


Bio-Oriented Synthesis and Molecular Docking Studies of 1,2,4-Triazole Based Derivatives as Potential Anti-Cancer Agents against HepG2 Cell Line


 ,
,  , , and
, , and
Abstract
1. Introduction
2. Results
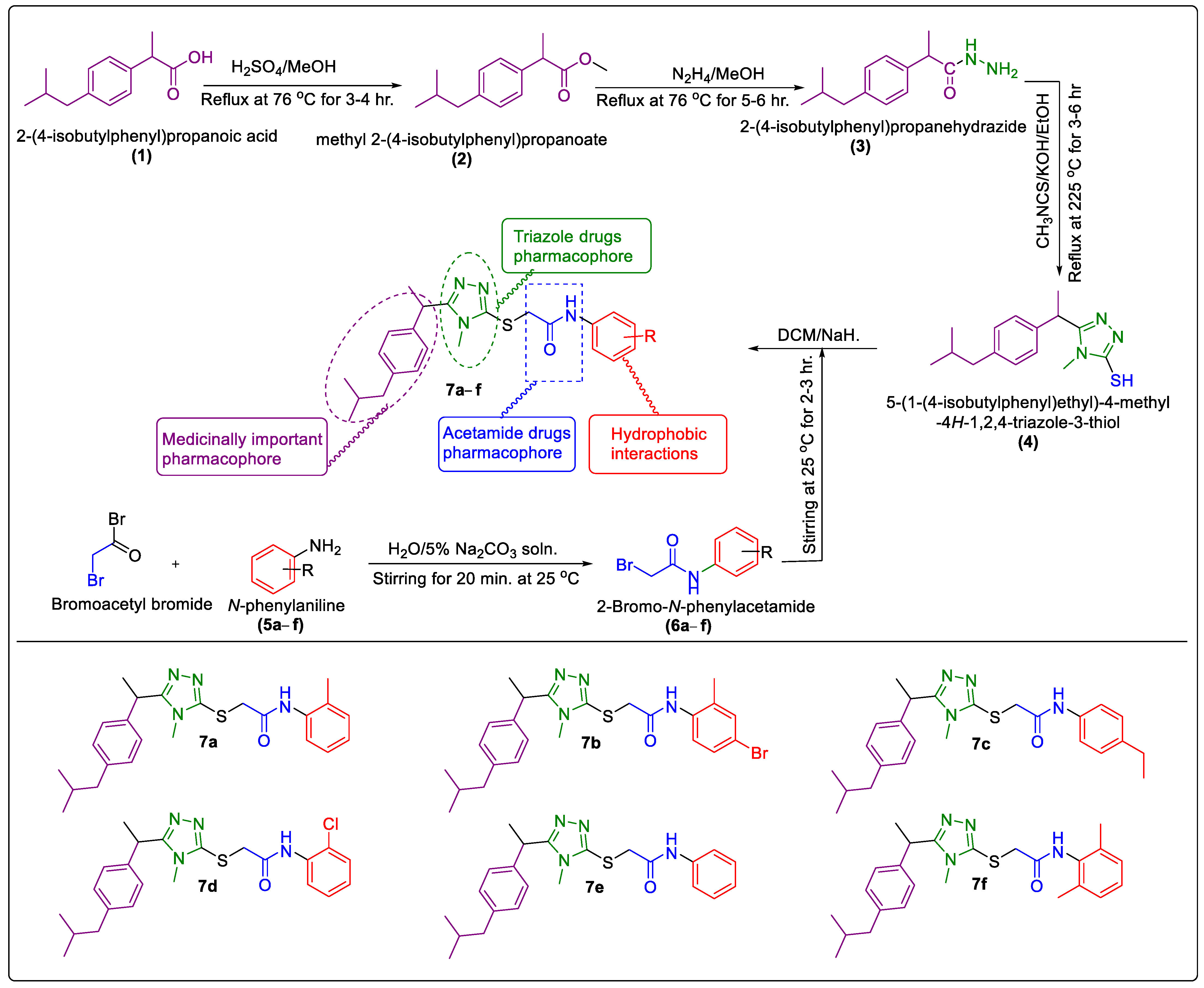
2.1. Chemistry
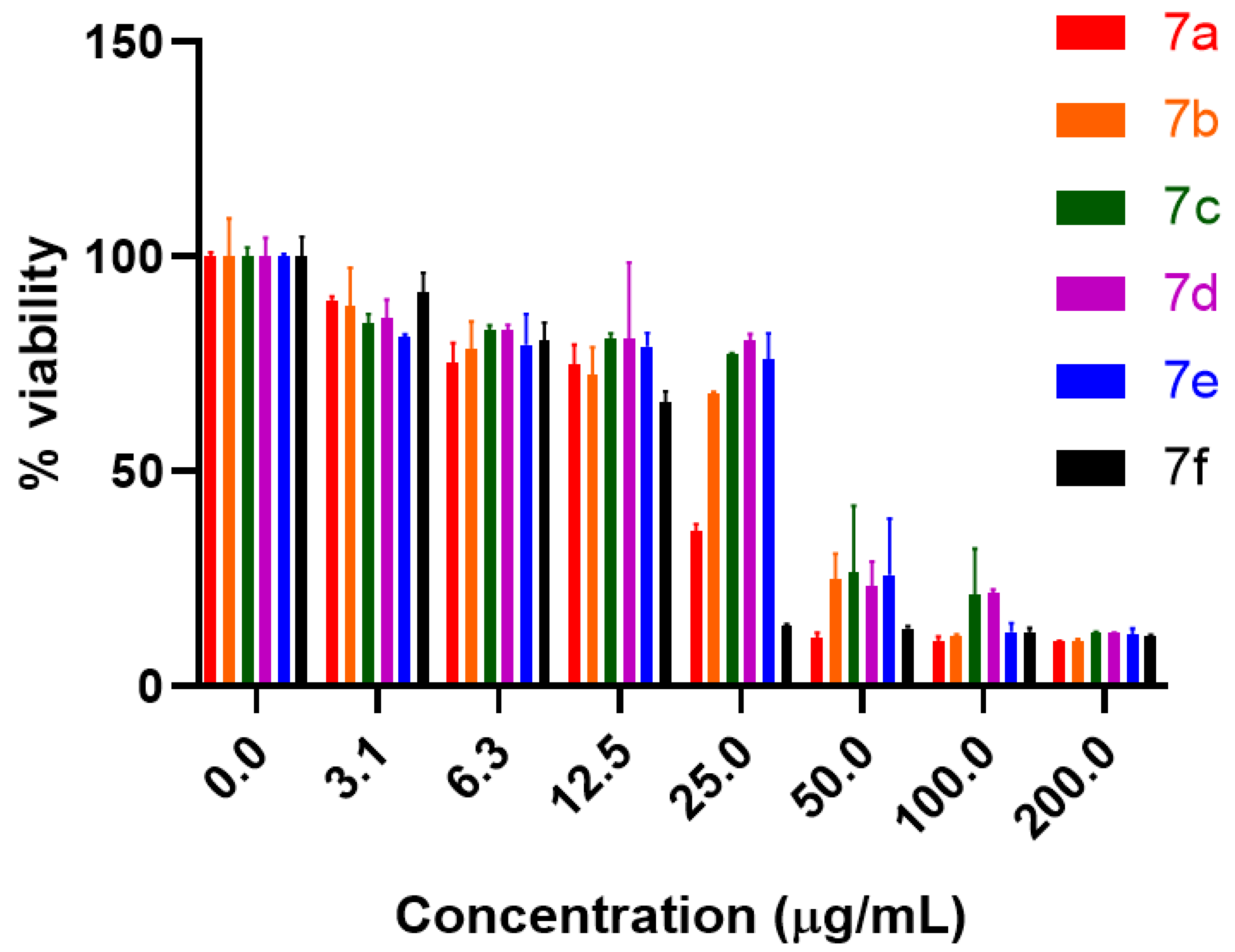
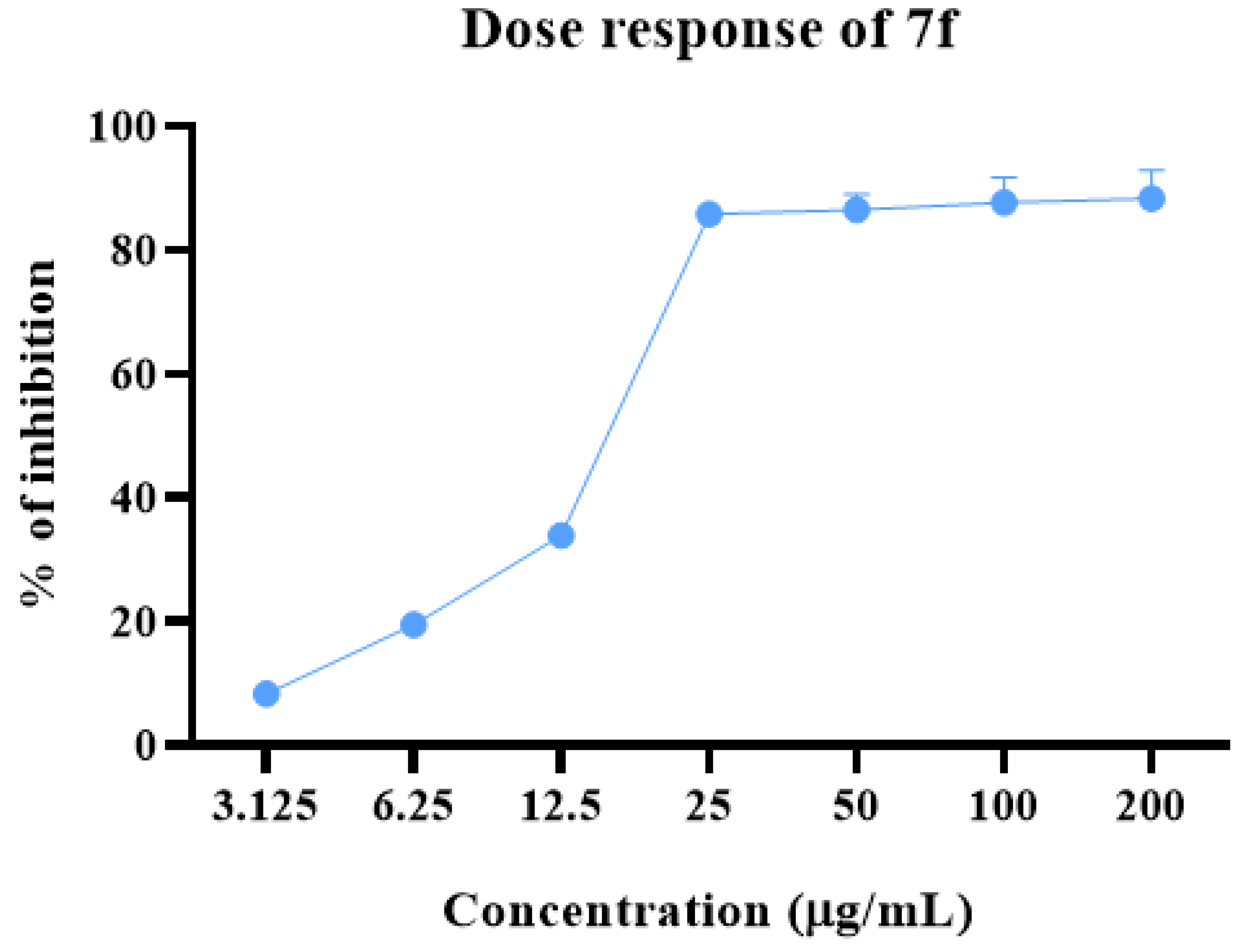
2.2. Anti-proliferative Potential
2.3. Hemolytic Activity Potential
2.4. Structure–Activity Relationship of 7a–f
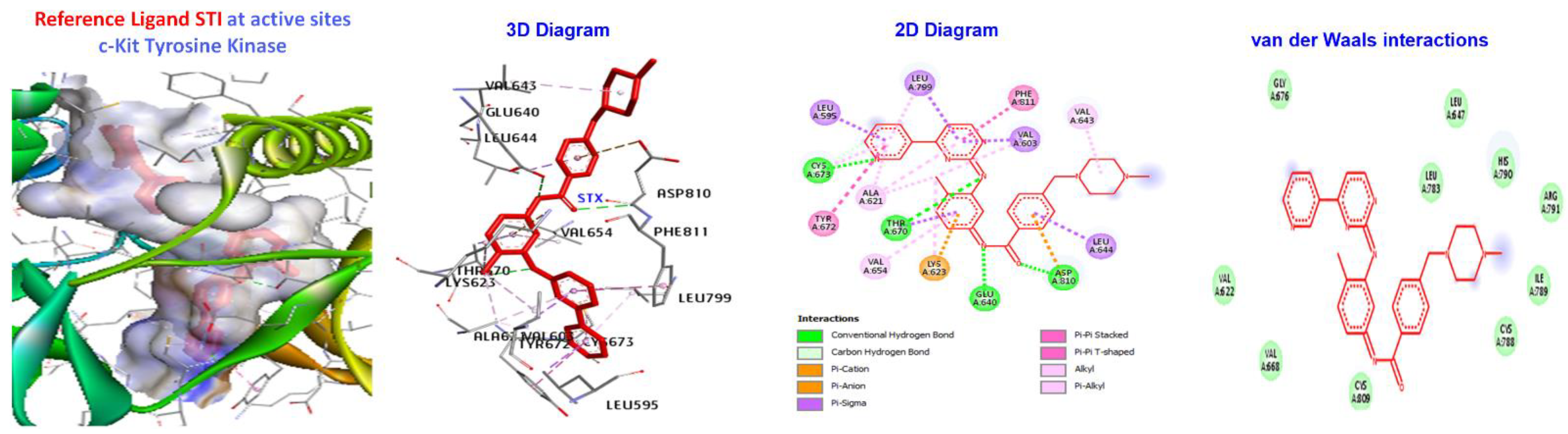
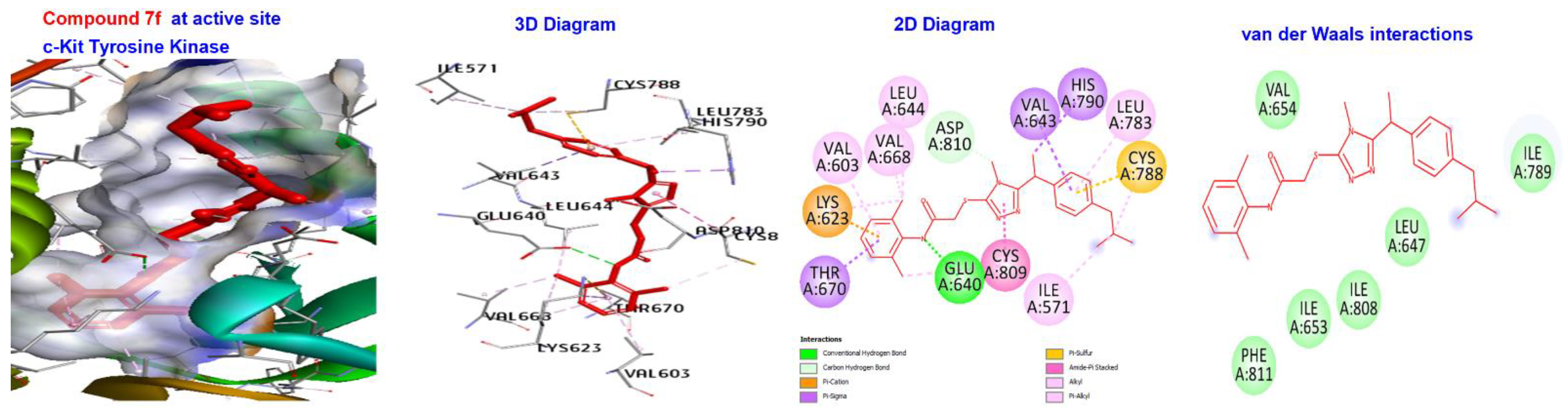
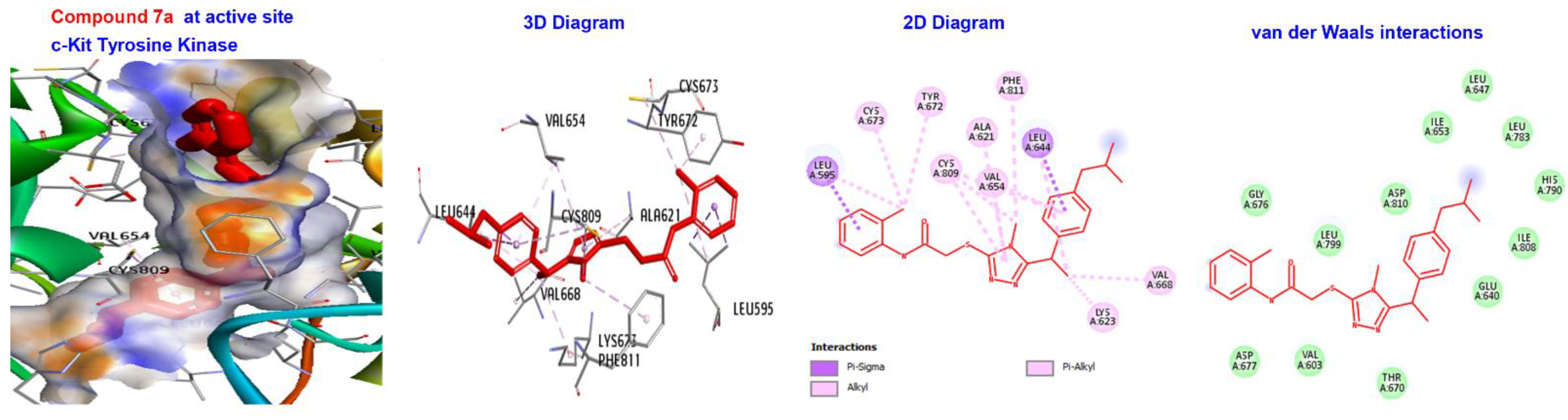
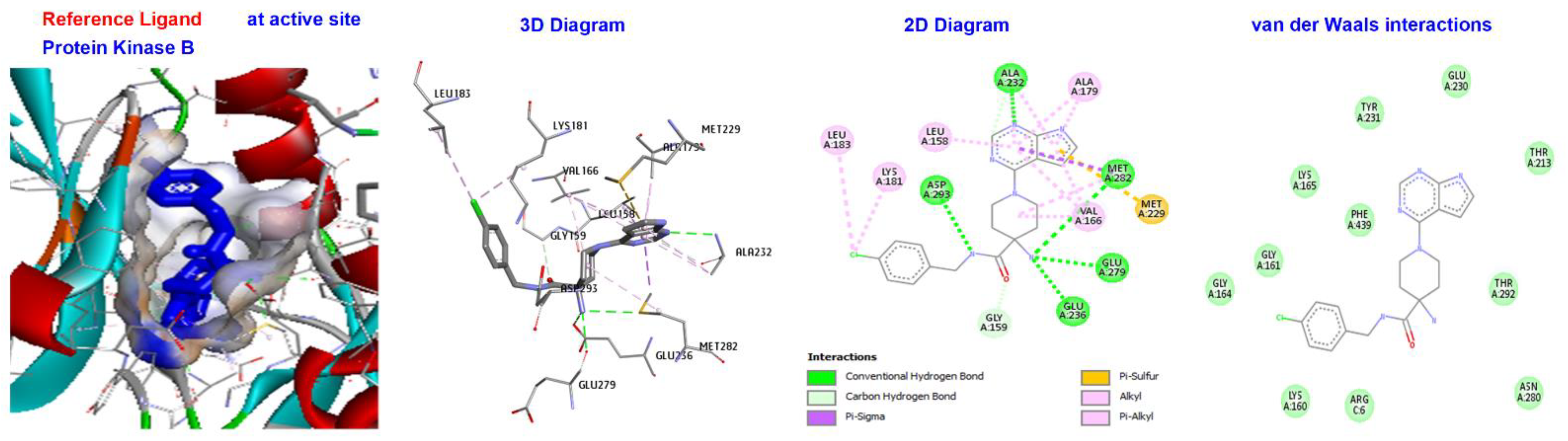
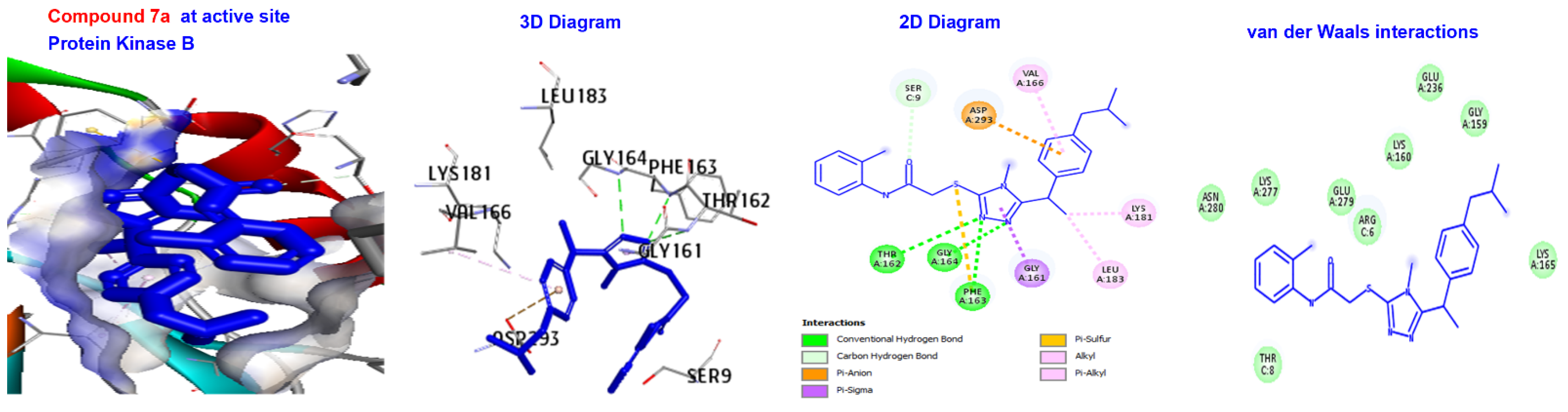
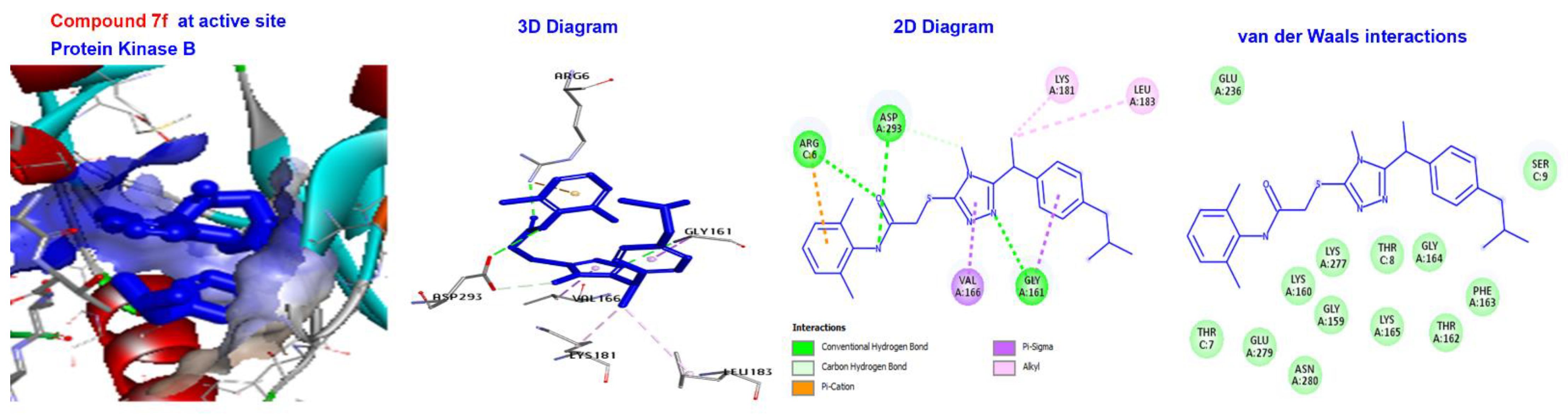
2.5. Molecular Docking
3. Materials and Methods
3.1. General
3.2. General Procedure for the Synthesis of Synthesized N-Arylated 5-Aryl-1,2,4-Triazole-Coupled Acetamide Scaffolds 7a–f
3.2.1. Synthesis of Methyl 2-(4-Isobutylphenyl)Propanoate (2)
3.2.2. Synthesis of 2-(4-Isobutylphenyl)Propanehydrazide (3)
3.2.3. Synthesis of 5-(1-(4- Isobutylphenyl)Ethyl)-1,2,4-Triazole -2-Thiol (4)
3.2.4. Synthesis of N–Aryl/Alkyl 2-Bromoroacetamides 6a–f
3.2.5. Synthesis of N-Arylated 5-(1-(4-Isobutylphenyl)Ethyl)-1,2,4-Triazole-2-yl- 2-Sulfanyl Coupled Acetamide Derivatives 7a–f
3.2.6. N-(2-Methylphenyl)-2-((5-(1-(4-isobutylphenyl)ethyl)-4-methyl-4H-1,2,4-triazol-3-yl)thio)Acetamide (7a)
3.2.7. N-(4-Bromo-2-Mthylphenyl)-2-((5-(1-(4-Isobutylphenyl)Ethyl)-4-Methyl-4H-1,2,4-Triazol-3-yl)Thio)Acetamide (7b)
3.2.8. N-(4-Ethylphenyl)-2-((5-(1-(4-Isobutylphenyl)Ethyl)-4-Methyl-4H-1,2,4-Triazol-3-yl)Thio)Acetamide (7c)
3.2.9. N-(2-Chlorophenyl)-2-((5-(1-(4-Isobutylphenyl)Ethyl)-4-Methyl-4H-1,2,4-Triazol-3-yl)Thio)Acetamide (7d)
3.2.10. N-(Phenyl)-2-((5-(1-(4-Isobutylphenyl)Ethyl)-4-Methyl-4H-1,2,4-Triazol-3-yl)Thio)Acetamide (7e)
3.2.11. N-(2,6-Dimethylphenyl)-2-((5-(1-(4-Isobutylphenyl)Ethyl)-4-Methyl-4H-1,2,4-Triazol-3-yl)Thio)Acetamide (7f)
3.3. Experimental Procedures for Biological Activities
3.3.1. Cell Culture and Treatment
3.3.2. Evaluation of Cell Viability
3.3.3. Hemolytic Activity Potential
3.4. Molecular Docking of Triazole-Coupled Acetamides
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Kamal, A.; Bharathi, E.V.; Reddy, J.S.; Ramaiah, M.J.; Dastagiri, D.; Reddy, M.K.; Viswanath, A.; Reddy, T.L.; Shaik, T.B.; Pushpavalli, S.; et al. Synthesis and biological evaluation of 3,5-diaryl isoxazoline/isoxazole linked 2,3-dihydroquinazolinone hybrids as anticancer agents. Eur. J. Med. Chem. 2011, 46, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, H.T.; Purohit, A.P.; Desai, S.C.; Jarwani, P.B. Retrospective analysis of histopathological spectrum of premalignant and malignant colorectal lesions. Cancer Res. Stat. Treat. 2021, 4, 472–478. [Google Scholar] [CrossRef]
