
In Silico and In Vitro Evaluation of the Mechanism of Action of Three VX809-Based Hybrid Derivatives as Correctors of the F508del CFTR Protein

 ,
,  ,
,  ,
,  , ,
, , 

Abstract
:1. Introduction
2. Results
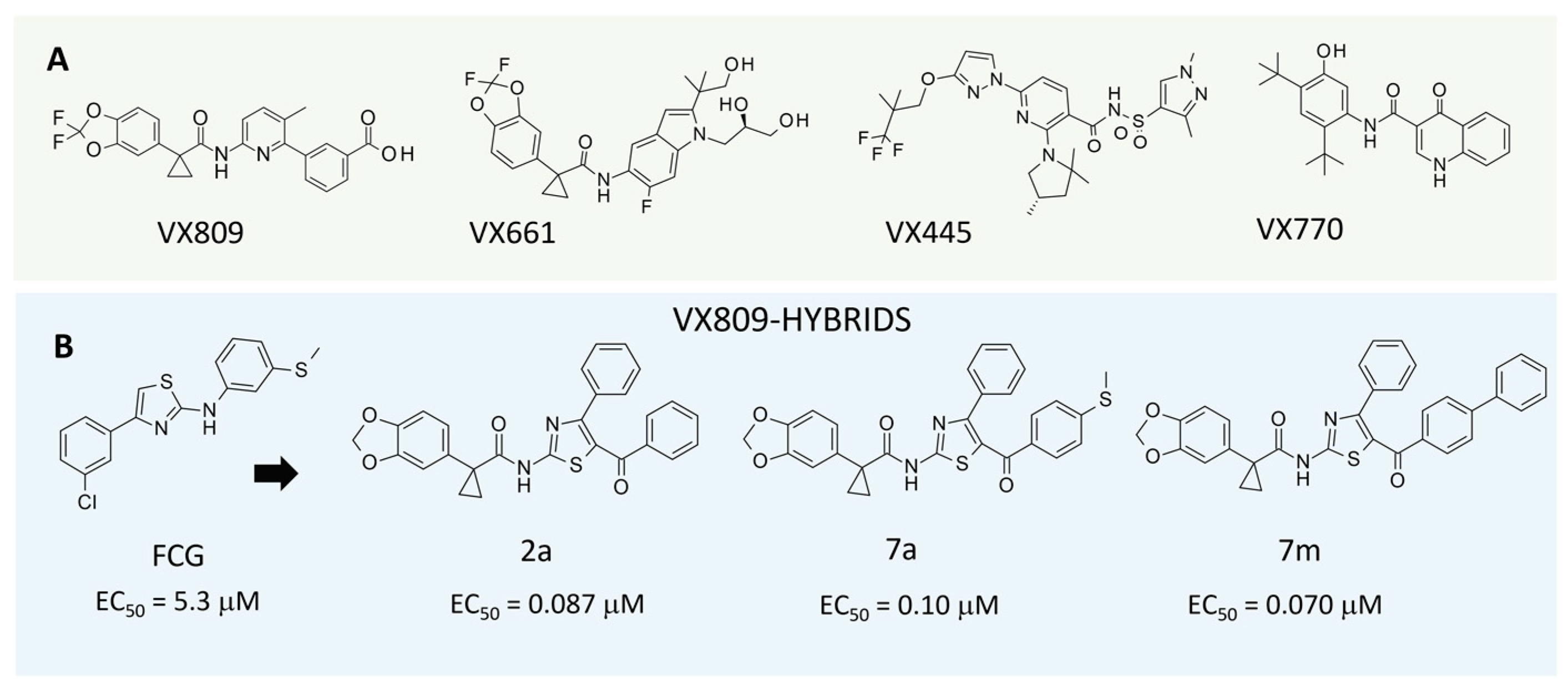
2.1. Computational Studies
2.1.1. In Silico Structure-Based Studies Assessment
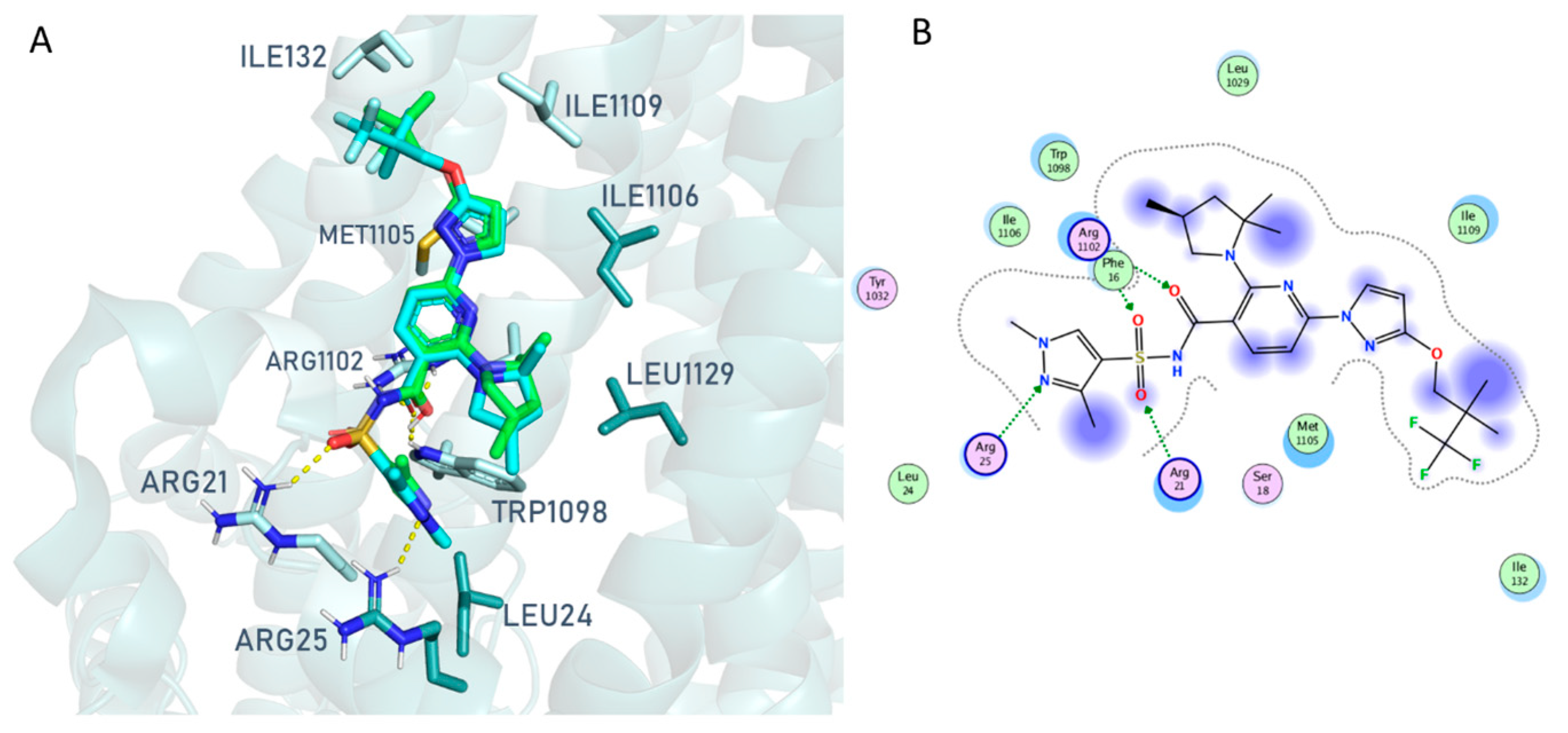
2.1.2. Molecular Docking Studies of Hybrids 2a, 7a, 7m
2.2. Biochemical Assays
2.2.1. Cytotoxicity of the Compounds under Analysis
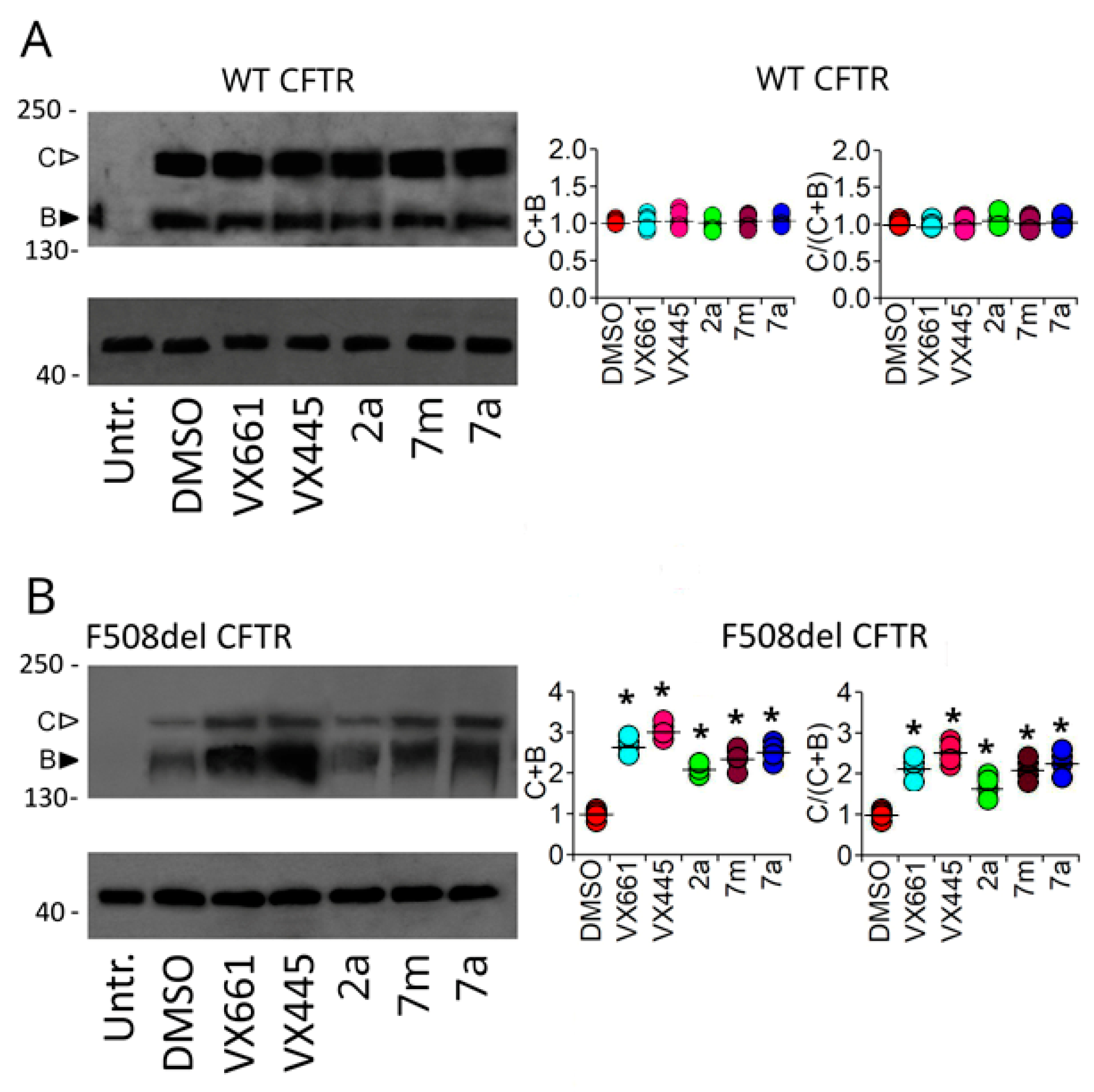
2.2.2. Assessing the Impact of Correctors on Full-Length WT and F508del CFTR Functional Expression
