

Drug Discovery Based on Oxygen and Nitrogen (Non-)Heterocyclic Compounds Developed @LAQV–REQUIMTE/Aveiro




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
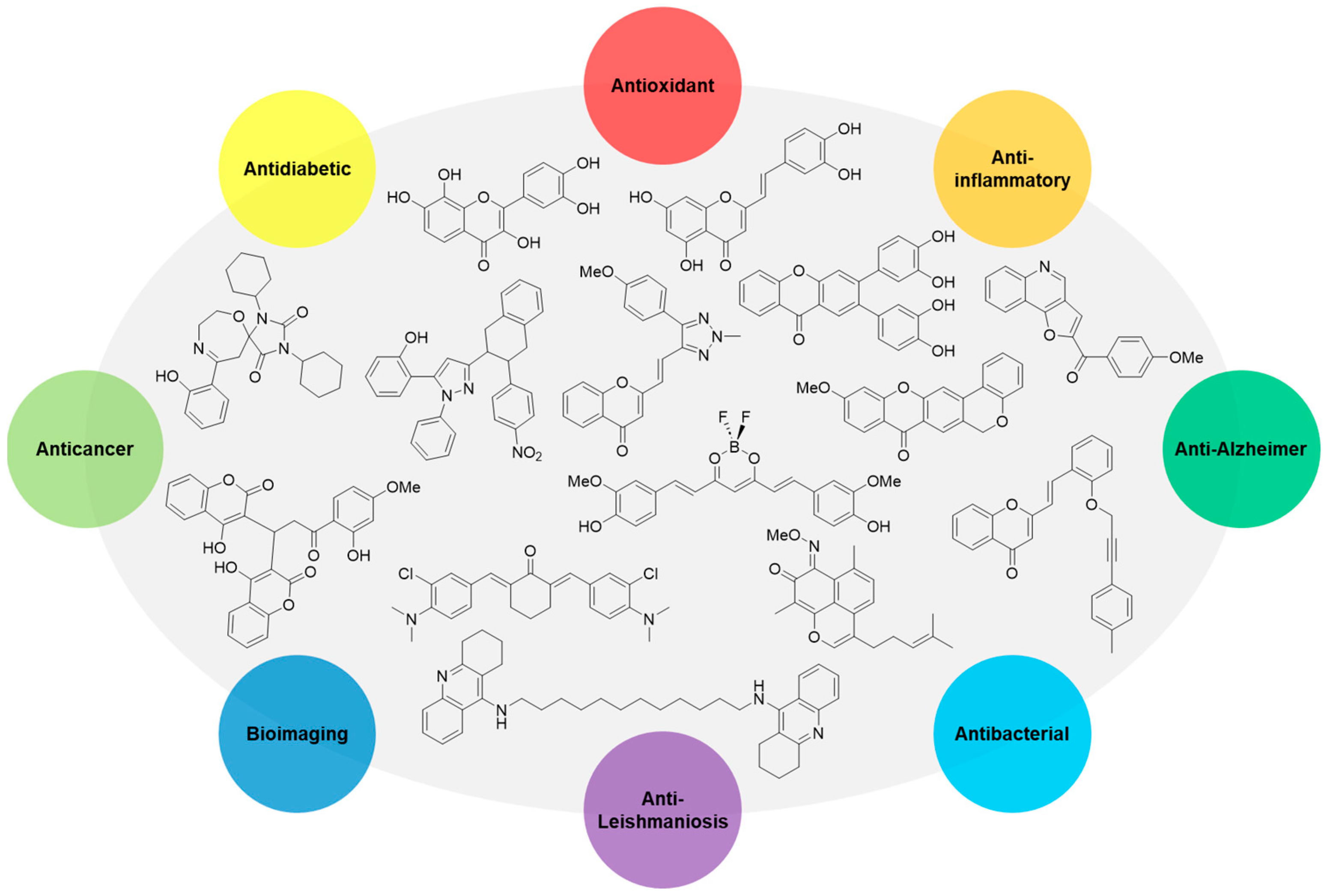
Abstract
:1. Introduction
2. Antioxidant Activity
2.1. ROS/RNS Scavenging Activity
2.2. Free Radicals Scavenging Activity
2.3. Enzymatic Inhibition Activity
3. Anti-Inflammatory Activity
3.1. Flavones
3.2. Xanthones
3.3. 2-Styrylchromones
4. Antidiabetic Activity
4.1. Flavonoids
4.1.1. Flavonols
4.1.2. Chlorinated Flavones
4.2. 2-(Styryl or 4-Arylbutadienyl)chromones
4.3. Xanthones
4.4. Pyrazoles
5. Anticancer Activity
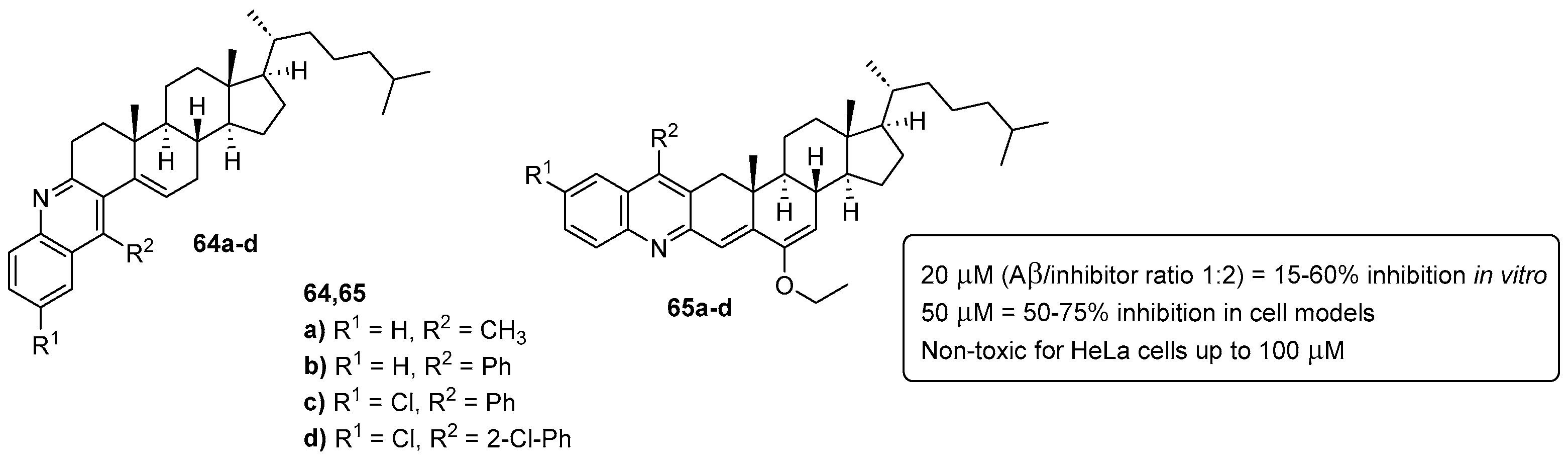
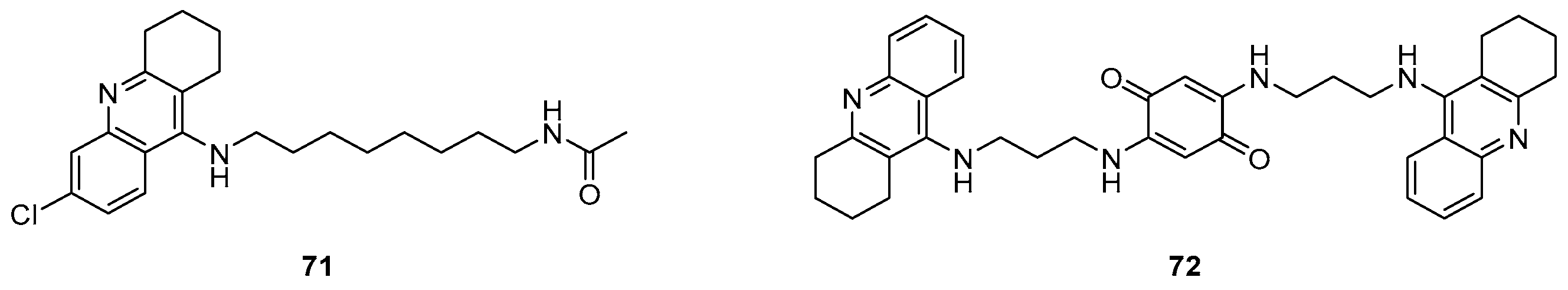
6. Anti-Alzheimer Activity
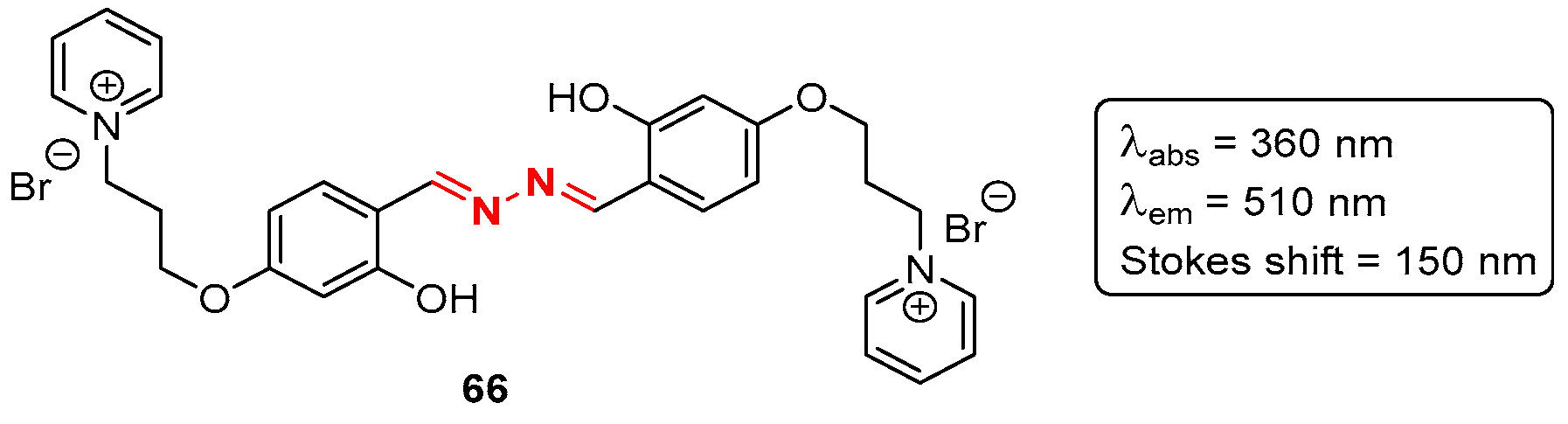
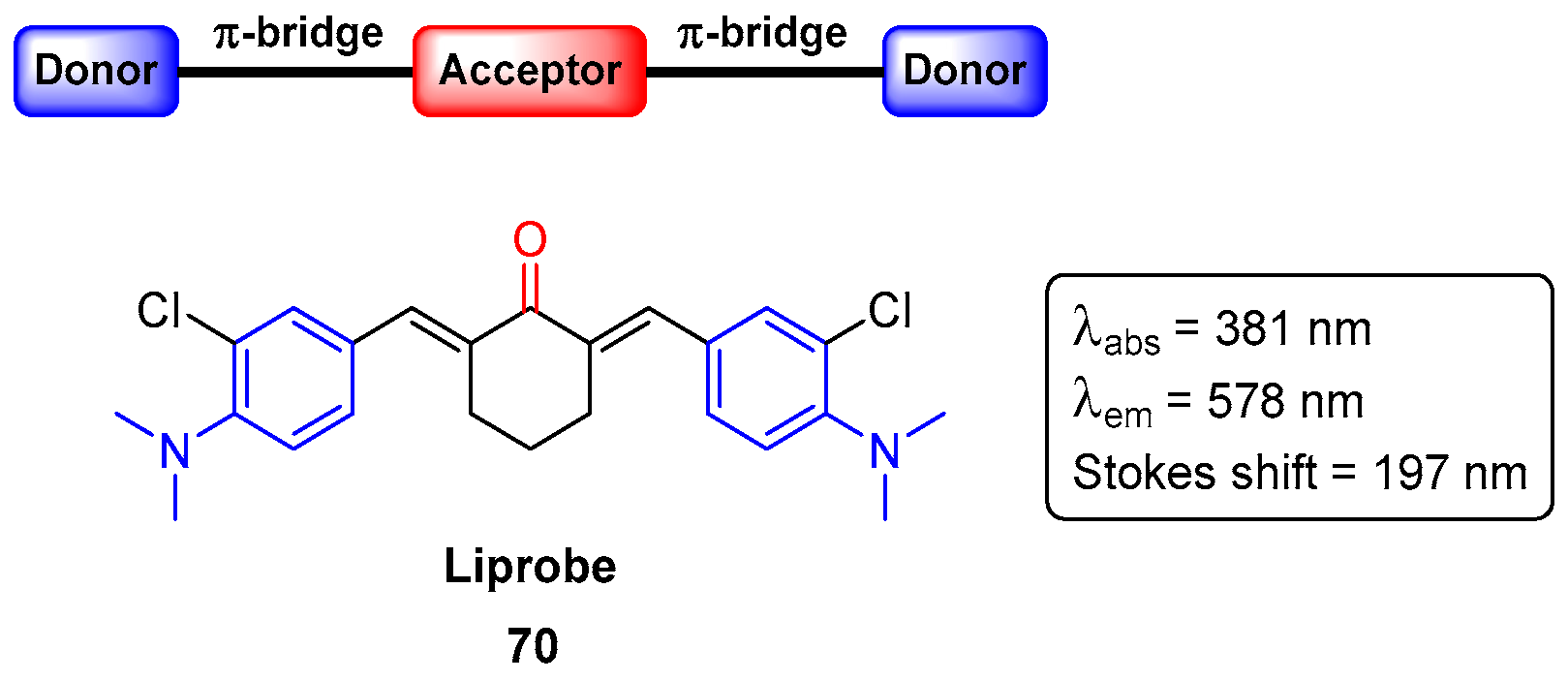
7. Bioimaging
8. Other Biological Activities
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef]
- Lombardino, J.G.; Lowe, J.A. The role of the medicinal chemist in drug discovery—Then and now. Nat. Rev. Drug Discov. 2004, 3, 853–862. [Google Scholar] [CrossRef]
- Moir, M.; Danon, J.J.; Reekie, T.A.; Kassiou, M. An overview of late-stage functionalization in today’s drug discovery. Expert Opin. Drug Discov. 2019, 14, 1137–1149. [Google Scholar] [CrossRef]
- Cernak, T.; Dykstra, K.D.; Tyagarajan, S.; Vachal, P.; Krska, S.W. The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev. 2016, 45, 546–576. [Google Scholar] [CrossRef] [PubMed]
- Heravi, M.M.; Zadsirjan, V. Prescribed drugs containing nitrogen heterocycles: An overview. RSC Adv. 2020, 10, 44247–44311. [Google Scholar] [CrossRef]
- Chaudhry, F.; Munir, R.; Malik, N. N-Heterocycles as Privileged Scaffolds in FDA Approved Different NMEs of 2021: A Review. Lett. Org. Chem. 2023, 20, 287–299. [Google Scholar] [CrossRef]