- Jang, J.-W.; Song, Y.; Kim, K.M.; Kim, J.-S.; Choi, E.K.; Kim, J.; Seo, H. Hepatocellular carcinoma-targeted drug discovery through image-based phenotypic screening in co-cultures of HCC cells with hepatocytes. BMC Cancer 2016, 16, 810. [Google Scholar] [CrossRef] [PubMed]
- Rumgay, H.; Arnold, M.; Ferlay, J.; Lesi, O.; Cabasag, C.J.; Vignat, J.; Laversanne, M.; McGlynn, K.A.; Soerjomataram, I. Global burden of primary liver cancer in 2020 and predictions to 2040. J. Hepatol. 2022, 77, 1598–1606. [Google Scholar] [CrossRef]
- Zheng, Z.; Liu, Q.; Kim, W.; Tharmalingam, N.; Fuchs, B.B.; Mylonakis, E. Antimicrobial activity of 1,3,4-oxadiazole derivatives against planktonic cells and biofilm of Staphylococcus aureus. Futur. Med. Chem. 2018, 10, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Chen, J.-S.; Chou, C.-H.; Wu, Y.-H.; Yang, M.-H.; Chu, S.-H.; Chao, Y.-S.; Chen, C.-N. CC-01 (chidamide plus celecoxib) modifies the tumor immune microenvironment and reduces tumor progression combined with immune checkpoint inhibitor. Sci. Rep. 2022, 12, 1100. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, B.; Wang, Y.; Wang, X.; Gou, S. Discovery of phthalazino[1,2-b]-quinazolinone derivatives as multi-target HDAC inhibitors for the treatment of hepatocellular carcinoma via activating the p53 signal pathway. Eur. J. Med. Chem. 2021, 229, 114058. [Google Scholar] [CrossRef]
- Chen, C.; Li, X.; Zhao, H.; Liu, M.; Du, J.; Zhang, J.; Yang, X.; Hou, X.; Fang, H. Discovery of DNA-Targeting HDAC Inhibitors with Potent Antitumor Efficacy In Vivo That Trigger Antitumor Immunity. J. Med. Chem. 2022, 65, 3667–3683. [Google Scholar] [CrossRef]
- Khan, I.; Ibrar, A.; Abbas, N.; Saeed, A. Recent advances in the structural library of functionalized quinazoline and quinazolinone scaffolds: Synthetic approaches and multifarious applications. Eur. J. Med. Chem. 2014, 76, 193–244. [Google Scholar] [CrossRef]
- Khwaza, V.; Mlala, S.; Oyedeji, O.; Aderibigbe, B. Pentacyclic Triterpenoids with Nitrogen-Containing Heterocyclic Moiety, Privileged Hybrids in Anticancer Drug Discovery. Molecules 2021, 26, 2401. [Google Scholar] [CrossRef] [PubMed]
- Gomha, S.M.; Abdel-Aziz, H.M.; El-Reedy, A.A.M. Facile Synthesis of Pyrazolo[3,4-c]pyrazoles Bearing Coumarine Ring as Anticancer Agents. J. Heterocycl. Chem. 2018, 55, 1960–1965. [Google Scholar] [CrossRef]
