein(a) Levels in Patients with Premature Coronary Heart Disease. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1995, 15, 458–464. [Google Scholar]
- Austin, A.; Warty, V.; Janosky, J.; Arslanian, S. The Relationship of Physical Fitness to Lipid and Lipoprotein(a) Levels in Adolescents with IDDm. Diabetes Care 1993, 16, 421–425. [Google Scholar] [CrossRef]
- Theodorou, A.A.; Panayiotou, G.; Volaklis, K.A.; Douda, H.T.; Paschalis, V.; Nikolaidis, M.G.; Smilios, I.; Toubekis, A.; Kyprianou, D.; Papadopoulos, I.; et al. Aerobic, resistance and combined training and detraining on body composition, muscle strength, lipid profile and inflammation in coronary artery disease patients. Res. Sport Med. 2016, 24, 171–184. [Google Scholar] [CrossRef]
- Ho, J.H.; Adam, S.; Liu, Y.; Azmi, S.; Dhage, S.; Syed, A.A.; Ammori, B.J.; Donn, R.; Heald, A.; Gibson, M.J.; et al. Effect of bariatric surgery on plasma levels of oxidised phospholipids, biomarkers of oxidised LDL and lipoprotein(a). J. Clin. Lipidol. 2021, 15, 320–331. [Google Scholar] [CrossRef]
- Durlach, V.; Bonnefont-Rousselot, D.; Boccara, F.; Varret, M.; Di-Filippo Charcosset, M.; Cariou, B.; Valero, R.; Charriere, S.; Farnier, M.; Morange, P.E.; et al. Lipoprotein(a): Pathophysiology, measurement, indication and treatment in cardiovascular disease. A consensus statement from the Nouvelle Société Francophone d’Athérosclérose (NSFA). Arch. Cardiovasc. Dis. 2021, 114, 828–847. [Google Scholar] [CrossRef]
- Burgess, S.; Ference, B.A.; Staley, J.R.; Freitag, D.F.; Mason, A.M.; Nielsen, S.F.; Willeit, P.; Young, R.; Surendran, P.; Karthikeyan, S.; et al. Association of LPA Variants with Risk of Coronary Disease and the Implications for Lipoprotein(a)-Lowering Therapies: A Mendelian Randomization Analysis. JAMA Cardiol. 2018, 3, 619–627. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Lipoprotein(a)-Lowering Therapy | Lipoprotein(a) Effect |
|---|---|
| Lipoprotein apheresis | Acute decrement of 70% to 80%. Regular apheresis can translate into a mean Lp(a) reduction between 25% and 40% [26]. |
| Statins | Mixed results from clinical trials. Statins may increase Lp(a) by an average of 8% to 24% [30,31]. |
| Niacin | Potential Lp(a)-lowering effect by 30% to 40% [37] |
| Ezetimibe | Neutral effect up to a modest 7% reduction in Lp(a) levels [43] |
Proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors:
|
|
| Fibrates | Potent reduction between 13% and 39% [52] |
| Lomitapide | Reduction 15% to 19% [55] |
| Mipomersen | Reduction 21% to 39% [57,58] |
| Cholesteryl ester transfer protein inhibitors | Reduction 24% to 36% [60,61,62] |
| Bempedoic acid | Neutral effect [67,68,69] |
| Bile acid sequestrants | Neutral effect [70] |
Antisense oligonucleotides:
|
|
Small interfering RNAs:
|
|
Sex hormone therapies:
| |
Thyroid hormone therapy
| |
| Growth hormone | Increase by 25% to 100% [94] |
| Antibodies against interleukin-6 (e.g., tocilizumab) | Reduction between 30% and 40% [98] |
| Protease inhibitors or antiretroviral therapy | Increase [100] |
| Low-saturated fat diets | Potential increase between 8% and 20% [102,103,104] |
| Low-carbohydrate, high-saturated fat diets and diets enriched with walnuts or pecans | Decrement by 15% and 6% to 15%, respectively [102,105] |
| Dietary supplements (L-carnitine, and coenzyme Q10) | Modest reduction between 10% and 30% [101,106] |
| Specific foods (coffee, tea, and alcoholic beverages, especially red wine) | Modest reduction between 10% and 30% [101,106] |
| Vitamin C | Neutral effect on plasma Lp(a) levels [107,108] |
| Bariatric surgery | Significant increase in Lp(a) plasma levels [111] |
| Lipoprotein(a) (Lp(a))-Lowering Therapy | Mechanism of Action |
|---|---|
| Statins | |
| Niacin | Decreases apo(a) production rate [39]. |
| Ezetimibe |
|
| PCSK9 inhibitors |
|
| Lomitapide | Decreases the number of apoB-containing lipoprotein particles secreted into the bloodstream, including Lp(a) particles [55]. |
| Mipomersen | Decreases the availability of apoB100 for Lp(a) assembly [59]. |
| CETP inhibitors | Mediate the transfer of cholesteryl esters from high-density lipoproteins to apoB100-containing particles, including VLDL and LDL, in exchange for triglycerides [60,61,62,63] |
| Bempedoic acid | Upregulates LDLRs and increases the clearance of LDL particles [66]. |
| Antisense oligonucleotides | Bind to its target complementary RNA sequence via base pairing, thereby leading to degradation of the apo(a) mRNA strand and reduced Lp(a) production [72]. |
| Small interfering RNAs | Double-stranded RNA molecules that dissociate once inside the cell, and the antisense strand is inserted into the RISC (RNA-Induced Silencing Complex). The antisense strand binds to its homologous target mRNA sequence, leading to its degradation. The antisense strand bound to the RISC forms a recyclable, stable complex, and, as a result, its action against target mRNA strands can be repeated [74]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koutsogianni, A.D.; Liamis, G.; Liberopoulos, E.; Adamidis, P.S.; Florentin, M. Effects of Lipid-Modifying and Other Drugs on Lipoprotein(a) Levels—Potent Clinical Implications. Pharmaceuticals 2023, 16, 750. https://doi.org/10.3390/ph16050750
Koutsogianni AD, Liamis G, Liberopoulos E, Adamidis PS, Florentin M. Effects of Lipid-Modifying and Other Drugs on Lipoprotein(a) Levels—Potent Clinical Implications. Pharmaceuticals. 2023; 16(5):750. https://doi.org/10.3390/ph16050750
Chicago/Turabian StyleKoutsogianni, Amalia Despoina, George Liamis, Evangelos Liberopoulos, Petros Spyridonas Adamidis, and Matilda Florentin. 2023. "Effects of Lipid-Modifying and Other Drugs on Lipoprotein(a) Levels—Potent Clinical Implications" Pharmaceuticals 16, no. 5: 750. https://doi.org/10.3390/ph16050750
APA StyleKoutsogianni, A. D., Liamis, G., Liberopoulos, E., Adamidis, P. S., & Florentin, M. (2023). Effects of Lipid-Modifying and Other Drugs on Lipoprotein(a) Levels—Potent Clinical Implications. Pharmaceuticals, 16(5), 750. https://doi.org/10.3390/ph16050750