

Microorganism-Derived Molecules as Enzyme Inhibitors to Target Alzheimer’s Diseases Pathways




Abstract
1. Introduction
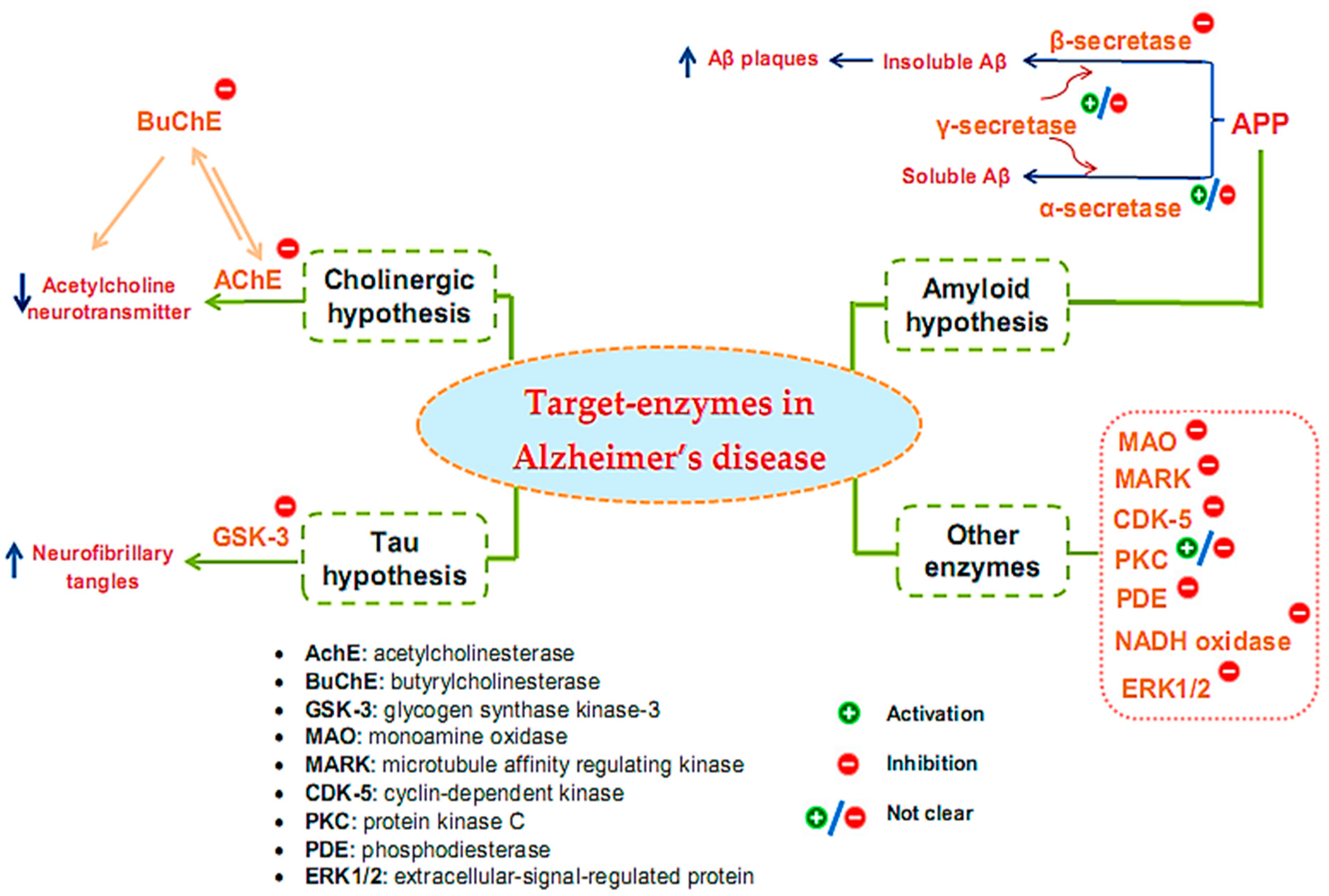
2. Overview of Enzyme-Associated Pathogenesis Mechanisms of Alzheimer’s Disease
3. Overview of Inhibitors on Target Enzyme for Alzheimer’s Treatment from Microorganisms
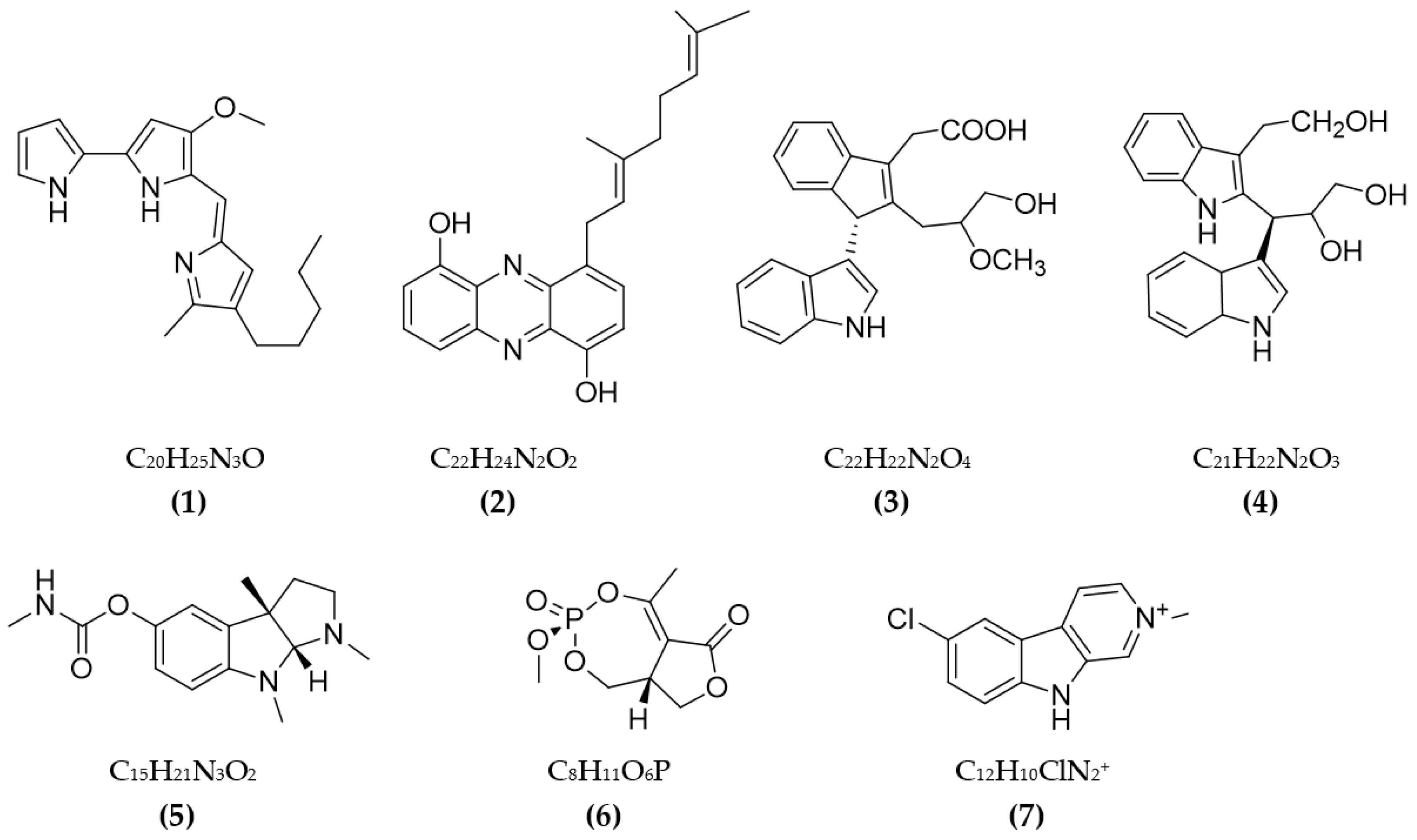
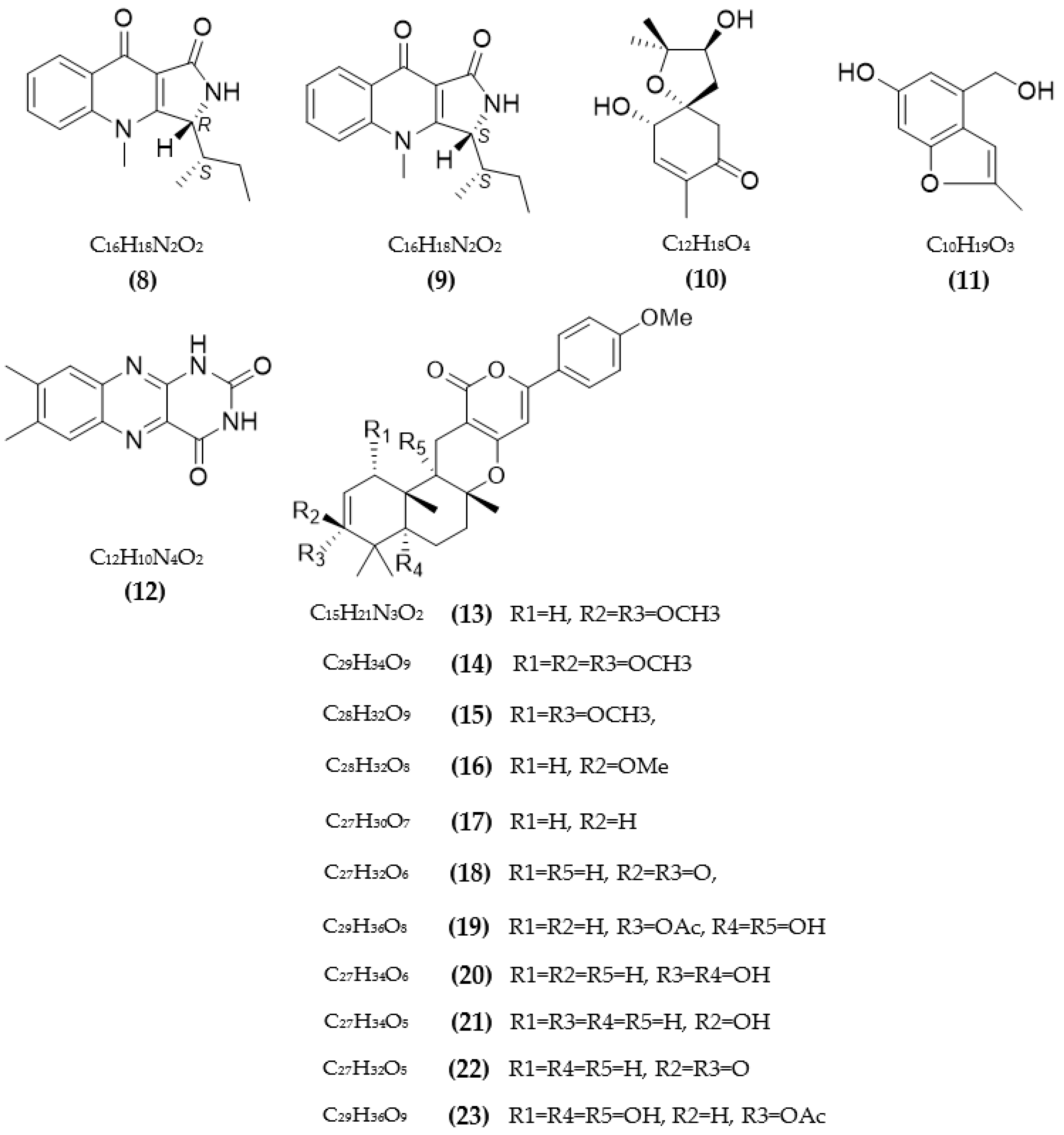
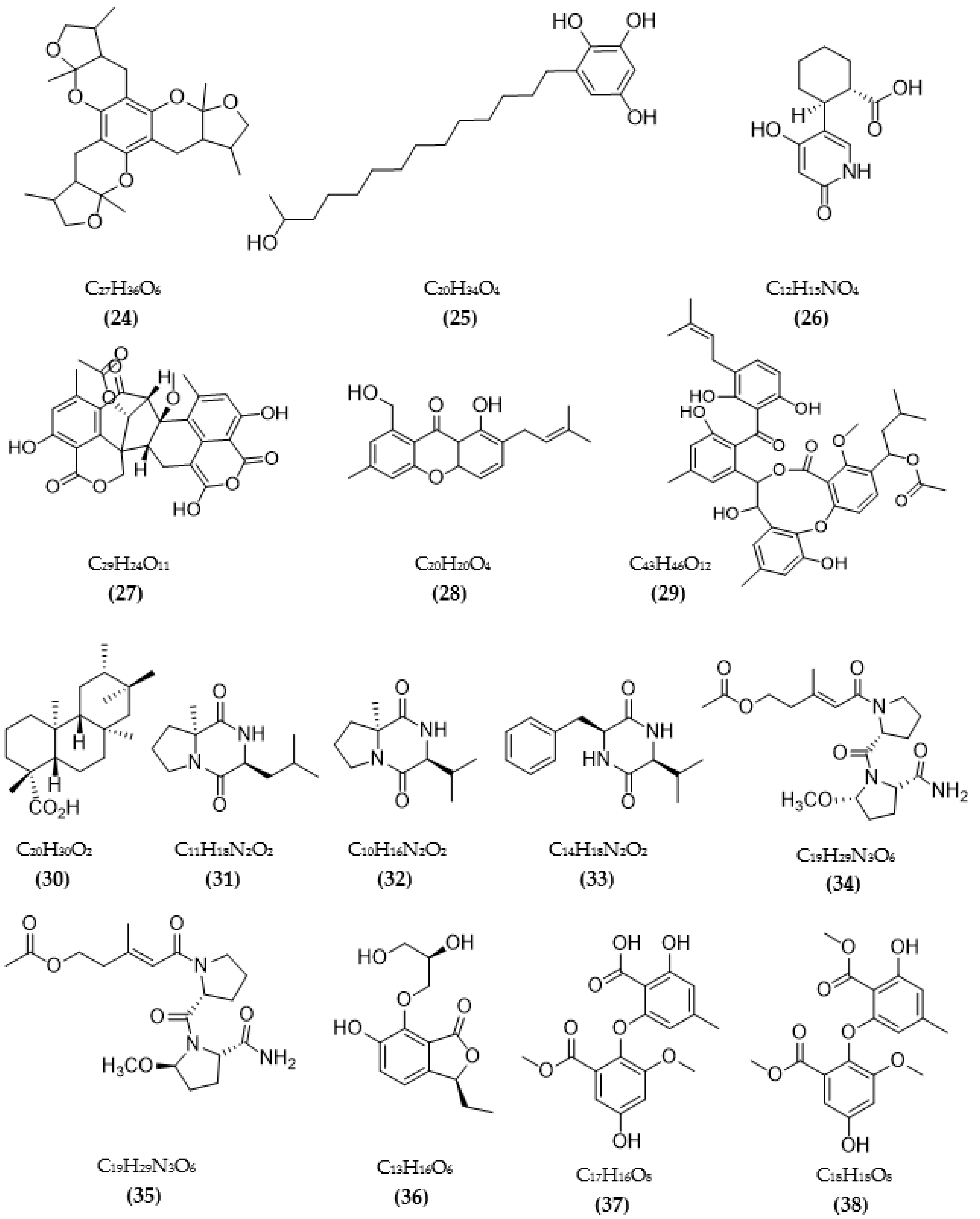
3.1. Inhibitors on AchE and BuChE from Microorganisms
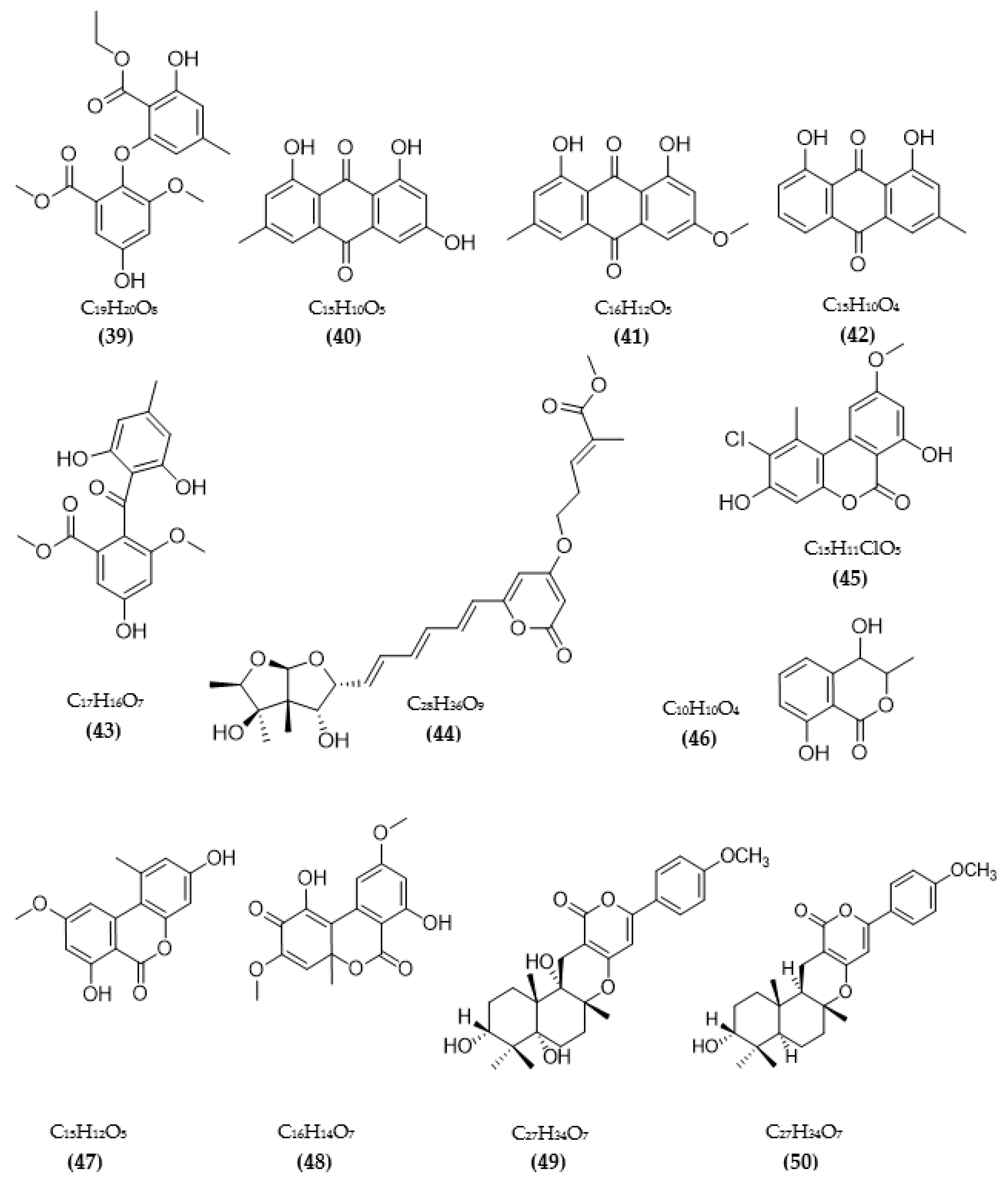
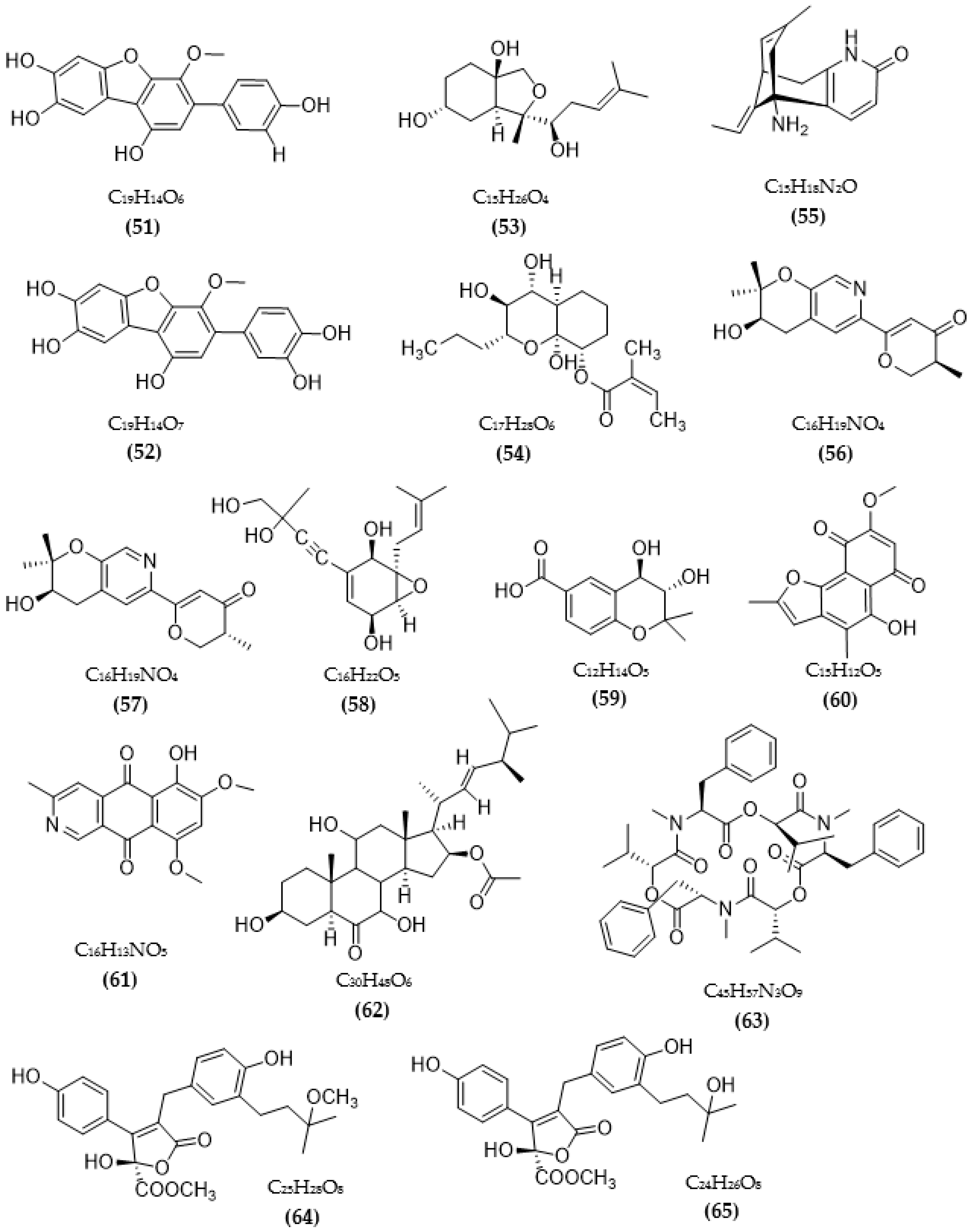
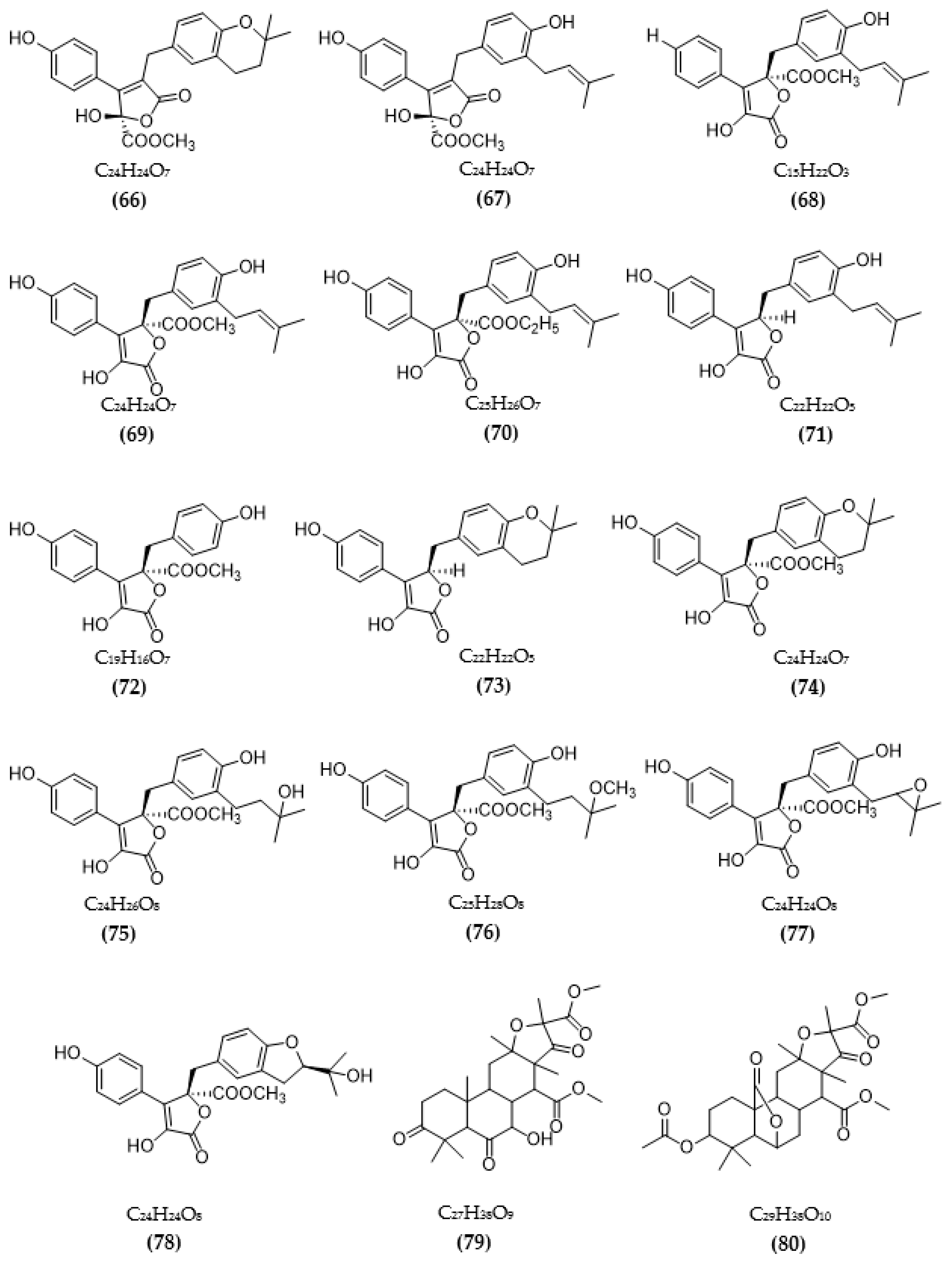
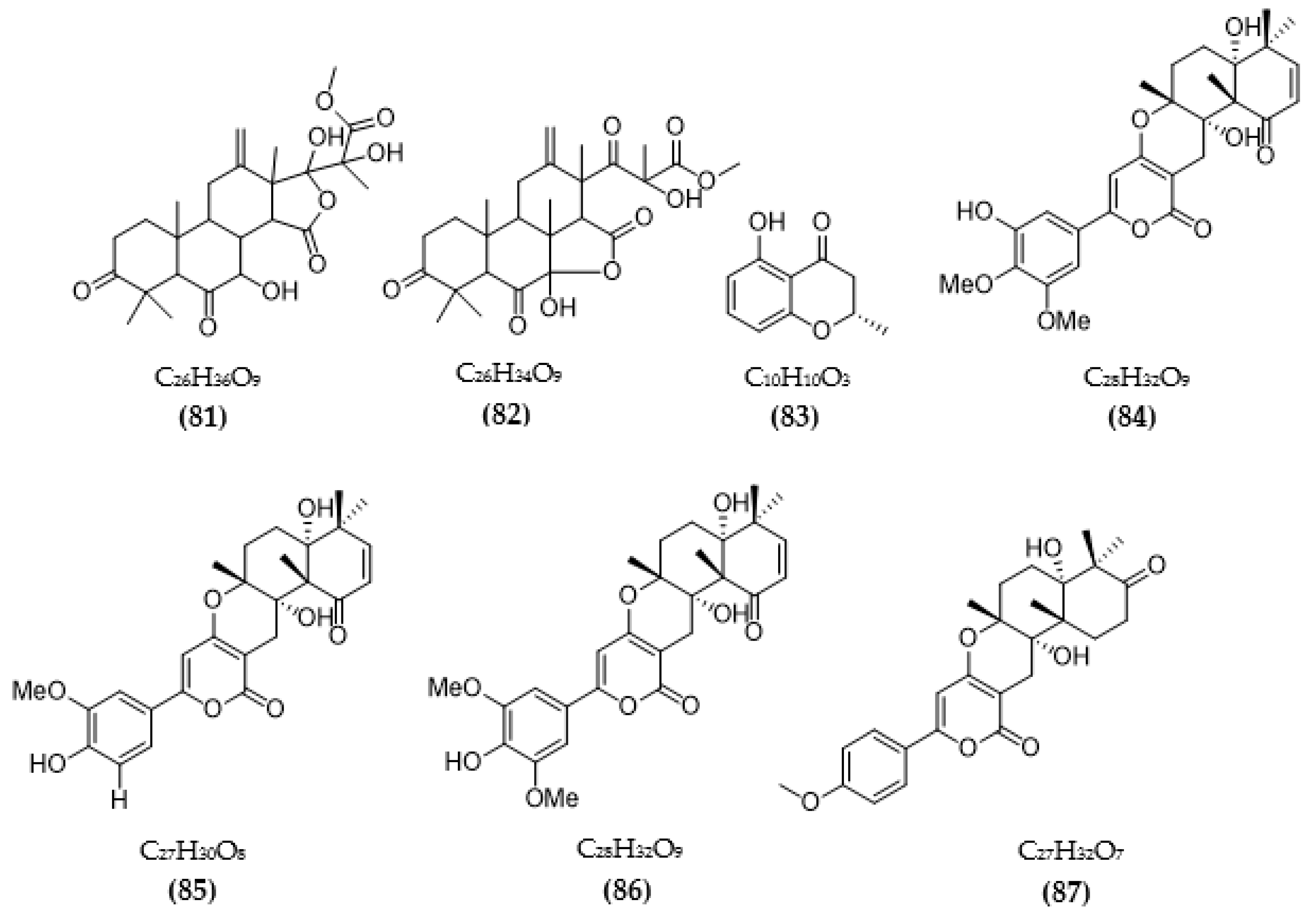
3.2. Inhibitors of Other Targeted Enzymes of AD from Microorganisms

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
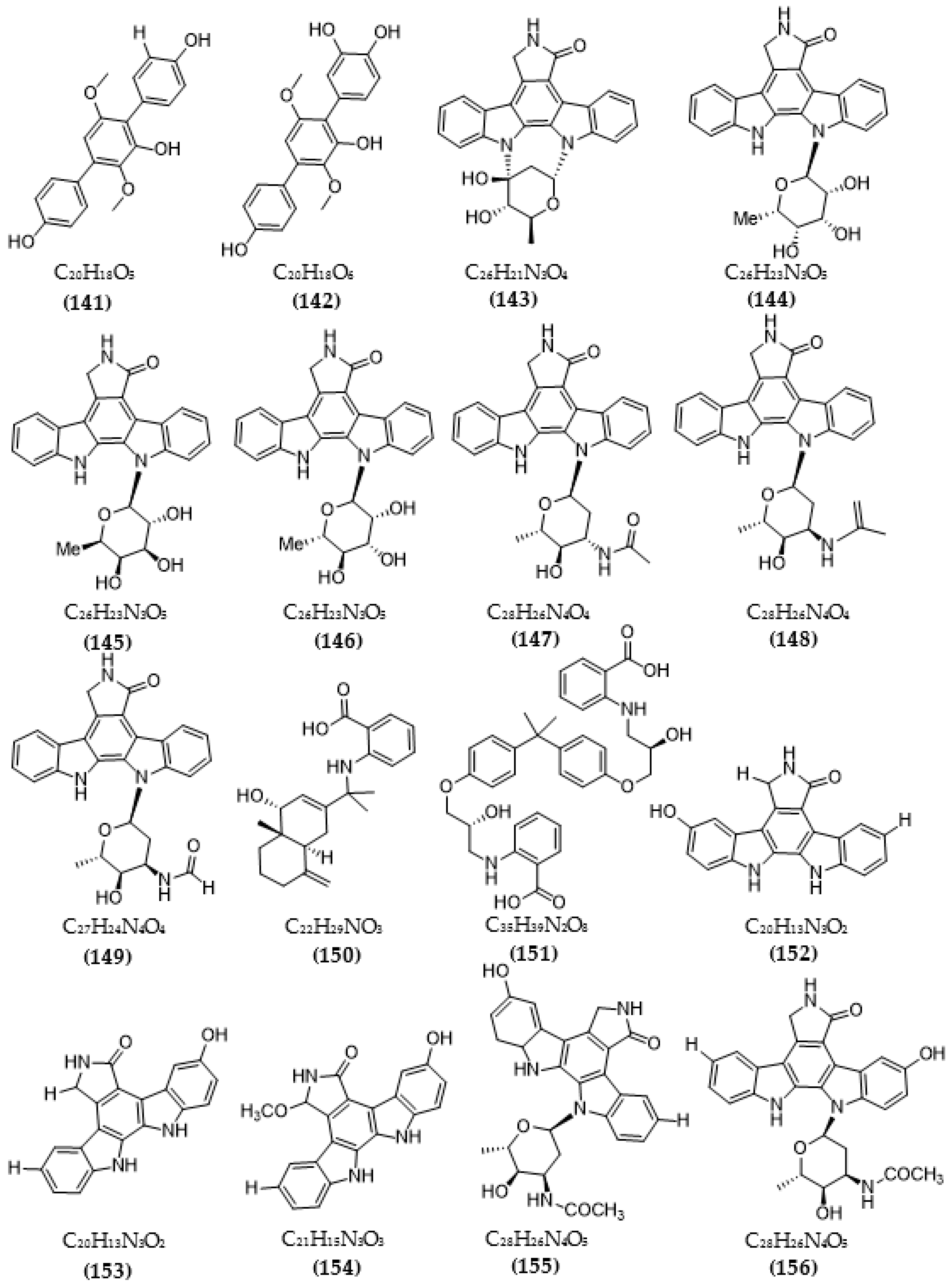{kind=link}
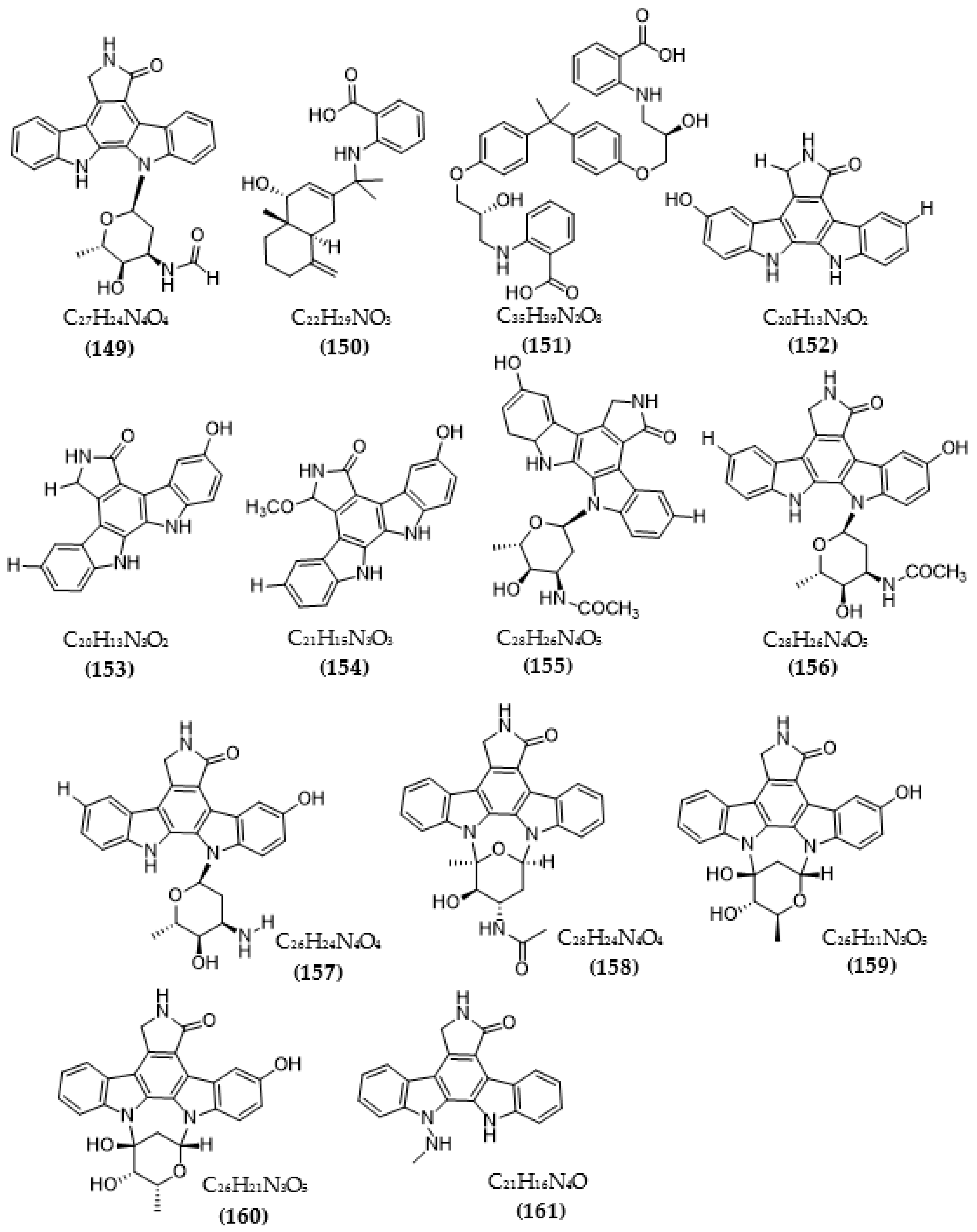{kind=link}
| Strain | Compound | Bioactivity (IC50) | Ref. |
|---|---|---|---|
| Anti-BACE1 | |||
| Cytospora rhizophorae | Extracts | <3.0 μg/mL | [97] |
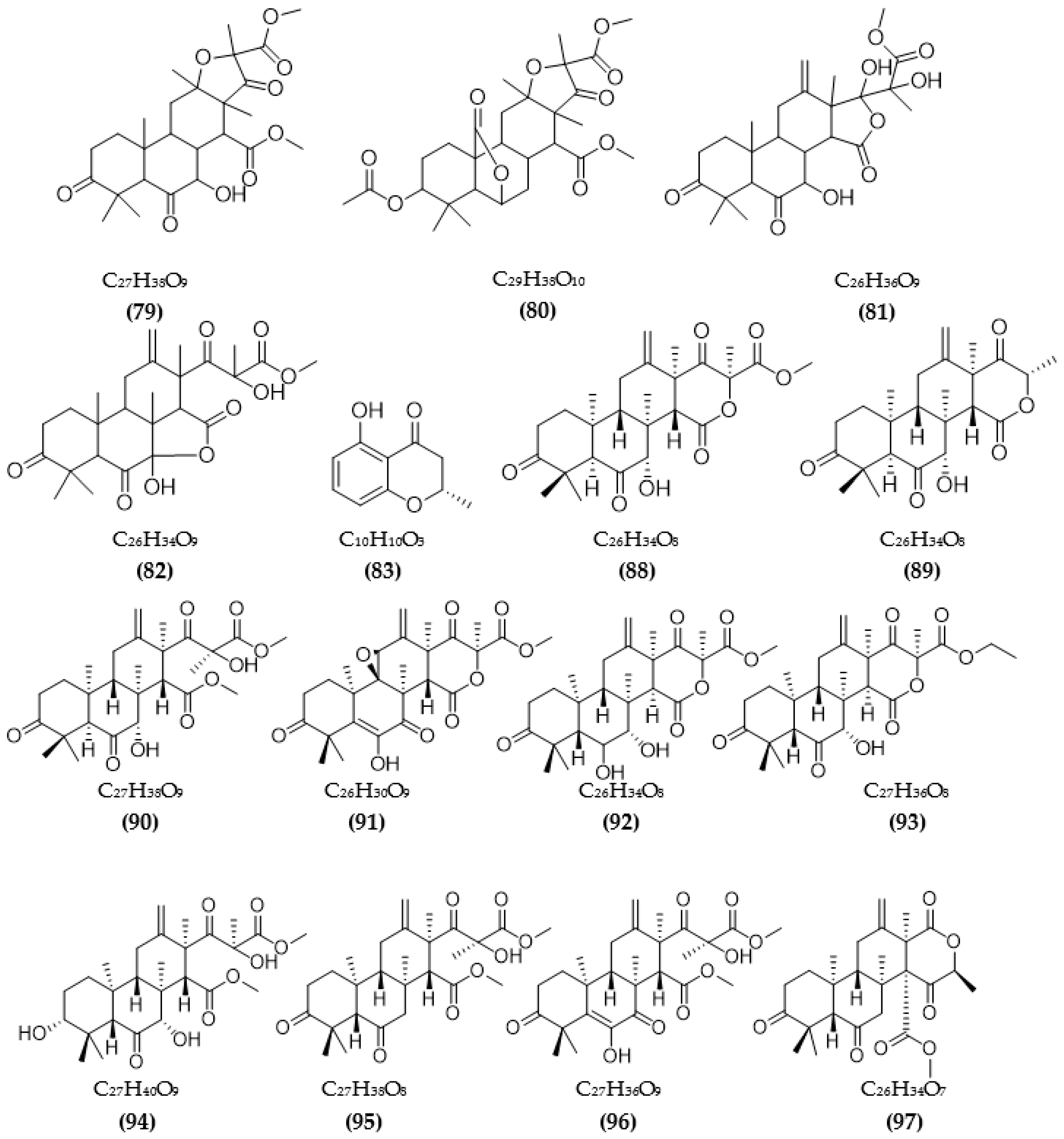
| Aspergillus terreus | Terreusterpene A (79) | 5.98 µM | [93] |
| Terreusterpene B (80) | 11.42 µM | ||
| Terreusterpene C (81) | 40 µM | ||
| Terreusterpene D (82) | 1.91 µM | ||
| Aspergillus terreus | Asperterpene E (88) | 13.3 μM | [98] |
| Asperterpene F (89) | 5.9 μM | ||
| Asperterpene J (90) | 31.7 μM | ||
| Asperterpene D (91) | >50 μM | ||
| Asperterpene G (92) | >50 μM | ||
| Asperterpene H (93) | >50 μM | ||
| Asperterpene I (94) | >50 μM | ||
| Asperterpene K (95) | >50 μM | ||
| Asperterpene L (96) | >50 μM | ||
| Asperterpene M (97) | >50 μM | ||
| Terretonin D (98) | >50 μM | ||
| Terretonin G (99) | >50 μM | ||
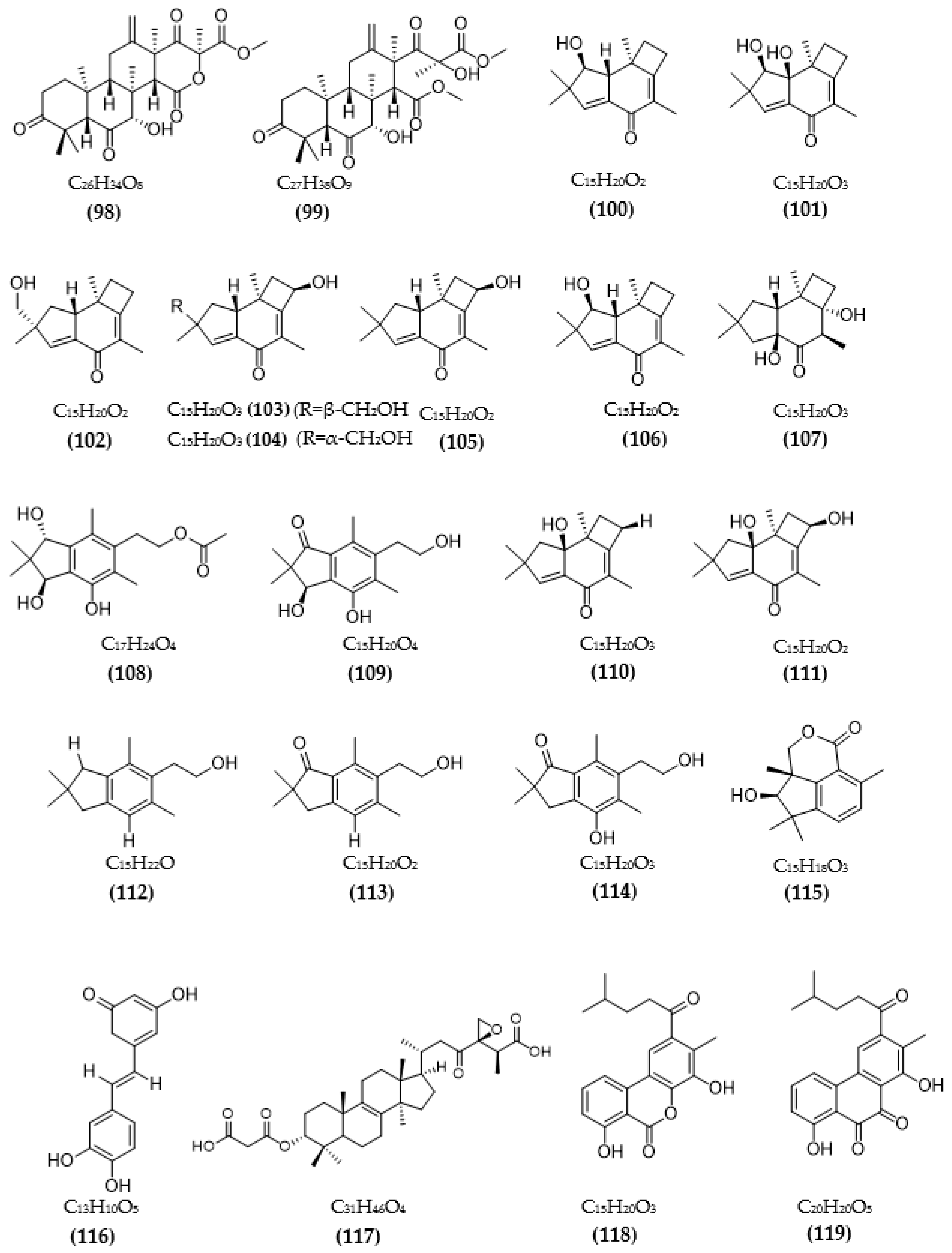
| Phomopsis sp. TJ507A | Phomophyllin A (100) | IR = 44% | [99] |
| Phomophyllin B (101) | IR = 35% | ||
| Phomophyllin C (102) | IR = 19% | ||
| Phomophyllin D (103) | IR = 40% | ||
| Phomophyllin E (104) | IR = 37% | ||
| Phomophyllin F (105) | IR = 38% | ||
| Phomophyllin G (106) | IR = 25% | ||
| Phomophyllin I (107) | IR = 39% | ||
| Phomophyllin L (108) | IR = 4% | ||
| Phomophyllin M (109) | IR = 5% | ||
| Granulone B (110) | IR = 6% | ||
| Radulone B (111) | IR = 39% | ||
| 2-(2,2,4,6-tetramethylindan-5-yl)ethanol (112) | IR < 1% | ||
| Pterosin Z (113) | IR < 1% | ||
| Onitin (114) | IR = 42% | ||
| 7-hydroxy-10-oxodehydrodihydrobotrydial (115) | IR = 40% | ||
| Phellinus linteus | Hispidin (116) | 4.9 μM | [100] |
| Daedalea sp. | Daedalol C (117) | 14.2 μM | [101] |
| Anti-MAO | |||
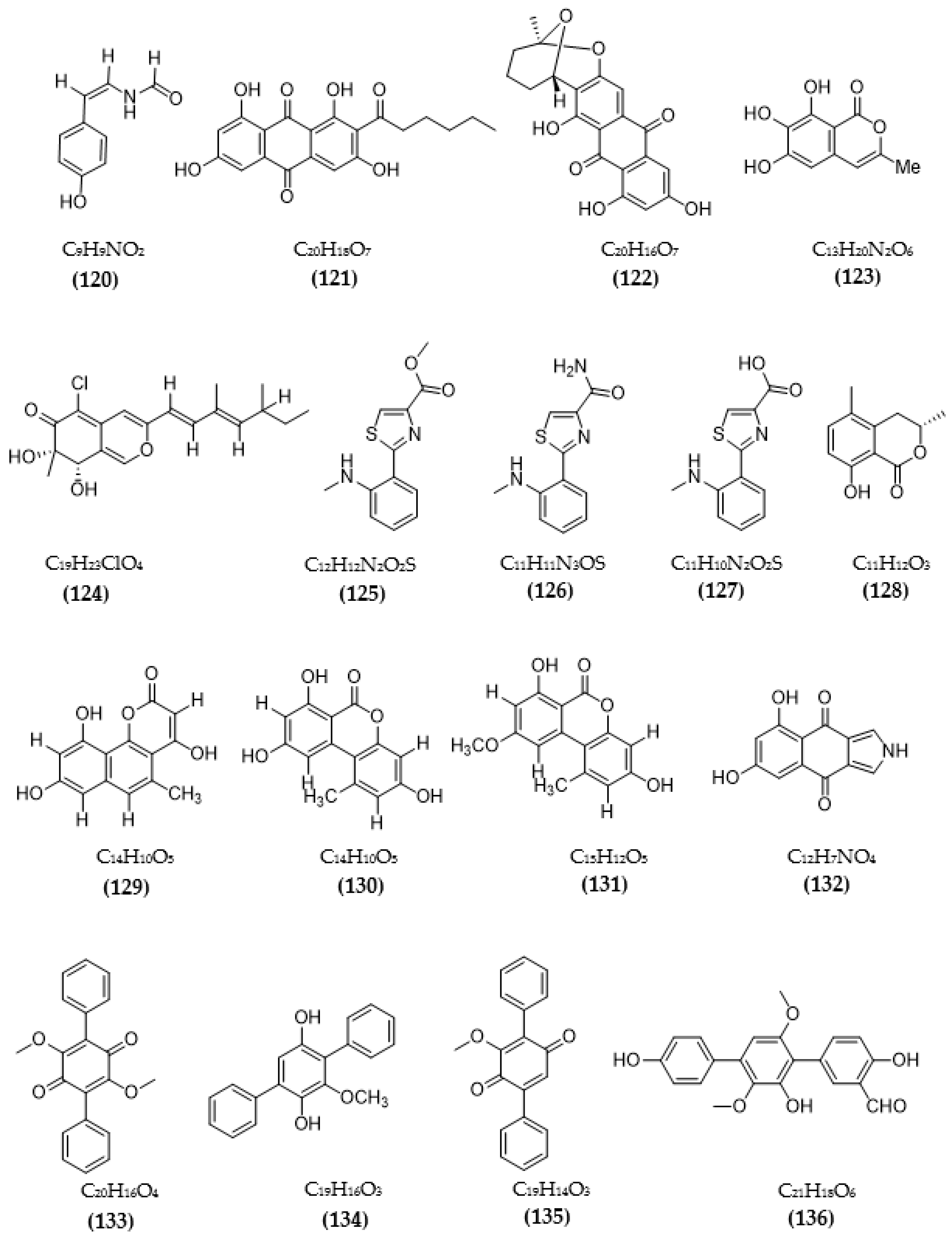
| Daldinia fissa | 5-hydroxy-2-methyl-chroman-4-on (83) | 13.9 μM (MAO-A) 3.2 μM/Ki:0.896 µM (MAO-B) | [94] |
| Streptomyces sp. CNQ-027 | 4,7-dihydroxy-3-methyl-2-(4-methyl-1-oxopentyl)-6H-dibenzo[b,d]pyran-6-one (118) | 6.47 μM (MAO-A) Ki = 0.573 µM 1.21 μM (MAO-B) Ki = 0.248 µM | [103] |
| 1,8-dihydroxy-2-methyl-3-(4-methyl-1-oxopentyl)-9,10-phenanthrenedione (119) | > 80 μM (MAO-A) 14.5 μM (MAO-B) | ||
| Talaromyces sp. LGT-2 | (Z)-N-(4-hydroxystyryl)formamide (120) | 61 μM | [106] |
| Emericella navahoensis | Norsolorinic acid (121) | 0.3 μM | [107] |
| Averufin (122) | 54.4 μM | ||
| 6, 7, 8-trihydroxy-3-methylisocoumarin (123) | 817.3 μM | ||
| Talaromyced luteus | TL-1 (124) | 6.6 μM | [108] |
| Streptomyces sp. | Anithiactin A (125) | 13 μM (MAO-A) Ki = 1.84 µM 183 μM (MAO-B) | [109] |
| Anithiactin B (126) | >85 μM (MAO-A) - (MAO-B) | ||
| Anithiactin C (127) | >170 μM (MAO-A) >170 μM (MAO-B) | ||
| Rosellinia corticium | (S)-5-methylmellein (128) | 5.31 μM (MAO-A) Ki = 2.45 μM 9.15 μM (MAO-B) | [110] |
| Anti-GSK3 | |||
| Aspergillus sp. | Pannorin (129) | 0.35 μM | [111] |
| Alternariol (130) | 0.13μM | ||
| Alternariol-9-methylether (131) | 0.20 μM | ||
| Biscogniauxia mediterranea | Biscogniauxone (132) | 8.04 μM | [112] |
| Anti- PDE | |||
| Phoma sp. | Betulinan A (133) | 44 μM | [114] |
| BTH-II0204-207:A (134) | 31 μM | ||
| Betulinan C (135) | 17 μM | ||
| Aspergillus sp. ITBBc1 | Sanshamycin A (136) | IR = 4.8% | [115] |
| Sanshamycin B (137) | IR = 6.7% | ||
| Sanshamycin C (138) | IR = 49.4% | ||
| Sanshamycin D (139) | IR = 12.4% | ||
| Sanshamycin E (140) | IR = 5.1% | ||
| Terphenyllin (141) | IR = 12.8% | ||
| 3-hydroxyterphenyllin (142) | IR = 23.2% | ||
| Anti-PKC | |||
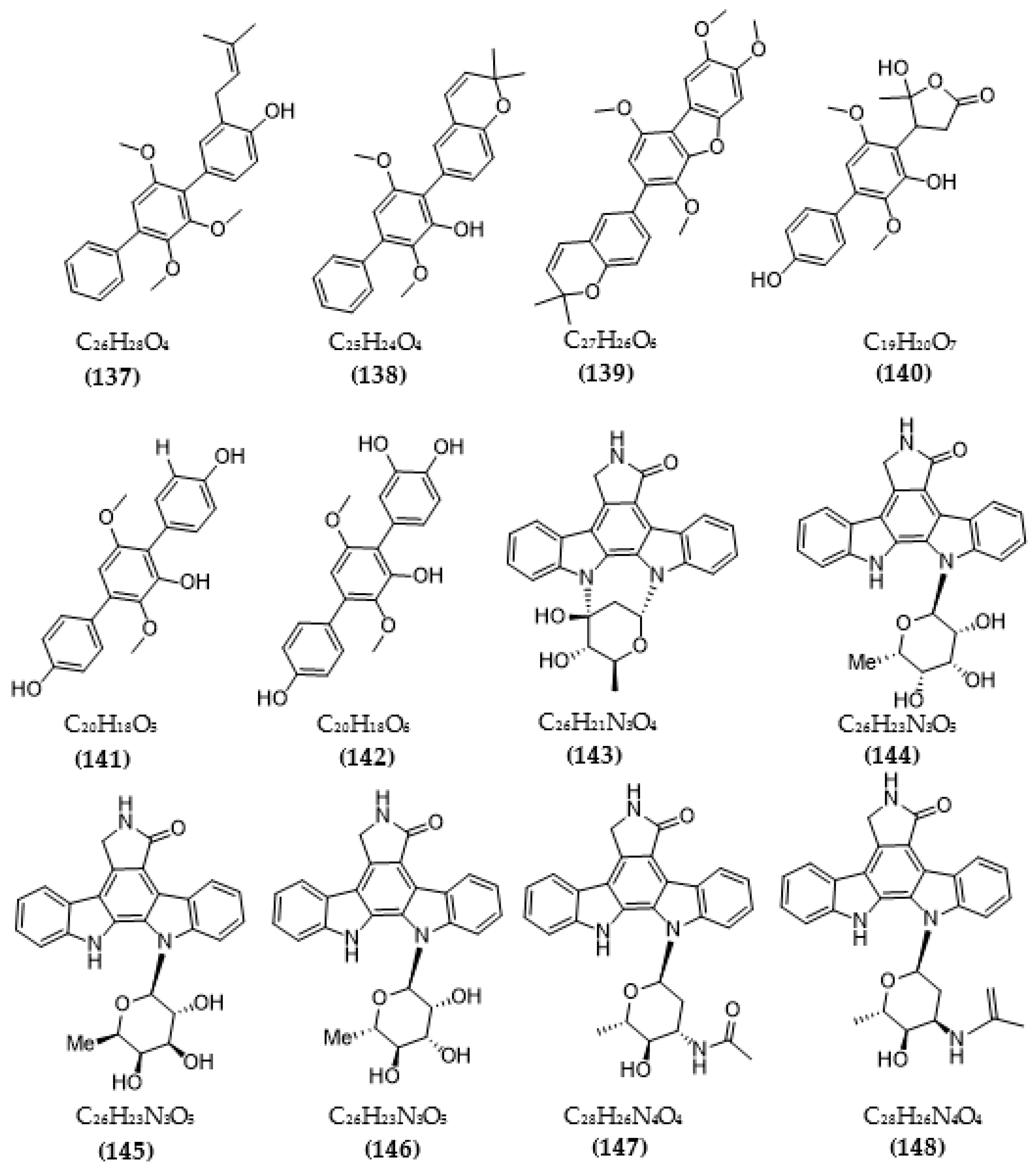
| Streptomyces sp. A65 | Streptocarbazoles C (143) | >20 µM | [116] |
| 30-epi-k252d (144) | 0.25 µM | ||
| 20,40-epi-k252d (145) | 0.35 µM | ||
| K252d (146) | 0.97 µM | ||
| Streptomyces sp. A68 | 3′-epi-N-Acetyl-holyrine A (147) | 0.17 µM | [117] |
| 3′-N-Acetyl-holyrine A (148) | 0.91 µM | ||
| 3′-N-Formyl-holyrine A (149) | 1.04 µM | ||
| Eudesm-4(15), 7-diene9α- hydroxy-11-amino-benzoicacid (150) | >20 µM | ||
| (9R, 22R)-bisphenol A bis (9, 22- hydroxy-10, 23-anthranilicacid-propyl) ether (151) | 1.32 µM | ||
| Streptomyces sp. DT-A61 | 9-hydroxyk252c (152) | 0.98 µM | [118] |
| 3-hydroxy-k252c (153) | 3.2 µM | ||
| 3-hydroxy-7- methoxy-k252c (154) | 1.4 µM | ||
| 9-hydroxy-3′-N-acetylholyrine A (155) | 0.097 µM | ||
| 3-hydroxy-3′-N-acetylholyrine A (156) | 0.46 µM | ||
| 3- hydroxyholyrine A (157) | 0.079 µM | ||
| 3′-O-demethyl-4′-N-demethyl-4′-N-acetyl-4′-epi-staurosporine (158) | 0.092 µM | ||
| Streptocarbazole D (159) | 2.1 µM | ||
| Streptocarbazole E (160) | 1.4 µM | ||
| Streptomyces sp. A22 | 12-N-methyl-k252c (161) | 1.84 μM | [119] |
4. Trends in Using Virtual Screening to Discover Potential Alzheimer’s Inhibitors from Microorganisms
