

Interpreting Iron Homeostasis in Congenital and Acquired Disorders

 , , , and
, , , and
Abstract
1. Introduction
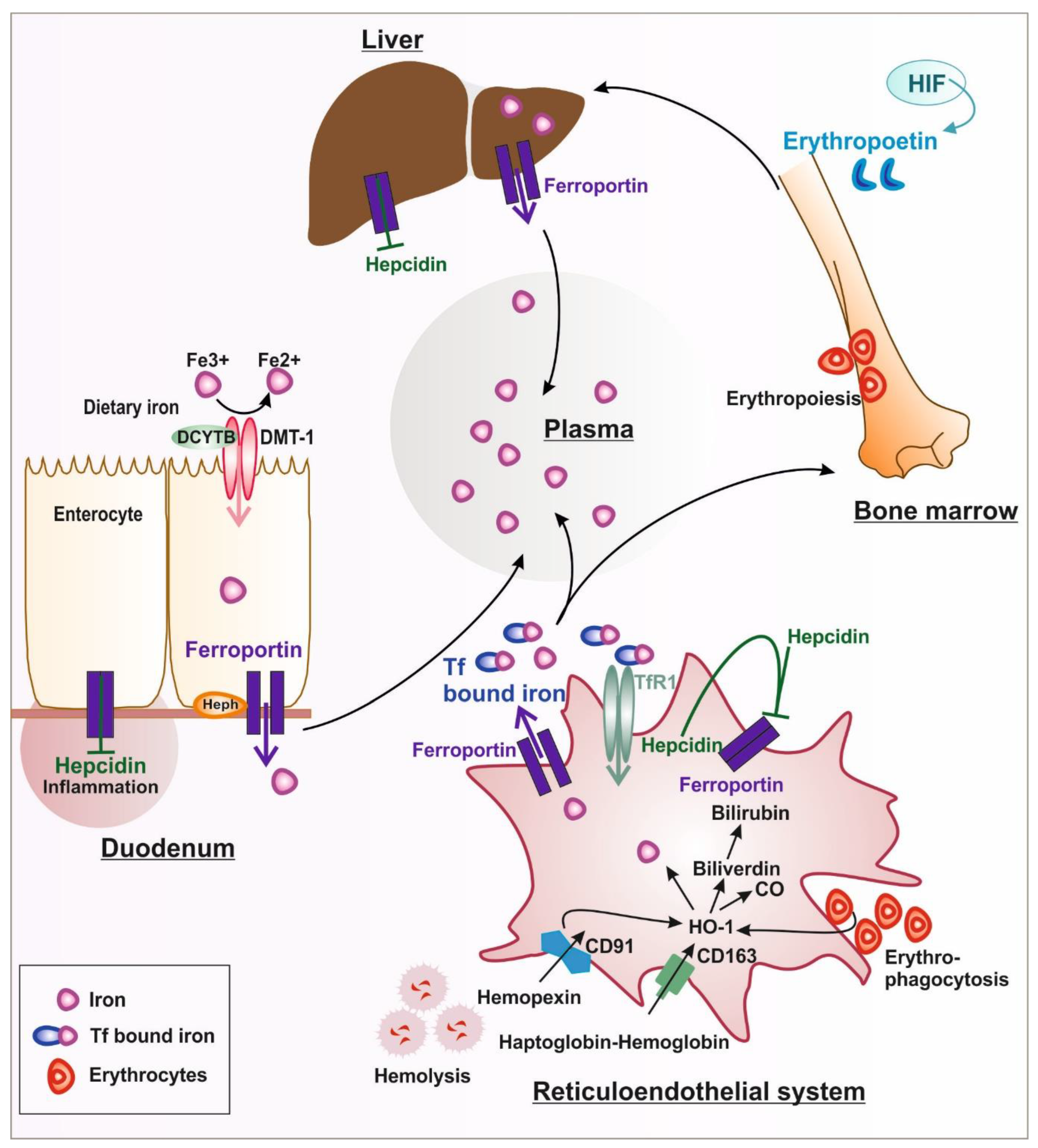
1.1. Systemic Iron Metabolism
1.1.1. Iron Uptake and Release
1.1.2. Iron Handling in Diseases Associated with Hemolysis
1.1.3. Adjustment of Iron Hemostasis According to Cellular Needs
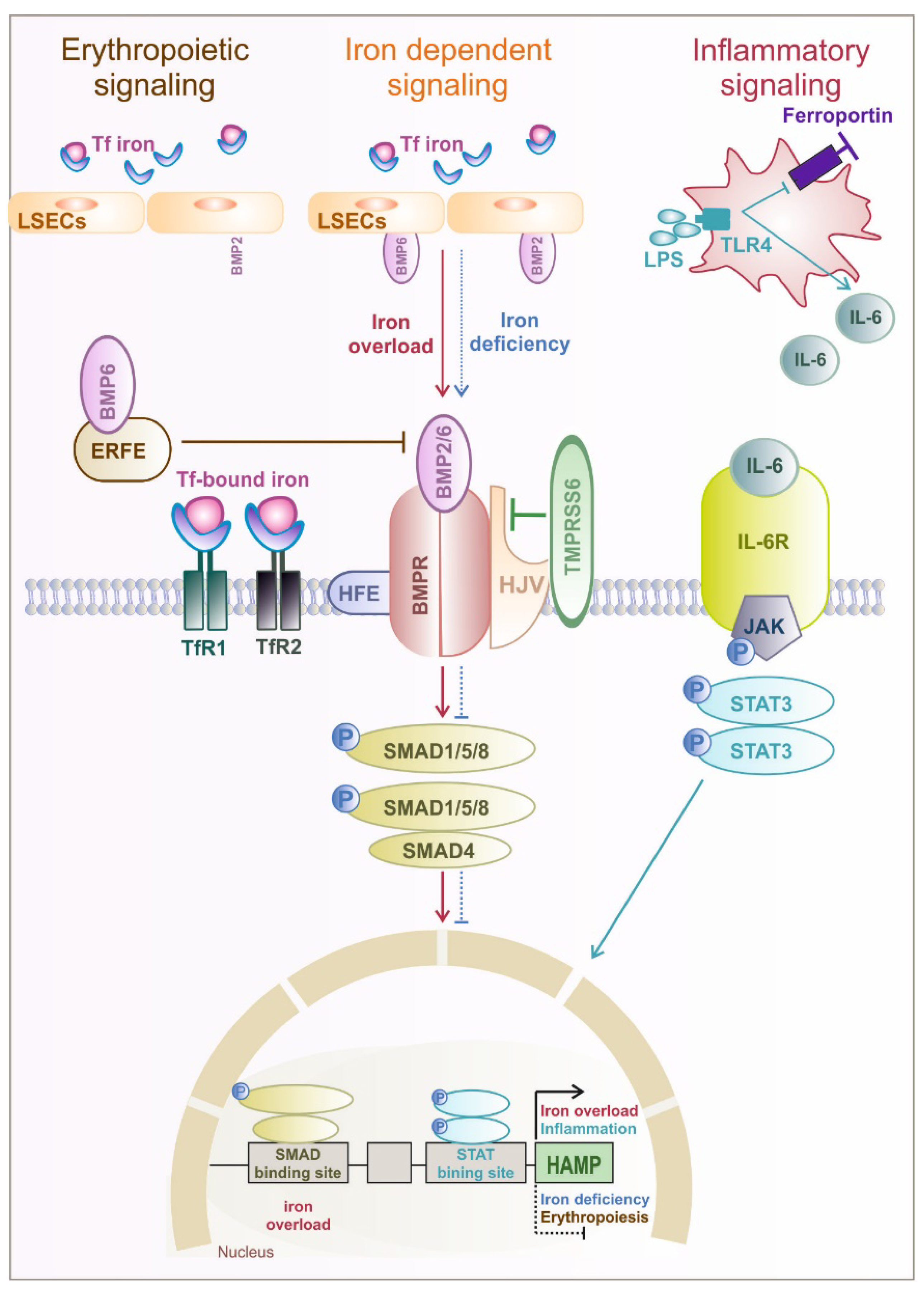
1.2. Hormonal Regulation of Systemic and Cellular Iron Homeostasis by Hepcidin
2. Iron Metabolism Imbalance
2.1. Iron Overload
2.1.1. Congenital Iron Overload without Anemia
2.1.2. Congenital Iron Overload with Anemia
Defect in Systemic Players of Iron Metabolism
Non-Transfusion-Dependent Thalassemia
Sideroblastic Anemia
2.1.3. Acquired Iron Overload
2.2. Iron and Cancer
2.2.1. Cancer Cell Iron Metabolism
2.2.2. Innate Immune Cells as Central Iron Regulators in the TME
2.3. Iron Deficiency
2.3.1. Acquired Absolute Iron Deficiency
2.3.2. Acquired Functional Iron Deficiency
2.3.3. Congenital Iron Deficiency
IRIDA (Iron Refractory Iron Deficiency Anemia)
3. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Muckenthaler, M.U.; Andrews, N.C. Balancing Acts: Molecular Control of Mammalian Iron Metabolism. Cell 2004, 117, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Katsarou, A.; Pantopoulos, K. Basics and principles of cellular and systemic iron homeostasis. Mol. Asp. Med. 2020, 75, 100866. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.C. The iron transporter DMT1. Int. J. Biochem. Cell Biol. 1999, 31, 991–994. [Google Scholar] [CrossRef]
- Vashchenko, G.; MacGillivray, R.T.A. Functional role of the putative iron ligands in the ferroxidase activity of recombinant human hephaestin. JBIC J. Biol. Inorg. Chem. 2012, 17, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Gkouvatsos, K.; Papanikolaou, G.; Pantopoulos, K. Regulation of iron transport and the role of transferrin. Biochim. Biophys. Acta (BBA) Gen. Subj. 2012, 1820, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Hepcidin and iron regulation, 10 years later. Blood 2011, 117, 4425–4433. [Google Scholar] [CrossRef]
- Corrons, J.V.; Casafont, L.B.; Frasnedo, E.F. Concise review: How do red blood cells born, live, and die? Ann. Hematol. 2021, 100, 2425–2433. [Google Scholar] [CrossRef]
- Effenberger-Neidnicht, K.; Hartmann, M. Mechanisms of Hemolysis During Sepsis. Inflammation 2018, 41, 1569–1581. [Google Scholar] [CrossRef]
- Burwick, R.M.; Rincon, M.; Beeraka, S.S.; Gupta, M.; Feinberg, B.B. Evaluation of Hemolysis as a Severe Feature of Preeclampsia. Hypertension 2018, 72, 460–465. [Google Scholar] [CrossRef]
- Kavanagh, P.L.; Fasipe, T.A.; Wun, T. Sickle Cell Disease: A Review. JAMA 2022, 328, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.; Henderson, A.C. Hemolytic Anemia: Evaluation and Differential Diagnosis. Am. Fam. Physician 2018, 98, 354–361. [Google Scholar] [PubMed]
- Luten, M.; Roerdinkholder-Stoelwinder, B.; Schaap, N.P.M.; De Grip, W.J.; Bos, H.J.; Bosman, G.J.C.G.M. Survival of red blood cells after transfusion: A comparison between red cells concentrates of different storage periods. Transfusion 2008, 48, 1478–1485. [Google Scholar] [CrossRef] [PubMed]
- Vinchi, F.; Costa da Silva, M.; Ingoglia, G.; Petrillo, S.; Brinkman, N.; Zuercher, A.; Cerwenka, A.; Tolosano, E.; Muckenthaler, M.U. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood 2016, 127, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.J.; Møller, H.J.; Moestrup, S.K. Hemoglobin and Heme Scavenger Receptors. Antioxid. Redox Signal. 2010, 12, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Hvidberg, V.; Maniecki, M.B.; Jacobsen, C.; Højrup, P.; Møller, H.J.; Moestrup, S.K. Identification of the receptor scavenging hemopexin-heme complexes. Blood 2005, 106, 2572–2579. [Google Scholar] [CrossRef]
- Kristiansen, M.; Graversen, J.H.; Jacobsen, C.; Sonne, O.; Hoffman, H.-J.; Law, S.K.A.; Moestrup, S.K. Identification of the haemoglobin scavenger receptor. Nature 2001, 409, 198–201. [Google Scholar] [CrossRef]
- Hider, R.C. Nature of nontransferrin-bound iron. Eur. J. Clin. Investig. 2002, 32, 50–54. [Google Scholar] [CrossRef]
