1. Introduction
In recent decades, antimicrobial resistance has become a global health-related concern worldwide, considering the increasing resistance of bacteria and fungi to commercial antibiotics and antifungals. With the continuous increase in microbial resistance in agriculture and medicine, the development of new antimicrobial agents has become an essential research task. Bacterial resistance to commercial antibiotics is also one of the reasons for the emergence of resistant bacterial biofilm communities that contribute to chronic infections. These structured communities (biofilms) of bacteria and fungi communicate via a quorum-sensing system and are embedded in an extracellular matrix that enables them to overcome the action of antimicrobial agents. Therefore, the detection of new antimicrobial agents that could end this emerging battleground is an overriding research challenge [
1]. Furthermore, in vitro cytotoxicity screening of new agents with antimicrobial potential must be an accompanying step in this research since it determines the potential safety of newly developed products and allows their possible medical use. Keeping in mind that oxidative stress (overproduction of free radicals without adequate neutralization) is held responsible for several chronic illnesses, including cancer, diabetes, and cardiovascular disease, the survey for compounds with antioxidant properties has become a trend of its own.
In this regard, plants are noteworthy, representing a rich source of diverse antioxidant and antimicrobial compounds, also known as specialized metabolites, such as tannins, flavonoids, alkaloids, and terpenoids [
2,
3,
4] that are not available in synthetic compound libraries [
5].
There are an estimated 250,000 plant species in the world, and only 5–15% have been tested as potentially pharmacologically useful [
6]. There is encouraging potential for the discovery of plant metabolites with useful pharmacological activities for the treatment of a variety of diseases in humans and animals.
Centaurium spicatum (L.) Fritch (syn.
Schenkia spicata L. Mansion) is a common Gentianaceae species of the Mediterranean region and Eastern Europe, where it is adapted to various saline soils and is capable of completing life cycles under very harsh environmental conditions [
7]. Spiked centaury (
Figure 1.) is a rich source of pharmacologically active specialized metabolites, including secoiridoid glucosides (sweroside, swertiamarin, and gentiopicrin) and phenolic compounds. Because of the bitterness of secoiridoid glucosides, these compounds are used in the preparation of some commercial beverages [
7]. The extract of
C. spicatum, along with other
Centaurium species such as
C. pulchellum, has been used in traditional medicine for the treatment of abdominal pain, hypertension, kidney and ureteral stones, renal colic, wounds, and diabetes [
8]. The compounds found in the methanol extract of
C. spicatum showed cytotoxic activities against three different types of human cancer cell lines, HeLa, THP-1, and HL-60 [
9]. Recent studies have shown that lisianthoside II, together with seven other secoiridoid glycosides from spiked centaury, may be a suitable basis for the development of new drugs against COVID-19 [
10].
It has been previously reported that the optimization of extraction conditions can affect the biological potential of extracts from plant species belonging to the Gentianaceae family [
11,
12,
13,
14]. Besides the extraction methods and conditions, such as type of solvent, solvent composition, temperature, extraction, time, and number of extraction steps [
15,
16], the composition of bioactive compounds in plant extracts depends on several other factors, such as plant physiological stage and geographical origin [
17].
The present study was conducted to determine the most efficient extraction solvent system on the comprehensive recovery of specialized metabolites from C. spicatum herbs, with the aim to optimize the pharmacological effects of extracts. In order to achieve this, a targeted metabolomics method was used to characterize nine extracts of spiked centaury using UHPLC/DAD/QqQMS/MS. The next objective was to screen these extracts for their bioactive properties, i.e., to evaluate their: (1) antimicrobial, (2) antibiofilm, (3) antioxidant, and (4) cytotoxic properties in a keratinocyte cell line (HaCaT). The data reported here could be of great importance for the facilitated use of C. spicatum extracts as food preservatives and nutraceuticals.
