
High-Density Lipoprotein in Metabolic Disorders and Beyond: An Exciting New World Full of Challenges and Opportunities

 , ,
, ,  and
and
Abstract

1. Introduction
2. HDL Proteome and Particle Functionality
3. HDL Lipidome and Particle Functionality
4. High-Density Lipoprotein and Plasma Triglyceride Levels
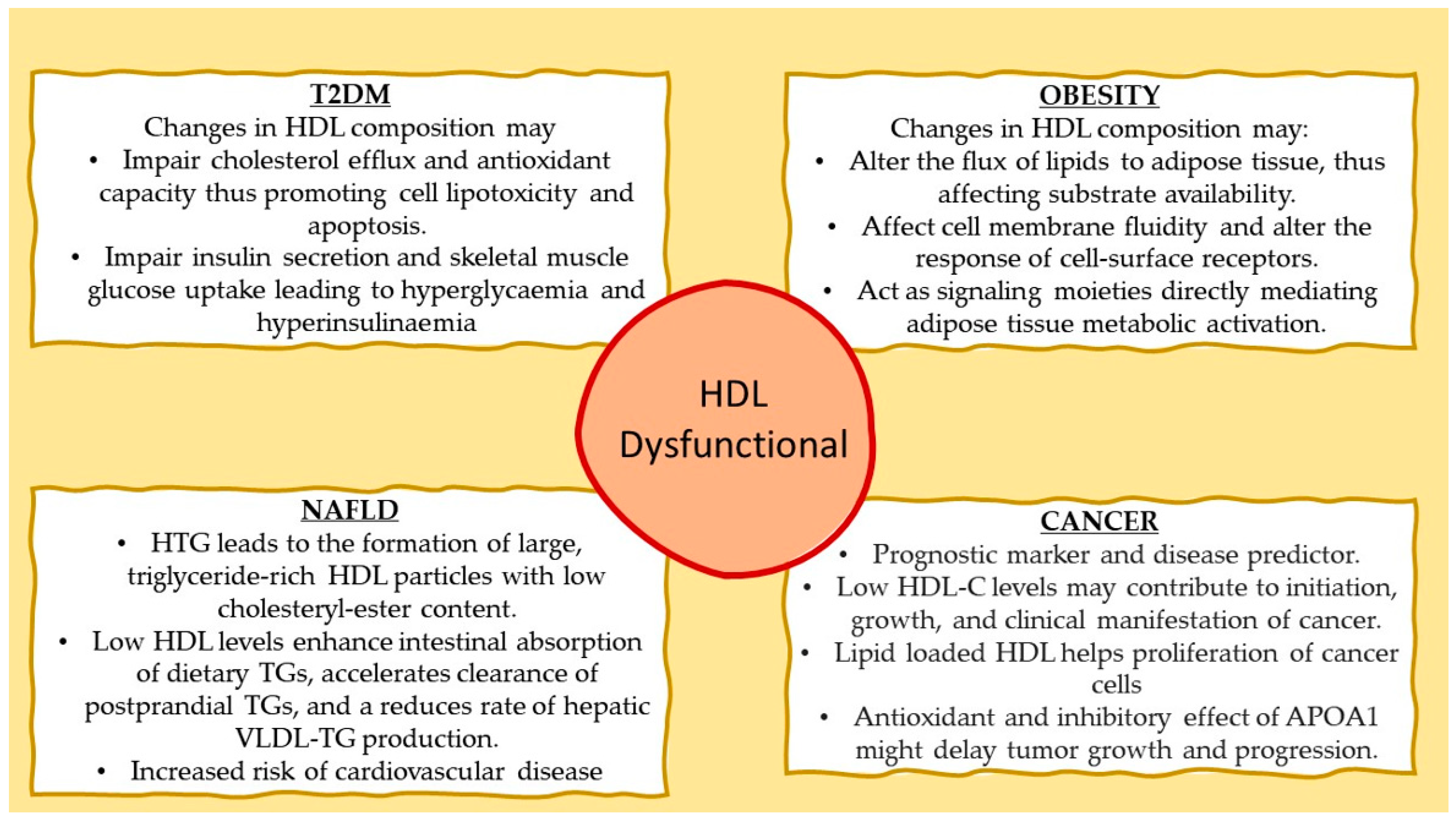
5. High-Density Lipoprotein in Type 2 Diabetes Mellitus
6. High-Density Lipoprotein and Adipose Tissue Metabolic Activity
7. High-Density Lipoprotein and Hepatic Triglyceride Deposition
8. High-Density Lipoprotein and Cancer
9. Current State of the Art in HDL Pharmaceuticals

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent | Trial Status | Clinical Outcomes | Reference and NCT |
|---|---|---|---|
| Obicetrapib | Active, recruiting | Significant increase in HDL-C levels from 24 mg/dL to a maximum of 65 mg/dL. Significant increase in APOA1 by 50–60%. Significant decrease in APOB levels by 30–50% and Lp(a) levels by 30%. | [75] NCT05202509 |
| Niaspan | Terminated (2012) | Significant increase in HDL-C levels by approximately 30%. Significant decrease in TG levels by 30%, LDL-C levels by 14%, (Lp(a)) levels by 32% and APOB levels by 9–39%. | [88] NCT00120289 |
| D-4F | Completed (2017) | Binds to oxidized lipids more effectively than endogenous APOA1 and lowers the HDL inflammatory index. | [81,82]. |
| CER-001 | Completed (2016) | Failed to demonstrate regression of atherosclerosis in patients with ACS already receiving statin therapy after 10 weekly infusions of CER-001. | [84] NCT02484378 |
| CSL-111 | Completed (2008) | Did not reduce atherosclerotic plaque volume and led to elevated levels of liver enzymes. Its development was discontinued. | [86] NCT00225719 |
| CSL-112 | Active, not recruiting | Well-tolerated in human trials. Increased HDL cholesterol efflux capacity. Currently assessing efficacy and safety. Results are expected by the end of 2023. | [87] NCT03473223 |
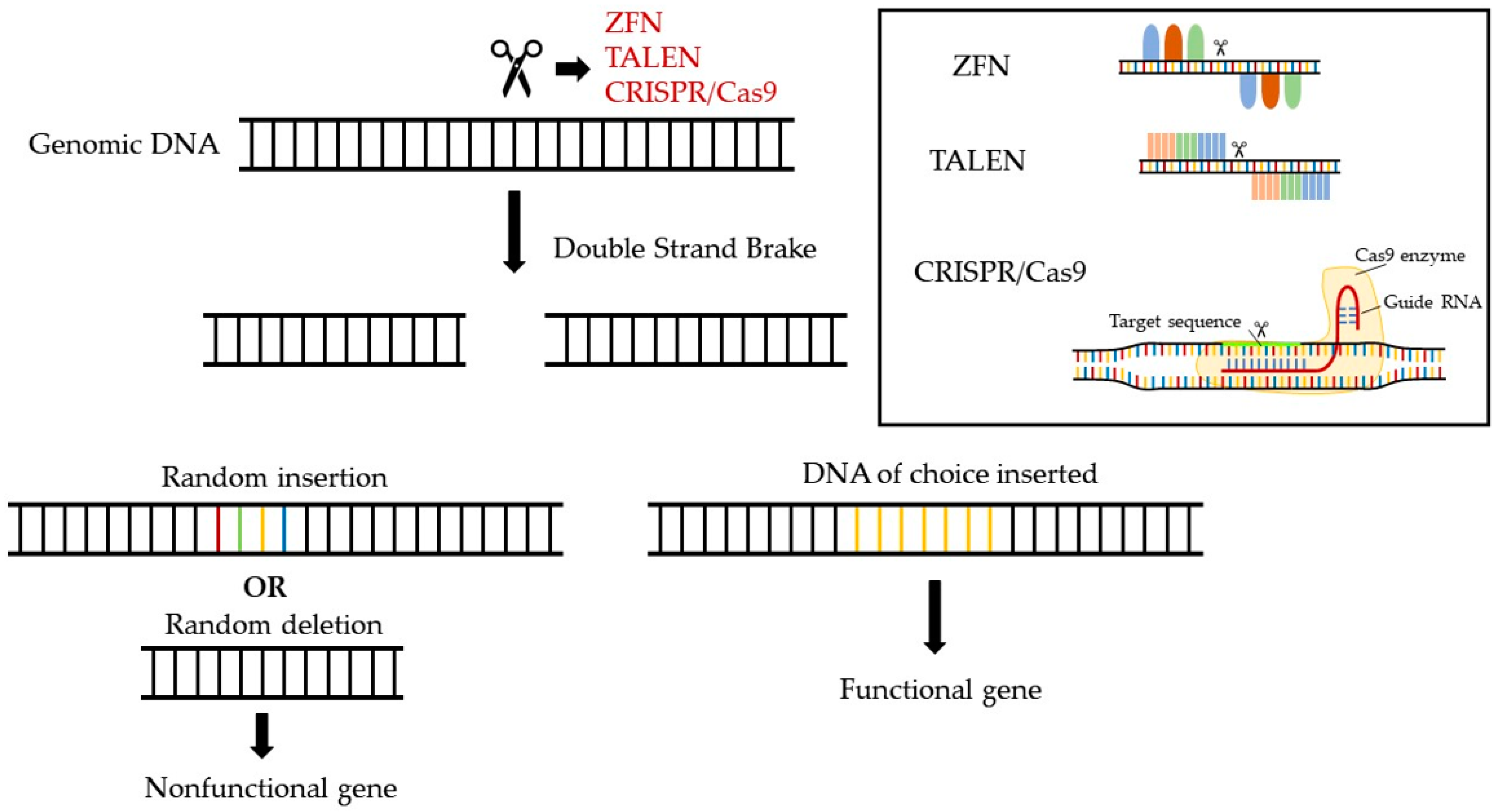
10. Opportunities for Novel Pharmaceuticals in the Gene-Editing Era
10.1. Examples of Gene-Editing-Based Pharmaceuticals in Cardiovascular Diseases
10.2. Challenges in the Development of Gene-Editing Pharmaceuticals
10.3. Opportunities for Gene-Editing Therapies for Treating Low and Dysfunctional HDL
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zvintzou, E.; Karampela, D.S.; Vakka, A.; Xepapadaki, E.; Karavia, E.A.; Hatziri, A.; Giannopoulou, P.C.; Kypreos, K.E. High Density Lipoprotein in Atherosclerosis and Coronary Heart Disease: Where Do We Stand Today? Vascul. Pharmacol. 2021, 141, 106928. [Google Scholar] [CrossRef] [PubMed]
- Bowe, B.; Xie, Y.; Xian, H.; Balasubramanian, S.; Zayed, M.A.; Al-Aly, Z. High Density Lipoprotein Cholesterol and the Risk of All-Cause Mortality among U.S. Veterans. Clin. J. Am. Soc. Nephrol. 2016, 11, 1784–1793. [Google Scholar] [CrossRef] [PubMed]
- Karavia, E.A.; Zvintzou, E.; Petropoulou, P.-I.; Xepapadaki, E.; Constantinou, C.; Kypreos, K.E. HDL Quality and Functionality: What can Proteins and Genes Predict? Expert Rev. Cardiovasc. Ther. 2014, 12, 521–532. [Google Scholar] [CrossRef]
- Tsompanidi, E.M.; Brinkmeier, M.S.; Fotiadou, E.H.; Giakoumi, S.M.; Kypreos, K.E. HDL Biogenesis and Functions: Role of HDL Quality and Quantity in Atherosclerosis. Atherosclerosis 2010, 208, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Kavo, A.E.; Rallidis, L.S.; Sakellaropoulos, G.C.; Lehr, S.; Hartwig, S.; Eckel, J.; Bozatzi, P.I.; Anastasiou-Nana, M.; Tsikrika, P.; Kypreos, K.E. Qualitative Characteristics of HDL in Young Patients of an Acute Myocardial Infarction. Atherosclerosis 2012, 220, 257–264. [Google Scholar] [CrossRef]
- Filou, S.; Lhomme, M.; Karavia, E.A.; Kalogeropoulou, C.; Theodoropoulos, V.; Zvintzou, E.; Sakellaropoulos, G.C.; Petropoulou, P.I.; Constantinou, C.; Kontush, A.; et al. Distinct Roles of Apolipoproteins A1 and e in the Modulation of High-Density Lipoprotein Composition and Function. Biochemistry 2016, 55, 3752–3762. [Google Scholar] [CrossRef]
