
Regulation of Hypoxic–Adenosinergic Signaling by Estrogen: Implications for Microvascular Injury


 ,
,
Abstract
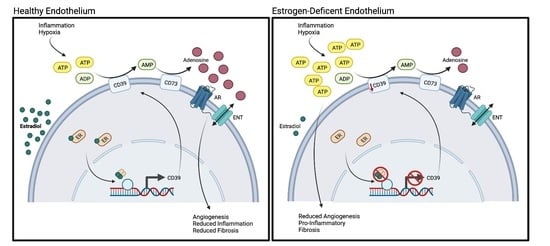
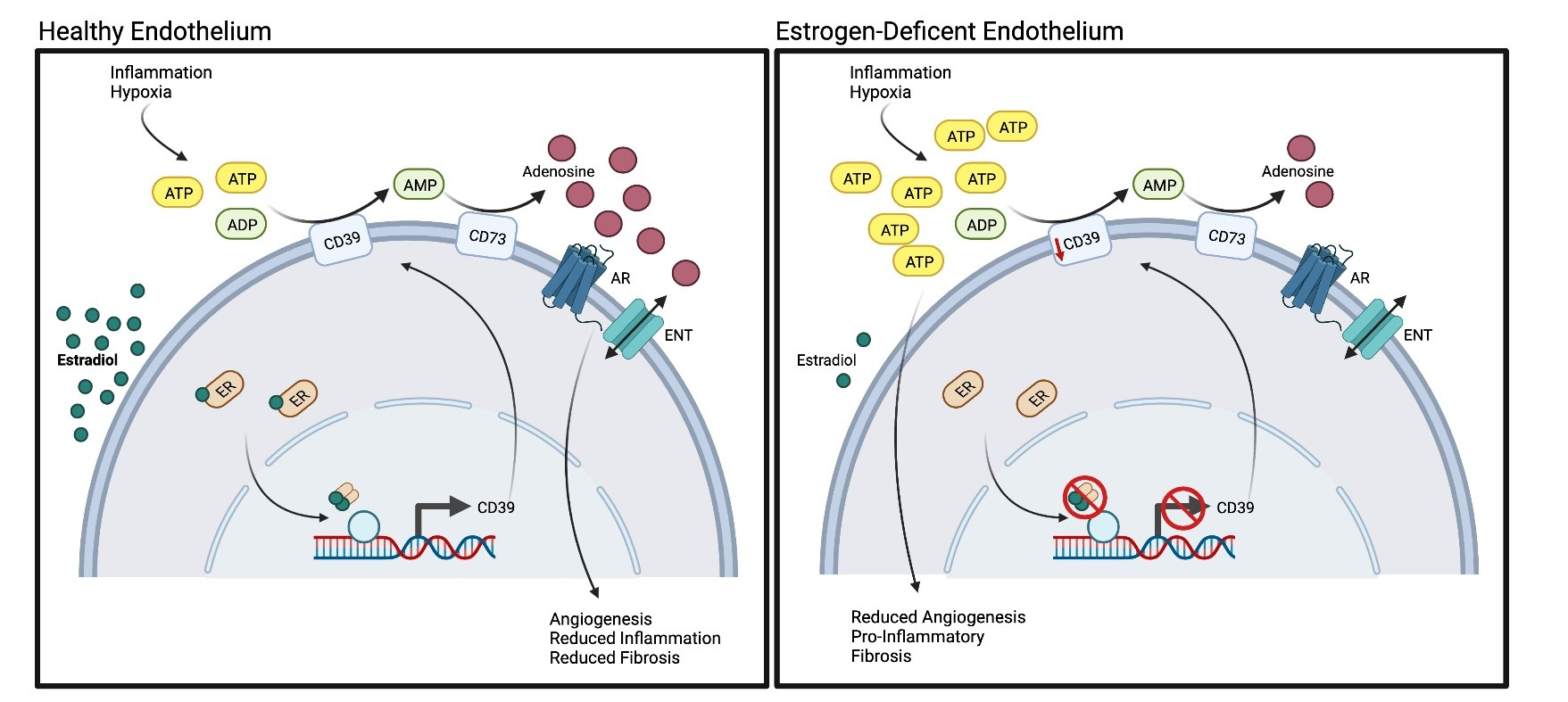
1. Introduction
2. Results
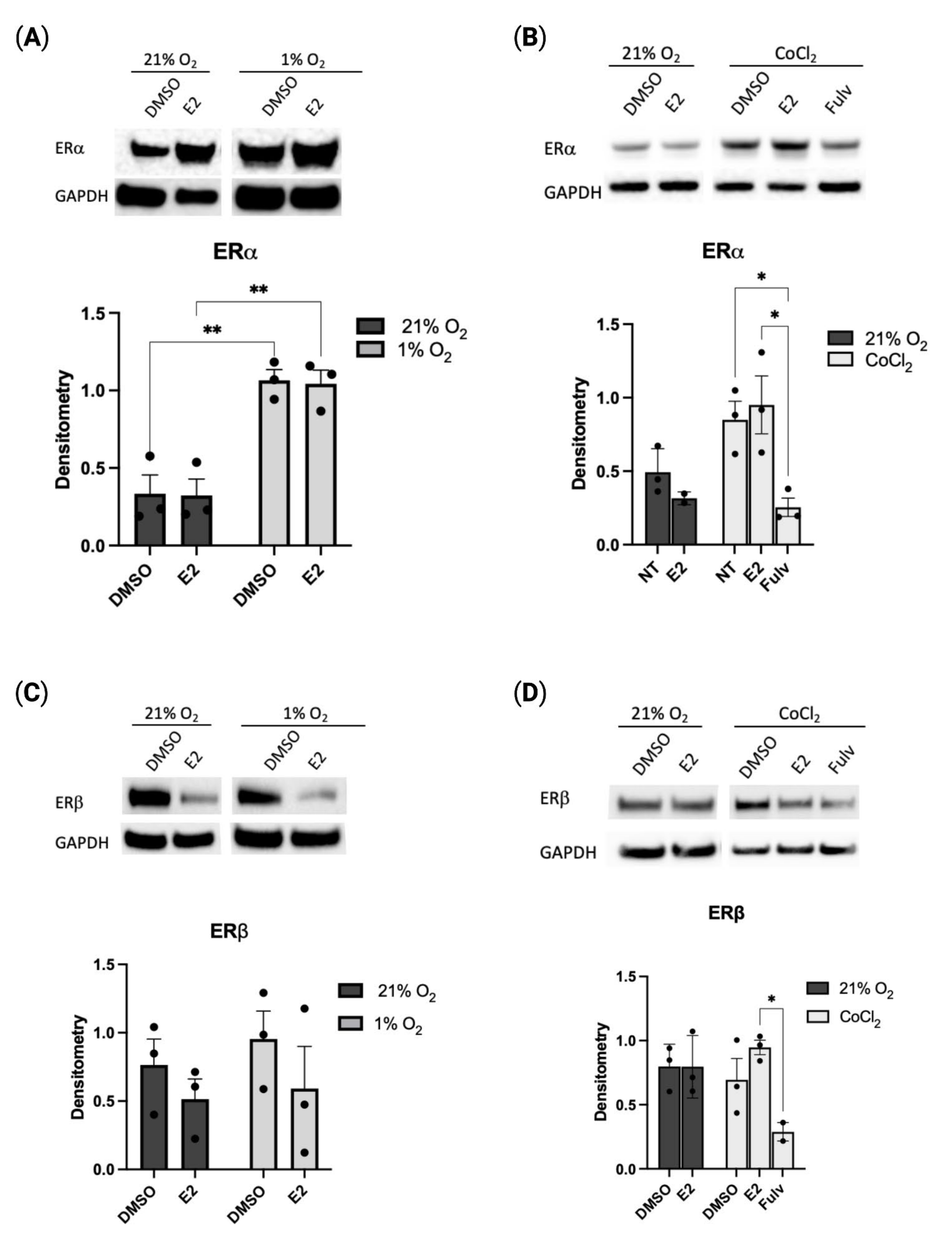
2.1. Hypoxia Regulates Estrogen Receptor Alpha Expression
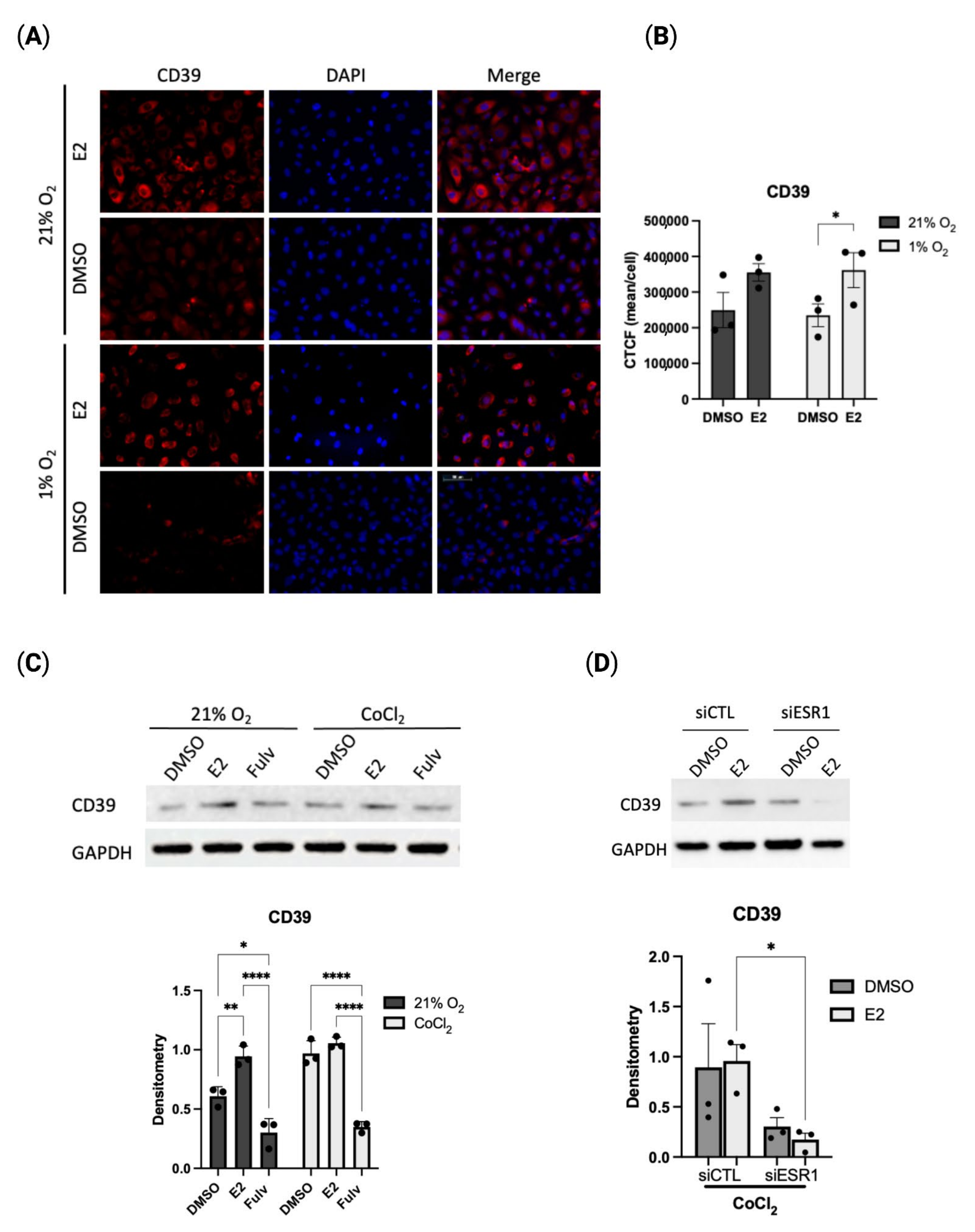
2.2. Estradiol Positively Regulates CD39 Levels in Hypoxia
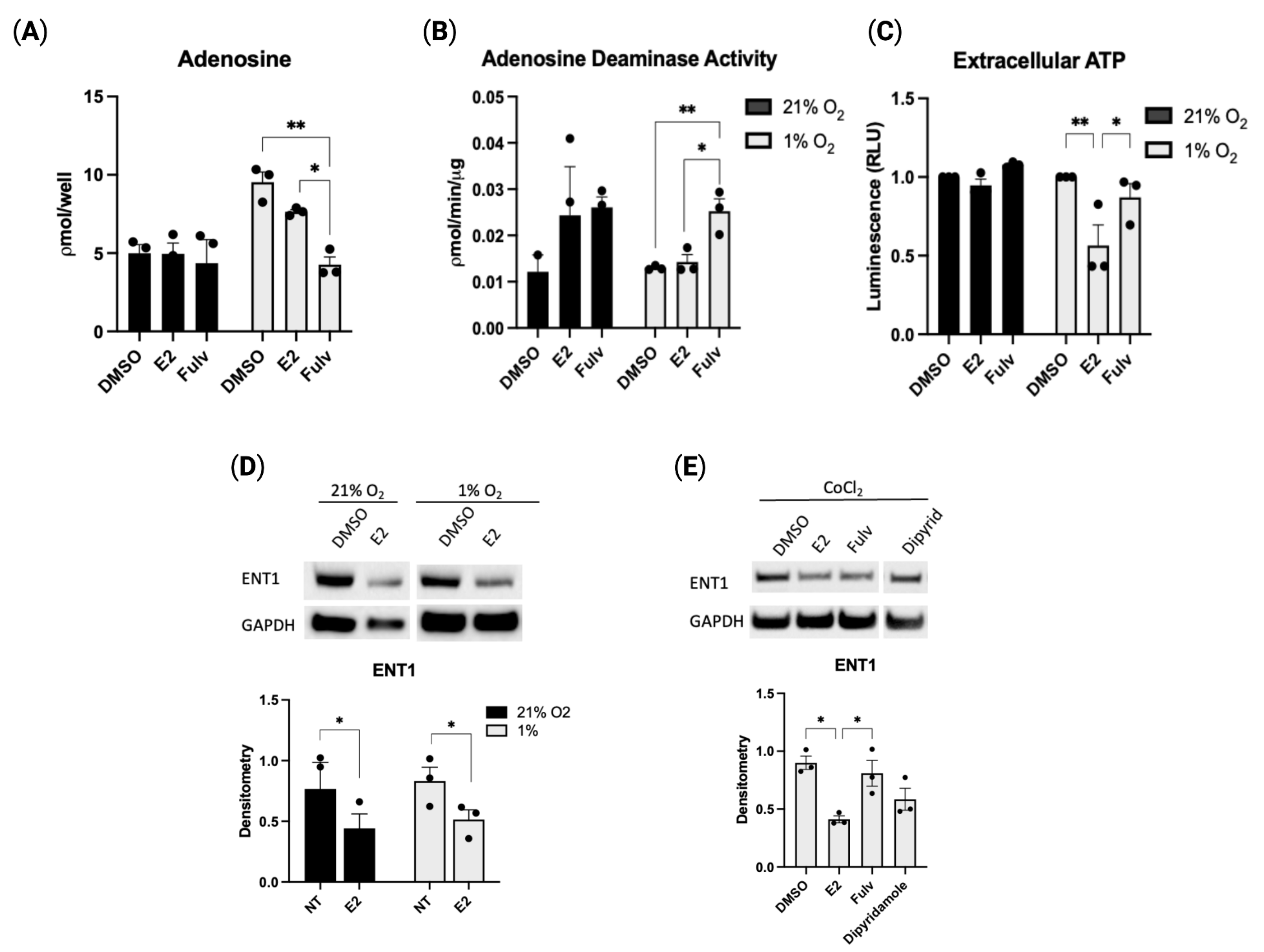
2.3. Estradiol-Mediated CD39 Activity Regulates Purinergic Signaling
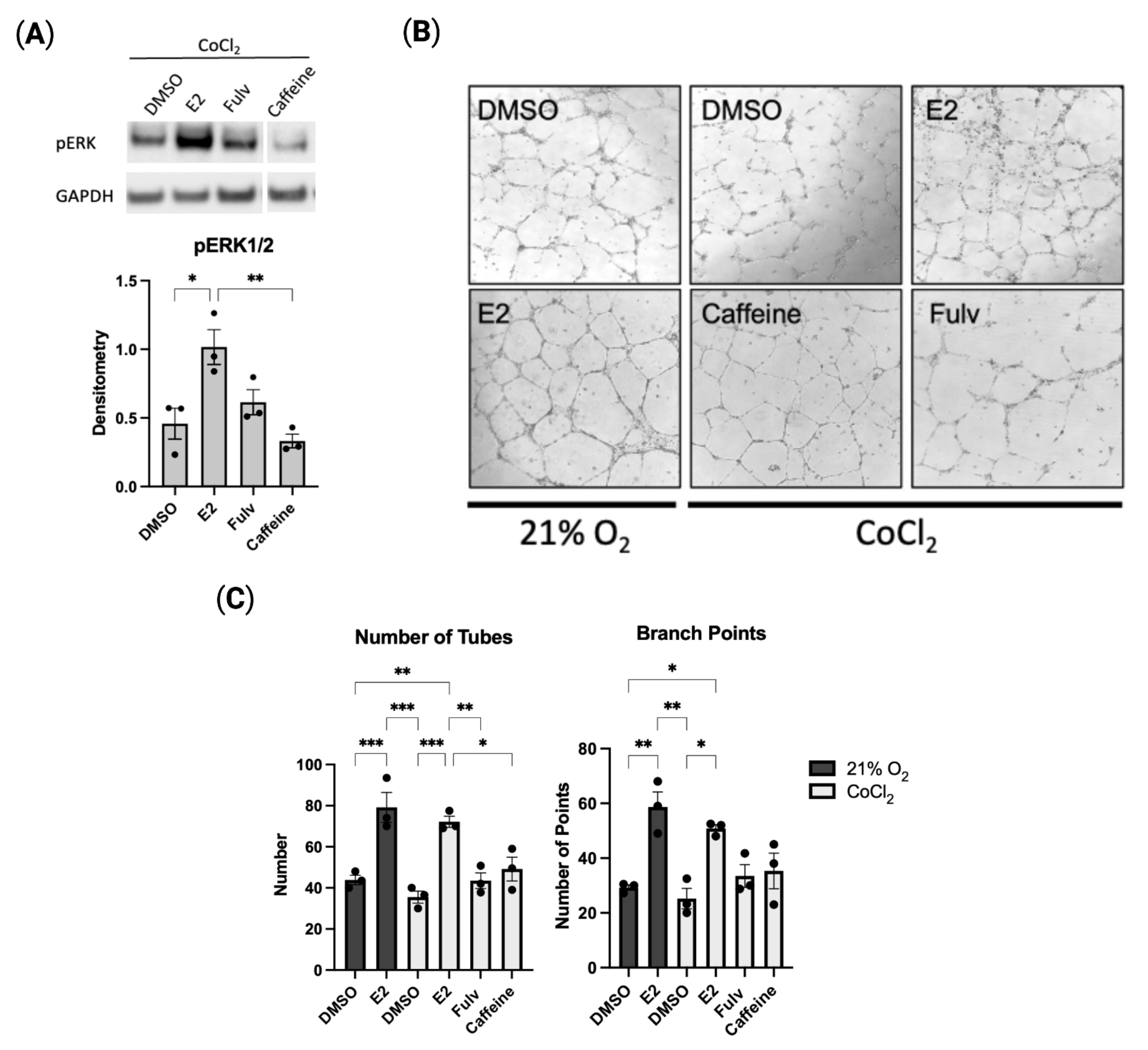
2.4. Estradiol Promotes In Vitro Angiogenesis in Hypoxic HUVECs
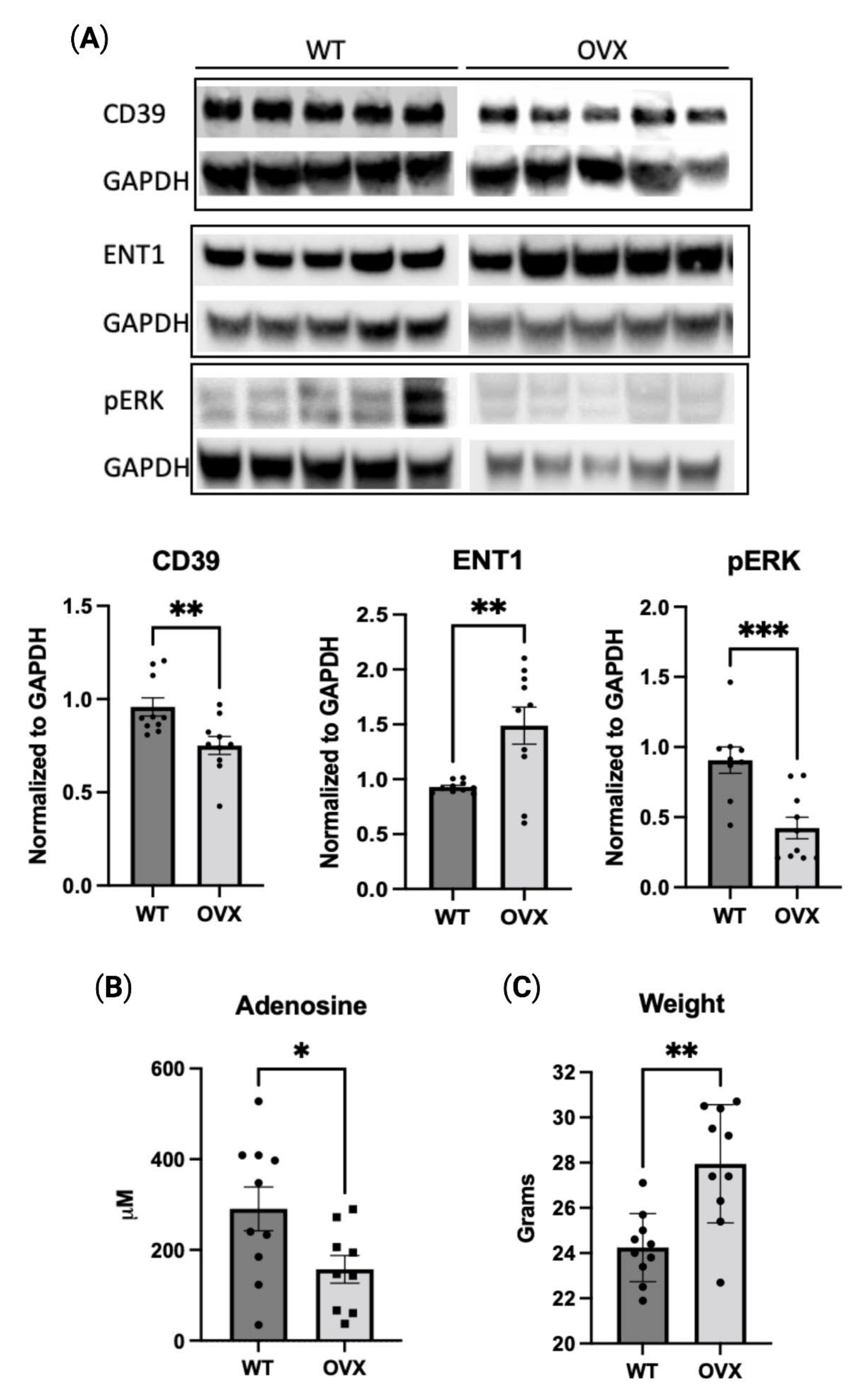
2.5. Estrogen Deficiency Reduces Purinergic Signaling In Vivo
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Patel, H.; Aggarwal, N.T.; Rao, A.; Bryant, E.; Sanghani, R.M.; Byrnes, M.; Kalra, D.; Dairaghi, L.; Braun, L.; Gabriel, S.; et al. Microvascular Disease and Small-Vessel Disease: The Nexus of Multiple Diseases of Women. J. Women’s Health 2020, 29, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Camici, P.G.; Crea, F. Coronary Microvascular Dysfunction. N. Engl. J. Med. 2007, 356, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Taqueti, V.R.; di Carli, M.F. Coronary Microvascular Disease Pathogenic Mechanisms and Therapeutic Options: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 2625. [Google Scholar] [CrossRef] [PubMed]
- Stanhewicz, A.E.; Wenner, M.M.; Stachenfeld, N.S. Sex differences in endothelial function important to vascular health and overall cardiovascular disease risk across the lifespan. Am. J. Physiol. Circ. Physiol. 2018, 315, H1569–H1588. [Google Scholar] [CrossRef] [PubMed]
