

CYP2C19 Contributes to THP-1-Cell-Derived M2 Macrophage Polarization by Producing 11,12- and 14,15-Epoxyeicosatrienoic Acid, Agonists of the PPARγ Receptor

 ,
,  ,
, 
Abstract
1. Introduction
2. Results
2.1. Cell Differentiation
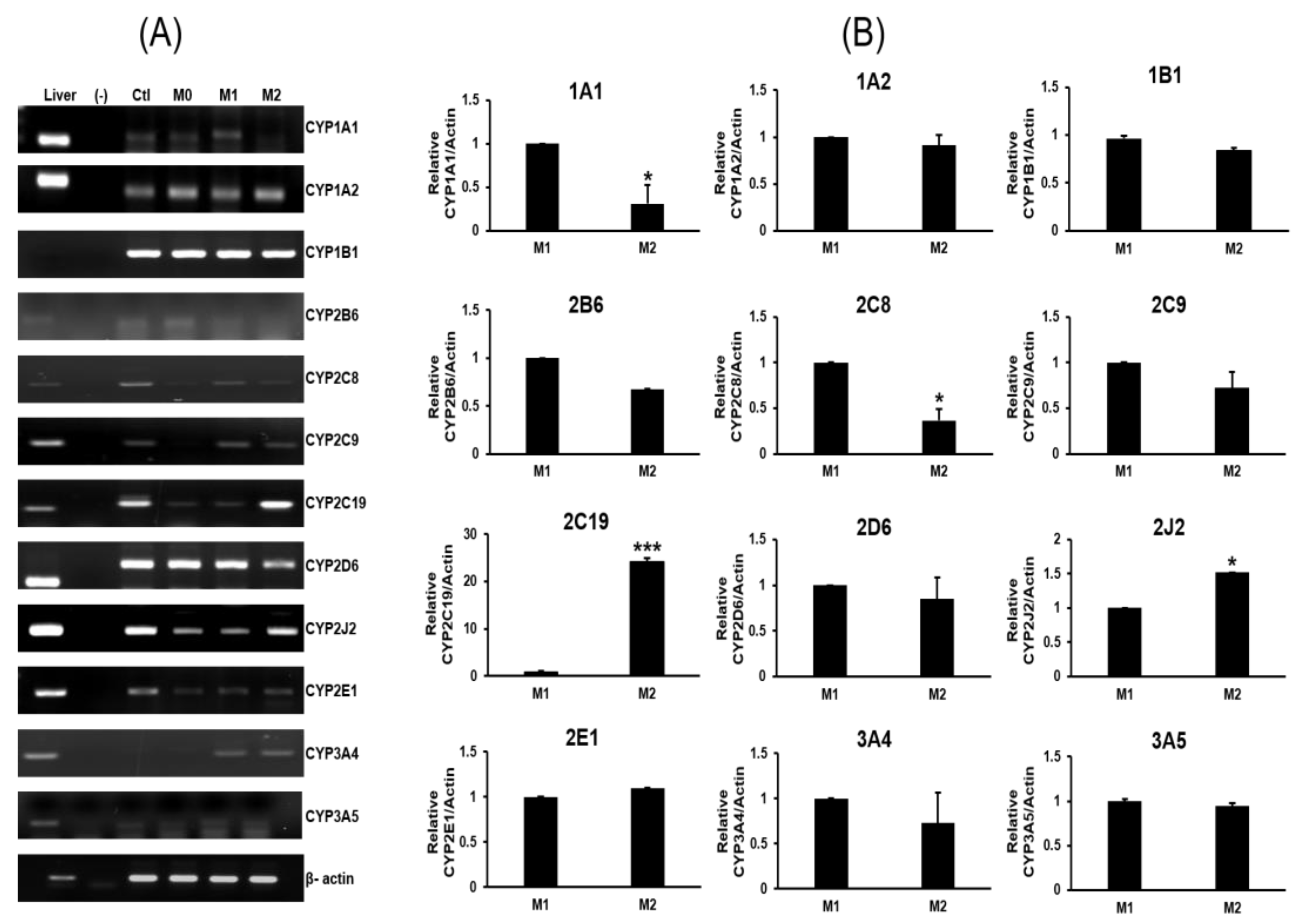
2.2. Expression Profiles of P450s
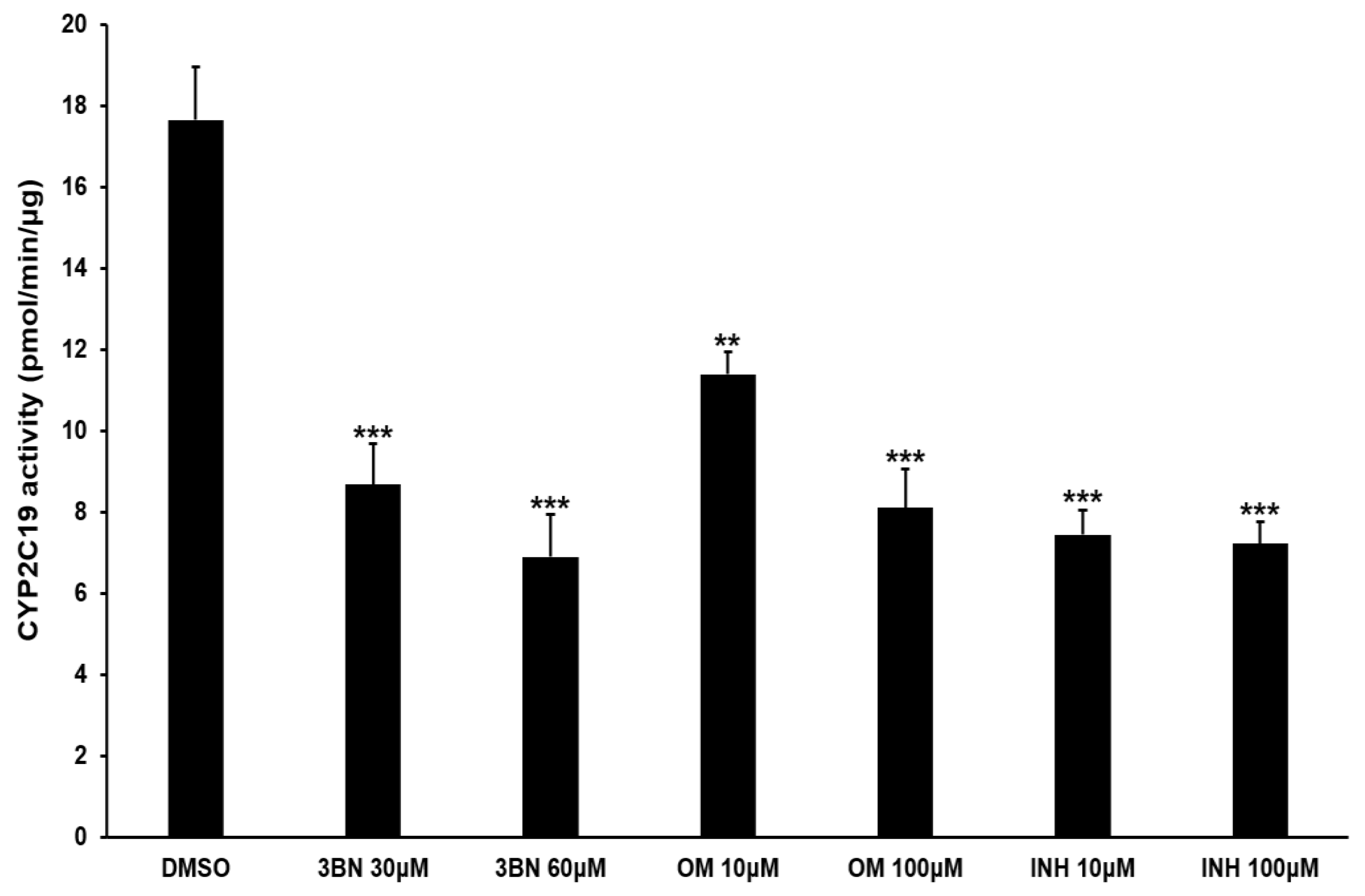
2.3. Analysis of CYP2C19 Acvitiy and Its Metabolites
2.4. PPAR Gamma Activation by CYP2C19 Metabolites and Their Relationship to M2 Marker Gene Expression
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. M1 and M2 Cell Culture and Cellular Differentiation
4.3. RNA Extraction and Reverse Transcription Polymerase Chain Reaction (RT-PCR)
4.4. Protein Extraction and Immunoblot Assay
4.5. CYP2C19 Enzyme Activity Assay
4.6. EET Assays
4.7. Binding Analysis of 11,12-EET and 14,15-EET to PPARγ
4.8. Cell Viability Assay
4.9. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Locati, M. Macrophage polarization comes of age. Immunity 2005, 23, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Russell, D.W. Clinical importance of the cytochromes P450. Lancet 2002, 360, 1155–1162. [Google Scholar] [CrossRef]
- Hakkola, J.; Bernasconi, C.; Coecke, S.; Richert, L.; Andersson, T.B.; Pelkonen, O. Cytochrome P450 Induction and Xeno-Sensing Receptors Pregnane X Receptor, Constitutive Androstane Receptor, Aryl Hydrocarbon Receptor and Peroxisome Proliferator-Activated Receptor α at the Crossroads of Toxicokinetics and Toxicodynamics. Basic. Clin. Pharmacol. Toxicol. 2018, 123 (Suppl. 5), 42–50. [Google Scholar] [CrossRef] [PubMed]
- Samokhvalov, V.; Vriend, J.; Jamieson, K.L.; Akhnokh, M.K.; Manne, R.; Falck, J.R.; Seubert, J.M. PPARγ signaling is required for mediating EETs protective effects in neonatal cardiomyocytes exposed to LPS. Front. Pharm. 2014, 5, 242. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Zhang, X.; Chen, C.; Wen, Z.; Hoopes, S.L.; Zeldin, D.C.; Wang, D.W. Cardiomyocyte-specific expression of CYP2J2 prevents development of cardiac remodelling induced by angiotensin II. Cardiovasc. Res. 2015, 105, 304–317. [Google Scholar] [CrossRef]