- Zvintzou, E.; Xepapadaki, E.; Kalogeropoulou, C.; Filou, S.; Kypreos, K.E. Pleiotropic Effects of Apolipoprotein A-Ⅱ on High-Density Lipoprotein Functionality, Adipose Tissue Metabolic Activity and Plasma Glucose Homeostasis. J. Biomed. Res. 2020, 34, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Zvintzou, E.; Lhomme, M.; Chasapi, S.; Filou, S.; Theodoropoulos, V.; Xapapadaki, E.; Kontush, A.; Spyroulias, G.; Tellis, C.; Tselepis, A.D.; et al. Pleiotropic Effects of Apolipoprotein C3 on HDL Functionality and Adipose Tissue Metabolic Activity. J. Lipid Res. 2017, 58, 1869–1883. [Google Scholar] [CrossRef]
- Davidson, W.S.; Hazlett, T.; Mantulin, W.W.; Jonas, A. The Role of Apolipoprotein AI Domains in Lipid Binding. Proc. Natl. Acad. Sci. USA 1996, 93, 13605–13610. [Google Scholar] [CrossRef]
- De Beer, M.C.; Durbin, D.M.; Cai, L.; Jonas, A.; De Beer, F.C.; Van der Westhuyzen, D.R. Apolipoprotein A-I Conformation Markedly Influences HDL Interaction with Scavenger Receptor BI. J. Lipid Res. 2001, 42, 309–313. [Google Scholar] [CrossRef]
- Zhao, Y.; Marcel, Y.L. Serum Albumin Is a Significant Intermediate in Cholesterol Transfer between Cells and Lipoproteins. Biochemistry 1996, 35, 7174–7180. [Google Scholar] [CrossRef] [PubMed]
- Marcel, Y.L.L.; Weech, P.K.K.; Nguyen, T.-D.D.; Milne, R.W.W.; McConathy, W.J.J. Apolipoproteins as the Basis for Heterogeneity in High-Density Lipoprotein2 and High-Density Lipoprotein3. Studies by Isoelectric Focusing on Agarose Films. Eur. J. Biochem. 1984, 143, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Catapano, A.L.; Pirillo, A.; Bonacina, F.; Norata, G.D. HDL in Innate and Adaptive Immunity. Cardiovasc. Res. 2014, 103, 372–383. [Google Scholar] [CrossRef]
- Constantinou, C.; Karavia, E.A.; Xepapadaki, E.; Petropoulou, P.I.; Papakosta, E.; Karavyraki, M.; Zvintzou, E.; Theodoropoulos, V.; Filou, S.; Hatziri, A.; et al. Advances in High-Density Lipoprotein Physiology: Surprises, Overturns, and Promises. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E1–E14. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A.; Lindahl, M.; Lhomme, M.; Calabresi, L.; Chapman, M.J.; Davidson, W.S. Structure of HDL: Particle Subclasses and Molecular Components. In Proceedings of the Handbook of Experimental Pharmacology; UMR-ICAN 1166; National Institute for Health and Medical Research (INSERM): Paris, France, 2015; Volume 224, pp. 3–51. [Google Scholar]
- Kontush, A.; Therond, P.; Zerrad, A.; Couturier, M.; Négre-Salvayre, A.; De Souza, J.A.; Chantepie, S.; Chapman, M.J. Preferential Sphingosine-1-Phosphate Enrichment and Sphingomyelin Depletion Are Key Features of Small Dense HDL3 Particles: Relevance to Antiapoptotic and Antioxidative Activities. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1843–1849. [Google Scholar] [CrossRef]
- Sato, K. Role of Sphingosine 1-Phosphate in Anti-Atherogenic Actions of High-Density Lipoprotein. World J. Biol. Chem. 2010, 1, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Sutter, I.; Klingenberg, R.; Othman, A.; Rohrer, L.; Landmesser, U.; Heg, D.; Rodondi, N.; Mach, F.; Windecker, S.; Matter, C.M.; et al. Decreased Phosphatidylcholine Plasmalogens—A Putative Novel Lipid Signature in Patients with Stable Coronary Artery Disease and Acute Myocardial Infarction. Atherosclerosis 2016, 246, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Argraves, K.M.; Sethi, A.A.; Gazzolo, P.J.; Wilkerson, B.A.; Remaley, A.T.; Tybjaerg-Hansen, A.; Nordestgaard, B.G.; Yeatts, S.D.; Nicholas, K.S.; Barth, J.L.; et al. S1P, Dihydro-S1P and C24:1-Ceramide Levels in the HDL-Containing Fraction of Serum Inversely Correlate with Occurrence of Ischemic Heart Disease. Lipids Health Dis. 2011, 10, 70. [Google Scholar] [CrossRef]
- Kontush, A.; Lhomme, M.; Chapman, M.J. Unraveling the Complexities of the HDL Lipidome. J. Lipid Res. 2013, 54, 2950–2963. [Google Scholar] [CrossRef]
- Zerrad-Saadi, A.; Therond, P.; Chantepie, S.; Couturier, M.; Rye, K.A.; Chapman, M.J.; Kontush, A. HDL3-Mediated Inactivation of LDL-Associated Phospholipid Hydroperoxides Is Determined by the Redox Status of Apolipoprotein A-I and HDL Particle Surface Lipid Rigidity: Relevance to Inflammation and Atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 2169–2175. [Google Scholar] [CrossRef]
- Hansel, B.; Giral, P.; Nobecourt, E.; Chantepie, S.; Bruckert, E.; Chapman, M.J.; Kontush, A. Metabolic Syndrome Is Associated with Elevated Oxidative Stress and Dysfunctional Dense High-Density Lipoprotein Particles Displaying Impaired Antioxidative Activity. J. Clin. Endocrinol. Metab. 2004, 89, 4963–4971. [Google Scholar] [CrossRef]