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cacabelos, R. Pharmacogenomics of Alzheimer’s and Parkinson’s diseases. Neurosci. Lett. 2020, 726, 133807. [Google Scholar] [CrossRef] [PubMed]
- Hajjo, R.; Sabbah, D.A.; Abusara, O.H.; Al Bawab, A.Q. A review of the recent advances in Alzheimer’s disease research and the utilization of network biology approaches for prioritizing diagnostics and therapeutics. Diagnostics 2022, 12, 2975. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, V.; Smith, S.T.; Mikobi, E.; Raji, M.A. Acetylcholinesterase inhibitors: Beneficial effects on comorbidities in patients with Alzheimer’s disease. Am. J. Alzheimers Dis. Other Demen. 2018, 33, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments in Alzheimer’s disease: An update. J. Cent. Nerv. Syst. Dis. 2020, 12, 1179573520907397. [Google Scholar] [CrossRef]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive review on Alzheimer’s disease: Causes and treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Liu, P.P.; Xie, Y.; Meng, X.Y.; Kang, J.S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Sig. Transduct. Target Ther. 2019, 4, 29. [Google Scholar] [CrossRef]
- McGirr, S.; Venegas, C.; Swaminathan, A. Alzheimer’s disease: A brief review. J. Exp. Neurol. 2020, 1, 89–98. [Google Scholar]
- Andrade, S.; Ramalho, M.J.; Loureiro, J.A.; Pereira, M.D.C. Natural compounds for Alzheimer’s disease therapy: A systematic review of preclinical and clinical studies. Int. J. Mol. Sci. 2019, 20, 2313. [Google Scholar] [CrossRef]
- Ma, Y.; Yang, M.W.; Li, X.W.; Yue, J.W.; Chen, J.Z.; Yang, M.W.; Huang, X.; Zhu, L.L.; Hong, F.F.; Yang, S.L. Therapeutic effects of natural drugs on Alzheimer’s disease. Front. Pharmacol. 2019, 10, 1355. [Google Scholar] [CrossRef] [PubMed]
- Ashrafian, H.; Zadeh, E.H.; Khan, R.H. Review on Alzheimer’s disease: Inhibition of amyloid beta and tau tangle formation. Int. J. Biol. Macromol. 2021, 167, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Eldar, F.H.; Martinez, A. GSK-3 inhibitors: Preclinical and clinical focus on CNS. Front. Mol. Neurosci. 2011, 4, 32. [Google Scholar]
- Zaki, A.G.; El-Sayed, S.R.; Elkodous, M.A.; El-Sayyad, G.S. Microbial acetylcholinesterase inhibitors for Alzheimer’s therapy: Recent trends on extraction, detection, irradiation-assisted production improvement and nano-structured drug delivery. Appl. Microbiol. Biotechnol. 2020, 104, 4717–4735. [Google Scholar] [CrossRef]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Prim. 2021, 7, 33. [Google Scholar] [CrossRef]
- Abubakar, M.B.; Sanusi, K.O.; Ugusman, A.; Mohamed, W.; Kamal, H.; Ibrahim, N.H.; Khoo, C.S.; Kumar, J. Alzheimer’s disease: An update and insights into pathophysiology. Front. Aging Neurosci. 2022, 14, 742408. [Google Scholar] [CrossRef]
- Kaloni, D.; Negi, A. A review on Alzheimer disease. Int. J. Neurodegener. Dis. 2019, 2, 10. [Google Scholar]
- Hashmi, A.; Srivastava, V.; Abul, K.S.; Kumar, M.D. Alzheimer’s disease: An insightful review on the future trends of the effective therapeutics. In Alzheimer’s Disease; IntechOpen: London, UK, 2022. [Google Scholar]
- Folch, J.; Ettcheto, M.; Petrov, D.; Abad, S.; Pedrós, I.; Marin, M.; Olloquequi, J.; Camins, A. Review of the advances in treatment for Alzheimer disease: Strategies for combating β-amyloid protein. Neurología 2018, 33, 47–58. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Khachaturian, A.S.; Vergallo, A.; Farlow, M.R.; Snyder, P.J.; Giacobini, E.; Khachaturian, Z.S. Revisiting the cholinergic hypothesis in Alzheimer’s disease: Emerging evidence from translational and clinical research. J. Prev. Alzheimers Dis. 2019, 6, 2–15. [Google Scholar]
- Karran, E.; De, S.B. The amyloid hypothesis in Alzheimer disease: New insights from new therapeutics. Nat. Rev. Drug Discov. 2022, 21, 306–318. [Google Scholar] [CrossRef]
- Anand, P.; Singh, B. A review on cholinesterase inhibitors for Alzheimer’s disease. Arch. Pharm. Res. 2013, 36, 375–399. [Google Scholar] [CrossRef] [PubMed]
- Marucci, G.; Buccioni, M.; Ben, D.D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2021, 190, 108352. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K. Cholinesterase inhibitors as Alzheimer’s therapeutics (Review). Mol. Med. Rep. 2019, 20, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.; Adem, A.; Sabbagh, M. New acetylcholinesterase inhibitors for Alzheimer’s disease. Int. J. Alzheimers Dis. 2012, 2012, 728983. [Google Scholar] [CrossRef] [PubMed]
- Moussa, P.N.M.; Abdin, S.M.; Omar, H.A.; Alniss, H.; Al-Tel, T.H. BACE1 inhibitors: Current status and future directions in treating Alzheimer’s disease. Med. Res. Rev. 2020, 40, 339–384. [Google Scholar] [CrossRef]
- Bazzari, F.H.; Bazzari, A.H. BACE1 inhibitors for Alzheimer’s disease: The past, present and any future. Molecules 2022, 27, 8823. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, D.; Sehgal, A.; Singh, S.; Sharma, N.; Zengin, G.; Andronie, C.F.L.; Toma, M.M.; Bungau, S.; Bumbu, A.G. Role of monoamine oxidase activity in Alzheimer’s disease: An insight into the therapeutic potential of inhibitors. Molecules 2021, 26, 3724. [Google Scholar] [CrossRef]
- Chen, X.Q.; Mobley, W.C. Exploring the pathogenesis of Alzheimer disease in basal forebrain cholinergic neurons: Converging insights from alternative hypotheses. Front. Neurosci. 2019, 13, 446. [Google Scholar] [CrossRef]
- Zhou, S.; Huang, G. The biological activities of butyrylcholinesterase inhibitors. Biomed. Pharmacother. 2022, 146, 112556. [Google Scholar] [CrossRef]