- Rocic, P.; Schwartzman, M.L. 20-HETE in the regulation of vascular and cardiac function. Pharm. 2018, 192, 74–87. [Google Scholar] [CrossRef]
- He, L.; Jhong, J.H.; Chen, Q.; Huang, K.Y.; Strittmatter, K.; Kreuzer, J.; DeRan, M.; Wu, X.; Lee, T.Y.; Slavov, N.; et al. Global characterization of macrophage polarization mechanisms and identification of M2-type polarization inhibitors. Cell. Rep. 2021, 37, 109955. [Google Scholar] [CrossRef]
- Liu, H.; Wang, M.; Jin, Z.; Sun, D.; Zhu, T.; Liu, X.; Tan, X.; Shi, G. FNDC5 induces M2 macrophage polarization and promotes hepatocellular carcinoma cell growth by affecting the PPARγ/NF-κB/NLRP3 pathway. Biochem. Biophys. Res. Commun. 2021, 582, 77–85. [Google Scholar] [CrossRef]
- Huang, J.T.; Welch, J.S.; Ricote, M.; Binder, C.J.; Willson, T.M.; Kelly, C.; Witztum, J.L.; Funk, C.D.; Conrad, D.; Glass, C.K. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature 1999, 400, 378–382. [Google Scholar] [CrossRef]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef]
- Jarrar, Y.B.; Lee, S.J. Molecular Functionality of Cytochrome P450 4 (CYP4) Genetic Polymorphisms and Their Clinical Implications. Int. J. Mol. Sci. 2019, 20, 4274. [Google Scholar] [CrossRef]
- Hokimoto, S.; Tabata, N.; Akasaka, T.; Arima, Y.; Kaikita, K.; Morita, K.; Kumagae, N.; Oniki, K.; Nakagawa, K.; Ogawa, H. Gender differences in impact of CYP2C19 polymorphism on development of coronary artery disease. J. Cardiovasc. Pharm. 2015, 65, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Peng, R.; Guo, Y.; Shen, L.; Zhao, S.; Xu, D. The role of 14,15-dihydroxyeicosatrienoic acid levels in inflammation and its relationship to lipoproteins. Lipids Health Dis. 2013, 12, 151. [Google Scholar] [CrossRef]
- Powell, W.S.; Rokach, J. Biosynthesis, biological effects, and receptors of hydroxyeicosatetraenoic acids (HETEs) and oxoeicosatetraenoic acids (oxo-ETEs) derived from arachidonic acid. Biochim. Biophys. Acta 2015, 1851, 340–355. [Google Scholar] [CrossRef]