- Brissot, P.; Ropert, M.; Le Lan, C.; Loréal, O. Non-transferrin bound iron: A key role in iron overload and iron toxicity. Biochim. Biophys. Acta (BBA) Gen. Subj. 2011, 1820, 403–410. [Google Scholar] [CrossRef]
- Mertens, C.; Marques, O.; Horvat, N.; Simonetti, M.; Muckenthaler, M.; Jung, M. The Macrophage Iron Signature in Health and Disease. Int. J. Mol. Sci. 2021, 22, 8457. [Google Scholar] [CrossRef]
- Vinchi, F.; De Franceschi, L.; Ghigo, A.; Townes, T.; Cimino, J.; Silengo, L.; Hirsch, E.; Altruda, F.; Tolosano, E. Hemopexin Therapy Improves Cardiovascular Function by Preventing Heme-Induced Endothelial Toxicity in Mouse Models of Hemolytic Diseases. Circulation 2013, 127, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Janciauskiene, S.; Vijayan, V.; Immenschuh, S. TLR4 Signaling by Heme and the Role of Heme-Binding Blood Proteins. Front. Immunol. 2020, 11, 1964. [Google Scholar] [CrossRef] [PubMed]
- Sudan, K.; Vijayan, V.; Madyaningrana, K.; Gueler, F.; Igarashi, K.; Foresti, R.; Motterlini, R.; Immenschuh, S. TLR4 activation alters labile heme levels to regulate BACH1 and heme oxygenase-1 expression in macrophages. Free. Radic. Biol. Med. 2019, 137, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.U.; Galy, B.; Hentze, M.W. Systemic Iron Homeostasis and the Iron-Responsive Element/Iron-Regulatory Protein (IRE/IRP) Regulatory Network. Annu. Rev. Nutr. 2008, 28, 197–213. [Google Scholar] [CrossRef]
- Sanchez, M.; Galy, B.; Schwanhaeusser, B.; Blake, J.; Bähr-Ivacevic, T.; Benes, V.; Selbach, M.; Muckenthaler, M.U.; Hentze, M.W. Iron regulatory protein-1 and -2: Transcriptome-wide definition of binding mRNAs and shaping of the cellular proteome by iron regulatory proteins. Blood 2011, 118, e168–e179. [Google Scholar] [CrossRef]
- Mastrogiannaki, M.; Matak, P.; Peyssonnaux, C. The gut in iron homeostasis: Role of HIF-2 under normal and pathological conditions. Blood 2013, 122, 885–892. [Google Scholar] [CrossRef]
- Schwartz, A.J.; Das, N.K.; Ramakrishnan, S.K.; Jain, C.; Jurkovic, M.T.; Wu, J.; Nemeth, E.; Lakhal-Littleton, S.; Colacino, J.A.; Shah, Y.M. Hepatic hepcidin/intestinal HIF-2α axis maintains iron absorption during iron deficiency and overload. J. Clin. Investig. 2018, 129, 336–348. [Google Scholar] [CrossRef]
- Park, C.H.; Valore, E.V.; Waring, A.J.; Ganz, T. Hepcidin, a Urinary Antimicrobial Peptide Synthesized in the Liver. J. Biol. Chem. 2001, 276, 7806–7810. [Google Scholar] [CrossRef]
- Koch, P.-S.; Olsavszky, V.; Ulbrich, F.; Sticht, C.; Demory, A.; Leibing, T.; Henzler, T.; Meyer, M.; Zierow, J.; Schneider, S.; et al. Angiocrine Bmp2 signaling in murine liver controls normal iron homeostasis. Blood 2017, 129, 415–419. [Google Scholar] [CrossRef]
- Canali, S.; Wang, C.-Y.; Zumbrennen-Bullough, K.B.; Bayer, A.; Babitt, J.L. Bone morphogenetic protein 2 controls iron homeostasis in mice independent of Bmp6. Am. J. Hematol. 2017, 92, 1204–1213. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, L.; Nai, A.; Dulja, A.; Pagani, A. Hepcidin and the BMP-SMAD pathway: An unexpected liaison. Vitam. Horm. 2019, 110, 71–99. [Google Scholar] [PubMed]
- Camaschella, C.; Nai, A.; Silvestri, L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica 2020, 105, 260–272. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to Tango: Regulation of Mammalian Iron Metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Ramos, E.; Kautz, L.; Rodriguez, R.; Hansen, M.; Gabayan, V.; Ginzburg, Y.; Roth, M.-P.; Nemeth, E.; Ganz, T. Evidence for distinct pathways of hepcidin regulation by acute and chronic iron loading in mice. Hepatology 2011, 53, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Feder, J.N.; Gnirke, A.; Thomas, W.; Tsuchihashi, Z.; Ruddy, D.A.; Basava, A.; Dormishian, F.; Domingo, R., Jr.; Ellis, M.C.; Fullan, A.; et al. A novel MHC class I–like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 1996, 13, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Pietrangelo, A.; Adams, P.C.; De Graaff, B.; McLaren, C.E.; Loréal, O. Haemochromatosis. Nat. Rev. Dis. Prim. 2018, 4, 18016. [Google Scholar] [CrossRef]