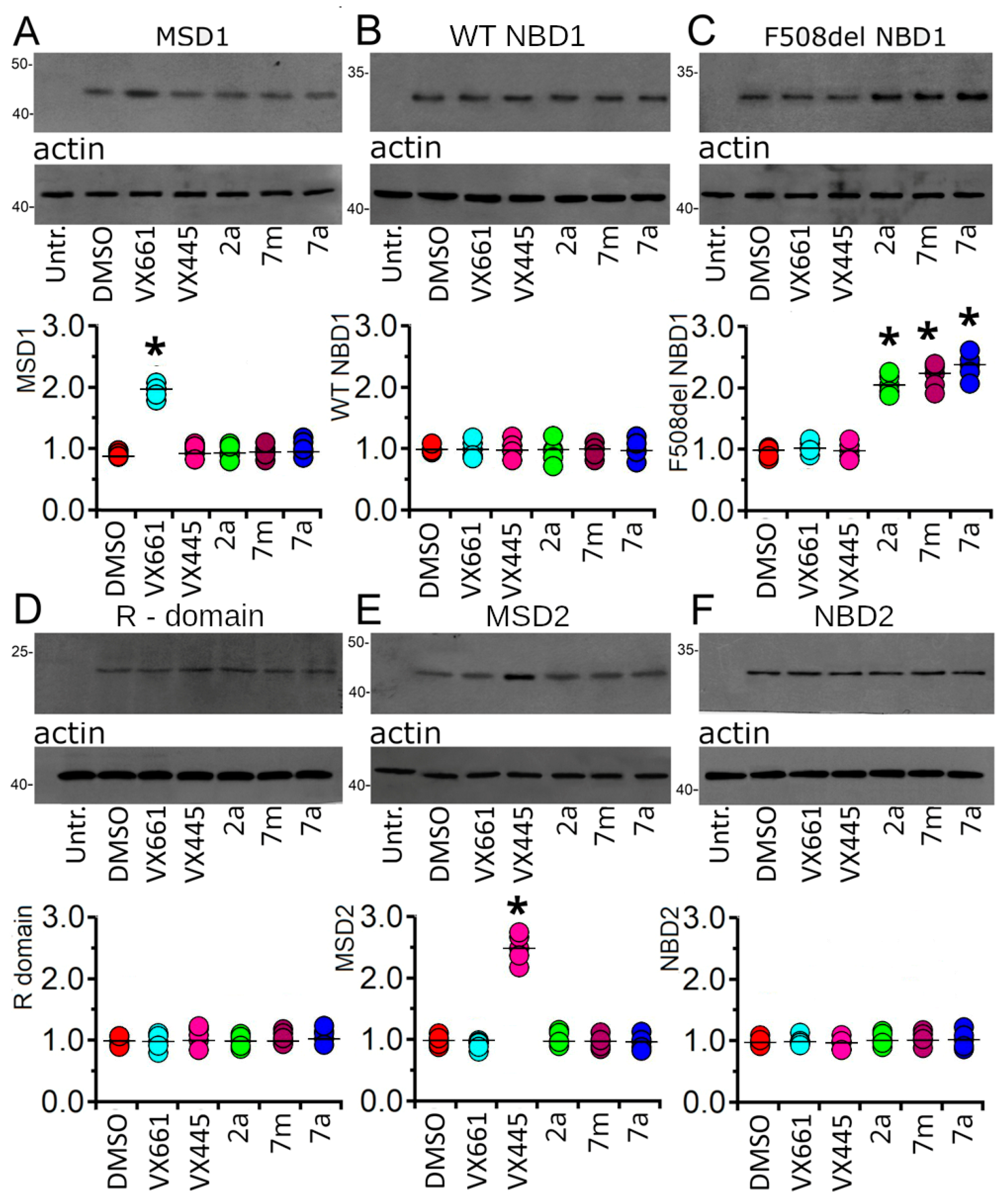
2.2.3. Assessing the Impact of Correctors on CFTR Single Domain Expression
2.2.4. Effect of Tested Compounds on the Stability of F508del NBD1
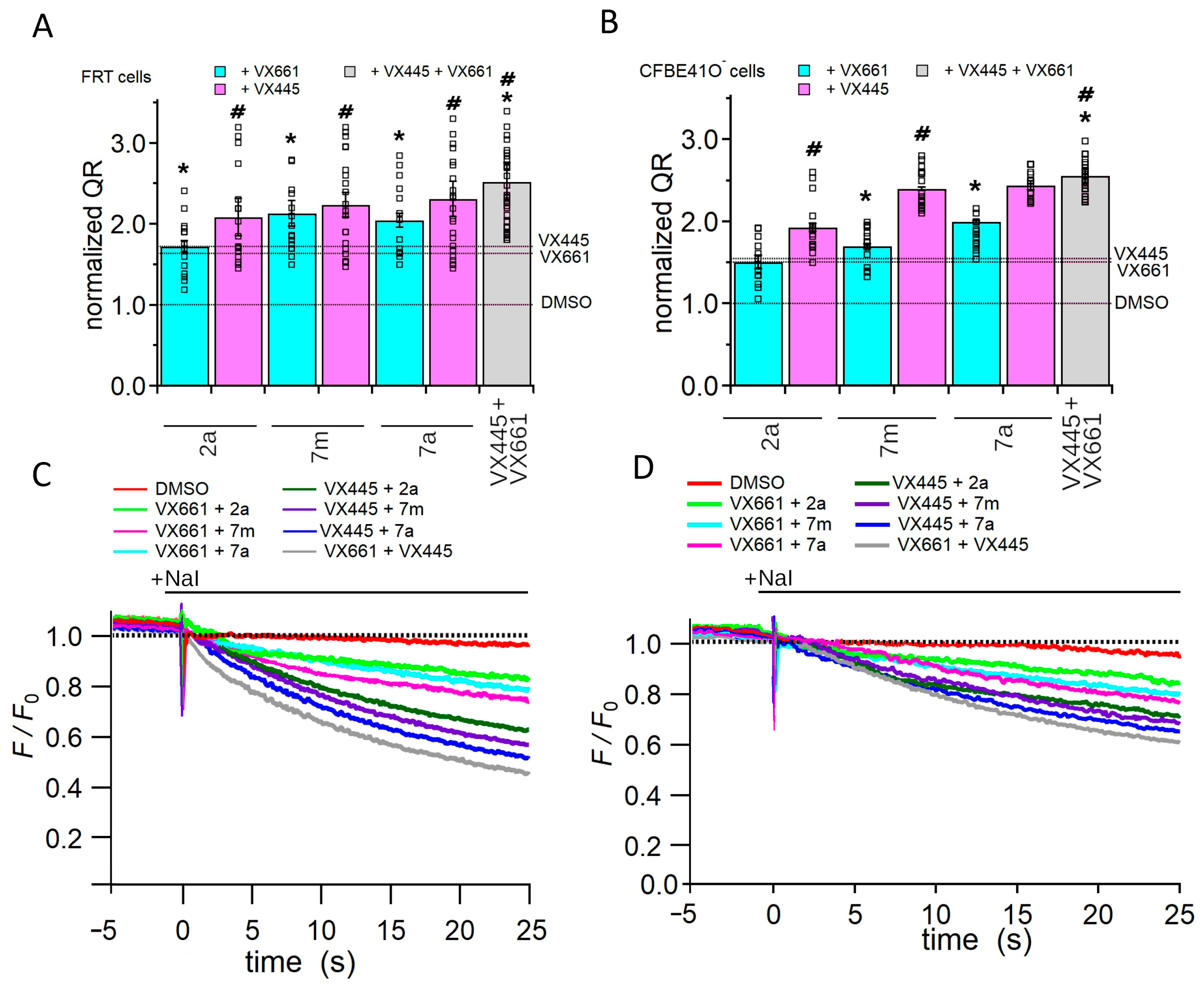
2.2.5. Assessing the Impact of Corrector Combinations on Full-Length F508del CFTR Function
3. Discussion
4. Materials and Methods
4.1. Computational Studies
4.2. Chemicals
4.3. Biochemical Assays
4.3.1. Cell Culture and Compound Toxicity Evaluation
4.3.2. Generation and Expression of CFTR Constructs
4.3.3. Western Blot
4.3.4. Cycloheximide Chase Assay
4.3.5. YFP Functional Assay
4.3.6. Data analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Riordan, J.R.; Rommens, J.M.; Kerem, B.-S.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.-L.; et al. Identification of the Cystic Fibrosis Gene: Cloning and Characterization of Complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Kerem, B.-S.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.-C. Identification of the Cystic Fibrosis Gene: Genetic Analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Castellani, C.; Assael, B.M. Cystic Fibrosis: A Clinical View. Cell. Mol. Life Sci. 2017, 74, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, P.; Seidler, U.; Kunzelmann, K. CFTR-beyond the Airways: Recent Findings on the Role of the CFTR Channel in the Pancreas, the Intestine and the Kidneys. J. Cyst. Fibros. 2023, 22, S17–S22. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, Z.; Csanády, L.; Gadsby, D.C.; Chen, J. Molecular Structure of the Human CFTR Ion Channel. Cell 2017, 169, 85–95.e8. [Google Scholar] [CrossRef] [PubMed]
- MIM 602421. Available online: http://www.genet.sickkids.on.ca/ (accessed on 15 September 2023).