- Delost, M.D.; Smith, D.T.; Anderson, B.J.; Njardarson, J.T. From Oxiranes to Oligomers: Architectures of U.S. FDA Approved Pharmaceuticals Containing Oxygen Heterocycles. J. Med. Chem. 2018, 61, 10996–11020. [Google Scholar] [CrossRef] [PubMed]
- Martins, T.; Silva, V.L.M.; Silva, A.M.S.; Lima, J.L.F.C.; Fernandes, E.; Ribeiro, D. Chalcones as Scavengers of HOCl and Inhibitors of Oxidative Burst: Structure-Activity Relationship Studies. Med. Chem. 2022, 18, 88–96. [Google Scholar] [CrossRef]
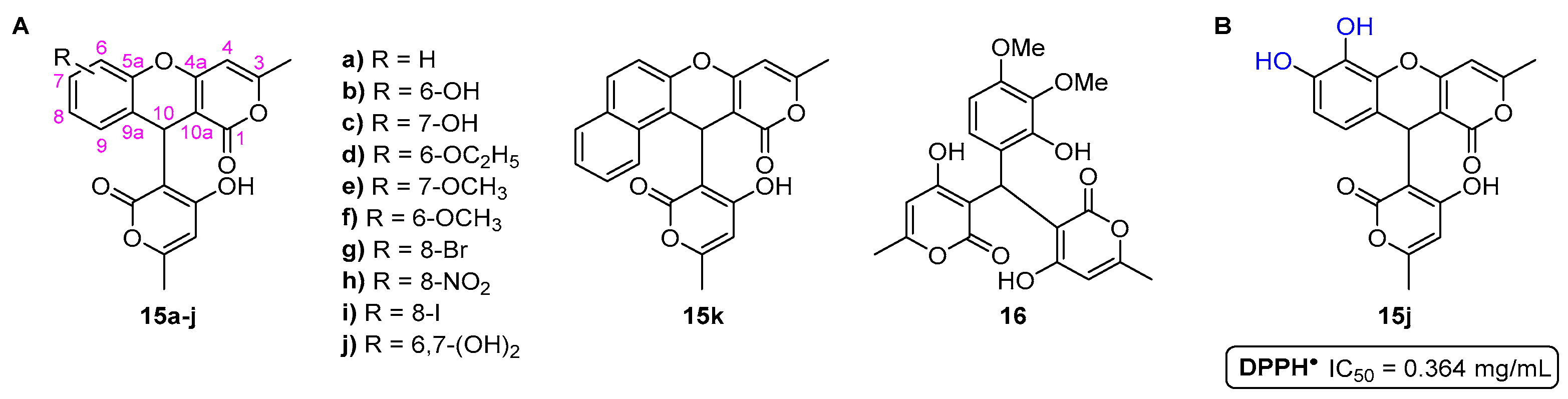
- Sousa, J.L.C.; Proença, C.; Freitas, M.; Fernandes, E.; Silva, A.M.S. New polyhydroxylated flavon-3-ols and 3-hydroxy-2-styrylchromones: Synthesis and ROS/RNS scavenging activities. Eur. J. Med. Chem. 2016, 119, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Proença, C.; Albuquerque, H.M.T.; Ribeiro, D.; Freitas, M.; Santos, C.M.M.; Silva, A.M.S.; Fernandes, E. Novel chromone and xanthone derivatives: Synthesis and ROS/RNS scavenging activities. Eur. J. Med. Chem. 2016, 115, 381–392. [Google Scholar] [CrossRef]
- Santos, C.M.M.; Freitas, M.; Ribeiro, D.; Gomes, A.; Silva, A.M.S.; Cavaleiro, J.A.S.; Fernandes, E. 2,3-Diarylxanthones as strong scavengers of reactive oxygen and nitrogen species: A structure-activity relationship study. Bioorganic Med. Chem. 2010, 18, 6776–6784. [Google Scholar] [CrossRef]
- Gomes, A.; Neuwirth, O.; Freitas, M.; Couto, D.; Ribeiro, D.; Figueiredo, A.G.P.R.; Silva, A.M.S.; Seixas, R.S.G.R.; Pinto, D.C.G.A.; Tomé, A.C.; et al. Synthesis and antioxidant properties of new chromone derivatives. Bioorganic Med. Chem. 2009, 17, 7218–7226. [Google Scholar] [CrossRef]