- Beale, A.L.; Meyer, P.; Marwick, T.H.; Lam, C.S.; Kaye, D.M. Sex Differences in Cardiovascular Pathophysiology: Why Women Are Overrepresented in Heart Failure with Preserved Ejection Fraction. Circulation 2018, 138, 198–205. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef]
- Bairey Merz, C.N.; Shaw, L.J.; Reis, S.E.; Bittner, V.; Kelsey, S.F.; Olson, M.; Johnson, D.B.; Pepine, C.J.; Mankad, S.; Sharaf, B.L.; et al. Insights from the NHLBI-sponsored Women’s Ischemia Syndrome Evaluation (WISE) study. Part II: Gender differences in presentation, diagnosis, and outcome with regard to gender-based pathophysiology of atherosclerosis and macrovascular and microvascular coronary disease. J. Am. Coll. Cardiol. 2006, 47, S21–S29. [Google Scholar]
- Sharma, K.; Kass, D.A. Heart failure with preserved ejection fraction: Mechanisms, clinical features, and therapies. Circ. Res. 2014, 115, 79–96. [Google Scholar] [CrossRef]
- Sickinghe, A.A.; Korporaal, S.J.A.; Ruijter, H.M.D.; Kessler, E.L. Estrogen Contributions to Microvascular Dysfunction Evolving to Heart Failure with Preserved Ejection Fraction. Front. Endocrinol. 2019, 10, 442. [Google Scholar] [CrossRef]
- Klinge, C.M. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001, 29, 1299–2905. [Google Scholar] [CrossRef]
- Chambliss, K.L.; Wu, Q.; Oltmann, S.; Konaniah, E.S.; Umetani, M.; Korach, K.S.; Thomas, G.D.; Mineo, C.; Yuhanna, I.S.; Kim, S.H.; et al. Non-nuclear estrogen receptor α signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J. Clin. Investig. 2010, 120, 2319–2330. [Google Scholar] [CrossRef]