- Muckenthaler, L.; Marques, O.; Colucci, S.; Kunz, J.B.; Fabrowski, P.; Bast, T.; Altamura, S.; Höchsmann, B.; Schrezenmeier, H.; Langlotz, M.; et al. Constitutional PIGA mutations cause a novel subtype of hemochromatosis in patients with neurologic dysfunction. Blood 2022, 139, 1418–1422. [Google Scholar] [CrossRef]
- Silvestri, L.; Pagani, A.; Nai, A.; De Domenico, I.; Kaplan, J.; Camaschella, C. The Serine Protease Matriptase-2 (TMPRSS6) Inhibits Hepcidin Activation by Cleaving Membrane Hemojuvelin. Cell Metab. 2008, 8, 502–511. [Google Scholar] [CrossRef]
- Corradini, E.; Rozier, M.; Meynard, D.; Odhiambo, A.; Lin, H.Y.; Feng, Q.; Migas, M.C.; Britton, R.S.; Babitt, J.L.; Fleming, R.E. Iron Regulation of Hepcidin Despite Attenuated Smad1,5,8 Signaling in Mice Without Transferrin Receptor 2 or Hfe. Gastroenterology 2011, 141, 1907–1914. [Google Scholar] [CrossRef]
- Wu, X.-G.; Wang, Y.; Wu, Q.; Cheng, W.H.; Liu, W.; Zhao, Y.; Mayeur, C.; Schmidt, P.J.; Yu, P.; Wang, F.; et al. HFE interacts with the BMP type I receptor ALK3 to regulate hepcidin expression. Blood 2014, 124, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Emrich, I.E.; Scheuer, A.; Wagenpfeil, S.; Ganz, T.; Heine, G.H. Increase of plasma erythroferrone levels during high-altitude exposure: A sub-analysis of the TOP OF HOMe study. Am. J. Hematol. 2021, 96, E179–E181. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-Y.; Babitt, J.L. Liver iron sensing and body iron homeostasis. Blood 2019, 133, 18–29. [Google Scholar] [CrossRef]
- Weiss, G.; Goodnough, L.T. Anemia of chronic disease. N. Engl. J. Med. 2005, 352, 1011–1023. [Google Scholar] [CrossRef]
- Cassat, J.E.; Skaar, E.P. Iron in Infection and Immunity. Cell Host Microbe 2013, 13, 509–519. [Google Scholar] [CrossRef]
- Marchetti, M.; De Bei, O.; Bettati, S.; Campanini, B.; Kovachka, S.; Gianquinto, E.; Spyrakis, F.; Ronda, L. Iron Metabolism at the Interface between Host and Pathogen: From Nutritional Immunity to Antibacterial Development. Int. J. Mol. Sci. 2020, 21, 2145. [Google Scholar] [CrossRef]
- Okonko, D.O.; Mandal, A.K.; Missouris, C.G.; Poole-Wilson, P.A. Disordered Iron Homeostasis in Chronic Heart Failure: Prevalence, Predictors, and Relation to Anemia, Exercise Capacity, and Survival. J. Am. Coll. Cardiol. 2011, 58, 1241–1251. [Google Scholar] [CrossRef]
- Klip, I.T.; Comin-Colet, J.; Voors, A.A.; Ponikowski, P.; Enjuanes, C.; Banasiak, W.; Lok, D.J.; Rosentryt, P.; Torrens, A.; Polonski, L.; et al. Iron deficiency in chronic heart failure: An international pooled analysis. Am. Heart J. 2013, 165, 575–582.e3. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Comin-Colet, J.; de Francisco, A.; Dignass, A.; Doehner, W.; Lam, C.S.; Macdougall, I.C.; Rogler, G.; Camaschella, C.; Kadir, R.; et al. Iron deficiency across chronic inflammatory conditions: International expert opinion on definition, diagnosis, and management. Am. J. Hematol. 2017, 92, 1068–1078. [Google Scholar] [CrossRef]
- Knutson, M.D. Non-transferrin-bound iron transporters. Free. Radic. Biol. Med. 2018, 133, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. Hereditary Hemochromatosis: Pathogenesis, Diagnosis, and Treatment. Gastroenterology 2010, 139, 393–408.e2. [Google Scholar] [CrossRef] [PubMed]
- Girelli, D.; Busti, F.; Brissot, P.; Cabantchik, I.; Muckenthaler, M.U.; Porto, G. Hemochromatosis classification: Update and recommendations by the BIOIRON Society. Blood 2022, 139, 3018–3029. [Google Scholar] [CrossRef]
- Altamura, S.; Marques, O.; Colucci, S.; Mertens, C.; Alikhanyan, K.; Muckenthaler, M.U. Regulation of iron homeostasis: Lessons from mouse models. Mol. Asp. Med. 2020, 75, 100872. [Google Scholar] [CrossRef] [PubMed]
- Altamura, S.; Kessler, R.; Gröne, H.-J.; Gretz, N.; Hentze, M.W.; Galy, B.; Muckenthaler, M.U. Resistance of Ferroportin to Hepcidin Binding causes Exocrine Pancreatic Failure and Fatal Iron Overload. Cell Metab. 2014, 20, 359–367. [Google Scholar] [CrossRef]
- Beutler, E. The significance of the 187G (H63D) mutation in hemochromatosis. Am. J. Hum. Genet. 1997, 61, 762–764. [Google Scholar] [CrossRef]