- Kaproń, B.; Czarnomysy, R.; Wysokiński, M.; Andrys, R.; Musilek, K.; Angeli, A.; Supuran, C.T.; Plech, T. 1,2,4-Triazole-based anticonvulsant agents with additional ROS scavenging activity are effective in a model of pharmacoresistant epilepsy. J. Enzym. Inhib. Med. Chem. 2020, 35, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-S.; Tian, H.; Zhao, D.-S.; Hu, D.-K.; Liu, X.-Y.; Jin, H.-W.; Song, G.-P.; Cui, Z.-N. Synthesis and bioactivity of pyrazole and triazole derivatives as potential PDE4 inhibitors. Bioorganic Med. Chem. Lett. 2016, 26, 3632–3635. [Google Scholar] [CrossRef]
- de Andrade, M.A.R.; Lima, B.J.S.; de Jesus, A.A.; Teixeira, L.D.A.C.; de Souza, C.R.S.; Fontes, R.B.; Mendonça, N.P.V.; Gonçalves, H.S.; Cesar, A.S.; Aragão, M.T. Burnout Syndrome in COVID-19: An analysis of physicians from the public and private health system in the state of Sergipe. Res. Soc. Dev. 2022, 11, e3911729602. [Google Scholar] [CrossRef]
- Kumar, S.; Khokra, S.L.; Yadav, A. Triazole analogues as potential pharmacological agents: A brief review. Futur. J. Pharm. Sci. 2021, 7, 106. [Google Scholar] [CrossRef]
- Han, S.; Zhang, F.-F.; Xie, X.; Chen, J.-Z. Design, synthesis, biological evaluation, and comparative docking study of 1,2,4-triazolones as CB1 receptor selective antagonists. Eur. J. Med. Chem. 2014, 74, 73–84. [Google Scholar] [CrossRef]
- Zhang, W.; Yuan, J. Poly(1-Vinyl-1,2,4-triazolium) Poly(Ionic Liquid)s: Synthesis and the Unique Behavior in Loading Metal Ions. Macromol. Rapid Commun. 2016, 37, 1124–1129. [Google Scholar] [CrossRef]
- Cao, X.; Wang, W.; Wang, S.; Bao, L. Asymmetric synthesis of novel triazole derivatives and their in vitro antiviral activity and mechanism of action. Eur. J. Med. Chem. 2017, 139, 718–725. [Google Scholar] [CrossRef]
- Hu, G.; Wang, C.; Xin, X.; Li, S.; Li, Z.; Zhao, Y.; Gong, P. Design, synthesis and biological evaluation of novel 2,4-diaminopyrimidine derivatives as potent antitumor agents. New J. Chem. 2019, 43, 10190–10202. [Google Scholar] [CrossRef]
- Ni, T.; Ding, Z.; Xie, F.; Hao, Y.; Bao, J.; Zhang, J.; Yu, S.; Jiang, Y.; Zhang, D. Design, Synthesis, and In Vitro and In Vivo Antifungal Activity of Novel Triazoles Containing Phenylethynyl Pyrazole Side Chains. Molecules 2022, 27, 3370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.-F.; Liu, X.; Zhang, S.; Pan, B.; Liu, M.-L. Ciprofloxacin derivatives and their antibacterial activities. Eur. J. Med. Chem. 2018, 146, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xu, L.; Zhu, L.; Zhao, Y.; Hu, T.; Yin, B.; Liu, Y.; Hou, Y. Design, synthesis and biological evaluation of novel pteridinone derivatives possessing a hydrazone moiety as potent PLK1 inhibitors. Bioorganic Med. Chem. Lett. 2020, 30, 127329. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, T.; Hameed, S.; Al-Masoudi, N.P.; Khan, K.M. Synthesis and anti-HIV activity of new chiral 1,2,4-triazoles and 1,3,4-thiadiazoles. Heteroat. Chem. 2007, 18, 316–322. [Google Scholar] [CrossRef]