- Wray, J.; Bishop-Bailey, D. Epoxygenases and peroxisome proliferator-activated receptors in mammalian vascular biology. Exp. Physiol. 2008, 93, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Razzak, Z.; Loyer, P.; Fautrel, A.; Gautier, J.C.; Corcos, L.; Turlin, B.; Beaune, P.; Guillouzo, A. Cytokines down-regulate expression of major cytochrome P-450 enzymes in adult human hepatocytes in primary culture. Mol. Pharm. 1993, 44, 707–715. [Google Scholar]
- Barker, C.W.; Fagan, J.B.; Pasco, D.S. Interleukin-1 beta suppresses the induction of P4501A1 and P4501A2 mRNAs in isolated hepatocytes. J. Biol. Chem. 1992, 267, 8050–8055. [Google Scholar] [CrossRef] [PubMed]
- Muntané-Relat, J.; Ourlin, J.C.; Domergue, J.; Maurel, P. Differential effects of cytokines on the inducible expression of CYP1A1, CYP1A2, and CYP3A4 in human hepatocytes in primary culture. Hepatology 1995, 22, 1143–1153. [Google Scholar] [CrossRef]
- Nakamura, M.; Imaoka, S.; Amano, F.; Funae, Y. P450 isoforms in a murine macrophage cell line, RAW264.7, and changes in the levels of P450 isoforms by treatment of cells with lipopolysaccharide and interferon-gamma. Biochim. Biophys. Acta 1998, 1385, 101–106. [Google Scholar] [CrossRef]
- Isali, I.; McClellan, P.; Shankar, E.; Gupta, S.; Jain, M.; Anderson, J.M.; Hijaz, A.; Akkus, O. Genipin guides and sustains the polarization of macrophages to the pro-regenerative M2 subtype via activation of the pSTAT6-PPAR-gamma pathway. Acta Biomater. 2021, 131, 198–210. [Google Scholar] [CrossRef]
- Batista-Gonzalez, A.; Vidal, R.; Criollo, A.; Carreño, L.J. New Insights on the Role of Lipid Metabolism in the Metabolic Reprogramming of Macrophages. Front. Immunol. 2019, 10, 2993. [Google Scholar] [CrossRef]
- Wu, H.M.; Ni, X.X.; Xu, Q.Y.; Wang, Q.; Li, X.Y.; Hua, J. Regulation of lipid-induced macrophage polarization through modulating peroxisome proliferator-activated receptor-gamma activity affects hepatic lipid metabolism via a Toll-like receptor 4/NF-κB signaling pathway. J. Gastroenterol. Hepatol. 2020, 35, 1998–2008. [Google Scholar] [CrossRef] [PubMed]