- Imai, M.; Kaczmarek, E.; Koziak, K.; Sévigny, J.; Goepfert, C.; Guckelberger, O.; Csizmadia, E.; Esch, J.S.A.; Robson, S.C. Suppression of ATP Diphosphohydrolase/CD39 in Human Vascular Endothelial Cells. Biochemistry 1999, 38, 13473–13479. [Google Scholar] [CrossRef]
- Burnstock, G. Purinergic Signaling in the Cardiovascular System. Circ. Res. 2017, 120, 207–228. [Google Scholar] [CrossRef]
- Löffler, M.; Morote-Garcia, J.C.; Eltzschig, S.A.; Coe, I.R.; Eltzschig, H.K. Physiological Roles of Vascular Nucleoside Transporters. Arter. Thromb. Vasc. Biol. 2007, 27, 1004–1013. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Sitkovsky, M.V.; Robson, S.C. Purinergic Signaling during Inflammation. N. Engl. J. Med. 2012, 367, 2322–2333. [Google Scholar] [CrossRef]
- Vuerich, M.; Harshe, R.; Frank, L.A.; Mukherjee, S.; Gromova, B.; Csizmadia, E.; Nasser, I.A.; Ma, Y.; Bonder, A.; Patwardhan, V.; et al. Altered aryl-hydrocarbon-receptor signalling affects regulatory and effector cell immunity in autoimmune hepatitis. J. Hepatol. 2020, 74, 48–57. [Google Scholar] [CrossRef]
- Frump, A.L.; Selej, M.; Wood, J.A.; Albrecht, M.; Yakubov, B.; Petrache, I.; Lahm, T. Hypoxia Upregulates Estrogen Receptor β in Pulmonary Artery Endothelial Cells in a HIF-1α–Dependent Manner. Am. J. Respir. Cell Mol. Biol. 2018, 59, 114–126. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Köhler, D.; Eckle, T.; Kong, T.; Robson, S.C.; Colgan, S.P. Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood 2009, 113, 224–232. [Google Scholar] [CrossRef]