- De Boeck, K.; Zolin, A.; Cuppens, H.; Olesen, H.V.; Viviani, L. The Relative Frequency of CFTR Mutation Classes in European Patients with Cystic Fibrosis. J. Cyst. Fibros. 2014, 13, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR Biology toward Combinatorial Pharmacotherapy: Expanded Classification of Cystic Fibrosis Mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef]
- Cheng, S.H.; Gregory, R.J.; Marshall, J.; Paul, S.; Souza, D.W.; White, G.A.; O’Riordan, C.R.; Smith, A.E. Defective Intracellular Transport and Processing of CFTR Is the Molecular Basis of Most Cystic Fibrosis. Cell 1990, 63, 827–834. [Google Scholar] [CrossRef]
- Lukacs, G.L.; Verkman, A.S. CFTR: Folding, Misfolding and Correcting the ΔF508 Conformational Defect. Trends Mol. Med. 2012, 18, 81–91. [Google Scholar] [CrossRef]
- Dalemans, W.; Barbry, P.; Champigny, G.; Jallat, S.; Dott, K.; Dreyer, D.; Crystal, R.G.; Pavirani, A.; Lecocq, J.P.; Lazdunski, M. Altered Chloride Ion Channel Kinetics Associated with the Delta F508 Cystic Fibrosis Mutation. Nature 1991, 354, 526–528. [Google Scholar] [CrossRef]
- Serohijos, A.W.R.; Hegedus, T.; Aleksandrov, A.A.; He, L.; Cui, L.; Dokholyan, N.V.; Riordan, J.R. Phenylalanine-508 Mediates a Cytoplasmic-Membrane Domain Contact in the CFTR 3D Structure Crucial to Assembly and Channel Function. Proc. Natl. Acad. Sci. USA 2008, 105, 3256–3261. [Google Scholar] [CrossRef]
- Mendoza, J.L.; Schmidt, A.; Li, Q.; Nuvaga, E.; Barrett, T.; Bridges, R.J.; Feranchak, A.P.; Brautigam, C.A.; Thomas, P.J. Requirements for Efficient Correction of ΔF508 CFTR Revealed by Analyses of Evolved Sequences. Cell 2012, 148, 164–174. [Google Scholar] [CrossRef]
- Okiyoneda, T.; Barrière, H.; Bagdány, M.; Rabeh, W.M.; Du, K.; Höhfeld, J.; Young, J.C.; Lukacs, G.L. Peripheral Protein Quality Control Removes Unfolded CFTR from the Plasma Membrane. Science 2010, 329, 805–810. [Google Scholar] [CrossRef]
- Swiatecka-Urban, A.; Brown, A.; Moreau-Marquis, S.; Renuka, J.; Coutermarsh, B.; Barnaby, R.; Karlson, K.H.; Flotte, T.R.; Fukuda, M.; Langford, G.M.; et al. The Short Apical Membrane Half-Life of Rescued ΔF508-Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Results from Accelerated Endocytosis of ΔF508-CFTR in Polarized Human Airway Epithelial Cells. J. Biol. Chem. 2005, 280, 36762–36772. [Google Scholar] [CrossRef] [PubMed]
- Lewis, H.A.; Zhao, X.; Wang, C.; Sauder, J.M.; Rooney, I.; Noland, B.W.; Lorimer, D.; Kearins, M.C.; Conners, K.; Condon, B.; et al. Impact of the ΔF508 Mutation in First Nucleotide-Binding Domain of Human Cystic Fibrosis Transmembrane Conductance Regulator on Domain Folding and Structure. J. Biol. Chem. 2005, 280, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Callebaut, I.; Eudes, R.; Mornon, J.-P.; Lehn, P. Nucleotide-Binding Domains of Human Cystic Fibrosis Transmembrane Conductance Regulator: Detailed Sequence Analysis and Three-Dimensional Modeling of the Heterodimer. Cell Mol. Life Sci. 2004, 61, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Eudes, R.; Lehn, P.; Férec, C.; Mornon, J.-P.; Callebaut, I. Nucleotide Binding Domains of Human CFTR: A Structural Classification of Critical Residues and Disease-Causing Mutations. Cell Mol. Life Sci. 2005, 62, 2112–2123. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Sharma, M.; Lukacs, G.L. The ΔF508 Cystic Fibrosis Mutation Impairs Domain-Domain Interactions and Arrests Post-Translational Folding of CFTR. Nat. Struct. Mol. Biol. 2005, 12, 17–25. [Google Scholar] [CrossRef]
- Pittman, J.E.; Ferkol, T.W. The Evolution of Cystic Fibrosis Care. Chest 2015, 148, 533–542. [Google Scholar] [CrossRef]
- Castaños, C. The Past 10 Years of Cystic Fibrosis Treatment: The Road to Cure. Lancet Respir. Med. 2023, 11, 864–865. [Google Scholar] [CrossRef]
- Elborn, J.S. Cystic Fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Kym, P.R.; Wang, X.; Pizzonero, M.; Van der Plas, S.E. Recent Progress in the Discovery and Development of Small-Molecule Modulators of CFTR. Prog. Med. Chem. 2018, 57, 235–276. [Google Scholar]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2020, 10, 1662. [Google Scholar] [CrossRef]