3. Materials and Methods
3.1. Plant Collection and Extractions
The aerial parts of
Centaurium spicatum (30–60 cm) were collected in July 2021 near the beach of Buljarice (geographic coordinates: 42.19324, 18.96843) in Montenegro. Plant materials were collected and identified by the botanist authors (J.B., D.S., and D.M.) by using the scientific botanical literature and plant morphological features. The plant species was deposited in our local institutional herbarium under voucher number CSBCG2021. Plants were air-dried and cut into small pieces. The samples (2 g) were crushed with liquid nitrogen and then extracted with 20 mL of the solvents listed in
Table 1. After 48 h at 4 °C, the samples were filtered through Whatman paper No. 4. The residue was re-extracted with an additional portion of solvent (20 mL) at 4 °C two more times for 24 h and 48 h, respectively. Totally, nine different extracts (Cs1-9) were prepared. Extracts Cs1, Cs6, Cs7, Cs8, and Cs9 were evaporated to dryness at 40 °C using a rotary vacuum evaporator (Büchi R-210), while the water-containing extracts (Cs2, Cs3, Cs4, and Cs5) were frozen at −20 °C and subsequently lyophilized (LH Leybold, Lyovac GT2, Frenkendorf). The extract yield was calculated using the following equation:
where m(ext) is the mass of the extract and m(pl) is the mass of the plant sample.
3.2. UHPLC/(±)HESI-QqQ-MS/MS Targeted Metabolomics Analysis
Separation and quantification of compounds in tested samples were performed using a Dionex Ultimate 3000 UHPLC system equipped with a TSQ Quantum Access Max triple-quadrupole (QqQ) mass spectrometer (ThermoFisher Scientific, Basel, Switzerland). Elution was performed at 30 °C on a Syncronis C18 column (100 mm × 2.1 mm) with a 1.7 μm particle size (ThermoFisher Scientific, Basel, Switzerland). The mobile phase consisted of (A) water with 0.1% formic acid (MS grade) and (B) acetonitrile with 0.1% formic acid (both MS grade), which were applied in the previously described gradient elution program [
44]: 5% B in the first 2.0 min, 2.0–14.0 min 5–95% B, 14.0–14.2 min from 95% to 5% B, and 5% B until the 20th min. The flow rate was set to 0.3 mL/min, and the injection volume was 5 μL. All analyses were performed in triplicate.
The parameters of the TSQ Quantum Access Max QQQ mass spectrometer equipped with heated electrospray ionization (HESI) source were the same as previously described in Božunović et al. [
18]: vaporizer temperature of 450 °C, spray voltage 4000 V, sheet gas (N2) pressure 50 AU, ion sweep gas pressure 0 AU and auxiliary gas pressure of 20 AU, capillary temperature at 320 °C, and skimmer offset 0 V. The MS data were acquired in negative ionization mode, in the m/z range from 100 to 1000. Multiple mass spectrometric scanning modes, including full scanning (FS), product ion scanning (PIS), and neutral loss scanning (NLS), were conducted for the qualitative analysis. Collision-induced fragmentation experiments were performed using argon as the collision gas, with collision energy set to 30 eV. A selected reaction monitoring (SRM) experiment for quantitative analysis was performed using two characteristic MS
2 fragments for each targeted compound (
Table S1), which were previously defined as dominant in PIS experiments [
45].
3.3. Antioxidant Activity
3.3.1. FRAP and ABTS Assays
Ferric-reducing antioxidant power (FRAP) and ABTS radical cation scavenging activity assays were performed as previously described in Božunović et al. [
18].
Briefly, the working FRAP reagent contained 300 mM Na-acetate buffer (pH = 3.6), 20 mM ferric chloride, and 10 mM ferric-2,4,6-tri(2-pyridil)-1,3,5-triazine (Fe3+-TPTZ) at 10:1:1 ratio. Fe3+-TPTZ was dissolved in 40 mM HCl solution. Each sample was tested in triplicate by combining 950 μL FRAP reagent and 50 μL of the sample, followed by 10 min incubation at room temperature. Absorbance at 593 nm was determined with Agilent 8453 spectrophotometer (Agilent Technologies, Waldbronn, Germany). Methanol solutions of gallic acid (GA) were used for the construction of the calibration curve, and the results are expressed as GA equivalents reducing activity (mmol GAE) per 100 mg−1 plant extract. All analyses are performed in triplicate.