- Barreto-Andrade, J.N.; de Fátima, L.A.; Campello, R.S.; Guedes, J.A.C.; de Freitas, H.S.; Okamoto, M.M. Estrogen Receptor 1 (ESR1) Enhances Slc2a4/GLUT4 Expression by a SP1 Cooperative Mechanism. Int. J. Med. Sci. 2018, 15, 1320. [Google Scholar] [CrossRef]
- Li, C.; Briggs, M.R.; Ahlborn, T.E.; Kraemer, F.B.; Liu, J. Requirement of Sp1 and Estrogen Receptor α Interaction in 17β-Estradiol-Mediated Transcriptional Activation of the Low Density Lipoprotein Receptor Gene Expression. Endocrinology 2001, 142, 1546–1553. [Google Scholar] [CrossRef]
- Grant, M.B.; Davis, M.I.; Caballero, S.; Feoktistov, I.; Biaggioni, I.; Belardinelli, L. Proliferation, migration, and ERK activation in human retinal endothelial cells through A(2B) adenosine receptor stimulation. Invest. Ophthalmol. Vis. Sci. 2001, 42, 2068–2073. [Google Scholar] [PubMed]
- Mitrović, N.; Zarić, M.; Drakulić, D.; Martinović, J.; Stanojlović, M.; Sévigny, J. 17β-Estradiol upregulates ecto-5’-nucleotidase (CD73) in hippocampal synaptosomes of female rats through action mediated by estrogen receptor-α and -β. Neuroscience 2016, 324, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Pochmann, D.; Böhmer, A.E.; Bruno, A.N.; Sarkis, J.J.F. Ecto-hydrolysis of adenine nucleotides in rat blood platelets are altered by ovariectomy. Platelets 2005, 16, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Huttinger, Z.M.; Milks, M.W.; Nickoli, M.S.; Aurand, W.L.; Long, L.C.; Wheeler, D.G.; Dwyer, K.M.; D’Apice, A.J.; Robson, S.C.; Cowan, P.J.; et al. Ectonucleotide Triphosphate Diphosphohydrolase-1 (CD39) Mediates Resistance to Occlusive Arterial Thrombus Formation after Vascular Injury in Mice. Am. J. Pathol. 