- Laselva, O.; Guerra, L.; Castellani, S.; Favia, M.; Di Gioia, S.; Conese, M. Small-Molecule Drugs for Cystic Fibrosis: Where Are We Now? Pulm. Pharmacol. Ther. 2022, 72, 102098. [Google Scholar] [CrossRef]
- Okiyoneda, T.; Veit, G.; Dekkers, J.F.; Bagdany, M.; Soya, N.; Xu, H.; Roldan, A.; Verkman, A.S.; Kurth, M.; Simon, A.; et al. Mechanism-Based Corrector Combination Restores ΔF508-CFTR Folding and Function. Nat. Chem. Biol. 2013, 9, 444–454. [Google Scholar] [CrossRef]
- Veit, G.; Xu, H.; Dreano, E.; Avramescu, R.G.; Bagdany, M.; Beitel, L.K.; Roldan, A.; Hancock, M.A.; Lay, C.; Li, W.; et al. Structure-Guided Combination Therapy to Potently Improve the Function of Mutant CFTRs. Nat. Med. 2018, 24, 1732–1742. [Google Scholar] [CrossRef]
- Farinha, C.M.; King-Underwood, J.; Sousa, M.; Correia, A.R.; Henriques, B.J.; Roxo-Rosa, M.; Da Paula, A.C.; Williams, J.; Hirst, S.; Gomes, C.M.; et al. Revertants, Low Temperature, and Correctors Reveal the Mechanism of F508del-CFTR Rescue by VX-809 and Suggest Multiple Agents for Full Correction. Chem. Biol. 2013, 20, 943–955. [Google Scholar] [CrossRef]
- Odolczyk, N.; Fritsch, J.; Norez, C.; Servel, N.; da Cunha, M.F.; Bitam, S.; Kupniewska, A.; Wiszniewski, L.; Colas, J.; Tarnowski, K.; et al. Discovery of Novel Potent ΔF508-CFTR Correctors That Target the Nucleotide Binding Domain. EMBO Mol. Med. 2013, 5, 1484–1501. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.C.; Moskowitz, S.M.; Brown, C.; Horsley, A.; Mall, M.A.; McKone, E.F.; Plant, B.J.; Prais, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; et al. VX-659–Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1599–1611. [Google Scholar] [CrossRef] [PubMed]
- Almughem, F.A.; Aldossary, A.M.; Tawfik, E.A.; Alomary, M.N.; Alharbi, W.S.; Alshahrani, M.Y.; Alshehri, A.A. Cystic Fibrosis: Overview of the Current Development Trends and Innovative Therapeutic Strategies. Pharmaceutics 2020, 12, 616. [Google Scholar] [CrossRef] [PubMed]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445–Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.; Girotti, S.; Pauro, F.; Leufkens, H.G.M.; Cipolli, M. The Impact of FDA and EMA Regulatory Decision-Making Process on the Access to CFTR Modulators for the Treatment of Cystic Fibrosis. Orphanet J. Rare Dis. 2022, 17, 188. [Google Scholar] [CrossRef] [PubMed]
- Sui, H.; Xu, X.; Su, Y.; Gong, Z.; Yao, M.; Liu, X.; Zhang, T.; Jiang, Z.; Bai, T.; Wang, J.; et al. Gene Therapy for Cystic Fibrosis: Challenges and Prospects. Front. Pharmacol. 2022, 13, 1015926. [Google Scholar] [CrossRef]
- Pedemonte, N.; Tomati, V.; Sondo, E.; Caci, E.; Millo, E.; Armirotti, A.; Damonte, G.; Zegarra-Moran, O.; Galietta, L.J.V. Dual Activity of Aminoarylthiazoles on the Trafficking and Gating Defects of the Cystic Fibrosis Transmembrane Conductance Regulator Chloride Channel Caused by Cystic Fibrosis Mutations. J. Biol. Chem. 2011, 286, 15215–15226. [Google Scholar] [CrossRef]
- Pesce, E.; Bellotti, M.; Liessi, N.; Guariento, S.; Damonte, G.; Cichero, E.; Galatini, A.; Salis, A.; Gianotti, A.; Pedemonte, N.; et al. Synthesis and Structure–Activity Relationship of Aminoarylthiazole Derivatives as Correctors of the Chloride Transport Defect in Cystic Fibrosis. Eur. J. Med. Chem. 2015, 99, 14–35. [Google Scholar] [CrossRef] [PubMed]
- Liessi, N.; Cichero, E.; Pesce, E.; Arkel, M.; Salis, A.; Tomati, V.; Paccagnella, M.; Damonte, G.; Tasso, B.; Galietta, L.J.V.; et al. Synthesis and Biological Evaluation of Novel Thiazole- VX-809 Hybrid Derivatives as F508del Correctors by QSAR-Based Filtering Tools. Eur. J. Med. Chem. 2018, 144, 179–200. [Google Scholar] [CrossRef]
- Brandas, C.; Ludovico, A.; Parodi, A.; Moran, O.; Millo, E.; Cichero, E.; Baroni, D. NBD2 Is Required for the Rescue of Mutant F508del CFTR by a Thiazole-Based Molecule: A Class II Corrector for the Multi-Drug Therapy of Cystic Fibrosis. Biomolecules 2021, 11, 1417. [Google Scholar] [CrossRef]