- Irfan, A.; Faiz, S.; Rasul, A.; Zafar, R.; Zahoor, A.F.; Kotwica-Mojzych, K.; Mojzych, M. Exploring the Synergistic Anticancer Potential of Benzofuran–Oxadiazoles and Triazoles: Improved Ultrasound- and Microwave-Assisted Synthesis, Molecular Docking, Hemolytic, Thrombolytic and Anticancer Evaluation of Furan-Based Molecules. Molecules 2022, 27, 1023. [Google Scholar] [CrossRef]
- Gul, S.; Rehman, A.U.; Abbasi, M.A.; Khan, K.M.; Nafeesa, K.; Siddiqa, A.; Akhtar, M.N.; Shahid, M.; Subhani, Z. Synthesis, antimicrobial evaluation and hemolytic activity of 2-[[5-alkyl/aralkyl substituted-1,3,4-oxadiazol-2-yl]thio]-N-[4-(4-morpholinyl)phenyl]acetamide derivatives. J. Saudi Chem. Soc. 2014, 21, S425–S433. [Google Scholar] [CrossRef]
- Khan, S.G.; Bokhari, T.H.; Anjum, F.; Akhter, N.; Rasool, S.; Shah, S.A.A.; Shahid, M.; Arshad, A. Synthesis, characterization, antibacterial, hemolytic and thrombolytic activity evaluation of 5-(3-chlorophenyl)-2-((N-(substituted)-2-acetamoyl) sulfanyl)-1, 3, 4-oxadiazole derivatives. Pak. J. Pharm. Sci. 2020, 33, 871–876. [Google Scholar]
- Mahmood, S.; Khan, S.G.; Rasul, A.; Christensen, J.B.; Abourehab, M.A.S. Ultrasound Assisted Synthesis and In Silico Modelling of 1,2,4-Triazole Coupled Acetamide Derivatives of 2-(4-Isobutyl phenyl)propanoic acid as Potential Anticancer Agents. Molecules 2022, 27, 7984. [Google Scholar] [CrossRef]
- Aziz-ur-Rehman; Khan, S.G.; Naqvi, S.A.R.; Ahmad, M.; Akhtar, N.; Bokhari, T.H.; Irfan, M.; Usman, A.; Batool, S.; Rasool, S. Synthesis, spectral analysis and biological evaluation of 2-{[(morpholin-4-yl) ethyl] thio}-5-phenyl/aryl-1, 3, 4-oxadiazole derivatives. Pak. J. Pharm. Sci. 2021, 34, 441–446. [Google Scholar] [CrossRef]
- Amewu, R.K.; Sakyi, P.O.; Osei-Safo, D.; Addae-Mensah, I. Synthetic and Naturally Occurring Heterocyclic Anticancer Compounds with Multiple Biological Targets. Molecules 2021, 26, 7134. [Google Scholar] [CrossRef]
- Zabiulla; Gulnaz, A.; Mohammed, Y.H.E.; Khanum, S.A. Design, synthesis and molecular docking of benzophenone conjugated with oxadiazole sulphur bridge pyrazole pharmacophores as anti-inflammatory and analgesic agents. Bioorganic Chem. 2019, 92, 103220. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Jamal, Q.; Iqbal, J.; Afreen, M.S.; Sandhu, M.Z.A.; Dar, E.; Farooq, U.; Mushtaq, M.F.; Arshad, N.; Iqbal, M.M. Synthesis of N-substituted acetamide derivatives of azinane-bearing 1,3,4-oxadiazole nucleus and screening for antibacterial activity. Trop. J. Pharm. Res. 2017, 16, 429. [Google Scholar] [CrossRef]