- Gomes, A.; Fernandes, E.; Silva, A.M.S.; Santos, C.M.M.; Pinto, D.C.G.A.; Cavaleiro, J.A.S.; Lima, J.L.F.C. 2-Styrylchromones: Novel strong scavengers of reactive oxygen and nitrogen species. Bioorganic Med. Chem. 2007, 15, 6027–6036. [Google Scholar] [CrossRef]
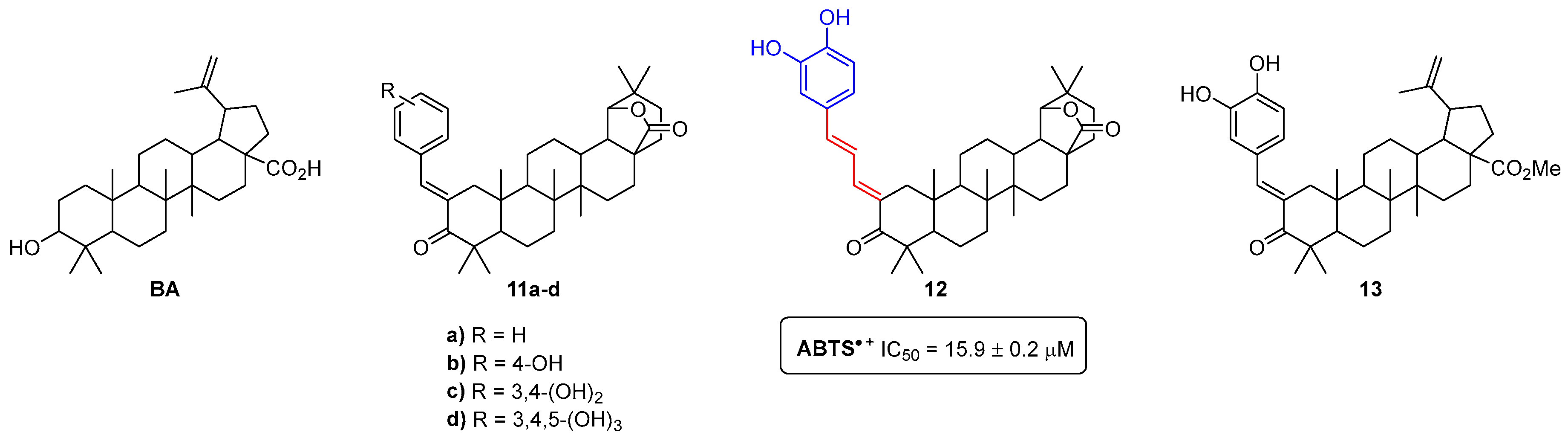
- Sousa, J.L.C.; Gonçalves, C.; Ferreira, R.M.; Cardoso, S.M.; Freire, C.S.R.; Silvestre, A.J.D.; Silva, A.M.S. Functionalization of betulinic acid with polyphenolic fragments for the development of new amphiphilic antioxidants. Antioxidants 2021, 10, 148. [Google Scholar] [CrossRef] [PubMed]
- Seca, A.M.L.; Leal, S.B.; Pinto, D.C.G.A.; Barreto, M.C.; Silva, A.M.S. Xanthenedione Derivatives, New Promising Antioxidant and Acetylcholinesterase Inhibitor Agents. Molecules 2014, 19, 8317–8333. [Google Scholar] [CrossRef] [PubMed]
- Saher, L.; Makhloufi-Chebli, M.; Dermeche, L.; Dermeche, S.; Boutemeur-Khedis, B.; Rabia, C.; Hamdi, M.; Silva, A.M.S. 10-(4-Hydroxy-6-methyl-2-oxo-2H-pyran-3-yl)-3-methyl-1H,10H-pyrano [4,3-b] chromen-1-ones from a pseudo-multicomponent reaction and evaluation of their antioxidant activity. Tetrahedron 2018, 74, 872–879. [Google Scholar] [CrossRef]
- Fernandes, E.; Carvalho, F.; Silva, A.M.S.; Santos, C.M.M.; Pinto, D.C.G.A.; Cavaleiro, J.A.S.; Bastos, M.D.L. 2-Styrylchromones as novel inhibitors of xanthine oxidase. A structure-activity study. J. Enzym. Inhib. Med. Chem. 2002, 17, 45–48. [Google Scholar] [CrossRef]
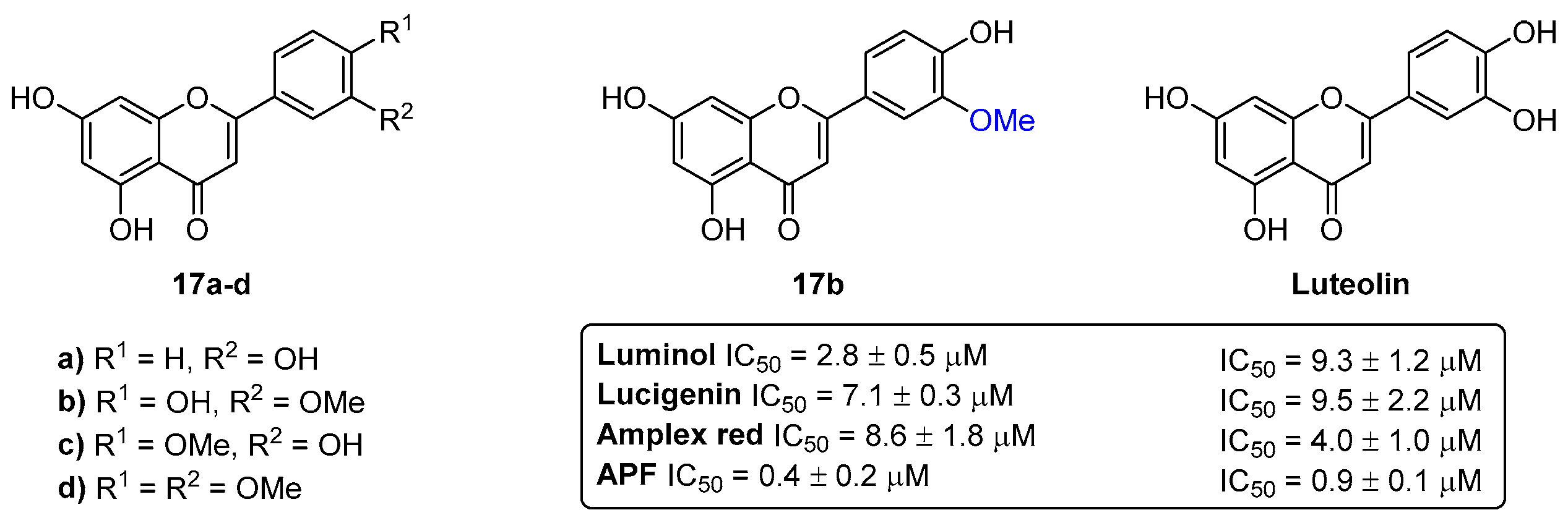
- Ribeiro, D.; Freitas, M.; Tomé, S.M.; Silva, A.M.S.; Porto, G.; Fernandes, E. Modulation of human neutrophils’ oxidative burst by flavonoids. Eur. J. Med. Chem. 2013, 67, 280–292. [Google Scholar] [CrossRef]