- Miličević, A.; Šinko, G. Evaluation of the key structural features of various butyrylcholinesterase inhibitors using simple molecular descriptors. Molecules 2022, 27, 6894. [Google Scholar] [CrossRef]
- Nordberg, A.; Ballard, C.; Bullock, R.; Darreh, S.T.; Somogyi, M. A review of butyrylcholinesterase as a therapeutic target in the treatment of Alzheimer’s disease. Prim. Care Companion CNS Disord. 2013, 15, PCC.12r01412. [Google Scholar] [CrossRef] [PubMed]
- Adeowo, F.Y.; Ejalonibu, M.A.; Elrashedy, A.A.; Lawal, M.M.; Kumalo, H.M. Multi-target approach for Alzheimer’s disease treatment: Computational biomolecular modeling of cholinesterase enzymes with a novel 4-N-phenylaminoquinoline derivative reveal promising potentials. J. Biomol. Struct. Dyn. 2021, 39, 3825–3841. [Google Scholar] [CrossRef]
- Harald, H.; Marsel, M.; Claudio, C.; Martin, R.F.; Ezio, G.; George, T.G.; Ara, S.K.; Andrea, V.; Enrica, C.; Peter, J.S.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar]
- Uddin, M.S.; Kabir, M.T.; Jeandet, P.; Mathew, B.; Ashraf, G.M.; Perveen, A.; Bin-Jumah, M.N.; Mousa, S.A.; Abdel-Daim, M.M. Novel anti-alzheimer’s therapeutic molecules targeting amyloid precursor protein processing. Oxid. Med. Cell Longev. 2020, 2020, 7039138. [Google Scholar] [CrossRef] [PubMed]
- Bui, T.T.; Nguyen, T.H. Natural product for the treatment of Alzheimer’s disease. J. Basic Clin. Physiol. Pharmacol. 2017, 28, 413–423. [Google Scholar] [CrossRef]
- Kim, N.; Lee, H.J. Target enzymes considered for the treatment of Alzheimer’s disease and Parkinson’s disease. Biomed. Res. Int. 2020, 2020, 2010728. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M.; Tang, J. Developing β-secretase inhibitors for treatment of Alzheimer’s disease. J. Neurochem. 2012, 120, 71–83. [Google Scholar] [CrossRef]
- Kwak, H.M.; Jeon, S.Y.; Sohng, B.H.; Kim, J.G.; Lee, J.M.; Lee, K.B.; Jeong, H.H.; Hur, J.M.; Kang, Y.H.; Song, K.S. Beta-secretase (BACE1) inhibitors from pomegranate (Punica granatum) husk. Arch. Pharm. Res. 2005, 28, 1328–1332. [Google Scholar] [CrossRef]
- Naushad, M.; Durairajan, S.S.K.; Bera, A.K.; Senapati, S.; Li, M. Natural compounds with anti-BACE1 activity as promising therapeutic drugs for treating Alzheimer’s disease. Planta Med. 2019, 85, 1316–1325. [Google Scholar]
- Kazuya, M. Chemical diversity of β-secretase inhibitors from natural resources. Nat. Prod. Commun. 2019, 14, 1934578X19894819. [Google Scholar]
- Maqbool, M.; Mobashir, M.; Hoda, N. Pivotal role of glycogen synthase kinase-3: A therapeutic target for Alzheimer’s disease. Eur. J. Med. Chem. 2016, 107, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Tantray, M.A.; Alam, M.S.; Hamid, H. Natural and synthetic bioactive inhibitors of glycogen synthase kinase. Eur. J. Med. Chem. 2017, 125, 464–477. [Google Scholar] [CrossRef]
- Shoaib, M.; Nasimul, H. A comprehensive review of monoamine oxidase inhibitors as anti-Alzheimer’s disease agents: A review. Eur. J. Med. Chem. 2020, 206, 112787. [Google Scholar]
- Ron, W.; Robert, H.; Marianne, G.H.; Timothy, E.; Susan, W. Antidepressants. In Rosen’s Emergency Medicine: Concepts and Clinical Practice, 10th ed.; Ron, M.W., Robert, S.H., Marianne, G.H., Eds.; Elsevier: Philadelphia, PA, USA, 2018. [Google Scholar]
- Cai, Z. Monoamine oxidase inhibitors: Promising therapeutic agents for Alzheimer’s disease (review). Mol. Med. Rep. 2014, 9, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, J.S.; Spruyt, M.A.; Brown, A.M.; Sahasrabudhe, S.R.; Blume, A.J.; Vitek, M.P.; Muenkel, H.A.; Sonnenberg, R.J. The release of Alzheimer’s disease beta amyloid peptide Is reduced by phorbol treatment. J. Biol. Chem. 1994, 269, 8376–8382. [Google Scholar] [CrossRef] [PubMed]
- Hung, A.Y.; Haass, C.; Nitsch, R.M.; Qiu, W.Q.; Citron, M.; Wurtman, R.J.; Growdon, J.H.; Selkoe, D.J. Activation of protein kinase C inhibits cellular production of the amyloid beta-protein. J. Biol. Chem. 1993, 268, 22959–22962. [Google Scholar] [CrossRef]
- Lordén, G.; Newton, A.C. Conventional protein kinase C in the brain: Repurposing cancer drugs for neurodegenerative treatment. Neuronal Signal. 2021, 5, NS20210036. [Google Scholar] [CrossRef]
- Alam, J.; Sharma, L. Potential enzymatic targets in Alzheimer’s: A comprehensive review. Curr. Drug Targets 2019, 20, 316–339. [Google Scholar] [CrossRef]
- Liu, S.L.; Wang, C.; Jiang, T.; Tan, L.; Xing, A.; Yu, J.T. The role of Cdk5 in Alzheimer’s disease. Mol. Neurobiol. 2016, 53, 4328–4342. [Google Scholar] [CrossRef]
- Kimura, T.; Ishiguro, K.; Hisanaga, S. Physiological and pathological phosphorylation of tau by Cdk5. Front. Mol. Neurosci. 2014, 7, 65. [Google Scholar] [CrossRef]
- Liu, F.; Su, Y.; Li, B.; Zhou, Y.; Ryder, J.; Gonzalez, D.P.; May, P.C.; Ni, B. Regulation of amyloid precursor protein (APP) phosphorylation and processing by p35/Cdk5 and p25/Cdk5. FEBS Lett. 2003, 547, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Yu, W.H.; Maloney, B.; Bailey, J.; Ma, J.; Marie, I.; Maurin, T.; Wang, L.; Figueroa, H.; Herman, M.; et al. Transcriptional regulation of beta-secretase by p25/cdk5 leads to enhanced amyloidogenic processing. Neuron 2008, 57, 680–690. [Google Scholar] [CrossRef] [PubMed]
- Toshiya, O.; Taro, S.; Akiko, A.; Sawako, S.; Koichi, M.I.; Kanae, A. Microtubule affinity–regulating kinase 4 with an Alzheimer’s disease-related mutation promotes tau accumulation and exacerbates neurodegeneration. J. Biol. Chem. 2020, 295, 17138–17147. [Google Scholar]