- Kontush, A.; Chapman, M.J.J. Antiatherogenic Function of HDL Particle Subpopulations: Focus on Antioxidative Activities. Curr.Opin.Lipidol. 2010, 21, 312–318. [Google Scholar] [CrossRef]
- Kontush, A.; De Faria, E.C.; Chantepie, S.; Chapman, M.J. A Normotriglyceridemic, Low HDL-Cholesterol Phenotype Is Characterised by Elevated Oxidative Stress and HDL Particles with Attenuated Antioxidative Activity. Atherosclerosis 2005, 182, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Xepapadaki, E.; Maulucci, G.; Constantinou, C.; Karavia, E.A.; Zvintzou, E.; Daniel, B.; Sasson, S.; Kypreos, K.E. Impact of Apolipoprotein A1- or Lecithin:Cholesterol Acyltransferase-Deficiency on White Adipose Tissue Metabolic Activity and Glucose Homeostasis in Mice. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1351–1360. [Google Scholar] [CrossRef]
- Wiktorowska-Owczarek, A.; Berezińska, M.; Nowak, J.Z. PUFAs: Structures, Metabolism and Functions. Adv. Clin. Exp. Med. 2015, 24, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Welty, F.K. How Do Elevated Triglycerides and Low HDL-Cholesterol Affect Inflammation and Atherothrombosis? Curr. Cardiol. Rep. 2013, 15, 400. [Google Scholar] [CrossRef]
- Lanktree, M.B.; Thériault, S.; Walsh, M.; Paré, G. HDL Cholesterol, LDL Cholesterol, and Triglycerides as Risk Factors for CKD: A Mendelian Randomization Study. Am. J. Kidney Dis. 2018, 71, 166–172. [Google Scholar] [CrossRef]
- Kypreos, K.E. ABCA1 Promotes the de Novo Biogenesis of Apolipoprotein CIII-Containing HDL Particles in Vivo and Modulates the Severity of Apolipoprotein CIII-Induced Hypertriglyceridemia. Biochemistry 2008, 47, 10491–10502. [Google Scholar] [CrossRef]
- Brooks-Wilson, A.; Marcil, M.; Clee, S.M.; Zhang, L.-H.; Roomp, K.; van Dam, M.; Yu, L.; Brewer, C.; Collins, J.A.; Molhuizen, H.O.F.; et al. Mutations in ABC1 in Tangier Disease and Familial High-Density Lipoprotein Deficiency. Nat. Genet. 1999, 22, 336–345. [Google Scholar] [CrossRef]
- Wang, C.-S.; Alaupovic, P.; Gregg, R.E.; Brewer, H.B. Studies on the Mechanism of Hypertriglyceridemia in Tangier Disease. Determination of Plasma Lipolytic Activities, K1 Values and Apolipoprotein Composition of the Major Lipoprotein Density Classes. Biochim. Biophys. Acta Lipids Lipid Metab. 1987, 920, 9–19. [Google Scholar] [CrossRef]
- Kolovou, G. Postprandial Hypertriglyceridaemia in Patients with Tangier Disease. J. Clin. Pathol. 2003, 56, 937–941. [Google Scholar] [CrossRef]
- Heinen, R.J.; Herbert, P.N.; Fredrickson, D.S.; Forte, T.; Lindgren, F.T. Properties of the Plasma Very Low and Low Density Lipoproteins in Tangier Disease. J. Clin. Investig. 1978, 61, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Xepapadaki, E.; Nikdima, I.; Sagiadinou, E.C.; Zvintzou, E.; Kypreos, K.E. HDL and Type 2 Diabetes: The Chicken or the Egg? Springer Science and Business Media: Berlin/Heidelberg, Germany, 2021; Volume 64, pp. 1917–1926. [Google Scholar]
- Diabetes. Available online: https://www.who.int/news-room/fact-sheets/detail/diabetes (accessed on 14 May 2023).
- IDF Diabetes Atlas|Tenth Edition. Available online: https://diabetesatlas.org/ (accessed on 14 May 2023).
- Gleason, M.M.; Medow, M.S.; Tulenko, T.N. Excess Membrane Cholesterol Alters Calcium Movements, Cytosolic Calcium Levels, and Membrane Fluidity in Arterial Smooth Muscle Cells. Circ. Res. 1991, 69, 216–227. [Google Scholar] [CrossRef]
- Hatziri, A.; Kalogeropoulou, C.; Xepapadaki, E.; Birli, E.; Karavia, E.A.; Papakosta, E.; Filou, S.; Constantinou, C.; Kypreos, K.E. Site-Specific Effects of Apolipoprotein E Expression on Diet-Induced Obesity and White Adipose Tissue Metabolic Activation. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Kypreos, K.E.; Karavia, E.A.; Constantinou, C.; Hatziri, A.; Kalogeropoulou, C.; Xepapadaki, E.; Zvintzou, E. Apolipoprotein E in Diet-Induced Obesity: A Paradigm Shift from Conventional Perception. J. Biomed. Res. 2018, 32, 183–190. [Google Scholar]
- McCullough, A.; Previs, S.F.; Dasarathy, J.; Lee, K.; Osme, A.; Kim, C.; Ilchenko, S.; Lorkowski, S.W.; Smith, J.D.; Dasarathy, S.; et al. HDL Flux Is Higher in Patients with Nonalcoholic Fatty Liver Disease. Am. J. Physiol. Metab. 2019, 317, E852–E862. [Google Scholar] [CrossRef]