- Ribeiro, D.; Freitas, M.; Tomé, S.M.; Silva, A.M.S.; Laufer, S.; Lima, J.L.F.C.; Fernandes, E. Flavonoids Inhibit COX-1 and COX-2 Enzymes and Cytokine/Chemokine Production in Human Whole Blood. Inflammation 2015, 38, 858–870. [Google Scholar] [CrossRef]
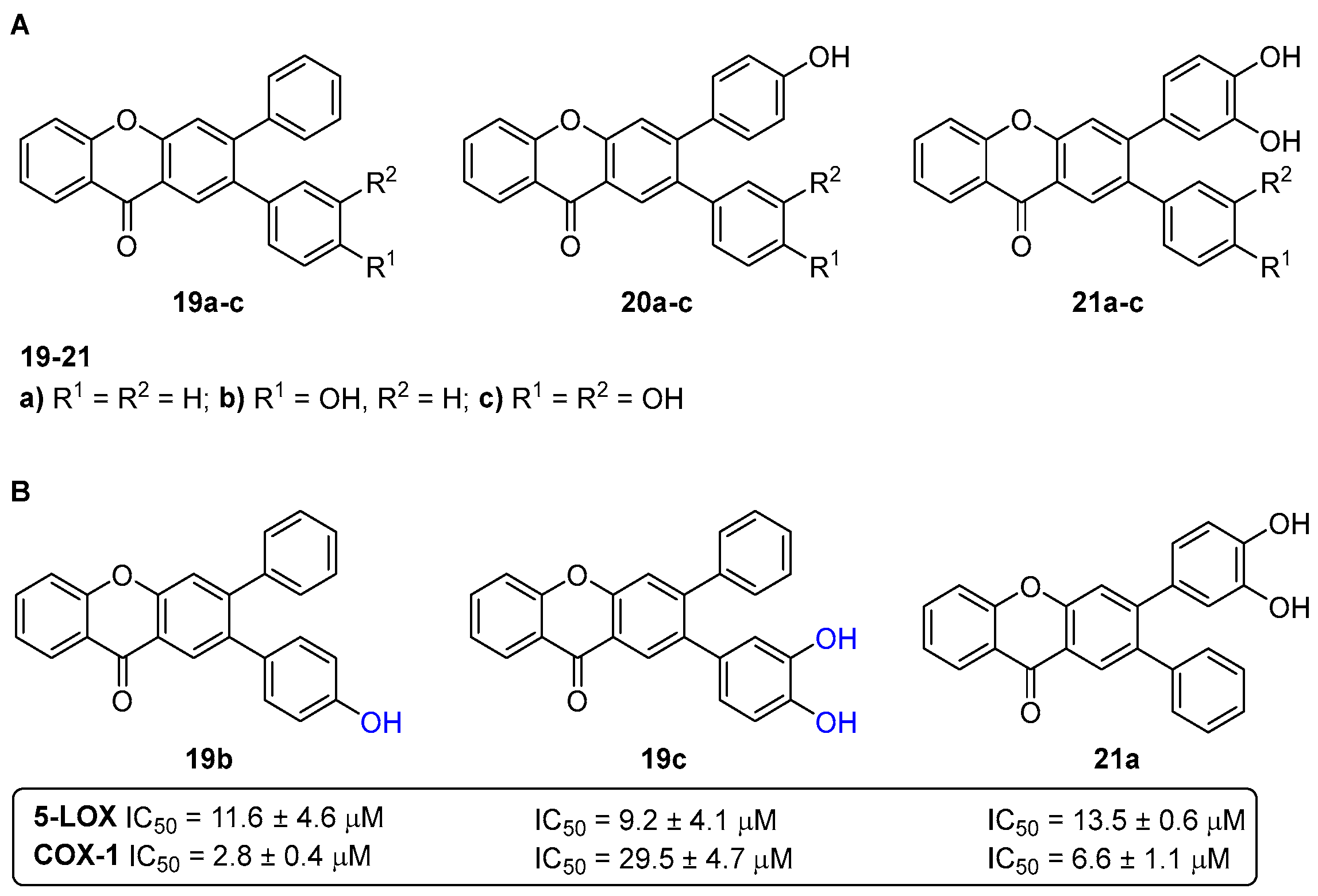
- Ribeiro, D.; Freitas, M.; Tomé, S.M.; Silva, A.M.S.; Porto, G.; Cabrita, E.J.; Marques, M.M.B.; Fernandes, E. Inhibition of LOX by flavonoids: A structure-activity relationship study. Eur. J. Med. Chem. 2014, 72, 137–145. [Google Scholar] [CrossRef]
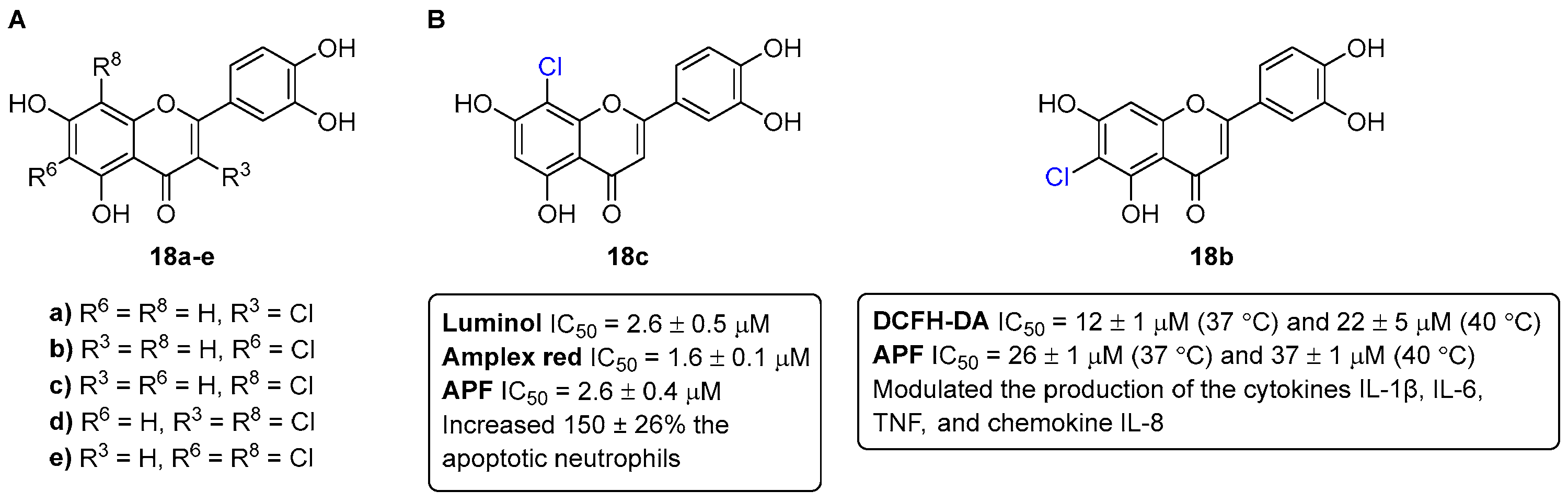
- Freitas, M.; Ribeiro, D.; Tomé, S.M.; Silva, A.M.S.; Fernandes, E. Synthesis of chlorinated flavonoids with anti-inflammatory and pro-apoptotic activities in human neutrophils. Eur. J. Med. Chem. 2014, 86, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Proença, C.; Ribeiro, D.; Soares, T.; Tomé, S.M.; Silva, A.M.S.; Lima, J.L.F.C.; Fernandes, E.; Freitas, M. Chlorinated Flavonoids Modulate the Inflammatory Process in Human Blood. Inflammation 2017, 40, 1155–1165. [Google Scholar] [CrossRef]
- Santos, C.M.M.; Ribeiro, D.; Silva, A.M.S.; Fernandes, E. 2,3-Diarylxanthones as potential inhibitors of arachidonic acid metabolic pathways. Inflammation 2017, 40, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.; Fernandes, E.; Silva, A.M.S.; Pinto, D.C.G.A.; Santos, C.M.M.; Cavaleiro, J.A.S.; Lima, J.L.F.C. Anti-inflammatory potential of 2-styrylchromones regarding their interference with arachidonic acid metabolic pathways. Biochem. Pharmacol. 2009, 78, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://idf.org/about-diabetes/diabetes-facts-figures/ (accessed on 15 November 2023).