- El-Sherbeni, A.A.; El-Kadi, A.O. Repurposing Resveratrol and Fluconazole To Modulate Human Cytochrome P450-Mediated Arachidonic Acid Metabolism. Mol. Pharm. 2016, 13, 1278–1288. [Google Scholar] [CrossRef] [PubMed]
- Akasaka, T.; Sueta, D.; Arima, Y.; Tabata, N.; Takashio, S.; Izumiya, Y.; Yamamoto, E.; Yamamuro, M.; Tsujita, K.; Kojima, S.; et al. Association of CYP2C19 variants and epoxyeicosatrienoic acids on patients with microvascular angina. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1409–H1415. [Google Scholar] [CrossRef]
- Mayer-Barber, K.D.; Andrade, B.B.; Oland, S.D.; Amaral, E.P.; Barber, D.L.; Gonzales, J.; Derrick, S.C.; Shi, R.; Kumar, N.P.; Wei, W.; et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 2014, 511, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Bafica, A.; Scanga, C.A.; Serhan, C.; Machado, F.; White, S.; Sher, A.; Aliberti, J. Host control of Mycobacterium tuberculosis is regulated by 5-lipoxygenase-dependent lipoxin production. J. Clin. Investig. 2005, 115, 1601–1606. [Google Scholar] [CrossRef]
- Chen, M.; Divangahi, M.; Gan, H.; Shin, D.S.; Hong, S.; Lee, D.M.; Serhan, C.N.; Behar, S.M.; Remold, H.G. Lipid mediators in innate immunity against tuberculosis: Opposing roles of PGE2 and LXA4 in the induction of macrophage death. J. Exp. Med. 2008, 205, 2791–2801. [Google Scholar] [CrossRef]
- Kulkarni, A.; Pineros, A.R.; Walsh, M.A.; Casimiro, I.; Ibrahim, S.; Hernandez-Perez, M.; Orr, K.S.; Glenn, L.; Nadler, J.L.; Morris, M.A.; et al. 12-Lipoxygenase governs the innate immune pathogenesis of islet inflammation and autoimmune diabetes. JCI Insight 2021, 6, e147812. [Google Scholar] [CrossRef]
- Kulkarni, A.; Nadler, J.L.; Mirmira, R.G.; Casimiro, I. Regulation of Tissue Inflammation by 12-Lipoxygenases. Biomolecules 2021, 11, 717. [Google Scholar] [CrossRef] [PubMed]
- Nagy, L.; Szanto, A.; Szatmari, I.; Széles, L. Nuclear hormone receptors enable macrophages and dendritic cells to sense their lipid environment and shape their immune response. Physiol. Rev. 2012, 92, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wang, X.; Li, Y.; Geng, X.; Jia, X.; Zhang, L.; Yang, H. Arachidonic Acid Metabolism Controls Macrophage Alternative Activation Through Regulating Oxidative Phosphorylation in PPARγ Dependent Manner. Front. Immunol. 2021, 12, 618501. [Google Scholar] [CrossRef]