- Kantartzis, K.; Rittig, K.; Cegan, A.; Machann, J.; Schick, F.; Balletshofer, B.; Fritsche, A.; Schleicher, E.; Häring, H.-U.; Stefan, N. Fatty Liver Is Independently Associated With Alterations in Circulating HDL2 and HDL3 Subfractions. Diabetes Care 2008, 31, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.; Watanabe, T.; Sakaue, T.; Lewis, G.F. Mechanisms of HDL Lowering in Insulin Resistant, Hypertriglyceridemic States: The Combined Effect of HDL Triglyceride Enrichment and Elevated Hepatic Lipase Activity. Clin. Biochem. 2003, 36, 421–429. [Google Scholar] [CrossRef]
- Fadaei, R.; Poustchi, H.; Meshkani, R.; Moradi, N.; Golmohammadi, T.; Merat, S. Impaired HDL Cholesterol Efflux Capacity in Patients with Non-Alcoholic Fatty Liver Disease Is Associated with Subclinical Atherosclerosis. Sci. Rep. 2018, 8, 11691. [Google Scholar] [CrossRef]
- van den Berg, E.H.; Gruppen, E.G.; Ebtehaj, S.; Bakker, S.J.L.; Tietge, U.J.F.; Dullaart, R.P.F. Cholesterol Efflux Capacity Is Impaired in Subjects with an Elevated Fatty Liver Index, a Proxy of Non-Alcoholic Fatty Liver Disease. Atherosclerosis 2018, 277, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Tushuizen, M.E.; Nieuwland, R.; Scheffer, P.G.; Sturk, A.; Heine, R.J.; Diamant, M. Two Consecutive High-Fat Meals Affect Endothelial-Dependent Vasodilation, Oxidative Stress and Cellular Microparticles in Healthy Men. J. Thromb. Haemost. 2006, 4, 1003–1010. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Lundman, P.; Harmer, J.A.; Cutri, B.; Griffiths, K.A.; Rye, K.-A.; Barter, P.J.; Celermajer, D.S. Consumption of Saturated Fat Impairs the Anti-Inflammatory Properties of High-Density Lipoproteins and Endothelial Function. J. Am. Coll. Cardiol. 2006, 48, 715–720. [Google Scholar] [CrossRef]
- Patel, S.; Puranik, R.; Nakhla, S.; Lundman, P.; Stocker, R.; Wang, X.S.; Lambert, G.; Rye, K.-A.; Barter, P.J.; Nicholls, S.J.; et al. Acute Hypertriglyceridaemia in Humans Increases the Triglyceride Content and Decreases the Anti-Inflammatory Capacity of High Density Lipoproteins. Atherosclerosis 2009, 204, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Karavia, E.A.; Papachristou, D.J.; Liopeta, K.; Triantaphyllidou, I.E.; Dimitrakopoulos, O.; Kypreos, K.E. Apolipoprotein A-I Modulates Processes Associated with Diet-Induced Nonalcoholic Fatty Liver Disease in Mice. Mol. Med. 2012, 18, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Karavia, E.A.; Papachristou, D.J.; Kotsikogianni, I.; Triantafyllidou, I.-E.; Kypreos, K.E. Lecithin/Cholesterol Acyltransferase Modulates Diet-Induced Hepatic Deposition of Triglycerides in Mice. J. Nutr. Biochem. 2013, 24, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, S.; Atzori, L.; Mereu, R.; Satta, G.; Macis, M.D.; Congia, M.; Tedde, A.; Desogus, A.; Muntoni, S. Serum Lipoproteins and Cancer. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 218–225. [Google Scholar] [CrossRef]
- Choi, Y.J.; Lee, D.H.; Han, K.D.; Shin, C.M.; Kim, N. Abdominal Obesity, Glucose Intolerance and Decreased High-Density Lipoprotein Cholesterol as Components of the Metabolic Syndrome Are Associated with the Development of Colorectal Cancer. Eur. J. Epidemiol. 2018, 33, 1077–1085. [Google Scholar] [CrossRef]
- Mihajlovic, M.; Gojkovic, T.; Vladimirov, S.; Miljkovic, M.; Stefanovic, A.; Vekic, J.; Zeljkovic, D.; Trifunovic, B.; Kotur-Stevuljevic, J.; Spasojevic-Kalimanovska, V.; et al. Changes in Lecithin: Cholesterol Acyltransferase, Cholesteryl Ester Transfer Protein and Paraoxonase-1 Activities in Patients with Colorectal Cancer. Clin. Biochem. 2019, 63, 32–38. [Google Scholar] [CrossRef]
- Stevanovic, M.; Vekic, J.; Bogavac-Stanojevic, N.; Janac, J.; Stjepanovic, Z.; Zeljkovic, D.; Trifunovic, B.; Spasojevic-Kalimanovska, V.; Zeljkovic, A. Significance of LDL and HDL Subclasses Characterization in the Assessment of Risk for Colorectal Cancer Development. Biochem. Med. 2018, 28, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Şahin, F.; Aslan, A.F. Relationship between Inflammatory and Biological Markers and Lung Cancer. J. Clin. Med. 2018, 7, 160. [Google Scholar] [CrossRef] [PubMed]