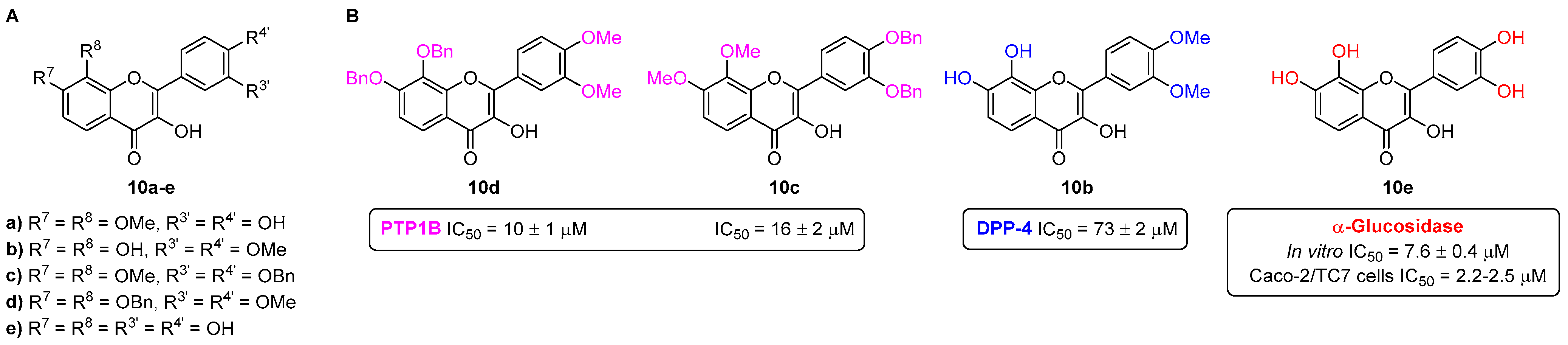
- Proença, C.; Freitas, M.; Ribeiro, D.; Sousa, J.L.C.; Carvalho, F.; Silva, A.M.S.; Fernandes, P.A.; Fernandes, E. Inhibition of protein tyrosine phosphatase 1B by flavonoids: A structure–activity relationship study. Food Chem. Toxicol. 2018, 111, 474–481. [Google Scholar] [CrossRef]
- Proença, C.; Freitas, M.; Ribeiro, D.; Tomé, S.M.; Araújo, A.N.; Silva, A.M.S.; Fernandes, P.A.; Fernandes, E. The dipeptidyl peptidase-4 inhibitory effect of flavonoids is hindered in protein rich environments. Food Funct. 2019, 10, 5718–5731. [Google Scholar] [CrossRef] [PubMed]
- Proença, C.; Oliveira, A.; Freitas, M.; Ribeiro, D.; Sousa, J.L.C.; Ramos, M.J.; Silva, A.M.S.; Fernandes, P.A.; Fernandes, E. Structural Specificity of Flavonoids in the Inhibition of Human Fructose 1,6-Bisphosphatase. J. Nat. Prod. 2020, 83, 1541–1552. [Google Scholar] [CrossRef]
- Proença, C.; Freitas, M.; Ribeiro, D.; Oliveira, E.F.T.; Sousa, J.L.C.; Tomé, S.M.; Ramos, M.J.; Silva, A.M.S.; Fernandes, P.A.; Fernandes, E. α-Glucosidase inhibition by flavonoids: An in vitro and in silico structure–activity relationship study. J. Enzym. Inhib. Med. Chem. 2017, 32, 1216–1228. [Google Scholar] [CrossRef]
- Proença, C.; Rufino, A.T.; Ferreira de Oliveira, J.M.P.; Freitas, M.; Fernandes, P.A.; Silva, A.M.S.; Fernandes, E. Inhibitory activity of flavonoids against human sucrase-isomaltase (α-glucosidase) activity in a Caco-2/TC7 cellular model. Food Funct. 2022, 13, 1108–1118. [Google Scholar] [CrossRef]
- Proença, C.; Freitas, M.; Ribeiro, D.; Tomé, S.M.; Oliveira, E.F.T.; Viegas, M.F.; Araújo, A.N.; Ramos, M.J.; Silva, A.M.S.; Fernandes, P.A.; et al. Evaluation of a flavonoids library for inhibition of pancreatic α-amylase towards a structure–activity relationship. J. Enzyme Inhib. Med. Chem. 2019, 34, 577–588. [Google Scholar] [CrossRef]
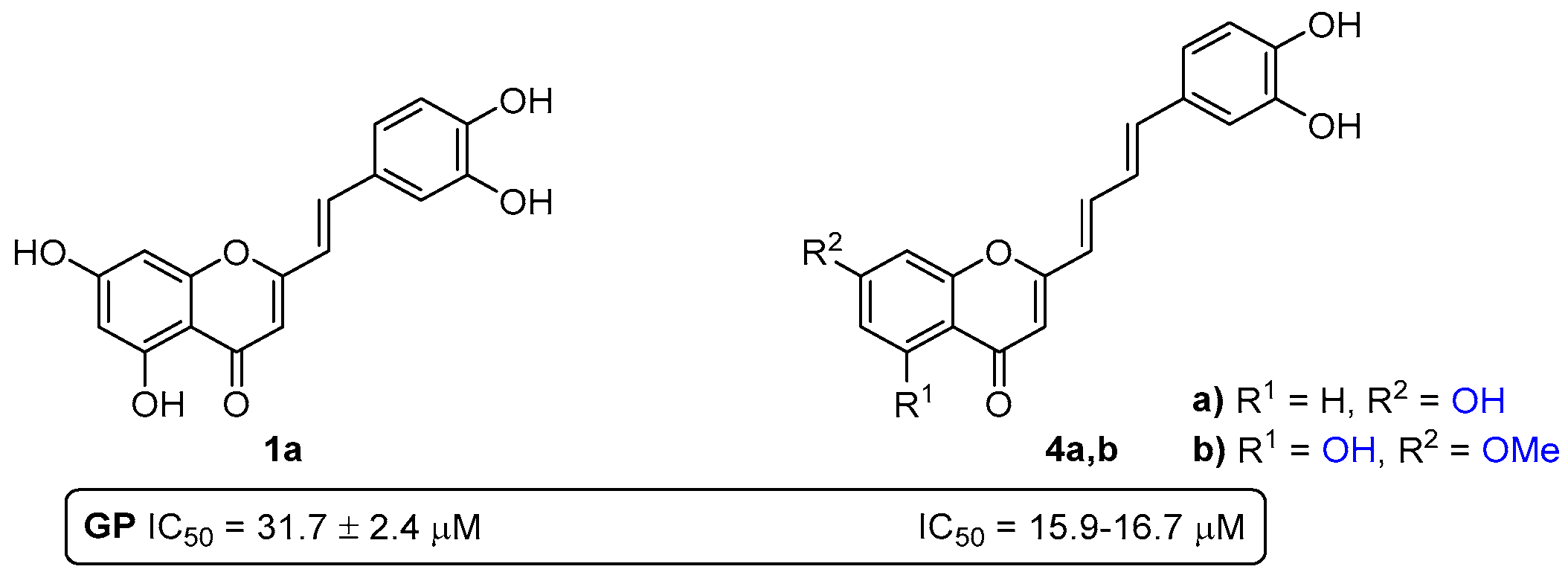
- Rocha, S.; Aniceto, N.; Guedes, R.C.; Albuquerque, H.M.T.; Silva, V.L.M.; Silva, A.M.S.; Corvo, M.L.; Fernandes, E.; Freitas, M. An In Silico and an In Vitro Inhibition Analysis of Glycogen Phosphorylase by Flavonoids, Styrylchromones, and Pyrazoles. Nutrients 2022, 14, 306. [Google Scholar] [CrossRef]
- Santos, C.M.M.; Proença, C.; Freitas, M.; Araújo, A.N.; Silva, A.M.S.; Fernandes, E. Inhibition of the carbohydrate-hydrolyzing enzymes α-amylase and α-glucosidase by hydroxylated xanthones. Food Funct. 2022, 13, 7930–7941. [Google Scholar] [CrossRef]
- Rocha, S.; Lucas, M.; Silva, V.L.M.; Gomes, P.M.O.; Silva, A.M.S.; Araújo, A.N.; Aniceto, N.; Guedes, R.C.; Corvo, M.L.; Fernandes, E.; et al. Pyrazoles as novel protein tyrosine phosphatase 1B (PTP1B) inhibitors: An in vitro and in silico study. Int. J. Biol. Macromol. 2021, 181, 1171–1182. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Talhi, O.; Jang, D.; Cerella, C.; Gaigneaux, A.; Kim, K.-W.; Lee, J.W.; Dicato, M.; Bachari, K.; Han, B.W.; et al. Cytostatic hydroxycoumarin OT52 induces ER/Golgi stress and STAT3 inhibition triggering non-canonical cell death and synergy with BH3 mimetics in lung cancer. Cancer Lett. 2018, 416, 94–108. [Google Scholar] [CrossRef]
- Mazumder, A.; Lee, J.-Y.; Talhi, O.; Cerella, C.; Chateauvieux, S.; Gaigneaux, A.; Hong, C.R.; Kang, H.J.; Lee, Y.; Kim, K.-W.; et al. Hydroxycoumarin OT-55 kills CML cells alone or in synergy with imatinib or Synribo: Involvement of ER stress and DAMP release. Cancer Lett. 2018, 438, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Talhi, O.; Schnekenburger, M.; Panning, J.; Pinto, D.G.C.; Fernandes, J.A.; Almeida Paz, F.A.; Jacob, C.; Diederich, M.; Silva, A.M.S. Bis(4-hydroxy-2H-chromen-2-one): Synthesis and effects on leukemic cell lines proliferation and NF-κB regulation. Bioorganic Med. Chem. 2014, 22, 3008–3015. [Google Scholar] [CrossRef] [PubMed]
- Valente, S.; Xu, Z.; Bana, E.; Zwergel, C.; Mai, A.; Jacob, C.; Meiser, P.; Bagrel, D.; Silva, A.M.S.; Kirsch, G. Reactivity of 4-Vinyl-2H-1-benzopyran-2-ones in Diels–Alder Cycloaddition Reactions: Access to Coumarin-Based Polycycles with Cdc25 Phosphatase-Inhibiting Activity. Eur. J. Org. Chem. 2013, 2013, 2869–2877. [Google Scholar] [CrossRef]