2012, 181, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Guckelberger, O.; Sun, X.F.; Sévigny, J.; Imai, M.; Kaczmarek, E.; Enjyoji, K.; Robson, S.C. Beneficial effects of CD39/ecto-nucleoside triphosphate diphosphohydrolase-1 in murine intestinal ischemia-reperfusion injury. Thromb Haemost. 2004, 91, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Duan, R.; Safe, S. Estrogen Induces Adenosine Deaminase Gene Expression in MCF-7 Human Breast Cancer Cells: Role of Estrogen Receptor-Sp1 Interactions. Endocrinology 1999, 140, 219–227. [Google Scholar] [CrossRef]
- Dubey, R.K.; Gillespie, D.G.; Jackson, E.K. A 2B Adenosine Receptors Stimulate Growth of Porcine and Rat Arterial Endothelial Cells. Hypertension 2002, 39, 530–535. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Ibla, J.C.; Furuta, G.T.; Leonard, M.O.; Jacobson, K.A.; Enjyoji, K.; Colgan, S.P. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: Role of ectonucleotidases and adenosine A2B receptors. J. Exp. Med. 2003, 198, 783–796. [Google Scholar] [CrossRef]
- Fang, Y.; Olah, M.E. Cyclic AMP-Dependent, Protein Kinase A-Independent Activation of Extracellular Signal-Regulated Kinase 1/2 Following Adenosine Receptor Stimulation in Human Umbilical Vein Endothelial Cells: Role of Exchange Protein Activated by cAMP 1 (Epac1). J. Pharmacol. Exp. Ther. 2007, 322, 1189–1200. [Google Scholar] [CrossRef]