- Zabłocka-Słowińska, K.; Płaczkowska, S.; Skórska, K.; Prescha, A.; Pawełczyk, K.; Porębska, I.; Kosacka, M.; Grajeta, H. Oxidative Stress in Lung Cancer Patients Is Associated with Altered Serum Markers of Lipid Metabolism. PLoS ONE 2019, 14, e0215246. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Z.L.; Wu, Y.T.; Wu, H.; Dai, W.; Arshad, B.; Xu, Z.; Li, H.; Wu, K.N.; Kong, L.Q. Status of Lipid and Lipoprotein in Female Breast Cancer Patients at Initial Diagnosis and during Chemotherapy. Lipids Health Dis. 2018, 17, 91. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhang, K.; Gong, F.; Jin, H. A Study on Changes and Clinical Significance of Blood Glucose, Blood Lipid and Inflammation in Patients with Ovarian Cancer. JBUON 2019, 24, 2325–2329. [Google Scholar]
- Li, D.; Zhou, L.; Ma, C.; Chen, W.; Zhang, Y.; Yu, S.; Wang, D.; Zou, Y.; Wu, J.; Qiu, L. Comparative Analysis of the Serum Proteome Profiles of Thyroid Cancer: An Initial Focus on the Lipid Profile. Oncol. Lett. 2019, 18, 3349–3357. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.Y.; Park, B.J.; Nam, J.H.; Kook, M.C. Effect of Helicobacter Pylori Eradication and High-Density Lipoprotein on the Risk of de Novo Gastric Cancer Development. Gastrointest. Endosc. 2019, 90, 448–456.e1. [Google Scholar] [CrossRef]
- Hatziri, A.; Lazaris, V.; Symeonidis, A.; Kypreos, K.E. Lipid and Lipoprotein Profile of Patients with Multiple Myeloma before and after First-Line Treatment. Atherosclerosis 2021, 331, e125. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, X.Q.; Lin, H.C.; Wang, D.S.; Wang, Z.Q.; Shao, Q.; Wang, F.H.; Yan, S.M.; Liang, J.Y.; Zeng, Z.L.; et al. Correlation between Immune Signature and High-Density Lipoprotein Cholesterol Level in Stage II/III Colorectal Cancer. Cancer Med. 2019, 8, 1209–1217. [Google Scholar] [CrossRef]
- Jafri, H.; Alsheikh-Ali, A.A.; Karas, R.H. Baseline and On-Treatment High-Density Lipoprotein Cholesterol and the Risk of Cancer in Randomized Controlled Trials of Lipid-Altering Therapy. J. Am. Coll. Cardiol. 2010, 55, 2846–2854. [Google Scholar] [CrossRef]
- Long, J.; Zhang, C.-J.; Zhu, N.; Du, K.; Yin, Y.-F.; Tan, X.; Liao, D.-F.; Qin, L. Lipid Metabolism and Carcinogenesis, Cancer Development. Am. J. Cancer Res. 2018, 8, 778. [Google Scholar]
- Georgila, K.; Vyrla, D.; Drakos, E. Apolipoprotein A-I (ApoA-I), Immunity, Inflammation and Cancer. Cancers 2019, 11, 1097. [Google Scholar] [CrossRef]
- Kalaivani, V.; Jaleel, A. Apolipoprotein(a), an Enigmatic Anti-Angiogenic Glycoprotein in Human Plasma: A Curse or Cure? Pharmacol. Res. 2020, 158, 104858. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Dannenberg, A.J. The Obese Adipose Tissue Microenvironment in Cancer Development and Progression. Nat. Rev. Endocrinol. 2019, 15, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Kuliszkiewicz-Janus, M.; Małecki, R.; Mohamed, A. Lipid Changes Occuring in the Course of Hematological Cancers. Cell. Mol. Biol. Lett. 2008, 13, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Yavasoglu, I.; Tombuloglu, M.; Kadikoylu, G.; Donmez, A.; Cagırgan, S.; Bolaman, Z. Cholesterol Levels in Patients with Multiple Myeloma. Ann. Hematol. 2008, 87, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Li, J.; Fu, H.; Liu, X.; Liu, P. Identification of High Serum Apolipoprotein A1 as a Favorable Prognostic Indicator in Patients with Multiple Myeloma. J. Cancer 2019, 10, 4852–4859. [Google Scholar] [CrossRef]
- Kyrgiou, M.; Kalliala, I.; Markozannes, G.; Gunter, M.J.; Paraskevaidis, E.; Gabra, H.; Martin-Hirsch, P.; Tsilidis, K.K. Adiposity and Cancer at Major Anatomical Sites: Umbrella Review of the Literature. BMJ 2017, 356, j477. [Google Scholar] [CrossRef]
- Steele, C.B.; Thomas, C.C.; Henley, S.J.; Massetti, G.M.; Galuska, D.A.; Agurs-Collins, T.; Puckett, M.; Richardson, L.C. Vital Signs: Trends in Incidence of Cancers Associated with Overweight and Obesity—United States, 2005–2014. MMWR Morb. Mortal. Wkly. Rep. 2017, 66, 1052–1058. [Google Scholar] [CrossRef]
- Kalogeropoulou, C.; Hatziri, A.; Xepapadaki, E.; Savvoulidou, O.; Karavia, E.A.; Zvintzou, E.; Constantinou, C.; Kypreos, K.E. Isoform and Tissue Dependent Impact of Apolipoprotein E on Adipose Tissue Metabolic Activation: The Role of Apolipoprotein A1. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158551. [Google Scholar] [CrossRef]