- Thongnest, S.; Chawengrum, P.; Keeratichamroen, S.; Lirdprapamongkol, K.; Eurtivong, C.; Boonsombat, J.; Kittakoop, P.; Svasti, J.; Ruchirawat, S. Vernodalidimer L, a sesquiterpene lactone dimer from Vernonia extensa and anti-tumor effects of vernodalin, vernolepin, and vernolide on HepG2 liver cancer cells. Bioorganic Chem. 2019, 92, 103197. [Google Scholar] [CrossRef] [PubMed]
- Shahzadi, I.; Parveen, B.; Ahmad, S.; Zahoor, A.F.; Rasul, A.; Zahid, F.M. In-vitro cytotoxic evaluation, hemolytic and thrombolytic potential of newly designed acefylline based hydrazones as potent anti-cancer agents against human lung cancer cell line (A549). Pak. J. Pharm. Sci. 2022, 35, 885–889. [Google Scholar] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [PubMed]
- Mol, C.D.; Dougan, D.R.; Schneider, T.R.; Skene, R.J.; Kraus, M.L.; Scheibe, D.N.; Snell, G.P.; Zou, H.; Sang, B.-C.; Wilson, K.P. Structural Basis for the Autoinhibition and STI-571 Inhibition of c-Kit Tyrosine Kinase. J. Biol. Chem. 2004, 279, 31655–31663. [Google Scholar] [CrossRef] [PubMed]
- McHardy, T.; Caldwell, J.J.; Cheung, K.-M.; Hunter, L.J.; Taylor, K.; Rowlands, M.; Ruddle, R.; Henley, A.; Brandon, A.D.H.; Valenti, M.; et al. Discovery of 4-Amino-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamides As Selective, Orally Active Inhibitors of Protein Kinase B (Akt). J. Med. Chem. 2010, 53, 2239–2249. [Google Scholar] [CrossRef] [PubMed]
- Elkins, J.M.; Santaguida, S.; Musacchio, A.; Knapp, S. Crystal Structure of Human Aurora B in Complex with INCENP and VX-680. J. Med. Chem. 2012, 55, 7841–7848. [Google Scholar] [CrossRef]
- Degorce, S.L.; Boyd, S.; Curwen, J.O.; Ducray, R.; Halsall, C.T.; Jones, C.D.; Lach, F.; Lenz, E.M.; Pass, M.; Pass, S.; et al. Discovery of a Potent, Selective, Orally Bioavailable, and Efficacious Novel 2-(Pyrazol-4-ylamino)-pyrimidine Inhibitor of the Insulin-like Growth Factor-1 Receptor (IGF-1R). J. Med. Chem. 2016, 59, 4859–4866. [Google Scholar] [CrossRef]
- Zhang, Q.; Sang, F.; Qian, J.; Lyu, S.; Wang, W.; Wang, Y.; Li, Q.; Du, L. Identification of novel potential PI3Kα inhibitors for cancer therapy. J. Biomol. Struct. Dyn. 2020, 39, 3721–3732. [Google Scholar] [CrossRef]
- Bai, L.; Zhou, H.; Xu, R.; Zhao, Y.; Chinnaswamy, K.; McEachern, D.; Chen, J.; Yang, C.-Y.; Liu, Z.; Wang, M.; et al. A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo. Cancer Cell 2019, 36, 498–511.e17. [Google Scholar] [CrossRef] [PubMed]
- Sardar, A.; Abid, O.-U.; Daud, S.; Fakhar-E-Alam, M.; Siddique, M.H.; Ashraf, M.; Shahid, W.; Ejaz, S.A.; Atif, M.; Ahmad, S.; et al. Design, synthesis, in vitro and in silico studies of naproxen derivatives as dual lipoxygenase and α-glucosidase inhibitors. J. Saudi Chem. Soc. 2022, 26, 101468. [Google Scholar] [CrossRef]