- Kong, T.; Westerman, K.A.; Faigle, M.; Eltzschig, H.K.; Colgan, S.P. HIF-dependent induction of adenosine A2B receptor in hypoxia. FASEB J. 2006, 20, 2242–2250. [Google Scholar] [CrossRef]
- Panjehpour, M.; Mohamadi, A.; Aghaei, M. Estrogen stimulates adenosine receptor expression subtypes in human breast cancer MCF-7 cell line. Res. Pharm. Sci. 2018, 13, 57–64. [Google Scholar] [CrossRef]
- Troncoso, F.; Herlitz, K.; Acurio, J.; Aguayo, C.; Guevara, K.; Castro, F.O.; Godoy, A.S.; Martin, S.S.; Escudero, C. Advantages in Wound Healing Process in Female Mice Require Upregulation A2A-Mediated Angiogenesis under the Stimulation of 17β-Estradiol. Int. J. Mol. Sci. 2020, 21, 7145. [Google Scholar] [CrossRef]
- Casanello, P.; Torres, A.; Sanhueza, F.; González, M.; Farías, M.; Gallardo, V.; Pastor-Anglada, M.; Martín, R.S.; Sobrevia, L. Equilibrative Nucleoside Transporter 1 Expression Is Downregulated by Hypoxia in Human Umbilical Vein Endothelium. Circ. Res. 2005, 97, 16–24. [Google Scholar] [CrossRef]
- Kaneko, M.; Hakuno, F.; Kamei, H.; Yamanaka, D.; Chida, K.; Minami, S.; Coe, I.R.; Takahashi, S.-I. Steroid hormones are novel nucleoside transport inhibitors by competition with nucleosides for their transporters. Biochem. Biophys. Res. Commun. 2014, 443, 505–510. [Google Scholar] [CrossRef]
- Minet, E.; Arnould, T.; Michel, G.; Roland, I.; Mottet, D.; Raes, M.; Remacle, J.; Michiels, C. ERK activation upon hypoxia: Involvement in HIF-1 activation. FEBS Lett. 2000, 468, 53–58. [Google Scholar] [CrossRef]