- Kypreos, K.E.; Bitzur, R.; Karavia, E.A.; Xepapadaki, E.; Panayiotakopoulos, G.; Constantinou, C. Pharmacological Management of Dyslipidemia in Atherosclerosis: Limitations, Challenges, and New Therapeutic Opportunities. Angiology 2019, 70, 197–209. [Google Scholar] [CrossRef]
- Bowman, L.; Hopewell, J.; Chen, F.; Wallendszus, K.; Stevens, W.; Collins, R.; Wiviott, S.; Cannon, C.; Braunwald, E.; Sammons, E.; et al. Effects of Anacetrapib in Patients with Atherosclerotic Vascular Disease. N. Engl. J. Med. 2017, 377, 1217–1227. [Google Scholar] [CrossRef]
- Hovingh, G.K.; Kastelein, J.J.P.; van Deventer, S.J.H.; Round, P.; Ford, J.; Saleheen, D.; Rader, D.J.; Brewer, H.B.; Barter, P.J. Cholesterol Ester Transfer Protein Inhibition by TA-8995 in Patients with Mild Dyslipidaemia (TULIP): A Randomised, Double-Blind, Placebo-Controlled Phase 2 Trial. Lancet 2015, 386, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Gordts, P.L.S.M.; Nora, C.; Yeang, C.; Witztum, J.L. Statin Therapy Increases Lipoprotein(a) Levels. Eur. Heart J. 2020, 41, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, D.; Huang, L.H.; Huang, H. Roles of Reconstituted High-Density Lipoprotein Nanoparticles in Cardiovascular Disease: A New Paradigm for Drug Discovery. Int. J. Mol. Sci. 2020, 21, 739. [Google Scholar] [CrossRef]
- Franceschini, G.; Sirtori, C.R.; Capurso, A. A-I(Milano) Apoprotein. Decreased High Density Lipoprotein Cholesterol Levels with Significant Lipoprotein Modifications and without Clinical Atherosclerosis in an Italian Family. J. Clin. Investig. 1980, 66, 892–900. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Puri, R.; Ballantyne, C.M.; Jukema, J.W.; Kastelein, J.J.P.; Koenig, W.; Wright, R.S.; Kallend, D.; Wijngaard, P.; Borgman, M.; et al. Effect of Infusion of High-Density Lipoprotein Mimetic Containing Recombinant Apolipoprotein A-I Milano on Coronary Disease in Patients with an Acute Coronary Syndrome in the MILANO-PILOT Trial: A Randomized Clinical Trial. JAMA Cardiol. 2018, 3, 806–814. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, W.; Stonik, J.A.; Murphy, A.; Demosky, S.J.; Sethi, A.A.; Moore, X.L.; Chin-Dusting, J.; Remaley, A.T.; Sviridov, D. Structure/Function Relationships of Apolipoprotein A-I Mimetic Peptides: Implications for Antiatherogenic Activities of High-Density Lipoprotein. Circ. Res. 2010, 107, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Van Lenten, B.J.; Wagner, A.C.; Jung, C.L.; Ruchala, P.; Waring, A.J.; Lehrer, R.I.; Watson, A.D.; Hama, S.; Navab, M.; Anantharamaiah, G.M.; et al. Anti-Inflammatory ApoA-I-Mimetic Peptides Bind Oxidized Lipids with Much Higher Affinity than Human ApoA-I. J. Lipid Res. 2008, 49, 2302–2311. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, R.L.; Movva, R.; Bloedon, L.A.T.; Duffy, D.; Norris, R.B.; Navab, M.; Fogelman, A.M.; Rader, D.J. Oral Apolipoprotein A-I Mimetic D-4F Lowers HDL-Inflammatory Index in High-Risk Patients: A First-in-Human Multiple-Dose, Randomized Controlled Trial. Clin. Transl. Sci. 2017, 10, 455–469. [Google Scholar] [CrossRef]
- Karjalainen, M.K.; Holmes, M.V.; Wang, Q.; Anufrieva, O.; Kähönen, M.; Lehtimäki, T.; Havulinna, A.S.; Kristiansson, K.; Salomaa, V.; Perola, M.; et al. Apolipoprotein A-I Concentrations and Risk of Coronary Artery Disease: A Mendelian Randomization Study. Atherosclerosis 2020, 299, 56–63. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Andrews, J.; Kastelein, J.J.P.; Merkely, B.; Nissen, S.E.; Ray, K.K.; Schwartz, G.G.; Worthley, S.G.; Keyserling, C.; Dasseux, J.L.; et al. Effect of Serial Infusions of CER-001, a Pre-β High-Density Lipoprotein Mimetic, on Coronary Atherosclerosis in Patients Following Acute Coronary Syndromes in the CER-001 Atherosclerosis Regression Acute Coronary Syndrome Trial: A Randomized Clinical Tria. JAMA Cardiol. 2018, 3, 815–822. [Google Scholar] [CrossRef]