- Nixon, A.M.; Neely, E.; Simpson, I.A.; Connor, J.R. The role of HFE genotype in macrophage phenotype. J. Neuroinflamm. 2018, 15, 30. [Google Scholar] [CrossRef]
- Buzzetti, E.; Parikh, P.M.; Gerussi, A.; Tsochatzis, E. Gender differences in liver disease and the drug-dose gender gap. Pharmacol. Res. 2017, 120, 97–108. [Google Scholar] [CrossRef]
- Adams, P.C.; Barton, J.C. Haemochromatosis. Lancet 2007, 370, 1855–1860. [Google Scholar] [CrossRef]
- Salgia, R.J.; Brown, K. Diagnosis and Management of Hereditary Hemochromatosis. Clin. Liver Dis. 2015, 19, 187–198. [Google Scholar] [CrossRef]
- Rombout-Sestrienkova, E.; Van Kraaij, M.G.J.; Koek, G.H. How we manage patients with hereditary haemochromatosis. Br. J. Haematol. 2016, 175, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Loréal, O.; Cavey, T.; Robin, F.; Kenawi, M.; Guggenbuhl, P.; Brissot, P. Iron as a Therapeutic Target in HFE-Related Hemochromatosis: Usual and Novel Aspects. Pharmaceuticals 2018, 11, 131. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C.; Pagani, A. Mendelian inheritance of anemia due to disturbed iron homeostasis. Semin. Hematol. 2021, 58, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Palmer, W.C.; Vishnu, P.; Sanchez, W.; Aqel, B.; Riegert-Johnson, D.; Seaman, L.A.K.; Bowman, A.W.; Rivera, C.E. Diagnosis and Management of Genetic Iron Overload Disorders. J. Gen. Intern. Med. 2018, 33, 2230–2236. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Bernard, D.G.; Brissot, E.; Loréal, O.; Troadec, M.-B. Rare anemias due to genetic iron metabolism defects. Mutat. Res. Mol. Mech. Mutagen. 2018, 777, 52–63. [Google Scholar] [CrossRef]
- Camaschella, C. How I manage patients with atypical microcytic anaemia. Br. J. Haematol. 2012, 160, 12–24. [Google Scholar] [CrossRef]
- Bardou-Jacquet, E.; Island, M.-L.; Jouanolle, A.-M.; Détivaud, L.; Fatih, N.; Ropert, M.; Brissot, E.; Mosser, A.; Maisonneuve, H.; Brissot, P.; et al. A novel N491S mutation in the human SLC11A2 gene impairs protein trafficking and in association with the G212V mutation leads to microcytic anemia and liver iron overload. Blood Cells Mol. Dis. 2011, 47, 243–248. [Google Scholar] [CrossRef]
- Iolascon, A.; D’Apolito, M.; Servedio, V.; Cimmino, F.; Piga, A.; Camaschella, C. Microcytic anemia and hepatic iron overload in a child with compound heterozygous mutations in DMT1 (SCL11A2). Blood 2006, 107, 349–354. [Google Scholar] [CrossRef]
- Hayashi, A.; Wada, Y.; Suzuki, T.; Shimizu, A. Studies on familial hypotransferrinemia: Unique clinical course and molecular pathology. Am. J. Hum. Genet. 1993, 53, 201–213. [Google Scholar]
- Craven, C.M.; Alexander, J.; Eldridge, M.; Kushner, J.P.; Bernstein, S.; Kaplan, J. Tissue distribution and clearance kinetics of non-transferrin-bound iron in the hypotransferrinemic mouse: A rodent model for hemochromatosis. Proc. Natl. Acad. Sci. USA 1987, 84, 3457–3461. [Google Scholar] [CrossRef]
- De Domenico, I.; Ward, D.M.; Di Patti, M.C.B.; Jeong, S.Y.; David, S.; Musci, G.; Kaplan, J. Ferroxidase activity is required for the stability of cell surface ferroportin in cells expressing GPI-ceruloplasmin. EMBO J. 2007, 26, 2823–2831. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Loréal, O. Iron metabolism and related genetic diseases: A cleared land, keeping mysteries. J. Hepatol. 2015, 64, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Musallam, K.M.; Cappellini, M.D. β-Thalassemias. N. Engl. J. Med. 2021, 384, 727–743. [Google Scholar] [CrossRef] [PubMed]
- Mettananda, S.; Higgs, D.R. Molecular Basis and Genetic Modifiers of Thalassemia. Hematol. Clin. N. Am. 2018, 32, 177–191. [Google Scholar] [CrossRef]
- Musallam, K.M.; Cappellini, M.D.; Viprakasit, V.; Kattamis, A.; Rivella, S.; Taher, A.T. Revisiting the non-transfusion-dependent (NTDT) vs. transfusion-dependent (TDT) thalassemia classification 10 years later. Am. J. Hematol. 2021, 96, E54–E56. [Google Scholar] [CrossRef]
- Musallam, K.M.; Cappellini, M.D.; Wood, J.C.; Taher, A.T. Iron overload in non-transfusion-dependent thalassemia: A clinical perspective. Blood Rev. 2012, 26 (Suppl. S1), S16–S19. [Google Scholar] [CrossRef]