- Mahajan, S.; Dkhar, H.K.; Chandra, V.; Dave, S.; Nanduri, R.; Janmeja, A.K.; Agrewala, J.N.; Gupta, P. Mycobacterium tuberculosis modulates macrophage lipid-sensing nuclear receptors PPARγ and TR4 for survival. J. Immunol. 2012, 188, 5593–5603. [Google Scholar] [CrossRef] [PubMed]
- Gijón, M.A.; Leslie, C.C. Regulation of arachidonic acid release and cytosolic phospholipase A2 activation. J. Leukoc. Biol. 1999, 65, 330–336. [Google Scholar] [CrossRef]
- Rajaram, M.V.; Brooks, M.N.; Morris, J.D.; Torrelles, J.B.; Azad, A.K.; Schlesinger, L.S. Mycobacterium tuberculosis activates human macrophage peroxisome proliferator-activated receptor gamma linking mannose receptor recognition to regulation of immune responses. J. Immunol. 2010, 185, 929–942. [Google Scholar] [CrossRef]
- Kim, C.E.; Park, H.Y.; Won, H.J.; Kim, M.; Kwon, B.; Lee, S.J.; Kim, D.H.; Shin, J.G.; Seo, S.K. Repression of PPARγ reduces the ABCG2-mediated efflux activity of M2 macrophages. Int. J. Biochem. Cell. Biol. 2021, 130, 105895. [Google Scholar] [CrossRef]
- Yi, M.; Shin, J.G.; Lee, S.J. Expression of CYP4V2 in human THP1 macrophages and its transcriptional regulation by peroxisome proliferator-activated receptor gamma. Toxicol. Appl. Pharm. 2017, 330, 100–106. [Google Scholar] [CrossRef]
- McKay, J.A.; Melvin, W.T.; Ah-See, A.K.; Ewen, S.W.; Greenlee, W.F.; Marcus, C.B.; Burke, M.D.; Murray, G.I. Expression of cytochrome P450 CYP1B1 in breast cancer. FEBS Lett. 1995, 374, 270–272. [Google Scholar] [CrossRef]
- Wei, Y.D.; Helleberg, H.; Rannug, U.; Rannug, A. Rapid and transient induction of CYP1A1 gene expression in human cells by the tryptophan photoproduct 6-formylindolo[3,2-b]carbazole. Chem. Biol. Interact. 1998, 110, 39–55. [Google Scholar] [CrossRef]