- Sutanto, F.; Konstantinidou, M.; Dömling, A. Covalent inhibitors: A rational approach to drug discovery. RSC Med. Chem. 2020, 11, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Gehringer, M.; Laufer, S.A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62, 5673–5724. [Google Scholar] [CrossRef]
- Shindo, N.; Ojida, A. Recent progress in covalent warheads for in vivo targeting of endogenous proteins. Bioorganic Med. Chem. 2021, 47, 116386. [Google Scholar] [CrossRef]
- Talhi, O.; Brodziak-Jarosz, L.; Panning, J.; Orlikova, B.; Zwergel, C.; Tzanova, T.; Philippot, S.; Pinto, D.C.G.A.; Paz, F.A.A.; Gerhäuser, C.; et al. One-Pot Synthesis of Benzopyran-4-ones with Cancer Preventive and Therapeutic Potential. Eur. J. Org. Chem. 2016, 2016, 965–975. [Google Scholar] [CrossRef]
- Bouhenna, M.M.; Orlikova, B.; Talhi, O.; Schram, B.; Pinto, D.C.G.A.; Taibi, N.; Bachari, K.; Diederich, M.; Silva, A.M.S.; Mameri, N. Anti-proliferative, Cytotoxic and NF-ĸB Inhibitory Properties of Spiro(Lactone-Cyclohexanone) Compounds in Human Leukemia. Anticancer Res. 2017, 37, 5225–5233. [Google Scholar]
- Lamia Hamdan, R.; Oualid, T.; Nadia, T.; Laetitia, D.; Caroline, D.; Artur, S.; Khaldoun, B.; Marie-Paule, V.; Florence, C.-C. Effects of Spiro-bisheterocycles on Proliferation and Apoptosis in Human Breast Cancer Cell Lines. Anticancer Res. 2016, 36, 6399. [Google Scholar]
- Naouri, A.; Djemoui, A.; Ouahrani, M.R.; Lahrech, M.B.; Lemouari, N.; Rocha, D.H.A.; Albuquerque, H.; Mendes, R.F.; Almeida Paz, F.A.; Helguero, L.A.; et al. Multicomponent and 1,3-dipolar cycloaddition synthesis of triazole- and isoxazole-acridinedione/xanthenedione heterocyclic hybrids: Cytotoxic effects on human cancer cells. J. Mol. Struct. 2020, 1217, 128325. [Google Scholar] [CrossRef]
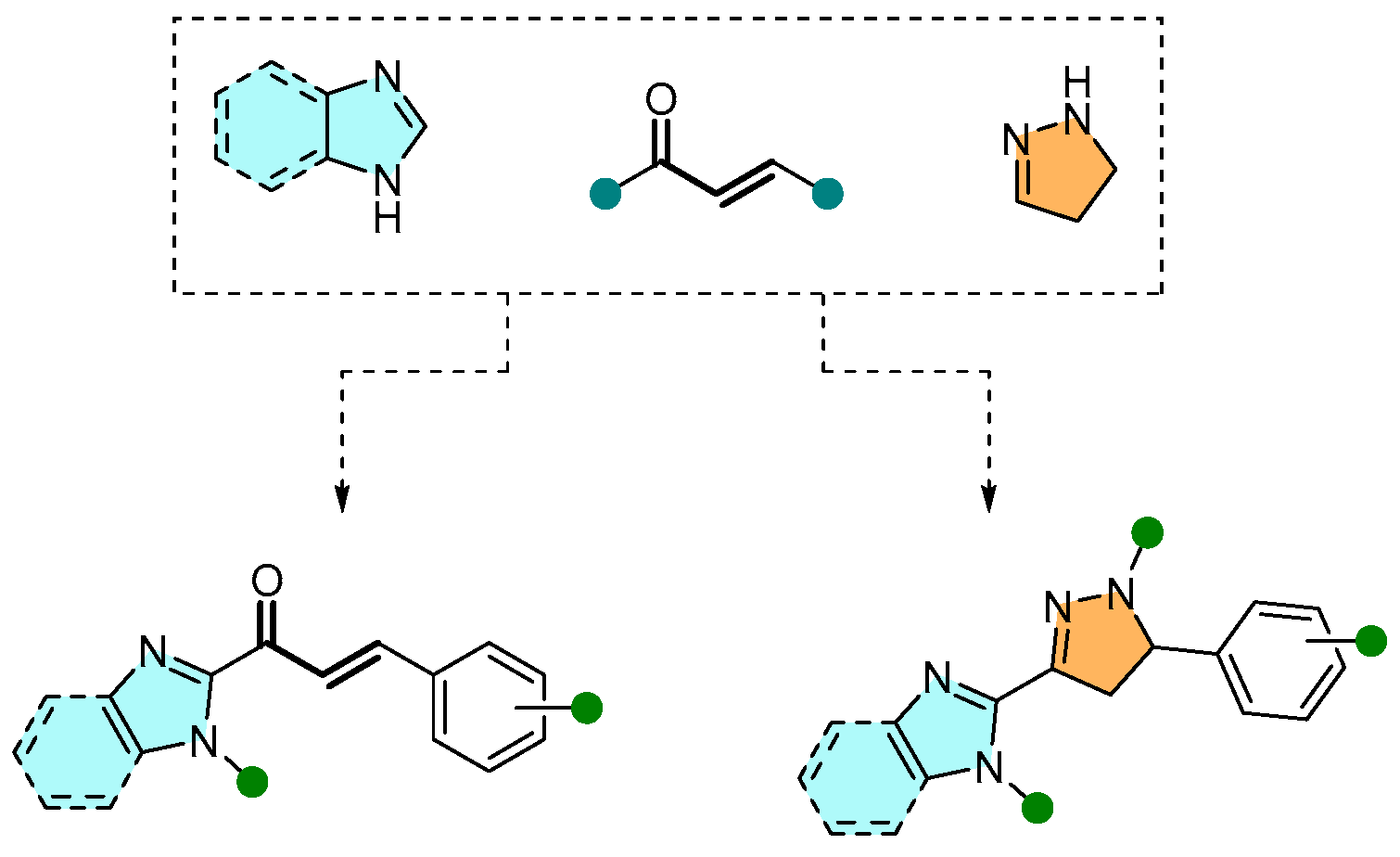
- Djemoui, A.; Naouri, A.; Ouahrani, M.R.; Djemoui, D.; Lahcene, S.; Lahrech, M.B.; Boukenna, L.; Albuquerque, H.M.T.; Saher, L.; Rocha, D.H.A.; et al. A step-by-step synthesis of triazole-benzimidazole-chalcone hybrids: Anticancer activity in human cells+. J. Mol. Struct. 2020, 1204, 127487. [Google Scholar] [CrossRef]
- Bozorov, K.; Zhao, J.; Aisa, H.A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorganic Med. Chem. 2019, 27, 3511–3531. [Google Scholar] [CrossRef] [PubMed]
- Saher, L.; Monteiro, F.L.; Rocha, D.H.A.; Albuquerque, H.M.T.; Silva, A.M.S.; Helguero, L.A. Selective Induction of DNA Damage and Cell Cycle Arrest Mediated by Chromone-Triazole Dyads Derivatives: Effects on Breast and Prostate Cancer Cells. Chem. Biodivers. 2023, 20, e202300251. [Google Scholar] [CrossRef] [PubMed]
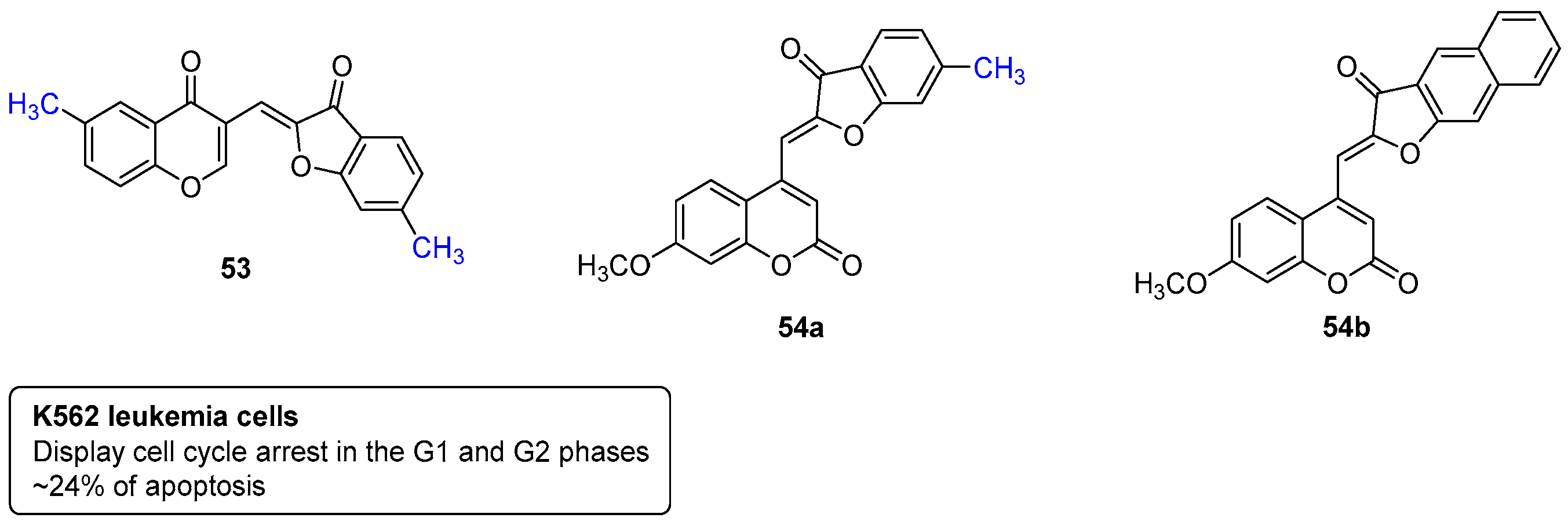
- Zwergel, C.; Valente, S.; Salvato, A.; Xu, Z.; Talhi, O.; Mai, A.; Silva, A.; Altucci, L.; Kirsch, G. Novel benzofuran–chromone and –coumarin derivatives: Synthesis and biological activity in K562 human leukemia cells. MedChemComm 2013, 4, 1571–1579. [Google Scholar] [CrossRef]
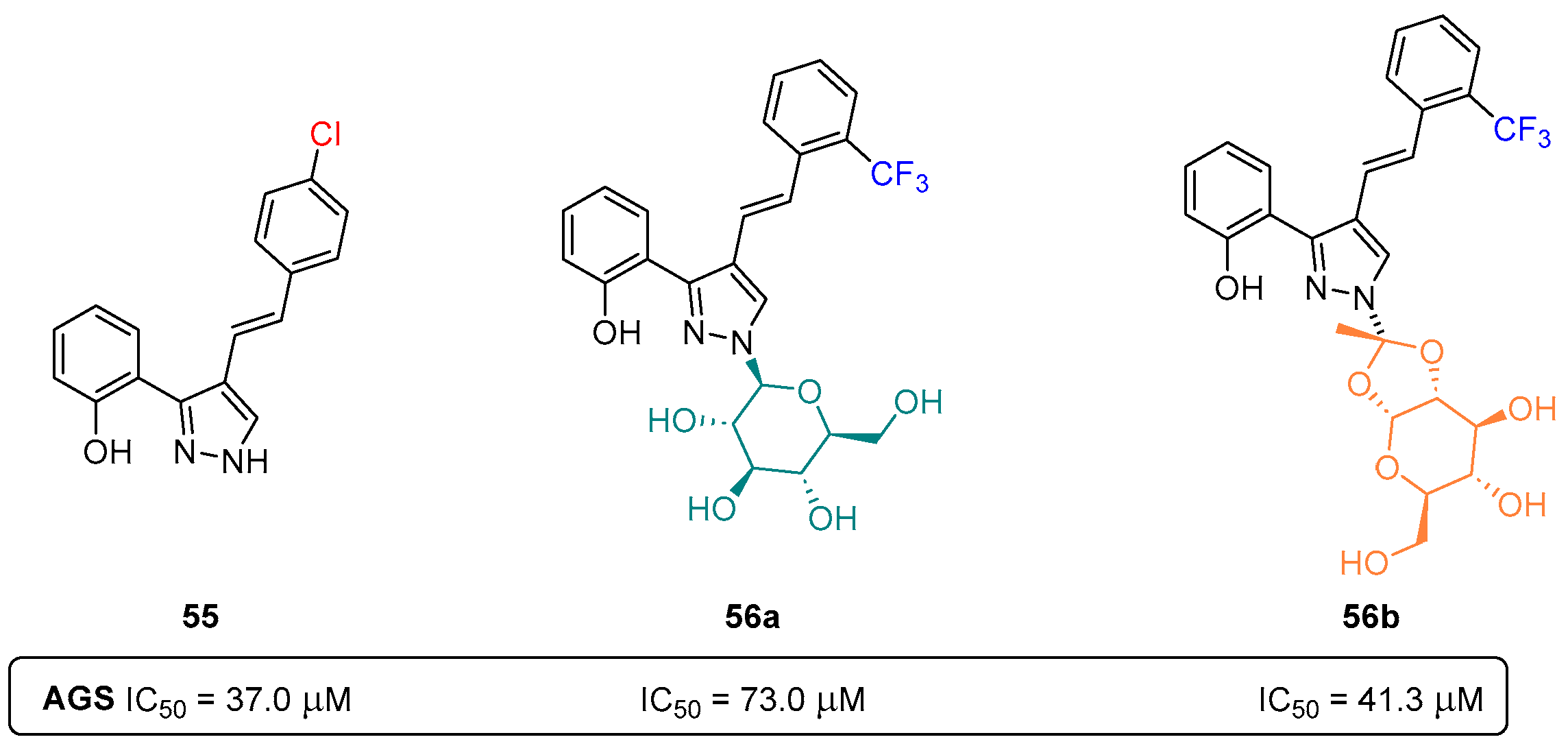
- Carreira, A.R.F.; Pereira, D.M.; Andrade, P.B.; Valentão, P.; Silva, A.M.S.; Braga, S.S.; Silva, V.L.M. Novel styrylpyrazole-glucosides and their dioxolo-bridged doppelgangers: Synthesis and cytotoxicity. New J. Chem. 2019, 43, 8299–8310. [Google Scholar] [CrossRef]
- Chouiter, M.I.; Boulebd, H.; Pereira, D.M.; Valentão, P.; Andrade, P.B.; Belfaitah, A.; Silva, A.M.S. New chalcone-type compounds and 2-pyrazoline derivatives: Synthesis and caspase-dependent anticancer activity. Future Med. Chem. 2020, 12, 493–509. [Google Scholar] [CrossRef] [PubMed]
- Saidi, L.; Rocha, D.H.A.; Talhi, O.; Bentarzi, Y.; Nedjar-Kolli, B.; Bachari, K.; Almeida Paz, F.A.; Helguero, L.A.; Silva, A.M.S. Synthesis of Benzophenones and in vitro Evaluation of Their Anticancer Potential in Breast and Prostate Cancer Cells. ChemMedChem 2019, 14, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- da Silva, A.F.F.; Seixas, R.S.G.R.; Silva, A.M.S.; Coimbra, J.; Fernandes, A.C.; Santos, J.P.; Matos, A.; Rino, J.; Santos, I.; Marques, F. Synthesis, characterization and biological evaluation of carboranylmethylbenzo[b]acridones as novel agents for boron neutron capture therapy. Org. Biomol. Chem. 2014, 12, 5201–5211. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, A.; Norton, N.; Fast, T.; Frölich, L.; Georges, J.; Holzapfel, D.; Kirabali, T.; Krolak-Salmon, P.; Rossini, P.M.; Ferretti, M.T.; et al. Global estimates on the number of persons across the Alzheimer’s disease continuum. Alzheimer’s Dement. 2023, 19, 658–670. [Google Scholar] [CrossRef]