- Taher, A.T.; Porter, J.; Viprakasit, V.; Kattamis, A.; Chuncharunee, S.; Sutcharitchan, P.; Siritanaratkul, N.; Galanello, R.; Karakas, Z.; Lawniczek, T.; et al. Deferasirox reduces iron overload significantly in nontransfusion-dependent thalassemia: 1-year results from a prospective, randomized, double-blind, placebo-controlled study. Blood 2012, 120, 970–977. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Tefferi, A. Myelodysplastic syndromes with ring sideroblasts (MDS-RS) and MDS/myeloproliferative neoplasm with RS and thrombocytosis (MDS/MPN-RS-T)—“2021 update on diagnosis, risk-stratification, and management”. Am. J. Hematol. 2021, 96, 379–394. [Google Scholar] [CrossRef]
- Gao, J.; Zhou, Q.; Wu, D.; Chen, L. Mitochondrial iron metabolism and its role in diseases. Clin. Chim. Acta 2020, 513, 6–12. [Google Scholar] [CrossRef]
- Long, Z.; Li, H.; Du, Y.; Han, B. Congenital sideroblastic anemia: Advances in gene mutations and pathophysiology. Gene 2018, 668, 182–189. [Google Scholar] [CrossRef]
- Bottomley, S.S.; Fleming, M.D. Sideroblastic anemia: Diagnosis and management. Hematol. Oncol. Clin. N. Am. 2014, 28, 653–670. [Google Scholar] [CrossRef]
- Porter, J.B.; de Witte, T.; Cappellini, M.D.; Gattermann, N. New insights into transfusion-related iron toxicity: Implications for the oncologist. Crit. Rev. Oncol. 2016, 99, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Adams-Graves, P.; Bronte-Jordan, L. Recent treatment guidelines for managing adult patients with sickle cell disease: Challenges in access to care, social issues, and adherence. Expert Rev. Hematol. 2016, 9, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Shander, A.; Cappellini, M.D.; Goodnough, L.T. Iron overload and toxicity: The hidden risk of multiple blood transfusions. Vox Sang. 2009, 97, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Rivella, S. Iron metabolism under conditions of ineffective erythropoiesis in β-thalassemia. Blood 2019, 133, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Borgna-Pignatti, C.; Marsella, M. Iron Chelation in Thalassemia Major. Clin. Ther. 2015, 37, 2866–2877. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. Mechanism of iron toxicity. Adv. Exp. Med. Biol. 2002, 509, 19–43. [Google Scholar]
- Zhang, H.; Zhabyeyev, P.; Wang, S.; Oudit, G.Y. Role of iron metabolism in heart failure: From iron deficiency to iron overload. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1865, 1925–1937. [Google Scholar] [CrossRef]
- Sikorska, K.; Bernat, A.; Wróblewska, A. Molecular pathogenesis and clinical consequences of iron overload in liver cirrhosis. Hepatobiliary Pancreat. Dis. Int. 2016, 15, 461–479. [Google Scholar] [CrossRef]
- Borgna-Pignatti, C.; Cappellini, M.D.; De Stefano, P.; Del Vecchio, G.C.; Forni, G.L.; Gamberini, M.R.; Ghilardi, R.; Piga, A.; Romeo, M.A.; Zhao, H.; et al. Cardiac morbidity and mortality in deferoxamine- or deferiprone-treated patients with thalassemia major. Blood 2006, 107, 3733–3737. [Google Scholar] [CrossRef]
- Neufeld, E.J. Oral chelators deferasirox and deferiprone for transfusional iron overload in thalassemia major: New data, new questions. Blood 2006, 107, 3436–3441. [Google Scholar] [CrossRef]
- Galanello, R. Deferiprone in the treatment of transfusion-dependent thalassemia: A review and perspective. Ther. Clin. Risk Manag. 2007, 3, 795–805. [Google Scholar]
- Hoffbrand, A.V.; Cohen, A.; Hershko, C. Role of deferiprone in chelation therapy for transfusional iron overload. Blood 2003, 102, 17–24. [Google Scholar] [CrossRef]
- Fradette, C.; Pichette, V.; Sicard, É.; Stilman, A.; Jayashankar, S.; Tsang, Y.C.; Spino, M.; Tricta, F. Effects of renal impairment on the pharmacokinetics of orally administered deferiprone. Br. J. Clin. Pharmacol. 2016, 82, 994–1001. [Google Scholar] [CrossRef]
- Galanello, R.; Agus, A.; Campus, S.; Danjou, F.; Giardina, P.J.; Grady, R.W. Combined iron chelation therapy. Ann. N. Y. Acad. Sci. 2010, 1202, 79–86. [Google Scholar] [CrossRef]