- Sheng, J.; Zhang, S.; Wu, L.; Kumar, G.; Liao, Y.; Fan, H. Inhibition of phosphodiesterase: A novel therapeutic target for the treatment of mild cognitive impairment and Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 1019187. [Google Scholar] [CrossRef]
- Ma, M.W.; Wang, J.; Zhang, Q.; Wang, R.; Krishnan, M.D.; Ratna, K.V.; Darrell, W.B. NADPH oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegener. 2017, 12, 7. [Google Scholar] [CrossRef]
- Mohammad, R.K.; Keyvan, Y.; Ayda, E.; Morteza, G.B. The role of ERK1/2 pathway in the pathophysiology of Alzheimer’s disease: An overview and update on new developments. Cell. Mol. Neurobiol. 2023, 43, 177–191. [Google Scholar]
- Muraleva, N.A.; Kolosova, N.G.; Stefanova, N.A. MEK1/2-ERK pathway alterations as a therapeutic target in sporadic Alzheimer’s disease: A study in senescence-accelerated OXYS rats. Antioxidants 2021, 10, 1058. [Google Scholar] [CrossRef]
- Atanasova, M.; Dimitrov, I.; Ivanov, S.; Georgiev, B.; Berkov, S.; Zheleva, D.D.; Doytchinova, I. Virtual screening and hit selection of natural compounds as acetylcholinesterase inhibitors. Molecules 2022, 27, 3139. [Google Scholar] [CrossRef]
- Alhazmi, H.A.; Albratty, M. An update on the novel and approved drugs for Alzheimer disease. Saudi Pharm. J. 2022, 30, 1755–1764. [Google Scholar] [CrossRef]
- Nguyen, V.B.; Wang, S.L.; Nguyen, A.D.; Phan, T.Q.; Techato, K.; Pradit, S. bioproduction of prodigiosin from fishery processing waste shrimp heads and evaluation of its potential bioactivities. Fishes 2021, 6, 30. [Google Scholar] [CrossRef]
- Ohlendorf, B.; Schulz, D.; Erhard, A.; Nagel, K.; Imhoff, J.F. Geranylphenazinediol, an acetylcholinesterase inhibitor produced by a Streptomyces species. J. Nat. Prod. 2012, 75, 1400–1404. [Google Scholar] [CrossRef] [PubMed]
- Li, J.L.; Huang, L.; Liu, J.; Song, Y.; Gao, J.; Jung, J.H.; Liu, Y.; Chen, G. Acetylcholinesterase inhibitory dimeric indole derivatives from the marine actinomycetes Rubrobacter radiotolerans. Fitoterapia 2015, 102, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Murao, S.; Hayashi, H. Physostigmine and N8-norphysostigmine, insecticidal compounds, from Streptomyces sp. Agric. Biol. Chem. 1986, 50, 523–524. [Google Scholar] [CrossRef]
- Kurokawa, T.; Suzuki, K.; Hayaoka, T.; Nakagawa, T.; Izawa, T.; Kobayashi, M.; Harada, N. Cyclophostin, acetylcholinesterase inhibitor from Streptomyces lavendulae. J. Antibiot. 1993, 46, 1315–1318. [Google Scholar] [CrossRef] [PubMed]
- Almasi, F.; Mohammadipanah, F.; Adhami, H.; Hamedi, J. Introduction of marine-derived Streptomyces sp. UTMC 1334 as a source of pyrrole derivatives with anti- acetylcholinesterase activity. J. Appl. Microbiol. 2018, 125, 1370–1382. [Google Scholar] [CrossRef]
- Becher, P.G.; Beuchat, J.; Gademann, K.; Juttner, F. Nostocarboline: Isolation and synthesis of a new cholinesterase inhibitor from Nostoc 78-12A. J. Nat. Prod. 2005, 68, 1793–1795. [Google Scholar] [CrossRef]
- Kim, W.; Song, N.; Yoo, C.D. Quinolactacins Al and A2, new acetylcholinesterase inhibitors from Penicillium citrinum in MeOH was further purified by reverse phase HPLC. J. Antibiot. 2001, 54, 831–835. [Google Scholar] [CrossRef]
- Alves, A.J.S.; Pereira, J.A.; Dethoup, T.; Cravo, S.; Mistry, S.; Silva, A.M.S.; Pinto, M.M.M.; Kijjoa, A. A new meroterpene, a new benzofuran derivative and other constituents from cultures of the marine sponge-associated fungus Acremonium persicinum KUFA 1007 and their anticholinesterase activities. Mar. Drugs 2019, 17, 379. [Google Scholar] [CrossRef]
- Omura, S.; Kuno, F.; Otoguro, K.; Sunazuka, T.; Shiomi, K.; Masuma, R.; Iwai, Y. Arisugacin, a novel and selective inhibitor of acetylcholinesterase from Penicillium sp. FO-4259. J. Antibiot. 1995, 48, 745–746. [Google Scholar] [CrossRef]
- Otoguro, K.; Shiomi, K.; Yamaguchi, Y.; Arai, N.; Sunazuka, T.; Masuma, R.; Iwai, Y.; Omura, S. Arisugacins C and D, Novel acetylcholinesterase inhibitors and their related novel metabolites produced by Penicillium sp. FO-4259-11. J. Antibiot. 2001, 53, 50–57. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, X.; Feng, S.; Jiang, G.; Luo, J.; Zhou, S. Five unique compounds: Xyloketals from mangrove fungus Xylaria sp. from the south china sea coast. J. Organomet. Chem. 2001, 66, 6252–6256. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, R.K.C.; Divakar, S.; Karanth, N.G.; Sattur, A. 14-(2’,3’,5’-Trihydroxyphenyl) tetradecan-2-ol, a novel acetylcholinesterase inhibitor from Chrysosporium sp. J. Antibiot. 2001, 54, 848–849. [Google Scholar]
- Paula, A.; Teles, C.; Takahashi, J.A. Paecilomide, a new acetylcholinesterase inhibitor from Paecilomyces lilacinus. Microbiol. Res. 2013, 168, 204–210. [Google Scholar]
- Wu, B.; Ohlendorf, B.; Oesker, V.; Wiese, J.; Malien, S.; Schmaljohann, R.; Imhoff, J.F. Acetylcholinesterase inhibitors from a marine fungus Talaromyces sp. strain LF458. Mar. Biotechnol. 2014, 17, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Dos, S.G.F.; Silva, L.G.; Pereira, O.G.; Souza, F.J.D.; Silva, A.L.; Rodrigues, F.E.; Takahashi, J.A. New AChE inhibitors from microbial transformation of trathechyloban-19-oic acid by Syncephalastrum racemosum. Bioorg. Chem. 2018, 79, 60–63. [Google Scholar]
- Silva, L.G.; Rocha, A.M.; Santos, G.F.; D’Silva, A.F.; Marriel, I.E.; Takahashi, J.A. Metabolic response of Aspergillus sydowii to OSMAC modulation produces acetylcholinesterase inhibitors. Phytochem. Lett. 2018, 24, 39–45. [Google Scholar] [CrossRef]
- Dai, Y.; Li, K.; She, J.; Zeng, Y.; Wang, H.; Liao, S.; Lin, X.; Yang, B.; Wang, J.; Tao, H.; et al. Lipopeptide epimers and a phthalide glycerol ether with AChE inhibitory activities from the marine-derived fungus Cochliobolus Lunatus SCSIO41401. Mar. Drugs 2020, 18, 547. [Google Scholar] [CrossRef]