- Alderawy MQ, A.; Alrubaie LA, R.; Sheri, F.H. Synthesis, Characterization of Ibuprofen N-Acyl-1, 3, 4-Oxadiazole Derivatives and Anti-cancer Activity against MCF-7 Cell Line. Syst. Rev. Pharm. 2020, 11, 681–689. [Google Scholar] [CrossRef]
- Shaikh, M.H.; Subhedar, D.D.; Nawale, L.; Sarkar, D.; Khan, F.A.K.; Sangshetti, J.N.; Shingate, B.B. 1,2,3-Triazole derivatives as antitubercular agents: Synthesis, biological evaluation and molecular docking study. MedChemComm 2015, 6, 1104–1116. [Google Scholar] [CrossRef]
- Abutaha, N.; Al-Mekhlafi, F.A.; Almutairi, B.O.; Wadaan, M.A. S-phase cell cycle arrest, and apoptotic potential of Echium arabicum phenolic fraction in hepatocellular carcinoma HepG2 cells. J. King Saud Univ.—Sci. 2021, 34, 101735. [Google Scholar] [CrossRef]
- Win, T.S.; Malik, A.A.; Prachayasittikul, V.S.; Wikberg, J.E.; Nantasenamat, C.; Shoombuatong, W. HemoPred: A web server for predicting the hemolytic activity of peptides. Futur. Med. Chem. 2017, 9, 275–291. [Google Scholar] [CrossRef]
- Le, P.T.; Cheng, H.; Ninkovic, S.; Plewe, M.; Huang, X.; Wang, H.; Bagrodia, S.; Sun, S.; Knighton, D.R.; Rogers, C.M.L.; et al. Design and synthesis of a novel pyrrolidinyl pyrido pyrimidinone derivative as a potent inhibitor of PI3Kα and mTOR. Bioorganic Med. Chem. Lett. 2012, 22, 5098–5103. [Google Scholar] [CrossRef]
- Thomsen, R.; Christensen, M.H. MolDock: A New Technique for High-Accuracy Molecular Docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Alkyl/Aryl | Cell Viability IC50 Value (µg/mL) | Hemolytic Activity (Mean% ± S.D) |
|---|---|---|---|
| 7a | 2-methyl phenyl | 20.667 | 2.46 ± 0.31 |
| 7b | 2-methyl-4-bromo phenyl | 33.565 | 2.43 ± 0.11 |
| 7c | 4-ethyl phenyl | 39.002 | 4.32 ± 0.24 |
| 7d | 2-chloro phenyl | 39.667 | 7.33 ± 0.42 |
| 7e | phenyl | 39.105 | 4.19 ± 0.02 |
| 7f | 2,6-dimethyl phenyl | 16.782 | 1.19 ± 0.02 |
| Sorafenib | 05.971 | ||
| PBS | 0.00 ± 0.0 | ||
| Triton-X-100 | 100 ± 0.0 |
| Concentration (µg/mL) | 7a | 7b | 7c | 7d | 7e | 7f |
|---|---|---|---|---|---|---|
| 200 | 89.40 ± 0.34 | 89.50 ± 0.06 | 87.51 ± 0.20 | 87.70 ± 0.18 | 88.00 ± 1.44 | 88.36 ± 0.31 |
| 100 | 88.17 ± 0.26 | 89.39 ± 0.91 | 78.82 ± 10.78 | 78.33 ± 0.78 | 87.34 ± 2.09 | 87.71 ± 1.34 |
| 50 | 75.18 ± 5.99 | 88.64 ± 1.11 | 73.66 ± 15.54 | 76.69 ± 5.63 | 74.24 ± 13.16 | 86.57 ± 0.56 |
| 25 | 63.98 ± 1.67 | 31.90 ± 0.30 | 22.82 ± 0.29 | 19.52 ± 1.44 | 24.01 ± 6.12 | 85.90 ± 0.30 |
| 12.5 | 27.70 ± 6.46 | 25.23 ± 4.67 | 19.14 ± 1.25 | 19.11 ± 17.71 | 20.99 ± 3.13 | 33.96 ± 2.47 |