- Shaw, J.A.; Bobik, A.; Murphy, A.; Kanellakis, P.; Blombery, P.; Mukhamedova, N.; Woollard, K.; Lyon, S.; Sviridov, D.; Dart, A.M. Infusion of Reconstituted High-Density Lipoprotein Leads to Acute Changes in Human Atherosclerotic Plaque. Circ. Res. 2008, 103, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.C.; Grégoire, J.; L’Allier, P.L.; Ibrahim, R.; Lespérance, J.; Heinonen, T.M.; Kouz, S.; Berry, C.; Basser, R.; Lavoie, M.A.; et al. Effects of Reconstituted High-Density Lipoprotein Infusions on Coronary Atherosclerosis: A Randomized Controlled Trial. J. Am. Med. Assoc. 2007, 297, 1675–1682. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.M.; Korjian, S.; Tricoci, P.; Daaboul, Y.; Yee, M.; Jain, P.; Alexander, J.H.; Steg, P.G.; Lincoff, A.M.; Kastelein, J.J.P.; et al. Safety and Tolerability of CSL112, a Reconstituted, Infusible, Plasma-Derived Apolipoprotein A-I, after Acute Myocardial Infarction: The AEGIS-I Trial (ApoA-I Event Reducing in Ischemic Syndromes I). Circulation 2016, 134, 1918–1930. [Google Scholar] [CrossRef] [PubMed]
- The AIM-HIGH Investigators. Niacin in Patients with Low HDL Cholesterol Levels Receiving Intensive Statin Therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef]
- Zeman, M.; Vecka, M.; Perlík, F.; Hromádka, R.; Staňková, B.; Tvrzická, E.; Žák, A. Niacin in the Treatment of Hyperlipidemias in Light of New Clinical Trials: Has Niacin Lost Its Place? Med. Sci. Monit. 2015, 21, 2156–2162. [Google Scholar] [CrossRef]
- Adli, M. The CRISPR Tool Kit for Genome Editing and Beyond. Nat. Commun. 2018, 9, 1911. [Google Scholar] [CrossRef]
- Doudna, J.A. The Promise and Challenge of Therapeutic Genome Editing. Nature 2020, 578, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Saha, K.; Sontheimer, E.J.; Brooks, P.J.; Dwinell, M.R.; Gersbach, C.A.; Liu, D.R.; Murray, S.A.; Tsai, S.Q.; Wilson, R.C.; Anderson, D.G.; et al. The NIH Somatic Cell Genome Editing Program. Nature 2021, 592, 195–204. [Google Scholar] [CrossRef]
- Pavlovic, K.; Tristán-Manzano, M.; Maldonado-Pérez, N.; Cortijo-Gutierrez, M.; Sánchez-Hernández, S.; Justicia-Lirio, P.; Carmona, M.D.; Herrera, C.; Martin, F.; Benabdellah, K. Using Gene Editing Approaches to Fine-Tune the Immune System. Front. Immunol. 2020, 11, 570672. [Google Scholar] [CrossRef]
- Yin, S.; Zhang, M.; Liu, Y.; Sun, X.; Guan, Y.; Chen, X.; Yang, L.; Huo, Y.; Yang, J.; Zhang, X.; et al. Engineering of Efficiency-Enhanced Cas9 and Base Editors with Improved Gene Therapy Efficacies. Mol. Ther. 2023, 31, 744–759. [Google Scholar] [CrossRef]
- Pecori, R.; Di Giorgio, S.; Paulo Lorenzo, J.; Nina Papavasiliou, F. Functions and Consequences of AID/APOBEC-Mediated DNA and RNA Deamination. Nat. Rev. Genet. 2022, 23, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Bauer, D.E.; Chiarle, R. Assessing and Advancing the Safety of CRISPR-Cas Tools: From DNA to RNA Editing. Nat. Commun. 2023, 14, 212. [Google Scholar] [CrossRef]
- Palaz, F.; Kalkan, A.K.; Can, Ö.; Demir, A.N.; Tozluyurt, A.; Özcan, A.; Ozsoz, M. CRISPR-Cas13 System as a Promising and Versatile Tool for Cancer Diagnosis, Therapy, and Research. ACS Synth. Biol. 2021, 10, 1245–1267. [Google Scholar] [CrossRef] [PubMed]
- Kordyś, M.; Sen, R.; Warkocki, Z. Applications of the Versatile CRISPR-Cas13 RNA Targeting System. WIREs RNA 2022, 13, e1694. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Tian, P.; Tan, T. CRISPR-Cas13 Technology Portfolio and Alliance with Other Genetic Tools. Biotechnol. Adv. 2022, 61, 108047. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Cohen, J.C.; Hobbs, H.H. Molecular Biology of PCSK9: Its Role in LDL Metabolism. Trends Biochem. Sci. 2007, 32, 71. [Google Scholar] [CrossRef]
- Palumbo, M.; Giammanco, A.; Purrello, F.; Pavanello, C.; Mombelli, G.; Di Pino, A.; Piro, S.; Cefalù, A.B.; Calabresi, L.; Averna, M.; et al. Effects of PCSK9 Inhibitors on HDL Cholesterol Efflux and Serum Cholesterol Loading Capacity in Familial Hypercholesterolemia Subjects: A Multi-Lipid-Center Real-World Evaluation. Front. Mol. Biosci. 2022, 9, 925587. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Su, J.; Liu, Y.; Jin, X.; Zhong, X.; Mo, L.; Wang, Q.; Deng, H.; Yang, Y. In Vivo PCSK9 Gene Editing Using an All-in-One Self-Cleavage AAV-CRISPR System. Mol. Ther. Methods Clin. Dev. 2021, 20, 652–659. [Google Scholar] [CrossRef]
- Lee, R.G.; Mazzola, A.M.; Braun, M.C.; Platt, C.; Vafai, S.B.; Kathiresan, S.; Rohde, E.; Bellinger, A.M.; Khera, A.V. Efficacy and Safety of an Investigational Single-Course CRISPR Base-Editing Therapy Targeting PCSK9 in Nonhuman Primate and Mouse Models. Circulation 2023, 147, 242–253. [Google Scholar] [CrossRef]