- Xiao, Y.; Liang, W.; Liu, D.; Zhang, Z.; Chang, J.; Zhu, D. Isolation and acetylcholinesterase inhibitory activity of asterric acid derivatives produced by Talaromyces aurantiacus FL15, an endophytic fungus from Huperzia serrata. 3 Biotech 2022, 12, 60. [Google Scholar] [CrossRef]
- Wang, M.; Sun, M.; Hao, H.; Lu, C. Avertoxins A−D, prenyl asteltoxin derivatives from Aspergillus versicolor Y10, an endophytic fungus of Huperzia serrata. J. Nat. Prod. 2015, 78, 3067–3070. [Google Scholar] [CrossRef]
- Meng, X.; Mao, Z.; Lou, J.; Xu, L.; Zhong, L.; Peng, Y.; Zhou, L.; Wang, M. Benzopyranones from the endophytic fungus Hyalodendriella sp. Ponipodef12 and their bioactivities. Molecules 2012, 17, 11303–11314. [Google Scholar] [CrossRef]
- Huang, X.; Sun, X.; Ding, B.; Lin, M.; Liu, L.; Huang, H.; She, Z. A new anti-acetylcholinesterase α -pyrone meroterpene, arigsugacin I, from mangrove endophytic fungus Penicillium sp. sk5GW1L of Kandelia candel. Planta Med. 2013, 79, 1572–1575. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.B.; Feng, X.J.; Xiao, Z.E.; Liu, L.; Li, H.X.; Ma, L.; Lu, Y.J.; Ju, J.H.; She, Z.G.; Lin, Y.C. Azaphilones and p-Terphenyls from the mangrove endophytic fungus Penicillium chermesinum (ZH4-E2) isolated from the South China sea. J. Nat. Prod. 2011, 74, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.W.; Yang, Z.D.; Li, X.F.; Sun, J.H.; Yang, L.J.; Zhang, X.G. Colletotrichine B, a new sesquiterpenoid from Colletotrichum gloeosporioides GT-7, a fungal endophyte of Uncaria rhynchophylla. Nat. Prod. Res. 2019, 33, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Biasetto, C.R.; Somensi, A.; Sordi, R.; Chapla, V.M.; Ebrahimi, S.N.; Silva, G.H.; Teles, H.L.; Bolzani, V.S.; Young, M.C.M.; Pfenning, L.H.; et al. The new koninginins T-U from Phomopsis stipata, an endophytic fungus isolated from Styrax camporum Pohl. Phytochem. Lett. 2020, 36, 106–110. [Google Scholar] [CrossRef]
- Wang, Y.; Zeng, Q.G.; Zhang, Z.B.; Yan, R.M.; Wang, L.Y.; Zhu, D. Isolation and characterization of endophytic huperzine A-producing fungi from Huperzia serrata. J. Ind. Microbiol. Biotechnol. 2011, 38, 1267–1278. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Guo, H.; Wu, Q.; Yuan, S.; Liu, L. two new picolinederived meroterpenoids with anti-acetylcholinesterase activity from ascidian-derived fungus Amphichorda felina. Molecules 2022, 27, 5076. [Google Scholar] [CrossRef]
- Chapla, V.M.; Honório, A.E.; Gubiani, J.R.; Vilela, A.F.L.; Young, M.C.M.; Cardoso, C.L.; Pavan, F.R.; Cicarelli, R.M.; Michel, P.F.P.; Bolzani, V.S.; et al. Acetylcholinesterase inhibition and antifungal activity of cyclohexanoids from the endophytic fungus Saccharicola sp. Phytochem. Lett. 2020, 39, 116–123. [Google Scholar] [CrossRef]
- Deng, C.; Liu, S.; Huang, C.; Pang, J.; Lin, Y. Secondary metabolites of a mangrove endophytic fungus Aspergillus terreus (no. GX7-3B) from the South China Sea. Mar. Drugs 2013, 11, 2616–2624. [Google Scholar] [CrossRef]
- Ling, K.H.; Yang, C.K.; Peng, F.T. Territrems, tremorgenic mycotoxins of Aspergillus terreus. Appl. Environ. Microbiol. 1979, 37, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Deng, S.; Li, G.; Zhang, Y.; Wang, L.; Wu, C.; Deng, Y. Butenolide derivatives from Aspergillus terreus selectively inhibit butyrylcholinesterase. Front. Chem. 2022, 10, 1063284. [Google Scholar] [CrossRef]
- Qi, C.; Qiao, Y.; Gao, W.; Liu, Q.; Zhou, Q.; Chen, C.; Lai, Y.; Xue, Y.; Zhang, J.; Li, D.; et al. New 3,5-dimethylorsellinic acid-based meroterpenoids with BACE1 and AchE inhibitory activities from Aspergillus terreus. Org. Biomol. Chem. 2018, 16, 9046–9052. [Google Scholar] [CrossRef]
- Jeong, G.S.; Kang, M.G.; Han, S.A.; Noh, J.I.; Park, J.E.; Nam, S.J.; Park, D.; Yee, S.T.; Kim, H. Selective inhibition of human monoamine oxidase B by 5-hydroxy-2-methyl-chroman-4-one isolated from an endogenous lichen fungus Daldinia fissa. J. Fungi 2021, 7, 84. [Google Scholar] [CrossRef] [PubMed]
- Nong, X.H.; Wang, Y.F.; Zhang, X.Y.; Zhou, M.P.; Xu, X.Y.; Qi, S.H. Territrem and butyrolactone derivatives from a marine-derived fungus Aspergillus terreus. Mar. Drugs 2014, 12, 6113–6124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Tanzi, R.E. Natural modulators of amyloid-beta precursor protein processing. Curr. Alzheimer Res. 2012. [Google Scholar]
- Harun, A.; James, R.M.; Lim, S.M.; Abdul, M.A.B.; Cole, A.L.; Ramasamy, K. BACE1 inhibitory activity of fungal endophytic extracts from Malaysian medicinal plants. BMC Complement. Altern. Med. 2011, 11, 79. [Google Scholar] [CrossRef]
- Qi, C.; Liu, M.; Zhou, Q.; Gao, W.; Chen, C.; Lai, Y.; Hu, Z.; Xue, Y.; Zhang, J.; Li, D.; et al. BACE1 inhibitory meroterpenoids from Aspergillus terreus. J. Nat. Prod. 2018, 81, 1937–1945. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Wu, Y.; Qiao, Y.; Guo, Y.; Wang, J.; Hu, Z.; Zhang, Q.; Li, X.; Huang, J.; Zhou, Q.; et al. Protoilludane, Illudalane, and botryane sesquiterpenoids from the endophytic fungus Phomopsis sp. TJ507A. J. Nat. Prod. 2018, 81, 1311–1320. [Google Scholar] [CrossRef]
- Park, I.H.; Jeon, S.Y.; Lee, H.J.; Kim, S.I.; Song, K.S. A beta-secretase (BACE1) inhibitor hispidin from the mycelial cultures of Phellinus linteus. Planta Med. 2004, 70, 143–146. [Google Scholar]
- Sorribas, A.; Jiménez, J.I.; Yoshida, W.Y.; Williams, P.G. Daedalols A-C, fungal-derived BACE1 inhibitors. Bioorg. Med. Chem. 2011, 19, 6581–6586. [Google Scholar] [CrossRef]