- Guo, S.; Casu, C.; Gardenghi, S.; Booten, S.; Aghajan, M.; Peralta, R.; Watt, A.; Freier, S.; Monia, B.P.; Rivella, S. Reducing TMPRSS6 ameliorates hemochromatosis and β-thalassemia in mice. J. Clin. Investig. 2013, 123, 1531–1541. [Google Scholar] [CrossRef]
- Manolova, V.; Nyffenegger, N.; Flace, A.; Altermatt, P.; Varol, A.; Doucerain, C.; Sundstrom, H.; Dürrenberger, F. Oral ferroportin inhibitor ameliorates ineffective erythropoiesis in a model of β-thalassemia. J. Clin. Investig. 2019, 130, 491–506. [Google Scholar] [CrossRef]
- Altamura, S.; Schaeper, U.; Dames, S.; Löffler, K.; Eisermann, M.; Frauendorf, C.; Müdder, K.; Neves, J.; Muckenthaler, M.U. SLN124, a GalNAc-siRNA Conjugate Targeting TMPRSS6, Efficiently Prevents Iron Overload in Hereditary Haemochromatosis Type 1. Hemasphere 2019, 3, e301. [Google Scholar] [CrossRef]
- Porter, J. Beyond transfusion therapy: New therapies in thalassemia including drugs, alternate donor transplant, and gene therapy. Hematology Am. Soc. Hematol. Educ. Program 2018, 2018, 361–370. [Google Scholar] [CrossRef]
- Torti, S.V.; Torti, F.M. Iron and Cancer: 2020 Vision. Cancer Res. 2020, 80, 5435–5448. [Google Scholar] [CrossRef]
- Edgren, G.; Reilly, M.; Hjalgrim, H.; Tran, T.N.; Rostgaard, K.; Adami, J.; Titlestad, K.; Shanwell, A.; Melbye, M.; Nyrén, O. Donation Frequency, Iron Loss, and Risk of Cancer Among Blood Donors. Gynecol. Oncol. 2008, 100, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Vostrejs, M.; Moran, P.L.; Seligman, P.A. Transferrin synthesis by small cell lung cancer cells acts as an autocrine regulator of cellular proliferation. J. Clin. Investig. 1988, 82, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Adachi, M.; Kai, K.; Yamaji, K.; Ide, T.; Noshiro, H.; Kawaguchi, A.; Aishima, S. Transferrin receptor 1 overexpression is associated with tumour de-differentiation and acts as a potential prognostic indicator of hepatocellular carcinoma. Histopathology 2019, 75, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.J.; Attwood, K.; Sharma, N.J.; Gross, K.W.; Smith, G.J.; Xu, B.; Kauffman, E.C. Transferrin receptor 1 upregulation in primary tumor and downregulation in benign kidney is associated with progression and mortality in renal cell carcinoma patients. Oncotarget 2017, 8, 107052–107075. [Google Scholar] [CrossRef]
- Liang, W.; Li, Q.; Ferrara, N. Metastatic growth instructed by neutrophil-derived transferrin. Proc. Natl. Acad. Sci. USA 2018, 115, 11060–11065. [Google Scholar] [CrossRef]
- Brookes, M.J.; Hughes, S.; Turner, F.E.; Reynolds, G.; Sharma, N.; Ismail, T.; Berx, G.; McKie, A.T.; Hotchin, N.; Anderson, G.J.; et al. Modulation of iron transport proteins in human colorectal carcinogenesis. Gut 2006, 55, 1449–1460. [Google Scholar] [CrossRef]
- Pinnix, Z.K.; Miller, L.D.; Wang, W.; D’Agostino, R.; Kute, T.; Willingham, M.C.; Hatcher, H.; Tesfay, L.; Sui, G.; Di, H.; et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci. Transl. Med. 2010, 2, 43ra56. [Google Scholar] [CrossRef]
- Zhou, Q.; Chen, J.; Feng, J.; Wang, J. E4BP4 promotes thyroid cancer proliferation by modulating iron homeostasis through repression of hepcidin. Cell Death Dis. 2018, 9, 987. [Google Scholar] [CrossRef]
- Toshiyama, R.; Konno, M.; Eguchi, H.; Asai, A.; Noda, T.; Koseki, J.; Asukai, K.; Ohashi, T.; Matsushita, K.; Iwagami, Y.; et al. Association of iron metabolic enzyme hepcidin expression levels with the prognosis of patients with pancreatic cancer. Oncol. Lett. 2018, 15, 8125–8133. [Google Scholar] [CrossRef]
- Tesfay, L.; Clausen, K.A.; Kim, J.W.; Hegde, P.; Wang, X.; Miller, L.D.; Deng, Z.; Blanchette, N.; Arvedson, T.; Miranti, C.K.; et al. Hepcidin Regulation in Prostate and Its Disruption in Prostate Cancer. Cancer Res. 2015, 75, 2254–2263. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef]
- Boutilier, A.; Elsawa, S. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef] [PubMed]
- Recalcati, S.; Locati, M.; Marini, A.; Santambrogio, P.; Zaninotto, F.; De Pizzol, M.; Zammataro, L.; Girelli, D.; Cairo, G. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur. J. Immunol. 2010, 40, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Shahdeo, D.; Roberts, A.; Kesarwani, V.; Horvat, M.; Chouhan, R.S.; Gandhi, S. Polymeric biocompatible iron oxide nanoparticles labeled with peptides for imaging in ovarian cancer. Biosci. Rep. 2022, 42, BSR20212622. [Google Scholar] [CrossRef] [PubMed]