| 6.25 | 21.44 ± 6.24 | 24.75 ± 4.62 | 17.35 ± 1.32 | 17.32 ± 1.40 | 20.52 ± 7.11 | 19.55 ± 4.07 |
| 3.125 | 11.57 + 8.85 | 10.33 + 0.96 | 15.40 ± 2.01 | 14.27 ± 4.27 | 18.65 ± 0.50 | 8.40 ± 4.54 |
| DMSO (-ve Control) | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Targets | Protein Kinase B (Akt) (PKB) | c-Kit Tyrosine Kinase (c-Kit) | Human Aurora B Kinase (AURKB) | Phosphatidylinositol 3-Kinase Alpha (PI3Kalpha) | Signal Transducer and Activator of Transcription 3 (STAT3) |
|---|---|---|---|---|---|
| PDB ID | 2X39 | 1T46 | 4AF3 | 4FA6 | 6NJS |
| Center of docking | X:43 | X:28 | X:21 | X:44 | X:13 |
| Coordinates | Y:31 | Y:26 | Y:-22 | Y:14 | Y:56 |
| Z:111 | Z:39 | Z:-10 | Z:31 | Z:0.32 | |
| Reference Ligand | X39 | STI | VX6 | 0TA | KQV |
| Ligands | Mol. Dock Score (Kcal/mol) | Mol. Dock Score (Kcal/mol) | Mol. Dock Score (Kcal/mol) | Mol. Dock Score (Kcal/mol) | Mol. Dock Score (Kcal/mol) |
| 7a 7b | −166.843 −166.371 | −173.411 −167.882 | −145.234 −138.33 | −139.389 −136.267 | −125.105 −120.348 |
| 7c | −162.234 | −167.814 | −137.943 | −134.596 | −118.623 |
| 7d | −154.675 | −158.747 | −132.083 | −124.135 | −107.246 |
| 7e | −156.207 | −161.394 | −136.421 | −131.706 | −113.82 |
| 7f | −170.066 | −176.749 | −149.617 | −149.36 | −125.441 |
| Reference Molecules | −130.624 | −181.533 | −144.231 | −112.819 | −197.521 |
| Ligand | (ACE) (kcal/mol) | Category | Types | Interacting Residues |
|---|---|---|---|---|
| 7a | −173.411 | H-bond | Alkyl | LEU595, LYS623, VAL654, LEU644, LEU595,LEU644, CYS673, CYS809, and VAL668. |
| Hydrophobic | Pi-alkyl | TYR672. | ||
| 7b | −167.882 | H-bond | Sulfur-X | CYS809. |
| Hydrophobic | C-alkyl Pi-alkyl | LEU595, VAL654, LEU644, CYS809, LEU595, ILE808, LEU644, AL654. TYR672, HIS790, and PHE811. | ||
| 7c | −167.814 | H-bond | Conventional | GLU640. |
| Hydrophobic | Pi-Alkyl | HIS790. | ||
| Hydrophobic | C-alkyl | VAL643, VAL603, LYS623, VAL668, LEU783, CYS788, LYS623, LEU644, VAL668, ALA621, and CYS788. | ||
| 7d | −158.747 | H-bond | Conventional | GLU640 and ASP810. |
| Hydrophobic | C-alkyl Pi-alkyl | ILE808. VAL603, VAL643, LEU783, CYS788, CYS809, LEU595, VAL603, VAL643, and LEU783. | ||
| 7e | −161.394 | H-bond | Conventional | CYS673, |
| Hydrophobic | C-alkyl Pi-alkyl | LEU595, VAL654, LEU644, CYS809, LYS623, LEU644, LEU644, TYR672, and VAL668. | ||
| 7f | −176.749 | H-bond | Conventional H-bond Pi-sigma | GLU640, ASP810, And HIS790. |
| Hydrophobic | Alkyl | VAL603, LYS623, VAL643, LEU783, CYS788, LYS623, LEU644, VAL668, CYS809, ILE571, and CYS788. | ||
| Reference Ligand | H-bond | Conventional | ALA232, GLU236, MET282, ASP293, GLU279, | |