- Horak, M.; Holubova, K.; Nepovimova, E.; Krusek, J.; Kaniakova, M.; Korabecny, J.; Vyklicky, L.; Kuca, K.; Stuchlik, A.; Ricny, J.; et al. The pharmacology of tacrine at N-methyl-d-aspartate receptors. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 75, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D. Evidence of Oxidative Stress in Alzheimer’s Disease Brain and Antioxidant Therapy. Ann. N. Y. Acad. Sci. 2008, 1147, 70–78. [Google Scholar] [CrossRef]
- Sharma, A.; Pachauri, V.; Flora, S.J.S. Advances in Multi-Functional Ligands and the Need for Metal-Related Pharmacology for the Management of Alzheimer Disease. Front. Pharmacol. 2018, 9, 1247. [Google Scholar] [CrossRef]
- Du, X.; Wang, X.; Geng, M. Alzheimer’s disease hypothesis and related therapies. Transl. Neurodegener. 2018, 7, 2. [Google Scholar] [CrossRef]
- Hamley, I.W. The Amyloid Beta Peptide: A Chemist’s Perspective. Role in Alzheimer’s and Fibrillization. Chem. Rev. 2012, 112, 5147–5192. [Google Scholar] [CrossRef]
- Malafaia, D.; Albuquerque, H.M.T.; Silva, A.M.S. Amyloid-β and tau aggregation dual-inhibitors: A synthetic and structure-activity relationship focused review. Eur. J. Med. Chem. 2021, 214, 113209. [Google Scholar] [CrossRef]
- Reitz, C. Alzheimer’s Disease and the Amyloid Cascade Hypothesis: A Critical Review. Int. J. Alzheimer’s Dis. 2012, 2012, 369808. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, T.; Shakeri, A.; Rao, P.P.N. Amyloid cascade in Alzheimer’s disease: Recent advances in medicinal chemistry. Eur. J. Med. Chem. 2016, 113, 258–272. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T.; Price, D.L.; DeLong, M.R. Alzheimer’s disease: A disorder of cortical cholinergic innervation. Science 1983, 219, 1184–1190. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, H. Reconsideration of Anticholinesterase Therapeutic Strategies against Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Gorecki, L.; Uliassi, E.; Bartolini, M.; Janockova, J.; Hrabinova, M.; Hepnarova, V.; Prchal, L.; Muckova, L.; Pejchal, J.; Karasova, J.Z.; et al. Phenothiazine-Tacrine Heterodimers: Pursuing Multitarget Directed Approach in Alzheimer’s Disease. ACS Chem. Neurosci. 2021, 12, 1698–1715. [Google Scholar] [CrossRef] [PubMed]
- Blaikie, L.; Kay, G.; Kong Thoo Lin, P. Current and emerging therapeutic targets of alzheimer’s disease for the design of multi-target directed ligands. MedChemComm 2019, 10, 2052–2072. [Google Scholar] [CrossRef] [PubMed]
- Malafaia, D.; Oliveira, A.; Fernandes, P.A.; Ramos, M.J.; Albuquerque, H.M.T.; Silva, A.M.S. Chromeno[3,4-b]xanthones as First-in-Class AChE and Aβ Aggregation Dual-Inhibitors. Int. J. Mol. Sci. 2021, 22, 4145. [Google Scholar] [CrossRef]
- Ferreira, J.P.S.; Cardoso, S.M.; Almeida Paz, F.A.; Silva, A.M.S.; Silva, V.L.M. Synthesis of 2-aroylfuro[3,2-c]quinolines from quinolone-based chalcones and evaluation of their antioxidant and anticholinesterase activities. New J. Chem. 2020, 44, 6501–6509. [Google Scholar] [CrossRef]
- Zhou, H.; Yang, Z.; Tian, X.; Chen, L.; Lee, S.; Huynh, T.; Ge, C.; Zhou, R. Lanosterol Disrupts the Aggregation of Amyloid-β Peptides. ACS Chem. Neurosci. 2019, 10, 4051–4060. [Google Scholar] [CrossRef]
- Liu, K.; Guo, T.L.; Chojnacki, J.; Lee, H.-G.; Wang, X.; Siedlak, S.L.; Rao, W.; Zhu, X.; Zhang, S. Bivalent Ligand Containing Curcumin and Cholesterol as a Fluorescence Probe for Aβ Plaques in Alzheimer’s Disease. ACS Chem. Neurosci. 2012, 3, 141–146. [Google Scholar] [CrossRef]
- Zhao, L.; Chen, X.-J.; Zhu, J.; Xi, Y.-B.; Yang, X.; Hu, L.-D.; Ouyang, H.; Patel, S.H.; Jin, X.; Lin, D.; et al. Lanosterol reverses protein aggregation in cataracts. Nature 2015, 523, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Yang, Z.; Zhou, R. Lanosterol Disrupts Aggregation of Human γD-Crystallin by Binding to the Hydrophobic Dimerization Interface. J. Am. Chem. Soc. 2018, 140, 8479–8486. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, X.-J.; Yang, Z.; Xi, Y.-B.; Wang, L.; Wu, Y.; Yan, Y.-B.; Rao, Y. Synthesis, Evaluation, and Structure–Activity Relationship Study of Lanosterol Derivatives To Reverse Mutant-Crystallin-Induced Protein Aggregation. J. Med. Chem. 2018, 61, 8693–8706. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, H.M.T.; Nunes da Silva, R.; Pereira, M.; Maia, A.; Guieu, S.; Soares, A.R.; Santos, C.M.M.; Vieira, S.I.; Silva, A.M.S. Steroid–Quinoline Hybrids for Disruption and Reversion of Protein Aggregation Processes. ACS Med. Chem. Lett. 2022, 13, 443–448. [Google Scholar] [CrossRef]