- Bregolat, N.F.; Ruetten, M.; Da Silva, M.C.; Aboouf, M.A.; Ademi, H.; von Büren, N.; Armbruster, J.; Stirn, M.; Altamura, S.; Marques, O.; et al. Iron- and erythropoietin-resistant anemia in a spontaneous breast cancer mouse model. Haematologica 2022, 107, 2454–2465. [Google Scholar] [CrossRef] [PubMed]
- Thielmann, C.M.; Costa da Silva, M.; Muley, T.; Meister, M.; Herpel, E.; Muckenthaler, M.U. Iron accumulation in tumor-associated macrophages marks an improved overall survival in patients with lung adenocarcinoma. Sci. Rep. 2019, 9, 11326. [Google Scholar] [CrossRef]
- Marques, O.; Porto, G.; Rêma, A.; Faria, F.; Cruz Paula, A.; Gomez-Lazaro, M.; Silva, P.; Martins da Silva, B.; Lopes, C. Local iron homeostasis in the breast ductal carcinoma microenvironment. BMC Cancer 2016, 16, 187. [Google Scholar] [CrossRef] [PubMed]
- Moestrup, S.K.; Møller, H.J. CD163: A regulated hemoglobin scavenger receptor with a role in the anti-inflammatory response. Ann. Med. 2004, 36, 347–354. [Google Scholar] [CrossRef]
- Mertens, C.; Mora, J.; Ören, B.; Grein, S.; Winslow, S.; Scholich, K.; Weigert, A.; Malmström, P.; Forsare, C.; Fernö, M.; et al. Macrophage-derived lipocalin-2 transports iron in the tumor microenvironment. Oncoimmunology 2017, 7, e1408751. [Google Scholar] [CrossRef]
- Kassebaum, N.J. The Global Burden of Anemia. Hematol. Clin. N. Am. 2016, 30, 247–308. [Google Scholar] [CrossRef]
- Pasricha, S.R.; Tye-Din, J.; Muckenthaler, M.U.; Swinkels, D.W. Iron deficiency. Lancet 2021, 397, 233–248. [Google Scholar] [CrossRef]
- Kautz, L.; Jung, G.; Du, X.; Gabayan, V.; Chapman, J.; Nasoff, M.; Nemeth, E.; Ganz, T. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of β-thalassemia. Blood 2015, 126, 2031–2037. [Google Scholar] [CrossRef]
- Stoffel, N.U.; Cercamondi, C.I.; Brittenham, G.; Zeder, C.; Geurts-Moespot, A.J.; Swinkels, D.W.; Moretti, D.; Zimmermann, M.B. Iron absorption from oral iron supplements given on consecutive versus alternate days and as single morning doses versus twice-daily split dosing in iron-depleted women: Two open-label, randomised controlled trials. Lancet Haematol. 2017, 4, e524–e533. [Google Scholar] [CrossRef]
- Moretti, D.; Goede, J.S.; Zeder, C.; Jiskra, M.; Chatzinakou, V.; Tjalsma, H.; Melse-Boonstra, A.; Brittenham, G.; Swinkels, D.W.; Zimmermann, M.B. Oral iron supplements increase hepcidin and decrease iron absorption from daily or twice-daily doses in iron-depleted young women. Blood 2015, 126, 1981–1989. [Google Scholar] [CrossRef]
- Elstrott, B.; Khan, L.; Olson, S.; Raghunathan, V.; Deloughery, T.; Shatzel, J.J. The role of iron repletion in adult iron deficiency anemia and other diseases. Eur. J. Haematol. 2019, 104, 153–161. [Google Scholar] [CrossRef]
- De Amicis, M.M.; Rimondi, A.; Elli, L.; Motta, I. Acquired refractory iron deficiency anemia. Mediterr. J. Hematol. Infect. Dis. 2021, 13, e2021028. [Google Scholar]
- Pisani, A.; Riccio, E.; Sabbatini, M.; Andreucci, M.; Del Rio, A.; Visciano, B. Effect of oral liposomal iron versus intravenous iron for treatment of iron deficiency anaemia in CKD patients: A randomized trial. Nephrol. Dial. Transplant. 2014, 30, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Antunes, C.V.D.A.; Nascimento, C.R.D.A.; Ribeiro, T.C.D.R.; Antunes, P.D.A.; Chebli, L.D.A.; Fava, L.M.G.; Malaguti, C.; Chebli, J.M.F. Treatment of iron deficiency anemia with liposomal iron in inflammatory bowel disease: Efficacy and impact on quality of life. Pharm. Weekbl. 2020, 42, 895–902. [Google Scholar]
- Auerbach, M.; Ballard, H. Clinical Use of Intravenous Iron: Administration, Efficacy, and Safety. Hematology Am. Soc. Hematol. Educ. Program 2010, 2010, 338–347. [Google Scholar] [CrossRef]
- Michael, B.; Coyne, D.W.; Fishbane, S.; Folkert, V.; Lynn, R.; Nissenson, A.R.; Agarwal, R.; Eschbach, J.W.; Fadem, S.Z.; Trout, J.R.; et al. Sodium ferric gluconate complex in hemodialysis patients: Adverse reactions compared to placebo and iron dextran. Kidney Int. 2002, 61, 1830–1839. [Google Scholar] [CrossRef] [PubMed]