- Parodi, A.; Righetti, G.; Pesce, E.; Salis, A.; Tomati, V.; Pastorino, C.; Tasso, B.; Benvenuti, M.; Damonte, G.; Pedemonte, N.; et al. Journey on VX-809-Based Hybrid Derivatives towards Drug-like F508del-CFTR Correctors: From Molecular Modeling to Chemical Synthesis and Biological Assays. Pharmaceuticals 2022, 15, 274. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Chen, J. Molecular Structures Reveal Synergistic Rescue of Δ508 CFTR by Trikafta Modulators. Science 2022, 378, 284–290. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Chen, J. Mechanism of CFTR Correction by Type I Folding Correctors. Cell 2022, 185, 158–168.e11. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, Z.; Levit, A.; Levring, J.; Touhara, K.K.; Shoichet, B.K.; Chen, J. Structural Identification of a Hotspot on CFTR for Potentiation. Science 2019, 364, 1184–1188. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, F.; Chen, J. Molecular Structure of the ATP-Bound, Phosphorylated Human CFTR. Proc. Natl. Acad. Sci. USA 2018, 115, 12757–12762. [Google Scholar] [CrossRef]
- Lester, A.; Sandman, M.; Herring, C.; Girard, C.; Dixon, B.; Ramsdell, H.; Reber, C.; Poulos, J.; Mitchell, A.; Spinney, A.; et al. Computational Exploration of Potential CFTR Binding Sites for Type I Corrector Drugs. Biochemistry 2023, 62, 2503–2515. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, D.; Caballero, J. Is It Reliable to Take the Molecular Docking Top Scoring Position as the Best Solution without Considering Available Structural Data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef] [PubMed]
- Cichero, E.; Calautti, A.; Francesconi, V.; Tonelli, M.; Schenone, S.; Fossa, P. Probing in Silico the Benzimidazole Privileged Scaffold for the Development of Drug-like Anti-RSV Agents. Pharmaceuticals 2021, 14, 1307. [Google Scholar] [CrossRef]
- Bell, E.W.; Zhang, Y. DockRMSD: An Open-Source Tool for Atom Mapping and RMSD Calculation of Symmetric Molecules through Graph Isomorphism. J. Cheminform 2019, 11, 40. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Corrector VX-809 Stabilizes the First Transmembrane Domain of CFTR. Biochem. Pharmacol. 2013, 86, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.Y.; Grove, D.E.; De La Rosa, O.; Houck, S.A.; Sopha, P.; Van Goor, F.; Hoffman, B.J.; Cyr, D.M. VX-809 Corrects Folding Defects in Cystic Fibrosis Transmembrane Conductance Regulator Protein through Action on Membrane-Spanning Domain 1. Mol. Biol. Cell 2013, 24, 3016–3024. [Google Scholar] [CrossRef]
- Amico, G.; Brandas, C.; Moran, O.; Baroni, D. Unravelling the Regions of Mutant F508del-CFTR More Susceptible to the Action of Four Cystic Fibrosis Correctors. Int. J. Mol. Sci. 2019, 20, 5463. [Google Scholar] [CrossRef]
- Bongiorno, R.; Ludovico, A.; Moran, O.; Baroni, D. Elexacaftor Mediates the Rescue of F508del CFTR Functional Expression Interacting with MSD2. Int. J. Mol. Sci. 2023, 24, 12838. [Google Scholar] [CrossRef]
- Griese, M.; Costa, S.; Linnemann, R.W.; Mall, M.A.; McKone, E.F.; Polineni, D.; Quon, B.S.; Ringshausen, F.C.; Taylor-Cousar, J.L.; Withers, N.J.; et al. Safety and Efficacy of Elexacaftor/Tezacaftor/Ivacaftor for 24 Weeks or Longer in People with Cystic Fibrosis and One or More F508del Alleles: Interim Results of an Open-Label Phase 3 Clinical Trial. Am. J. Respir. Crit. Care Med. 2021, 203, 381–385. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and Safety of the Elexacaftor plus Tezacaftor plus Ivacaftor Combination Regimen in People with Cystic Fibrosis Homozygous for the F508del Mutation: A Double-Blind, Randomised, Phase 3 Trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef] [PubMed]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor–Tezacaftor–Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group ULC Molecular Operating Environment (MOE2019.01) 2021. Available online: http://www.chemcomp.com/ (accessed on 21 September 2023).
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Abbotto, E.; Casini, B.; Piacente, F.; Scarano, N.; Cerri, E.; Tonelli, M.; Astigiano, C.; Millo, E.; Sturla, L.; Bruzzone, S.; et al. Novel Thiazole-Based SIRT2 Inhibitors Discovered via Molecular Modelling Studies and Enzymatic Assays. Pharmaceuticals 2023, 16, 1316. [Google Scholar] [CrossRef]
- Scarano, N.; Abbotto, E.; Musumeci, F.; Salis, A.; Brullo, C.; Fossa, P.; Schenone, S.; Bruzzone, S.; Cichero, E. Virtual Screening Combined with Enzymatic Assays to Guide the Discovery of Novel SIRT2 Inhibitors. Int. J. Mol. Sci. 2023, 24, 9363. [Google Scholar] [CrossRef]