- Macé, K.; Bowman, E.D.; Vautravers, P.; Shields, P.G.; Harris, C.C.; Pfeifer, A.M. Characterisation of xenobiotic-metabolising enzyme expression in human bronchial mucosa and peripheral lung tissues. Eur. J. Cancer 1998, 34, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Nitto, T.; Inoue, T.; Node, K. Expression of the cytochrome P450 epoxygenase CYP2J2 in human monocytic leukocytes. Life Sci. 2008, 83, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Ozog, M.A.; Bernier, S.M.; Bates, D.C.; Chatterjee, B.; Lo, C.W.; Naus, C.C. The complex of ciliary neurotrophic factor-ciliary neurotrophic factor receptor alpha up-regulates connexin43 and intercellular coupling in astrocytes via the Janus tyrosine kinase/signal transducer and activator of transcription pathway. Mol. Biol. Cell. 2004, 15, 4761–4774. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Liang, X.; Zheng, M.; Zhu, Z.; Zhu, G.; Yang, J.; Chen, Y. Expression of c-kit and Slug correlates with invasion and metastasis of salivary adenoid cystic carcinoma. Oral. Oncol. 2010, 46, 311–316. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Lee, S.J.; Buhler, D.R. Functional properties of a rainbow trout CYP3A27 expressed by recombinant baculovirus in insect cells. Drug. Metab. Dispos. 2002, 30, 1406–1412. [Google Scholar] [CrossRef]
- Chistyakov, D.V.; Astakhova, A.A.; Goriainov, S.V.; Sergeeva, M.G. Comparison of PPAR Ligands as Modulators of Resolution of Inflammation, via Their Influence on Cytokines and Oxylipins Release in Astrocytes. Int. J. Mol. Sci. 2020, 21, 9577. [Google Scholar] [CrossRef]
- Ishiyama, M.; Tominaga, H.; Shiga, M.; Sasamoto, K.; Ohkura, Y.; Ueno, K. A combined assay of cell viability and in vitro cytotoxicity with a highly water-soluble tetrazolium salt, neutral red and crystal violet. Biol. Pharm. Bull. 1996, 19, 1518–1520. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene and Primer a | Sequence (5′→3′) | References |
|---|---|---|
| CYP1B1-FP CYP1B1-RP | AACTGTCCATCAGGTGAGGT TAAGGAAGTATACCAGAAGGC | [39] |
| CYP1A1-FP CYP1A1-RP | CTTCATCCTGGAGACCTTCC AAGACCTCCCAGCGGGCAA | [40] |
| CYP1A2-FP CYP1A2-RP | GGAGGCCTTCATCCTGGAGA TCTCCCACTTGGCCAGGACT | [41] |
| CYP2C8-FP CYP2C8-RP | TTCATGCCTTTCTCAGCAGG ATTTGTGCAGTGACCTGAAC | [41] |