- Kim, S.C.; Boese, A.C.; Moore, M.H.; Cleland, R.M.; Chang, L.; Delafontaine, P.; Yin, K.-J.; Lee, J.-P.; Hamblin, M.H. Rapid estrogen receptor-α signaling mediated by ERK activation regulates vascular tone in male and ovary-intact female mice. Am. J. Physiol. Circ. Physiol. 2018, 314, H330–H342. [Google Scholar] [CrossRef]
- Rose’Meyer, R.B.; Mellick, A.S.; Garnham, B.G.; Harrison, G.J.; Massa, H.M.; Griffiths, L.R. The measurement of adenosine and estrogen receptor expression in rat brains following ovariectomy using quantitative PCR analysis. Brain Res. Protoc. 2003, 11, 9–18. [Google Scholar] [CrossRef]
- Manson, J.A.E.; Chlebowski, R.T.; Stefanick, M.L.; Aragaki, A.K.; Rossouw, J.E.; Prentice, R.L.; Anderson, G.; Howard, B.W.; Thomson, C.A.; La Croix, A.Z.; et al. The Women’s Health Initiative Hormone Therapy Trials: Update and Overview of Health Outcomes During the Intervention and Post-Stopping Phases. J. Am. Med. Assoc. 2013, 310, 1353. [Google Scholar] [CrossRef]
- Bastid, J.; Cottalorda-Regairaz, A.; Alberici, G.; Bonnefoy, N.; Eliaou, J.F.; Bensussan, A. ENTPD1/CD39 is a promising therapeutic target in oncology. Oncogene 2013, 32, 1743–1751. [Google Scholar] [CrossRef]
- Augustin, R.C.; Leone, R.D.; Naing, A.; Fong, L.; Bao, R.; Luke, J.J. Next steps for clinical translation of adenosine pathway inhibition in cancer immunotherapy. J. Immunother. Cancer 2022, 10, e004089. [Google Scholar] [CrossRef]
- Haskó, G.; Linden, J.; Cronstein, B.; Pacher, P. Adenosine receptors: Therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug Discov. 2008, 7, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Allahham, M.; Lerman, A.; Atar, D.; Birnbaum, Y. Why Not Dipyridamole: A Review of Current Guidelines and Re-evaluation of Utility in the Modern Era. Cardiovasc. Drugs Ther. 2021, 36, 525–532. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Forward | Reverse |