ABTS radical cations were obtained by combining equal volumes of 7 mM ABTS (2,20-azinobis(3-ethylbenzothiazoline-6-sulphonate)) and 2.45 mM potassium persulfate, after which the reagent was incubated in the dark at room temperature for 12 h. The solution was diluted with 80% ethanol until an absorbance of 0.7 ± 0.02 at 734 nm was obtained and used as the ABTS test reagent. Reaction mixtures containing 30 μL of sample and 970 μL of ABTS test reagent were incubated for 10 min at room temperature, and absorbance at 734 nm was subsequently measured. ABTS radical scavenging activity (%) was calculated using the formula:
where Asample represents the absorbance of the solution when the sample/reference compound has been added; Acontrol represents absorbance of the ABTS
+ solution without extract added. Methanol solutions of gallic acid (GA) were used for the calibration curve construction and the results are presented as mmol GAE per 100 mg
−1 plant extract. All analyses were performed in triplicate.
3.3.2. EPR Determination of Scavenging Activity toward DPPH Radicals
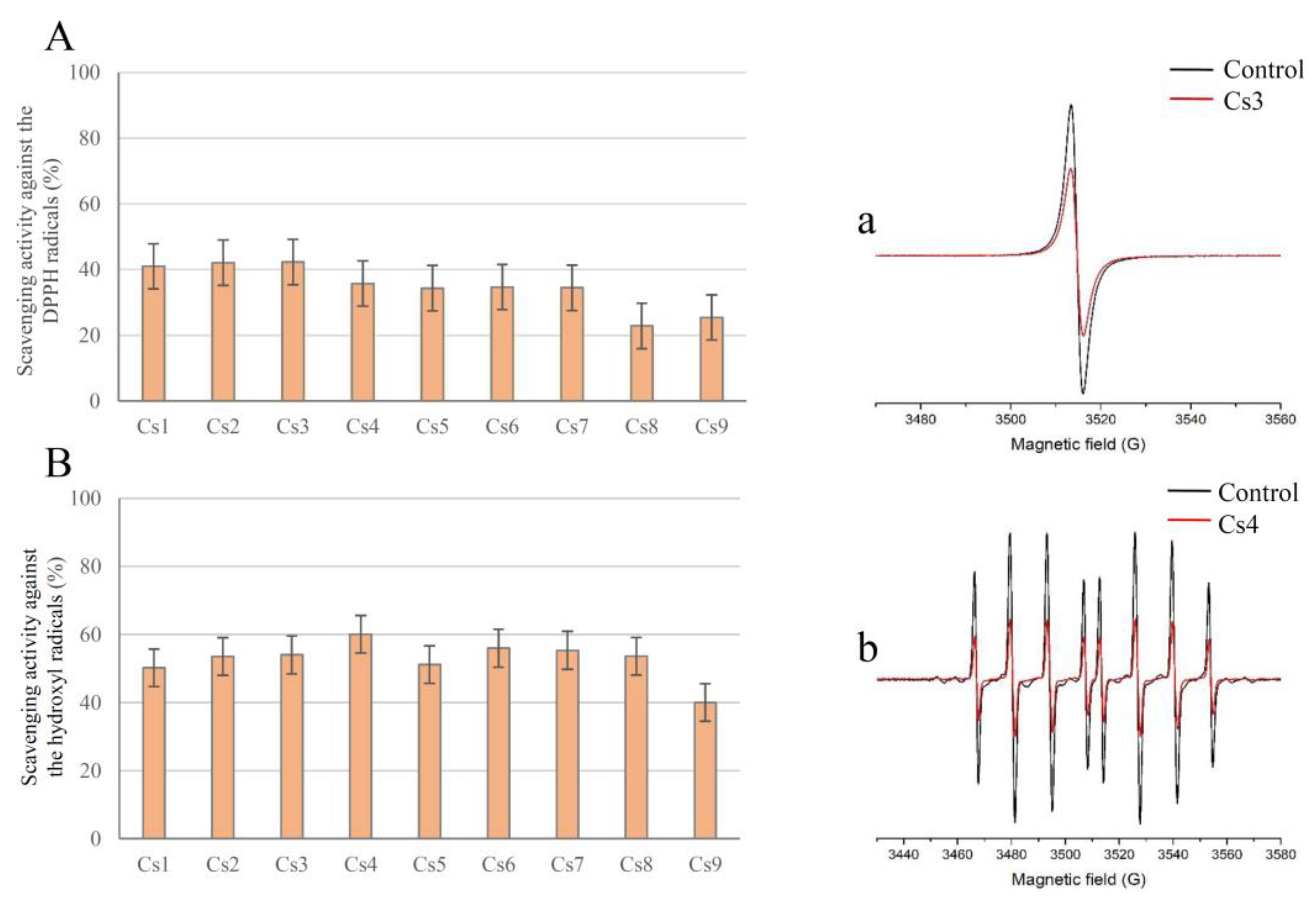
The interaction of DPPH free radicals with antioxidant compounds in the extracts was evaluated by measuring the intensity of the DPPH EPR signal. The DPPH radical is EPR active, which makes this compound suitable for EPR assays. The distinctive shape of the DPPH EPR spectra enables the determination of the initial radical concentration and the ability of spiked centaury extracts to reduce the amount of DPPH in the system. Each extract dissolved in water (29 µL) was mixed with 3.2 mM DPPH solution (1 µL) and transferred to a 1 mm diameter Teflon tube. After 2 min, EPR spectra were recorded with the following parameters: Center field 3510 G, microwave power 10 mW, microwave frequency 9.85 GHz, modulation frequency 100 kHz, and modulation amplitude 2 G.
The anti-DPPH activity (AA) of the tested extracts was calculated using the formula [
29]:
where Ic and Ia refer to the double integral values of the control and samples, respectively, determined from the EPR spectra (using Xepr software). The calculated an-ti-DPPH activities of the extracts are shown in
Figure 2.
3.3.3. EPR Determination of the Scavenging Activity toward the Hydroxyl Radicals
To determine the capacity of