- Mithun, S.R. Natural monoamine oxidase inhibitors: A review. J. Pharm. Res. 2010, 3, 482–485. [Google Scholar]
- Lee, H.W.; Choi, H.; Nam, S.J.; Fenical, W.; Kim, H. Potent inhibition of monoamine oxidase B by a piloquinone from marine-derived Streptomyces sp. CNQ-027. J. Microbiol. Biotechnol. 2017, 27, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Hong, A.; Tu, L.C.; Yang, I.; Lim, K.M.; Nam, S.J. Marine natural products with monoamine oxidase (MAO) inhibitory activity. Pharm. Biol. 2020, 58, 716–720. [Google Scholar] [CrossRef] [PubMed]
- Chaurasiya, N.D.; Leon, F.; Muhammad, I.; Tekwani, B.L. Natural products inhibitors of monoamine oxidases—Potential new drug leads for neuroprotection, neurological disorders, and neuroblastoma. Molecules 2022, 27, 4297. [Google Scholar] [CrossRef]
- Zhao, Q.H.; Yang, Z.D.; Shu, Z.M.; Wang, Y.G.; Wang, M.G. Secondary metabolites and biological activities of Talaromyces sp. LGT-2, an endophytic fungus from Tripterygium wilfordii. Iran J. Pharm. Res. 2016, 15, 453–457. [Google Scholar] [PubMed]
- Yamazaki, M.; Satoh, Y.; Maebayashi, Y.; Horie, Y. Monoamine oxidase inhibitors from a fungus, Emericella navahoensis. Chem. Pharm. Bull. 1988, 36, 670–675. [Google Scholar] [CrossRef]
- Satoh, Y.; Yamazaki, M. Studies on the monoamine oxidase (MAO) inhibitory potency of TL-1, isolated from a fungus, Talaromyces luteus. Chem. Pharm. Bull. 1989, 37, 206–207. [Google Scholar] [CrossRef]
- Lee, H.W.; Jung, W.K.; Kim, H.J.; Jeong, Y.S.; Nam, S.J.; Kang, H.; Kim, H. Inhibition of monoamine oxidase by anithiactins from Streptomyces sp. J. Microbiol. Biotechnol. 2015, 25, 1425–1428. [Google Scholar] [CrossRef]
- Jeong, G.S.; Lee, E.Y.; Kang, M.G.; Nam, S.J.; Park, D.; Kim, H. (S)-5-Methylmellein isolated from an endogenous lichen fungus Rosellinia corticium as a potent inhibitor of human monoamine oxidase A. Processes 2022, 10, 166. [Google Scholar] [CrossRef]
- Wiese, J.; Imhoff, J.F.; Gulder, T.A.M.; Labes, A.; Schmaljohann, R. Marine fungi as producers of benzocoumarins, a new class of inhibitors of glycogen-synthase-kinase 3β. Mar. Drugs 2016, 14, 200. [Google Scholar] [CrossRef]
- Wu, B.; Wiese, J.; Schmaljohann, R.; Imhoff, J. Biscogniauxone, a new isopyrrolonaphthoquinone compound from the fungus Biscogniauxia mediterranea isolated from deep-sea Sediments. Mar. Drugs 2016, 14, 204. [Google Scholar] [CrossRef]
- Crocetti, L.; Floresta, G.; Cilibrizzi, A.; Giovannoni, M.P. An overview of PDE4 inhibitors in clinical trials: 2010 to early 2022. Molecules 2022, 27, 4964. [Google Scholar] [CrossRef] [PubMed]
- El-Elimat, T.; Figueroa, M.; Raja, H.A.; Graf, T.N.; Adcock, A.F.; Kroll, D.J.; Day, C.S.; Wani, M.C.; Pearce, C.J.; Oberlies, N.H. Benzoquinones and terphenyl compounds as phosphodiesterase-4B inhibitors from a fungus of the order Chaetothyriales (MSX 47445). J. Nat. Prod. 2013, 76, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Abulaizi, A.; Huang, L.; Xiong, Z.; Zhang, S.; Liu, T.; Wang, R. Discovery of p-terphenyl metabolites as potential phosphodiesterase PDE4D inhibitors from the coral-associated fungus Aspergillus sp. ITBBc1. Mar. Drugs 2022, 20, 679. [Google Scholar] [CrossRef]
- Zhou, B.; Qin, L.L.; Ding, W.J.; Ma, Z.J. Cytotoxic indolocarbazoles alkaloids from the Streptomyces sp. A65. Tetrahedron 2018, 74, 726–730. [Google Scholar] [CrossRef]
- Qin, L.L.; Zhou, B.; Ding, W.; Ma, Z. Bioactive metabolites from marine-derived Streptomyces sp. A68 and its Rifampicin resistant mutant strain R-M1. Phytochem. Lett. 2018, 23, 46–51. [Google Scholar] [CrossRef]
- Wang, J.N.; Zhang, H.J.; Li, J.Q.; Ding, W.J.; Ma, Z.J. Bioactive indolocarbazoles from the marine-derived Streptomyces sp. DT-A61. J. Nat. Prod. 2018, 81, 949–956. [Google Scholar] [CrossRef]
- Cheng, X.; Zhou, B.; Liu, H.; Huo, C.; Ding, W. One new indolocarbazole alkaloid from the Streptomyces sp. A22. Nat. Prod. Res. 2018, 32, 2583–2588. [Google Scholar] [CrossRef]
- Uddin, M.S.; Al Mamun, A.; Kabir, M.T.; Ashraf, G.M.; Bin, J.M.N.; Abdel, D.M.M. Multi-target drug candidates for multifactorial Alzheimer’s disease: AChE and NMDAR as molecular targets. Mol. Neurobiol. 2021, 58, 281–303. [Google Scholar] [CrossRef]
- Wang, N.; Qiu, P.; Cui, W.; Yan, X.; Zhang, B.; He, S. Recent advances in multi-target anti-Alzheimer disease compounds (2013 up to the present). Curr. Med. Chem. 2019, 26, 5684–5710. [Google Scholar] [CrossRef]
- Gong, C.X.; Dai, C.L.; Liu, F.; Iqbal, K. Multi-targets: An unconventional drug development strategy for Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 837649. [Google Scholar] [CrossRef]
- Dubey, S.K.; Ram, M.S.; Krishna, K.V.; Saha, R.N.; Singhvi, G.; Agrawal, M.; Ajazuddin, S.S.; Saraf, S.; Alexander, A. Recent expansions on cellular models to uncover the scientific barriers towards drug development for Alzheimer’s disease. Cell. Mol. Neurobiol. 2019, 39, 181–209. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.; Mailankody, S. Research and development spending to bring a single cancer drug to market and revenues after approval. JAMA Int. Med. 2017, 177, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Miles, J.A.; Ross, B.P. Recent advances in virtual screening for cholinesterase inhibitors. ACS Chem. Neurosci. 2021, 12, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Ioppolo, A.; Eccles, M.; Groth, D.; Verdile, G.; Agostino, M. Evaluation of virtual screening strategies for the identification of γ-secretase inhibitors and modulators. Molecules 2022, 27, 176. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhao, J.; Wang, Z.; Guo, P.; Liu, A.; Du, G. Identification of multi-target anti-AD chemical constituents from traditional chinese medicine formulae by integrating virtual screening and in vitro validation. Front. Pharmacol. 2021, 12, 709607. [Google Scholar] [CrossRef]