| STI | −181.533 | Other | C-H bond | MET229, GLY159, |
| Hydrophobic | C-alkyl | VAL166, LEU158, ALA179, LYS181, and LEU183. |
| Ligand | (ACE) (kcal/mol) | Category | Types | Interacting Residues |
|---|---|---|---|---|
| 7a | −166.843 | H-bond | Conventional C-H bond | GLY164. SER9. |
| Other Hydrophobic | Pi-sulfur C-alkyl | PHE163. VAL166, LYS181, and EU183. | ||
| 7b | −166.371 | H-bond | Conventional | CYS809. |
| Hydrophobic | C-alkyl | LYS181, VAL166, and LEU296. | ||
| 7c | −162.234 | Hydrophobic | Alkyl | LYS181, VAL166, and LEU183. |
| 7d | −154.675 | H-bond | Conventional | GLY161, LEU158. |
| Hydrophobic | C-H bond C-alkyl Pi-alkyl | ASP293. VAL166, LYS181. PHE239, and PHE439. | ||
| 7e | −156.207 | H-bond | C-H bond | CYS673. |
| Hydrophobic | Alkyl | LEU296. | ||
| 7f | −170.066 | H-bond | Conventional C-H bond | GLY161, ASP293, and ARG6. ASP293. |
| Hydrophobic | C-alkyl | LYS181 and LEU183. | ||
| Reference Ligand | H-bond | Conventional | ALAA232, ASPA293, META282, GLUA279, GLUA236 | |
| X39 | −130.624 | Other | C-H bond | MET229, GLY159, |
| Hydrophobic | C-alkyl | VAL166, LEU A183, LEU A158, ALA179, LYS and A181. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akhter, N.; Batool, S.; Khan, S.G.; Rasool, N.; Anjum, F.; Rasul, A.; Adem, Ş.; Mahmood, S.; Rehman, A.u.; Nisa, M.u.; et al. Bio-Oriented Synthesis and Molecular Docking Studies of 1,2,4-Triazole Based Derivatives as Potential Anti-Cancer Agents against HepG2 Cell Line. Pharmaceuticals 2023, 16, 211. https://doi.org/10.3390/ph16020211
Akhter N, Batool S, Khan SG, Rasool N, Anjum F, Rasul A, Adem Ş, Mahmood S, Rehman Au, Nisa Mu, et al. Bio-Oriented Synthesis and Molecular Docking Studies of 1,2,4-Triazole Based Derivatives as Potential Anti-Cancer Agents against HepG2 Cell Line. Pharmaceuticals. 2023; 16(2):211. https://doi.org/10.3390/ph16020211
Chicago/Turabian StyleAkhter, Naheed, Sidra Batool, Samreen Gul Khan, Nasir Rasool, Fozia Anjum, Azhar Rasul, Şevki Adem, Sadaf Mahmood, Aziz ur Rehman, Mehr un Nisa, and et al. 2023. "Bio-Oriented Synthesis and Molecular Docking Studies of 1,2,4-Triazole Based Derivatives as Potential Anti-Cancer Agents against HepG2 Cell Line" Pharmaceuticals 16, no. 2: 211. https://doi.org/10.3390/ph16020211
APA StyleAkhter, N., Batool, S., Khan, S. G., Rasool, N., Anjum, F., Rasul, A., Adem, Ş., Mahmood, S., Rehman, A. u., Nisa, M. u., Razzaq, Z., Christensen, J. B., Abourehab, M. A. S., Shah, S. A. A., & Imran, S. (2023). Bio-Oriented Synthesis and Molecular Docking Studies of 1,2,4-Triazole Based Derivatives as Potential Anti-Cancer Agents against HepG2 Cell Line. Pharmaceuticals, 16(2), 211. https://doi.org/10.3390/ph16020211