C. spicatum extracts to remove hydroxyl radicals in the system, the solution consisting of the extract in the presence of a Fenton reaction with spin trap DEPMPO was used [
46]. This spin trap was selected for its well-known suitable selectivity and long DEPMPO/OH spin-adduct half-life. Briefly, the reaction mixture (29 µL in total) consisting of sample the extract (26 µL), H
2O
2 (2 µL, final concentration 0.35 mM), and DEPMPO (1 µL; final concentration 3.5 mM) was transferred to the gas-permeable Teflon tube, after which FeSO
4 (1 µL; final concentration 0.15 mM) was added immediately. Thereafter, EPR spectra were recorded 2 min later with the following experimental settings: center field 3500 G, microwave power 10 mW, microwave frequency 9.85 GHz, modulation frequency 100 kHz, modulation amplitude 1 G. The control experiments in both DPPH and
•OH radical scavenging activity tests have been made by replacing the sample with the same amount of solvent.
The anti-OH activity (AB) of the tested extracts was calculated using the formula [
29]:
where Ic and Ia refer to the double integral values of the control and samples, respectively, determined from the EPR spectra (using Xepr software). The calculated anti-OH activities of the extracts are shown in
Figure 2.
3.4. Antibacterial and Antifungal Activities
The following Gram (+) bacteria:
Staphylococcus aureus (ATCC 11632),
Bacillus cereus (food isolate), and
Listeria monocytogenes (NCTC 7973), and the Gram (−) bacteria
Escherichia coli (ATCC 25922),
Enterobacter cloacae (ATCC 35030), and
Salmonella Typhimurium (ATCC 13311) were tested using microdilution method in a 96-well microtiter plate. Antifungal activity of extracts was evaluated against the following micromycetes:
Aspergillus fumigatus (ATCC 9197), Aspergillus versicolor (ATCC 11730),
Penicillium funiculosum (ATCC 36839),
Penicillium verrucosum var. cyclopium (food isolate),
Trichoderma viride (IAM 5061). The tested microorganisms are deposited in the Mycological Laboratory, Department of Plant Physiology, Institute for Biological Research “Sinisa Stanković”- National Institute of the Republic of Serbia, University of Belgrade, Serbia. Minimal inhibitory concentrations (MIC) and minimal bactericidal concentrations (MBC) of the tested extracts were determined using the modified CLSI 2009 protocol [
47].