- Nandha, D.E.; Anantha, K.D.; Krishnasamy, G.; Ramesh, K.; Devaraj, S. Screening of potential drug for Alzheimer’s disease: A computational study with GSK-3 β inhibition through virtual screening, docking, and molecular dynamics simulation. J. Biomol. Struct. Dyn. 2021, 39, 7065–7079. [Google Scholar]
- Chandra, B.T.M.; Rajesh, S.S.; Bhaskar, B.V.; Devi, S.; Rammohan, A.; Sivaraman, T.; Rajendra, W. Molecular docking, moleculardynamics simulation, biological evaluation and 2D QSAR analysis of flavonoids from Syzygium alternifolium as potent anti-Helicobacter pylori agents. RSC Adv. 2017, 7, 18277–18292. [Google Scholar] [CrossRef]
- Tran, T.P.T.; Thanh, Q.B.; Phan, T.Q.; Nguyen, C.B.; Tran, V.L.; Tran, V.C.; Nguyen, L.C.; Nguyen, V.T.; Tran, V.S.; Nguyen, T.A.N. Isolation, semi-synthesis, docking-based prediction, and bioassay-based activity of Dolichandrone spathacea iridoids: Newcatalpol derivatives as glucosidase inhibitors. RSC Adv. 2021, 11, 11959–11975. [Google Scholar]
- Ding, Y.; Fang, Y.; Moreno, J.; Ramanujam, J.; Jarrell, M.; Brylinski, M. Assessing the similarity of ligand binding conformations with the contact mode score. Comput. Biol. Chem. 2016, 64, 403–413. [Google Scholar] [CrossRef]
- Trinh, T.H.T.; Wang, S.-L.; Nguyen, V.B.; Phan, T.Q.; Doan, M.D.; Tran, T.P.H.; Nguyen, T.H.; Le, T.A.H.; Ton, T.Q.; Nguyen, A.D. Novel nematocidal compounds from shrimp shell wastes valorized by Bacillus velezensis RB.EK7 against black pepper nematodes. Agronomy 2022, 12, 2300. [Google Scholar] [CrossRef]
- Walters, W.P.; Wang, R. New trends in virtual screening. J. Chem. Inf. Model. 2020, 60, 4109–4111. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.S.F.L.; Olanda, C.G.; Fokoue, H.H.; Sant’Anna, C.M.R. Virtual screening techniques in drug discovery: Review and recent applications. Curr. Top Med. Chem. 2019, 19, 1751–1767. [Google Scholar]














| Strain | Compound | Bioactivity (IC50) | Ref. | |
|---|---|---|---|---|
| AchE | BuChE | |||
| Bacteria | ||||
| Serratia marcescens CC17 | Prodigiosin (1) | 640 µg/mL | ND | [62] |
| Actinobacteria | ||||
| Streptomyces sp. LB173 | Geranyl-phenazine-diol (2) | 2.62 µM | ND | [63] |
| Rubrobacter radiotolerans | 2-(2-(3-hydroxy-1-(1H-indol-3-yl)-2methoxypropyl) -1H-indol-3-yl) acetic acid (3) | 11.8 µM | ND | [64] |
| 3-(3-(2-hydroxyethyl)-1H-indol-2-yl) (4) | 13.5 µM | ND | ||
| Streptomyces sp. AH-14 | Physostigmine (5) | 41 μM | ND | [65] |
| Streptomyces lavendulae NK901093 | Cyclophostin (6) | 7.6 × M | ND | [66] |
| Streptomyces sp. UTMC 1334 | Pyrrole derivatives (extract) | 360 µg/mL | ND | [67] |
| Cyanobacteria | ||||
| Nostoc 78-12A | Nostocarboline (7) | ND | 13.2 μM | [68] |
| Strain | Compound | Bioactivity (IC50) | Ref. | |
|---|---|---|---|---|
| AchE | BuChE | |||
| Penicillium citrinum | Quinolactacin A1 (8) | 280 µM | - | [69] |
| Quinolactacin A2 (9) | 19.8 µM | 650 µM | ||
| Acremonium persicinum KUFA 1007 | Acremine S (10) | - | - | [70] |
| Acremine T (11) | - | - | ||
| Lumichrome (12) | 12.24 µM | - | ||
| Penicillium sp. FO-4259 | Arisugacin (13) | 1 nM | >18,000 nM | [71] |
| Territrem B (14) | 7.6 nM | >20,000 nM | ||
| Territrem C (15) | 6.8 nM | >26,000 nM | ||
| Penicillium sp. FO-4259 | Arisugacin A (16) | 0.001 nM | >21 nM | [72] |
| Arisugacin B (17) | 0.02 nM | >516 nM | ||
| Arisugacin C (18) | 2.5 nM | >30 nM | ||
| Arisugacin D (19) | 3.5 nM | >30 nM | ||
| Arisugacin E (20) | >100 nM | >30 nM | ||
| Arisugacin F (21) | >100 nM | >30 nM | ||
| Arisugacin G (22) | >100 nM | >30 nM | ||
| Arisugacin H (23) | >100 nM | >30 nM | ||
| Xylaria sp. | Xyloketal A (24) | 1.5 × 10−6 mol/L | ND | [73] |
| Chrysosporium sp. | 14-(2′,3′,5′- Trihydroxyphenyl) tetradecan-2-ol (25) | 197 µM | ND | [74] |
| 195 µM | ND | |||
| 231 µM | ND | |||
| Paecilomyces lilacinus | Paecilomide (26) | IR = 57.5% | ND | [75] |
| Talaromyces sp. LF458 | Talaromycesone A (27) | 7.49 μM | ND | [76] |
| Talaroxanthenone (28) | 1.61 μM | ND | ||
| AS-186c (29) | 2.60 μM | ND | ||
| Syncephalastrum racemosum | Trachyloban-19-oic acid (30) | >1 µM | ND | [77] |
| Aspergillus sydowii | Cyclo-(l-leu-l-pro) (31) | 1.24 μmol/mL | ND | [78] |
| Cyclo-(l-val-l-pro) (32) | 1.06 μmol/mL | ND | ||
| Cyclo-(l-phe-l-val) (33) | 1.13 μmol/mL | ND | ||
| Cochliobolus lunatus SCSIO4140 | Sinulariapeptide A (34) | 1.8 µM | ND | [79] |
| Sinulariapeptide B (35) | 1.3 µM | ND | ||
| Phthalide glycerol (36) | 2.5 µM | ND | ||
| Talaromyces aurantiacus FL15 | Asterric acid (37) | 66.7 µM | >100 µM | [80] |
| Methyl asterrate (38) | 23.3 µM | >100 µM | ||
| Ethyl asterrate (39) | 20.1 µM | >100 µM | ||
| Emodin (40) | >100 µM | >100 µM | ||
| Physcion (41) | >100 µM | >100 µM | ||
| Chrysophanol (42) | >100 µM | >100 µM | ||
| Sulochrin (43) | >100 µM | >100 µM | ||
| Aspergillus versicolor Y10 | Avertoxin B (44) | 14.9 μM | ND | [81] |
| Hyalodendriella sp. Ponipodef12 | Palmariol B (45) | 115.31 µg/mL | ND | [82] |
| 4-hydroxymellein (46) | 116.05 µg/mL | ND | ||
| Alternariol 9-methyl ether (47) | 135.52 µg/mL | ND | ||
| Botrallin (48) | 103.7 µg/mL | ND | ||
| Penicillium sp. sk5GW1L | Arigsugacin I (49) | 0.64 µM | ND | [83] |
| Arigsugacin F (50) | 0.37 µM | ND | ||
| Territrem B (14) | 7.03 µM | ND | ||