- Blumenstein, I.; Shanbhag, S.; Langguth, P.; Kalra, P.A.; Zoller, H.; Lim, W. Newer formulations of intravenous iron: A review of their chemistry and key safety aspects–hypersensitivity, hypophosphatemia, and cardiovascular safety. Expert Opin. Drug Saf. 2021, 20, 757–769. [Google Scholar] [CrossRef]
- Zoller, H.; Schaefer, B.; Glodny, B. Iron-induced hypophosphatemia: An emerging complication. Curr. Opin. Nephrol. Hypertens. 2017, 26, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Schouten, B.J.; Hunt, P.J.; Livesey, J.H.; Frampton, C.M.; Soule, S.G. FGF23 Elevation and Hypophosphatemia after Intravenous Iron Polymaltose: A Prospective Study. J. Clin. Endocrinol. Metab. 2009, 94, 2332–2337. [Google Scholar] [CrossRef] [PubMed]
- Glaspy, J.A.; Wolf, M.; Strauss, W.E. Intravenous iron-induced hypophosphatemia: An emerging syndrome. Adv. Ther. 2021, 38, 3531–3549. [Google Scholar] [CrossRef]
- Ganz, T. Anemia of Inflammation. N. Engl. J. Med. 2019, 381, 1148–1157. [Google Scholar] [CrossRef]
- Kim, A.; Fung, E.; Parikh, S.G.; Valore, E.V.; Gabayan, V.; Nemeth, E.; Ganz, T. A mouse model of anemia of inflammation: Complex pathogenesis with partial dependence on hepcidin. Blood 2014, 123, 1129–1136. [Google Scholar] [CrossRef]
- Sheetz, M.; Barrington, P.; Callies, S.; Berg, P.; McColm, J.; Marbury, T.; Decker, B.; Dyas, G.L.; Truhlar, S.M.; Benschop, R.; et al. Targeting the hepcidin–ferroportin pathway in anaemia of chronic kidney disease. Br. J. Clin. Pharmacol. 2019, 85, 935–948. [Google Scholar] [CrossRef]
- De Falco, L.; Sanchez, M.; Silvestri, L.; Kannengiesser, C.; Muckenthaler, M.; Iolascon, A.; Gouya, L.; Camaschella, C.; Beaumont, C. Iron refractory iron deficiency anemia. Haematologica 2013, 98, 845–853. [Google Scholar] [CrossRef]
- Poggiali, E.; Andreozzi, F.; Nava, I.; Consonni, D.; Graziadei, G.; Cappellini, M.D. The role of TMPRSS6 polymorphisms in iron deficiency anemia partially responsive to oral iron treatment. Am. J. Hematol. 2014, 90, 306–309. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Disease | Serum Iron | Serum Ferritin | Transferrin Saturation | Hepcidin | ||
|---|---|---|---|---|---|---|
| Iron overload | Congenital | Hemochromatosis | ↑ | ↑ | ↑ | ↓ |
| Thalassemia | ↑ | ↑ | ↑ | ↓ | ||
| Sideroblastic anemia | ↑ | ↑ | ↑ | ↓ | ||
| Acquired | Transfusion iron overload | ↑ | ↑ | ↑ | ↑ | |
| Iron deficiency | Congenital | IRIDA | N/↓ | N/↓ | ↓ | ↑ |
| Acquired | Absolute iron deficiency | N/↓ | ↓ | ↓ | ↓ | |
| Functional iron deficiency | N | ↑ | ↓ | ↑ | ||
| Novel Classification | Molecular Presentation |
|---|---|
| HFE-related | C282Y/C282Y C282Y/other rare HFE pathogenic variants H63D |
| Non-HFE-related | HJV-related (G320V, I222N, D249H, Q312X) HAMP-related (93delG, R56X, C70R, G71D) TFR2-related (V162del, A77D, Y250X, E60X) SLC40A1-related (A77D, I152F and L233P) |
| Digenic | HFE and/or non-HFE |
| Molecularly undefined | Molecular characterization not available |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scaramellini, N.; Fischer, D.; Agarvas, A.R.; Motta, I.; Muckenthaler, M.U.; Mertens, C. Interpreting Iron Homeostasis in Congenital and Acquired Disorders. Pharmaceuticals 2023, 16, 329. https://doi.org/10.3390/ph16030329
Scaramellini N, Fischer D, Agarvas AR, Motta I, Muckenthaler MU, Mertens C. Interpreting Iron Homeostasis in Congenital and Acquired Disorders. Pharmaceuticals. 2023; 16(3):329. https://doi.org/10.3390/ph16030329
Chicago/Turabian StyleScaramellini, Natalia, Dania Fischer, Anand R. Agarvas, Irene Motta, Martina U. Muckenthaler, and Christina Mertens. 2023. "Interpreting Iron Homeostasis in Congenital and Acquired Disorders" Pharmaceuticals 16, no. 3: 329. https://doi.org/10.3390/ph16030329
APA StyleScaramellini, N., Fischer, D., Agarvas, A. R., Motta, I., Muckenthaler, M. U., & Mertens, C. (2023). Interpreting Iron Homeostasis in Congenital and Acquired Disorders. Pharmaceuticals, 16(3), 329. https://doi.org/10.3390/ph16030329