A modified microdilution technique was employed to examine the extracts’ antibacterial activity [
47]. In the Luria broth medium, bacterial species were grown for 24 h at 37 °C. With sterile 0.85% saline that contained 0.1% Tween 80 (
v/
v), the fungal spores were collected from the surface of the agar plates that were previously inoculated with microfungi and grown for 21 days at 28 °C. Sterile saline was used to adjust the bacterial cells and fungal spore suspension to a concentration of roughly 1.0 × 10
5 CFU in a final volume of 100 μL per well. For later use, the inocula were kept in storage at 4 °C. To ensure there was no contamination and to confirm the validity of the inoculum, dilutions of the inocula were cultured on Mueller-Hinton agar for bacteria and solid malt agar for fungi.
A serial dilution procedure was used to determine the minimum inhibitory concentration (MIC) in 96-well microtiter plates. The studied extracts were dissolved in 30% EtOH (20 mg/mL) and added to broth malt medium (for fungi) or Luria broth medium (for bacteria) with the inocula. The microplates were incubated for 24 h at 37 °C for bacteria and 72 h at 28 °C for fungi. The following day, 30 μL of INT (p-iodonitrotetrazolium violet) solution 0.2 mg/mL was added to the tested bacteria-containing medium, and the plates were placed back in the incubator for at least 30 min to guarantee a sufficient color reaction (Tsukatani et al., 2012). A clear solution or a distinct decrease in color response were indicators of growth inhibition. MICs were established as the lowest concentrations at which there was no discernible growth (under the binocular microscope). By serially subcultivating a 2 μL sample onto microtiter plates with 100 μL of broth in each well and then incubating the plates for 24 h or 72 h at 28 °C or 37 °C, the minimum bactericidal concentrations (MBCs) and minimum fungicidal concentrations (MFCs) were found. MBC/MFC stood for the lowest concentration with no discernible growth and represented a 99.5% killing of the original inoculum. The experiments were repeated in triplicate for each concentration used. E211 (sodium benzoate) and E244 (potassium metabisulfite) were used as positive controls. A 30% EtOH-negative control was employed. MIC and MBC/MFC values were expressed in mg/mL.
3.5. Anticandidal Assay
For the determination of minimum inhibitory (MIC) and minimum fungicidal concentrations (MFC), the microdilution method with some modification was used (EUCAST 2002). The following strains were used: Candida albicans ATCC 10231, C. albicans 475/15, C. albicans 1/6/15, C. parapsilosis ATCC 22019, C. krusei H1/16, and C. tropicalis ATCC 750. Yeast cultures were adjusted to McFarland 0.5 using sterile PBS (Phosphate-buffered saline). The 96-well microtiter plates containing serially diluted tested extracts (0.02–8.00 mg/mL) in liquid broth were incubated at 37 °C for 24 h. After incubation, the MIC and MFC were determined. The lowest concentrations without microscopically observed growth were considered MIC. For microscopic determination of growth, a Nikon Eclipse TS2 inverted microscope (Amsterdam, The Netherlands) was used, and fungal growth in the wells of 96-well microtiter plates was examined in comparison with the control (untreated yeast cells). MFC values were determined as concentrations without visible growth after serial subcultivation of 10 µL of the samples at 37 °C for 24 h. Ketoconazole (SigmaAldrich, Steinheim, Germany) was used as a positive control.
3.6. Antibiofilm Activity
C. albicans 475/15 and
C. parapsilosis ATCC 22019 were incubated with MIC and sub-MIC of the selected extracts, Cs2 and Cs6, respectively, in YPD (Yeast Extract–Peptone–Dextrose) medium at 37 °C for 24 h. After incubation, the plate was washed twice with sterile PBS and fixed with methanol for 10 min. The methanol was removed, and the plate was air-dried. The residual biofilm was stained with 0.1% crystal violet (Sigma Aldrich, Steinheim, Germany) for 30 min. Subsequently, the plate was washed with water and air-dried, and the residual stain was dissolved with 96% ethanol (Zorka, Sabac, Serbia). Absorbance was measured at 620 nm on a Multiskan™ FC microplate photometer (Thermo Scientific™, Waltham, MA, USA), and results are presented as inhibition of biofilm formation (%) according to the equation:
where A620control refers to the absorbance of the solution of untreated candida biofilm and A620sample refers to the absorbance of the solution of treated candida biofilm.