| Penicillium chermesinum (ZH4-E2) | 3″-deoxy-6′ -O-desmethylcandidusin B (51) | 7.8 μM | ND | [84] |
| 6′-O-desmethylcandidusin B (52) | 5.2 μM | ND | ||
| Colletotrichum gloeosporioides | Colletotrichine B (53) | 38 µg/mL | ND | [85] |
| Phomopsis stipata | Koninginin T (54) | 10.0 μg | ND | [86] |
| Aspergillus flavus LF40 | Huperzine A (55) | 0.6 μM | ND | [87] |
| Amphichorda felina | Amphichoterpenoid D (56) | 12.5 µM | ND | [88] |
| Amphichoterpenoid E (57) | 11.6 µM | ND | ||
| Saccharicola sp. | Speciosin U (58) | 0.026 mg/mL | ND | [89] |
| Trans-3,4-dihydro-3,4-dihydroxy-anofinic acid (59) | 0.053 mg/mL | ND | ||
| Aspergillus terreus | Anhydrojavanicin (60) | 2.01 µM | ND | [90] |
| 8-O-methylbostrycoidin (61) | 6.71 µM | ND | ||
| NGA0187 (62) | 1.89 µM | ND | ||
| Beauvericin (63) | 3.09 µM | ND | ||
| Aspergillus terreus | Territrem B (14) | 7.6 μM | ND | [91] |
| Aspergillus terreus SGP-1 | Asperteretal J (64) | IR = 11.2% | IR = 15.6% | [92] |
| Asperteretal K (65) | IR = 7.7% | IR = 3.2% | ||
| Flavipesolide B (66) | IR = 19.6% | IR = 22.5% | ||
| Butyrolactone VIII (67) | IR = 11.9% | IR = 67.3% Ki = 23.6 µM | ||
| Versicolactone B (68) | IR = 10.4% | IR = 64.4% Ki = 38.2 µM | ||
| Butyrolactone I (69) | - | IR = 68% Ki = 19.3 µM | ||
| Butyrolactone VII (70) | IR = 11.7% | IR = 76% Ki = 12.3 µM | ||
| 3-hydroxy-5-[[4-hydroxy-3-(3-methyl-2-buten-1-yl) phenyl] methyl] -4- (4-hydroxyphenyl)-2(5H)-furanone (71) | IR = 14.2% | IR = 60.9% Ki = 35.7 µM | ||
| Butyrolactone II (72) | IR = 9.6% | IR = 19.9% | ||
| 5-[(3,4-dihydro-2,2-dimethyl-2H-1-benzopyran-6-yl)methyl]-3-hydroxy-4-(4-hydroxyphenyl)-2(5H)-furanone (73) | IR = 23.2% | IR = 33.3% | ||
| Aspernolide A (74) | - | IR = 25.4% | ||
| Aspernolide B (75) | IR = 2.2% | IR = 13.4% | ||
| Aspernolide C (76) | IR = 3.9% | IR = 22.5% | ||
| Butyrolactone III (77) | - | - | ||
| Butytolactone IV (78) | IR = 13.3% | IR = 33.5% | ||
| Aspergillus terreus | Terreusterpene A (79) | >40 µM | ND | [93] |
| Terreusterpene B (80) | >40 µM | ND | ||
| Terreusterpene C (81) | >40 µM | ND | ||
| Terreusterpene D (82) | 8.86 µM | ND | ||
| Daldinia fissa | 5-hydroxy-2-methyl-chroman-4-one (83) | >40 µM | >40 µM | [94] |
| Aspergillus Terreus | Territrem D (84) | 4.2 nM | ND | [95] |
| Territrem E (85) | 4.5 nM | ND | ||
| Territrem B (14) | 4.2 nM | ND | ||
| Territrem C (86) | 20.1 nM | ND | ||
| Arisugacin A (16) | 11.9 nM | ND | ||
| Arisugacin H (23) | 5700 nM | ND | ||
| Terreulactone C (87) | 50 nM | ND | ||
| Strain | Enz. | Inhibitor | Experimental Activity | DS (kcal/mol) | RMSD (Å) | Linkage (Bonds) | Amino Acid Interaction | Ref. |
|---|---|---|---|---|---|---|---|---|
| S.marcescens CC17 | AChE from Electrophorus electricus (Structure: DOI:10.2210/pdb1GQR/pdb) | Cation-prodigiosin | 640 µg/mL | −12.3 | 1.35 | 6 | Asp326, Asp326, Asp393, Asp393, Lys325, Asp393 | [62] |
| Neutral-prodigiosin | −11.1 | 1.75 | 3 | Trp84, Trp84, Gly118 | ||||
| A. felina | AChE from Electrophorus electricus (PDB ID: 1QTI) | Amphichoterpenoid D | 12.5 µM | −9.3 | ND | 3 | Arg289, Phe288 | [88] |
| Amphichoterpenoid E | 11.6 µM | −9.3 | ND | 3 | Arg28, Tyr121 | |||
| (+)Amphichoterpenoids A | ND | −7.9 | ND | 2 | Leu305, Glu306 | |||
| (−)Amphichoterpenoids A | ND | −6.8 | ND | 0 | None | |||
| A.sterreus SGP-1 | BChE from equine serum (EC 3.1.1.8) | Butyrolactone I | 35.5 µM | −41.28 | ND | >7 | Gly116, Trp82, Trp231, Leu286, Val288, Phe398, Pro285, His438, | [92] |
| Butyrolactone VII | 18.4 µM | −48.85 | ND | >7 | Gly116, Trp82, Trp231, Leu286, Val288, Phe398, His438, Ala328, | |||
| D. fissa | MAO-A from human recombinant (PDB ID:2Z5X) | 5-hydroxy-2-methyl-chroman-4-one | 13.9 μM | −6.1 | ND | ND | - | [94] |
| MAO-B from human recombinant (PDB ID: 4A79) | 3.2 μM | −7.3 | ND | ND | Cys172 | |||
| R. corticium | MAO-A from human recombinant (PDB ID: 2Z5X) | (S)-5-methylmellein | 5.31 μM | −6.8 | ND | ND | Phe208 | [110] |
| MAO-B from human recombinant (PDB ID: 3PO7) | 9.15 μM | −6.4 | ND | ND | - | |||
| MAO-A from human recombinant (PDB ID: 2Z5X) | (R)-5-methylmellein | ND | −6.6 | ND | ND | Asn181 | ||
| MAO-B from human recombinant (PDB ID: 3PO7) | ND | −5.2 | ND | ND | - | |||
| Phoma sp. | PDE4B from human | Betulinan A | 44 μM | −8.071 | ND | ND | Phe446 | [114] |
| BTH-II0204-207:A | 31 μM | −8.277 | ND | ND | Phe446 | |||
| Betulinan C | 17 μM | −8.732 | ND | ND | Gln443, Phe446 | |||
| B.veleznesis RB.EK7 | AChE Electrophorus electricus (Structure: DOI:10.2210/pdb1GQR/pdb) | Thymine | ND | −7.0 | 1.35 | 4 | Asp182, Lys51, Asn183, Trp179 | [132] |
| Hexahydropyrrolo [1,2-a]pyra-zine-1,4-dione | ND | −6.89 | 1.02 | 3 | Met175, Phe35, Lys51 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.H.; Wang, S.-L.; Nguyen, V.B. Microorganism-Derived Molecules as Enzyme Inhibitors to Target Alzheimer’s Diseases Pathways. Pharmaceuticals 2023, 16, 580. https://doi.org/10.3390/ph16040580
Nguyen TH, Wang S-L, Nguyen VB. Microorganism-Derived Molecules as Enzyme Inhibitors to Target Alzheimer’s Diseases Pathways. Pharmaceuticals. 2023; 16(4):580. https://doi.org/10.3390/ph16040580
Chicago/Turabian StyleNguyen, Thi Hanh, San-Lang Wang, and Van Bon Nguyen. 2023. "Microorganism-Derived Molecules as Enzyme Inhibitors to Target Alzheimer’s Diseases Pathways" Pharmaceuticals 16, no. 4: 580. https://doi.org/10.3390/ph16040580
APA StyleNguyen, T. H., Wang, S.-L., & Nguyen, V. B. (2023). Microorganism-Derived Molecules as Enzyme Inhibitors to Target Alzheimer’s Diseases Pathways. Pharmaceuticals, 16(4), 580. https://doi.org/10.3390/ph16040580