| CYP2C9-FP CYP2C9-RP | TTCATGCCTTTCTCAGCAGG TTGCACAGTGAAACATAGGA | [41] |
| CYP2C19-FP CYP2C19-RP | TTCATGCCTTTCTCAGCAGG ACAGATAGTGAAATTTGGAC | [41] |
| CYP2E1-FP CYP2E1-RP | TGCCATCAAGGATAGGCAAG AATGCTGCAAAATGGCACAC | [41] |
| CYP2B6-FP CYP2B6-RP | ACACAGTGAATTCAGCCACC TGGTGTGTTGGGTGACAATG | [41] |
| CYP2D6-FP CYP2D6-RP | CCTGTGCATAGTGGTGGCTG GCTTCTCCCAGACGGCCTCA | [41] |
| CYP2J2-FP CYP2J2-RP | GGACTCTCCTACTGGGCACT CTCCGA AGGTGATGGAGCAA | [42] |
| CYP3A4-FP CYP3A4-RP | TGACCCAAAGTACTGGACAG CTATTCACAAAGTAATTTGAGG | [41] |
| CYP3A5-FP CYP3A5-RP | TGACCCAAAGTACTGGACAG TGAAGAAGTCCTTGCGTGTC | [41] |
| GAPDH-FP GAPDH-RP | AATGCATCCTGCACCACCAA GTAGCCATATTC ATTGTCAT | [43] |
| β-actin-FP β-actin-RP | GGCGGCACCACCATGTACCCT AGGGGCCGGACTCGTCATACT | [44] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, H.Y.; Ahn, S.; Cho, Y.-S.; Seo, S.-K.; Kim, D.H.; Shin, J.-G.; Lee, S.-J. CYP2C19 Contributes to THP-1-Cell-Derived M2 Macrophage Polarization by Producing 11,12- and 14,15-Epoxyeicosatrienoic Acid, Agonists of the PPARγ Receptor. Pharmaceuticals 2023, 16, 593. https://doi.org/10.3390/ph16040593
Cho HY, Ahn S, Cho Y-S, Seo S-K, Kim DH, Shin J-G, Lee S-J. CYP2C19 Contributes to THP-1-Cell-Derived M2 Macrophage Polarization by Producing 11,12- and 14,15-Epoxyeicosatrienoic Acid, Agonists of the PPARγ Receptor. Pharmaceuticals. 2023; 16(4):593. https://doi.org/10.3390/ph16040593
Chicago/Turabian StyleCho, Hee Young, Sangzin Ahn, Yong-Soon Cho, Su-Kil Seo, Dong Hyun Kim, Jae-Gook Shin, and Su-Jun Lee. 2023. "CYP2C19 Contributes to THP-1-Cell-Derived M2 Macrophage Polarization by Producing 11,12- and 14,15-Epoxyeicosatrienoic Acid, Agonists of the PPARγ Receptor" Pharmaceuticals 16, no. 4: 593. https://doi.org/10.3390/ph16040593
APA StyleCho, H. Y., Ahn, S., Cho, Y.-S., Seo, S.-K., Kim, D. H., Shin, J.-G., & Lee, S.-J. (2023). CYP2C19 Contributes to THP-1-Cell-Derived M2 Macrophage Polarization by Producing 11,12- and 14,15-Epoxyeicosatrienoic Acid, Agonists of the PPARγ Receptor. Pharmaceuticals, 16(4), 593. https://doi.org/10.3390/ph16040593