3.7. Anti-Hyphal Activity
C. albicans 475/15 was incubated with tested extracts for 4 h at 37 °C using YPD supplemented with 10% FBS (FBS, Gibco, Life Technologies Ltd., U.K.). After incubation, fungal cells were examined with a microscope (Nikon Eclipse TS2, Amsterdam, Netherlands). The number of cells that grew in the yeast or hyphal form was determined, and the percentage of hyphae was calculated according to Ivanov et al. [
48] by the following equation:
With n(hyphae) representing the number of cells growing in the hyphal morphology and n(total) representing the total number of Candida cells observed under a microscope.
3.8. Cytotoxicity toward HaCaT Cells
The cytotoxic effect of the extracts was determined using a spontaneously immortalized keratinocyte cell line (HaCaT, AddexBio T0020001) in the crystal violet assay, as previously described by Stojković et al. [
49] with some modifications. HaCaT cells were grown in high-glucose Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS, Gibco, Life Technologies Ltd., U.K.), 2 mM L-glutamine, and 1% antibiotic-antimycotic (Gibco, Life Technologies Corporation, Carlsbad, CA, USA) at 37 °C in a 5% CO
2 incubator. Cells (1.7 × 104 cells/well) were seeded into a 96-well microtiter plate with an adhesive bottom. After 24 h, the medium was removed, and the cells were treated with different concentrations of the extracts for the next 24 h. Subsequently, the medium was removed, and cells were washed twice with PBS pH 7.4 (Sigma-Aldrich, St. Louis, MI, USA), after which they were stained with 0.4% crystal violet (CV, Sigma-Aldrich, St. Louis, MI, USA) staining solution for 20 min. The CV staining solution was removed, and the cells were washed in a stream of tap water and air-dried at room temperature. The absorbance of the dye dissolved in methanol was measured at 570 nm (OD570) in a microplate reader. The results were expressed as IC50 value indicating 50% cell viability compared to the untreated control. The criterion for grading the cytotoxicity of the extracts in the HaCaT cell line was as follows: IC50 ≤ 20 µg/mL = strongly cytotoxic, IC50 between 21 and 200 µg/mL = moderately cytotoxic, IC50 between 201 and 400 µg/mL = weakly cytotoxic, and IC50 > 401 µg/mL = no cytotoxicity. The solvent was used as a negative control.
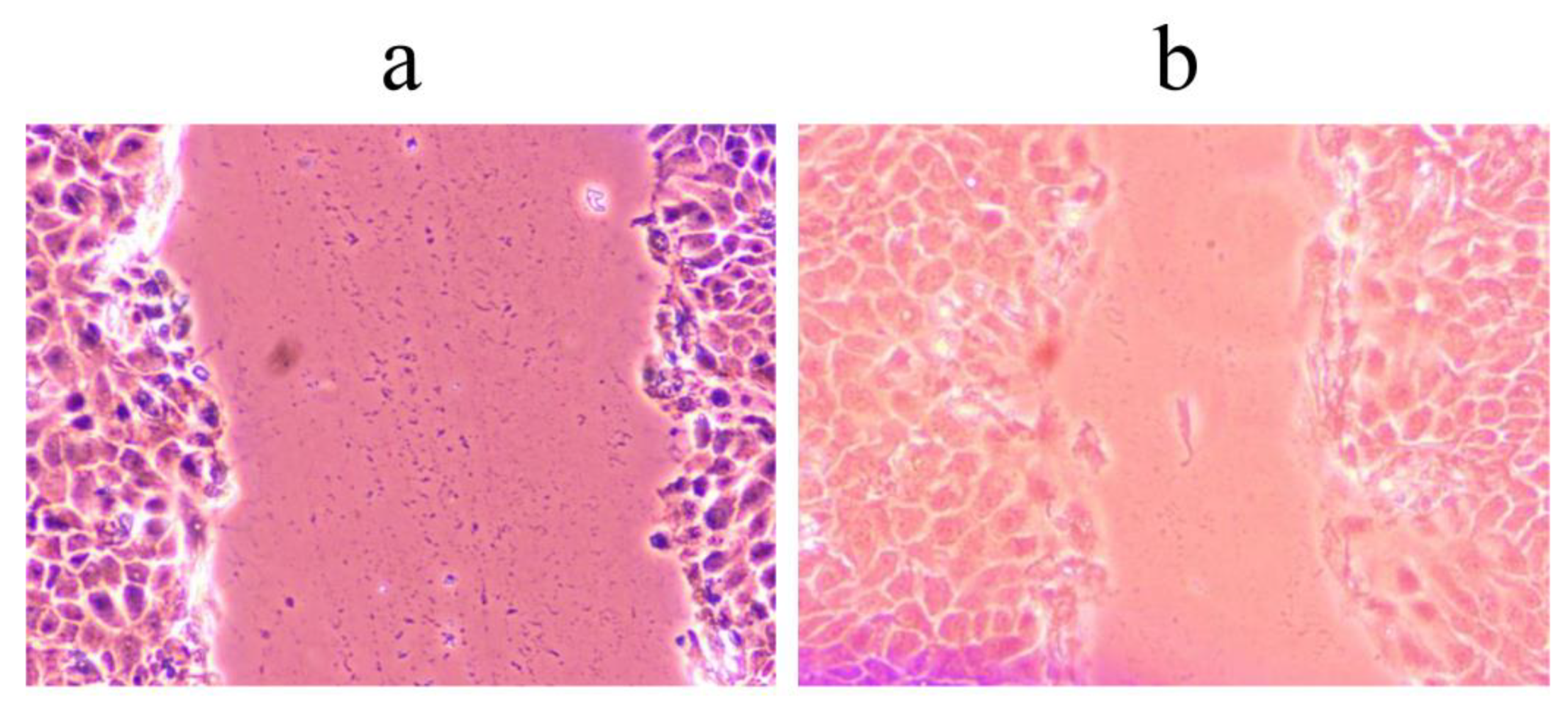