- Galietta, L.J.V.; Haggie, P.M.; Verkman, A.S. Green Fluorescent Protein-based Halide Indicators with Improved Chloride and Iodide Affinities. FEBS Lett. 2001, 499, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Galietta, L.V.J.; Jayaraman, S.; Verkman, A.S. Cell-Based Assay for High-Throughput Quantitative Screening of CFTR Chloride Transport Agonists. Am. J. Physiol. Cell Physiol. 2001, 281, C1734–C1742. [Google Scholar] [CrossRef]
- Louis, K.S.; Siegel, A.C. Cell Viability Analysis Using Trypan Blue: Manual and Automated Methods. In Mammalian Cell Viability: Methods and Protocols; Stoddart, M.J., Ed.; Humana Press: Totowa, NJ, USA, 2011; Volume 740, pp. 7–12. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB Code | Feature | Resolution (Å) | Release | Atom Count | Reference |
|---|---|---|---|---|---|
| 8EIQ | ELEXACAFTOR, TEZACAFTOR. IVACAFTOR | 3.00 | 2022 | 9578 | [40] |
| 8EIO | ELEXACAFTOR, LUMACAFTOR | 2.80 | 2022 | 9550 | [40] |
| 8EIG | ELEXACAFTOR | 3.60 | 2022 | 9249 | [40] |
| 7SVR | LUMACAFTOR NO COFACTOR | 3.90 | 2022 | 7841 | [41] |
| 7SVD | COFACTOR ATP (ADENOSINE-5′-TRIPHOSPHATE) | 2.70 | 2022 | 9635 | [41] |
| 7SV7 | TEZACAFTOR NO COFACTOR | 3.80 | 2022 | 9673 | [41] |
| 6O2P | IVACAFTOR COFACTOR ATP (ADENOSINE-5′-TRIPHOSPHATE) | 3.30 | 2019 | 9767 | [42] |
| 6O1V | GLPG1837 COFACTOR ATP (ADENOSINE-5′-TRIPHOSPHATE) | 3.20 | 2019 | 9711 | [42] |
| 6MSM | COFACTOR ATP (ADENOSINE-5′-TRIPHOSPHATE) | 3.20 | 2018 | 9703 | [43] |
| 5UAK | NO COFACTOR | 3.87 | 2017 | 9232 | [5] |
| 8EIG-VX445 Binding Site (Pocket A) | 8EIO-VX809 Binding Site (Pocket B) | 8EIQ-VX661 Binding Site (Pocket B) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Compounds | CL1 S Value | CL1 Population | CLN | CL1 S Value | CL1 Population | CLN | CL1 S Value | CL1 Population | CLN |
| VX-445 | −6.5860 | 8 | 2 | −7.3970 | 8 | 2 | −7.9867 | 3 | 5 |
| VX-809 | −5.4008 | 5 | 3 | −8.7534 | 5 | 2 | −9.0052 | 6 | 2 |
| VX-661 | −5.8536 | 8 | 3 | −9.7202 | 9 | 2 | −9.9823 | 8 | 2 |
| 2a | −4.4228 | 3 | 5 | −7.8760 | 7 | 3 | −8.3534 | 6 | 2 |
| 7a | −4.6130 | 4 | 4 | −8.5604 | 6 | 2 | −8.5864 | 4 | 4 |
| 7m | −5.5739 | 5 | 4 | −10.3685 | 6 | 3 | −8.8892 | 6 | 4 |
| HEK-t | FRT | CFBE41O− | ||||
|---|---|---|---|---|---|---|
| Compound | TD50 (µM) | TDMax (µM) | TD50 (µM) | TDMax (µM) | TD50 (µM) | TDMax (µM) |
| VX661 | 27.15 ± 17.88 | 56.42 ± 0.30 | 11.09 ± 3.22 | 23.59 ± 0.08 | 7.51 ± 2.22 | 15.62 ± 0.06 |
| VX445 | 9.60 ± 2.25 | 20.75 ± 0.04 | 4.29 ± 0.32 | 10.79 ± 0.01 | 11.75 ± 3.42 | 23.60 ± 0.07 |
| 2a | 2.44 ± 1.03 | 5.78 ± 0.04 | 9.24 ± 2.25 | 20.56 ±0.06 | 10.11 ± 1.80 | 14.44 ± 0.06 |
| 7m | 1. 40 ± 0.20 | 5.86 ± 0.01 | 5.76 ± 0.89 | 13.68 ± 0.03 | 9.78 ± 3.35 | 29.51 ± 0.10 |
| 7a | 1.59 ± 0.36 | 5.82 ± 0.01 | 6.13 ± 2.75 | 14.70 ± 0.07 | 11.97 ± 2.87 | 25.49 ± 0.08 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baroni, D.; Scarano, N.; Ludovico, A.; Brandas, C.; Parodi, A.; Lunaccio, D.; Fossa, P.; Moran, O.; Cichero, E.; Millo, E. In Silico and In Vitro Evaluation of the Mechanism of Action of Three VX809-Based Hybrid Derivatives as Correctors of the F508del CFTR Protein. Pharmaceuticals 2023, 16, 1702. https://doi.org/10.3390/ph16121702
Baroni D, Scarano N, Ludovico A, Brandas C, Parodi A, Lunaccio D, Fossa P, Moran O, Cichero E, Millo E. In Silico and In Vitro Evaluation of the Mechanism of Action of Three VX809-Based Hybrid Derivatives as Correctors of the F508del CFTR Protein. Pharmaceuticals. 2023; 16(12):1702. https://doi.org/10.3390/ph16121702
Chicago/Turabian StyleBaroni, Debora, Naomi Scarano, Alessandra Ludovico, Chiara Brandas, Alice Parodi, Dario Lunaccio, Paola Fossa, Oscar Moran, Elena Cichero, and Enrico Millo. 2023. "In Silico and In Vitro Evaluation of the Mechanism of Action of Three VX809-Based Hybrid Derivatives as Correctors of the F508del CFTR Protein" Pharmaceuticals 16, no. 12: 1702. https://doi.org/10.3390/ph16121702
APA StyleBaroni, D., Scarano, N., Ludovico, A., Brandas, C., Parodi, A., Lunaccio, D., Fossa, P., Moran, O., Cichero, E., & Millo, E. (2023). In Silico and In Vitro Evaluation of the Mechanism of Action of Three VX809-Based Hybrid Derivatives as Correctors of the F508del CFTR Protein. Pharmaceuticals, 16(12), 1702. https://doi.org/10.3390/ph16121702