|---|---|---|
| CD39 | 5′ CTGATTCCTGGGAGCACATC 3′ | 5′ GACATAGGTGGAGTGGGAGAG 3′ |
| ESR1 | 5′ GAAAGGTGGGATACGAAAAGACC 3′ | 5′ GCTGTTCTTCTTAGAGCGTTTGA 3′ |
| SP1 | 5′ TTGAAAAAGGAGTTGGTGGC 3′ | 5′ TGCTGGTTCTGTAAGTTGGG 3′ |
| GAPDH | 5′ GGAGCGAGATCCCTCCAAAAT 3′ | 5′ GCCTGTTGTCATACTTCTCATGG 3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cassavaugh, J.; Qureshi, N.; Csizmadia, E.; Longhi, M.S.; Matyal, R.; Robson, S.C. Regulation of Hypoxic–Adenosinergic Signaling by Estrogen: Implications for Microvascular Injury. Pharmaceuticals 2023, 16, 422. https://doi.org/10.3390/ph16030422
Cassavaugh J, Qureshi N, Csizmadia E, Longhi MS, Matyal R, Robson SC. Regulation of Hypoxic–Adenosinergic Signaling by Estrogen: Implications for Microvascular Injury. Pharmaceuticals. 2023; 16(3):422. https://doi.org/10.3390/ph16030422
Chicago/Turabian StyleCassavaugh, Jessica, Nada Qureshi, Eva Csizmadia, Maria Serena Longhi, Robina Matyal, and Simon C. Robson. 2023. "Regulation of Hypoxic–Adenosinergic Signaling by Estrogen: Implications for Microvascular Injury" Pharmaceuticals 16, no. 3: 422. https://doi.org/10.3390/ph16030422
APA StyleCassavaugh, J., Qureshi, N., Csizmadia, E., Longhi, M. S., Matyal, R., & Robson, S. C. (2023). Regulation of Hypoxic–Adenosinergic Signaling by Estrogen: Implications for Microvascular Injury. Pharmaceuticals, 16(3), 422. https://doi.org/10.3390/ph16030422