3.9. Wound Scratch Healing Assay
The assay was performed as described by Đorđevski et al. [
49] with some modifications. HaCaT cells were grown until reaching 85% confluence. The cell monolayer was scratched with a 200 μL sterile tip. Floating cells were washed, and cells were incubated in reduced DMEM supplemented with 1% FBS, 2 mM L-glutamine, and 1% antibiotic-antimycotic, containing 400 µg/mL of preparations. Cell migration was monitored using Nikon Eclipse TS2 (Amsterdam, the Netherlands) 48 h after wound preparation and treatment. The untreated control was used to measure wound closure under these conditions and without the addition of preparations. Results were presented as the percentage of wound closure during exposure to the tested extracts.
4. Conclusions
This study was the first to investigate the biological potential and chemical composition of C. spicatum as influenced by different extraction solvent systems. All nine C. spicatum extracts obtained within this study exerted various biological activities, including antimicrobial, antioxidant, anticandidal, antibiofilm, and wound healing. These biological activities can be altered and even enhanced by appropriate extraction procedures. In this study, we showed that among flavonoids, rutin was the most abundant compound in all analyzed extracts, and among iridoid compounds, sweroside predominated. The results indicate that ethanol-water-based extraction procedures (Cs2, Cs3, and Cs4) were the most efficient in isolating polyphenols. Consequently, the mentioned extracts proved to be the most suitable antioxidants. Moreover, the ethanol:water extracts Cs3 and Cs4 showed the best wound healing potential. Furthermore, Cs2 extract inhibited almost 40% of the preformed biofilm of C. parapsilossis, while butanol extract Cs6 was more efficient in reducing the ability of yeast cells to form hyphae. Considering the obtained results, we can conclude that solvent composition may be altered in a way to favor the extraction of desired compounds, or groups of compounds, from polyphenol- and iridoid-rich C. spicatum, depending on the biological activity needed to be exerted. Further in vivo studies are needed to confirm these results in order to make this plant commercially exploitable at higher levels.


 ,
, 



{kind=link}
{kind=link}
{kind=link}