- Basu, A.; Bebu, I.; Jenkins, A.J.; Stoner, J.A.; Zhang, Y.; Klein, R.L.; Lopes-Virella, M.F.; Garvey, W.T.; Budoff, M.J.; Alaupovic, P.; et al. Serum Apolipoproteins and Apolipoprotein-Defined Lipoprotein Subclasses: A Hypothesis-Generating Prospective Study of Cardiovascular Events in T1D. J. Lipid Res. 2019, 60, 1432–1439. [Google Scholar] [CrossRef]
- Kanter, J.E.; Shao, B.; Kramer, F.; Barnhart, S.; Shimizu-Albergine, M.; Vaisar, T.; Graham, M.J.; Crooke, R.M.; Manuel, C.R.; Haeusler, R.A.; et al. Increased Apolipoprotein C3 Drives Cardiovascular Risk in Type 1 Diabetes. J. Clin. Investig. 2019, 129, 4165–4179. [Google Scholar] [CrossRef]
- Jansson Sigfrids, F.; Stechemesser, L.; Dahlström, E.H.; Forsblom, C.M.; Harjutsalo, V.; Weitgasser, R.; Taskinen, M.; Groop, P. Apolipoprotein C-III Predicts Cardiovascular Events and Mortality in Individuals with Type 1 Diabetes and Albuminuria. J. Intern. Med. 2022, 291, 338–349. [Google Scholar] [CrossRef]
- Calcaterra, I.; Lupoli, R.; Di Minno, A.; Di Minno, M.N.D. Volanesorsen to Treat Severe Hypertriglyceridaemia: A Pooled Analysis of Randomized Controlled Trials. Eur. J. Clin. Investig. 2022, 52, e13841. [Google Scholar] [CrossRef]
- Guo, M.; Xu, Y.; Dong, Z.; Zhou, Z.; Cong, N.; Gao, M.; Huang, W.; Wang, Y.; Liu, G.; Xian, X. Inactivation of ApoC3 by CRISPR/Cas9 Protects Against Atherosclerosis in Hamsters. Circ. Res. 2020, 127, 1456–1458. [Google Scholar] [CrossRef] [PubMed]
- Zha, Y.; Lu, Y.; Zhang, T.; Yan, K.; Zhuang, W.; Liang, J.; Cheng, Y.; Wang, Y. CRISPR/Cas9-Mediated Knockout of APOC3 Stabilizes Plasma Lipids and Inhibits Atherosclerosis in Rabbits. Lipids Health Dis. 2021, 20, 180. [Google Scholar] [CrossRef]
- Luo, F.; Das, A.; Khetarpal, S.A.; Fang, Z.; Zelniker, T.A.; Rosenson, R.S.; Qamar, A. ANGPTL3 Inhibition, Dyslipidemia, and Cardiovascular Diseases. Trends Cardiovasc. Med. 2023; Epub ahead of print. [Google Scholar] [CrossRef]
- Mohamed, F.; Mansfield, B.S.; Raal, F.J. ANGPTL3 as a Drug Target in Hyperlipidemia and Atherosclerosis. Curr. Atheroscler. Rep. 2022, 24, 959. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Glass, Z.; Chen, J.; Haas, M.; Jin, X.; Zhao, X.; Rui, X.; Ye, Z.; Li, Y.; Zhang, F.; et al. Lipid Nanoparticle-Mediated Codelivery of Cas9 MRNA and Single-Guide RNA Achieves Liver-Specific in Vivo Genome Editing of Angptl3. Proc. Natl. Acad. Sci. USA 2021, 118, e2020401118. [Google Scholar] [CrossRef] [PubMed]
- Raguram, A.; Banskota, S.; Liu, D.R. Therapeutic In Vivo Delivery of Gene Editing Agents. Cell 2022, 185, 2806–2827. [Google Scholar] [CrossRef]
- Geller, A.S.; Polisecki, E.Y.; Diffenderfer, M.R.; Asztalos, B.F.; Karathanasis, S.K.; Hegele, R.A.; Schaefer, E.J. Genetic and Secondary Causes of Severe HDL Deficiency and Cardiovascular Disease. J. Lipid Res. 2018, 59, 2421–2435. [Google Scholar] [CrossRef]
- Iacob, A.O.; Choudhury, R.P. Targeting HDL-Cholesterol to Reduce Residual Cardiovascular Risk. Curr. Opin. Lipidol. 2012, 23, 172–174. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zvintzou, E.; Xepapadaki, E.; Skroubis, G.; Mparnia, V.; Giannatou, K.; Benabdellah, K.; Kypreos, K.E. High-Density Lipoprotein in Metabolic Disorders and Beyond: An Exciting New World Full of Challenges and Opportunities. Pharmaceuticals 2023, 16, 855. https://doi.org/10.3390/ph16060855
Zvintzou E, Xepapadaki E, Skroubis G, Mparnia V, Giannatou K, Benabdellah K, Kypreos KE. High-Density Lipoprotein in Metabolic Disorders and Beyond: An Exciting New World Full of Challenges and Opportunities. Pharmaceuticals. 2023; 16(6):855. https://doi.org/10.3390/ph16060855
Chicago/Turabian StyleZvintzou, Evangelia, Eva Xepapadaki, George Skroubis, Victoria Mparnia, Katerina Giannatou, Karim Benabdellah, and Kyriakos E. Kypreos. 2023. "High-Density Lipoprotein in Metabolic Disorders and Beyond: An Exciting New World Full of Challenges and Opportunities" Pharmaceuticals 16, no. 6: 855. https://doi.org/10.3390/ph16060855
APA StyleZvintzou, E., Xepapadaki, E., Skroubis, G., Mparnia, V., Giannatou, K., Benabdellah, K., & Kypreos, K. E. (2023). High-Density Lipoprotein in Metabolic Disorders and Beyond: An Exciting New World Full of Challenges and Opportunities. Pharmaceuticals, 16(6), 855. https://doi.org/10.3390/ph16060855