- Pereira, M.; Tomé, D.; Domingues, A.S.; Varanda, A.S.; Paulo, C.; Santos, M.A.S.; Soares, A.R. A Fluorescence-Based Sensor Assay that Monitors General Protein Aggregation in Human Cells. Biotechnol. J. 2018, 13, 1700676. [Google Scholar] [CrossRef]
- Hong, Y.; Meng, L.; Chen, S.; Leung, C.W.T.; Da, L.-T.; Faisal, M.; Silva, D.-A.; Liu, J.; Lam, J.W.Y.; Huang, X.; et al. Monitoring and Inhibition of Insulin Fibrillation by a Small Organic Fluorogen with Aggregation-Induced Emission Characteristics. J. Am. Chem. Soc. 2012, 134, 1680–1689. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Gandini, A.; Prati, F.; Uliassi, E. From Companion Diagnostics to Theranostics: A New Avenue for Alzheimer’s Disease? J. Med. Chem. 2016, 59, 7759–7770. [Google Scholar] [CrossRef]
- Aliyan, A.; Cook, N.P.; Martí, A.A. Interrogating Amyloid Aggregates using Fluorescent Probes. Chem. Rev. 2019, 119, 11819–11856. [Google Scholar] [CrossRef]
- Nunes da Silva, R.; Costa, C.C.; Santos, M.J.G.; Alves, M.Q.; Braga, S.S.; Vieira, S.I.; Rocha, J.; Silva, A.M.S.; Guieu, S. Fluorescent Light-up Probe for the Detection of Protein Aggregates. Chem. Asian J. 2019, 14, 859–863. [Google Scholar] [CrossRef]
- Brul, S.; Nussbaum, J.; Dielbandhoesing, S.K. Fluorescent probes for wall porosity and membrane integrity in filamentous fungi. J. Microbiol. Methods 1997, 28, 169–178. [Google Scholar] [CrossRef]
- Ferreira, J.R.M.; Alves, M.; Sousa, B.; Vieira, S.I.; Silva, A.M.S.; Guieu, S.; Cunha, Â.; Nunes da Silva, R. Curcumin-based molecular probes for fluorescence imaging of fungi. Org. Biomol. Chem. 2023, 21, 1531–1536. [Google Scholar] [CrossRef]
- Gross, D.A.; Silver, D.L. Cytosolic lipid droplets: From mechanisms of fat storage to disease. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 304–326. [Google Scholar] [CrossRef] [PubMed]
- Soares da Silva, A.M.; André Guieu, S.J.; Matias Celestino Gomes da Rocha, J.C.; Moreira Pinto Vieira, S.I.; da Cruz E Silva, O.; Dos Santos Dias, R.A.; Frade Ruivo, R. Fluorescent Compounds, Production Methods and Uses Thereof. WO/2017/182945, 26 October 2017. [Google Scholar]
- Vieira, S.I.; Nunes da Silva, R.; Alves, M.; Dias, R.A.; Meireles Sousa, A.M.; Camões, F.; Maia, A.; Almeida, M.; Rocha, J.; Silva, A.M.; et al. Liprobe, a vital dye for lipid aggregates detection in imaging and high-content screens. Front. Photonics 2022, 3, 963778. [Google Scholar] [CrossRef]
- Oryan, A.; Akbari, M. Worldwide risk factors in leishmaniasis. Asian Pac. J. Trop. Med. 2016, 9, 925–932. [Google Scholar] [CrossRef] [PubMed]
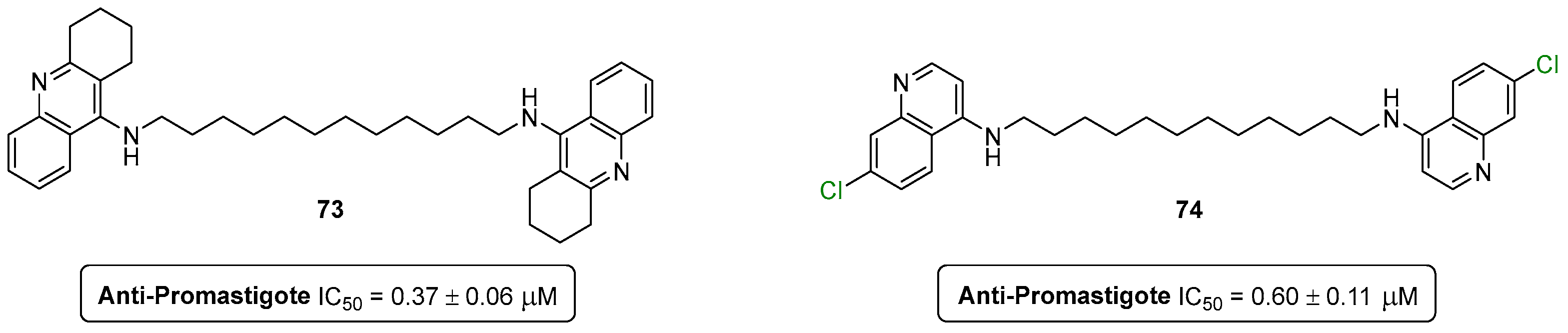
- Silva, C.F.M.; Leão, T.; Dias, F.; Tomás, A.M.; Pinto, D.C.G.A.; Oliveira, E.F.T.; Oliveira, A.; Fernandes, P.A.; Silva, A.M.S. Structure & ndash; Activity Relationship Studies of 9-Alkylamino-1,2,3,4-tetrahydroacridines against Leishmania (Leishmania) infantum Promastigotes. Pharmaceutics 2023, 15, 669. [Google Scholar]
- Dhanda, G.; Acharya, Y.; Haldar, J. Antibiotic Adjuvants: A Versatile Approach to Combat Antibiotic Resistance. ACS Omega 2023, 8, 10757–10783. [Google Scholar] [CrossRef]
- Almeida, M.C.; da Costa, P.M.; Sousa, E.; Resende, D.I.S.P. Emerging Target-Directed Approaches for the Treatment and Diagnosis of Microbial Infections. J. Med. Chem. 2023, 66, 32–70. [Google Scholar] [CrossRef] [PubMed]
- Souza, L.G.d.S.; Almeida, M.C.S.; Lemos, T.L.G.; Ribeiro, P.R.V.; de Brito, E.S.; Silva, V.L.M.; Silva, A.M.S.; Braz-Filho, R.; Costa, J.G.M.; Rodrigues, F.F.G.; et al. Synthesis, antibacterial and cytotoxic activities of new biflorin-based hydrazones and oximes. Bioorganic Med. Chem. Lett. 2016, 26, 435–439. [Google Scholar] [CrossRef]
- Zhang, M.; Fu, M.; Hu, Q. Advances in Human Norovirus Vaccine Research. Vaccines 2021, 9, 732. [Google Scholar] [CrossRef]
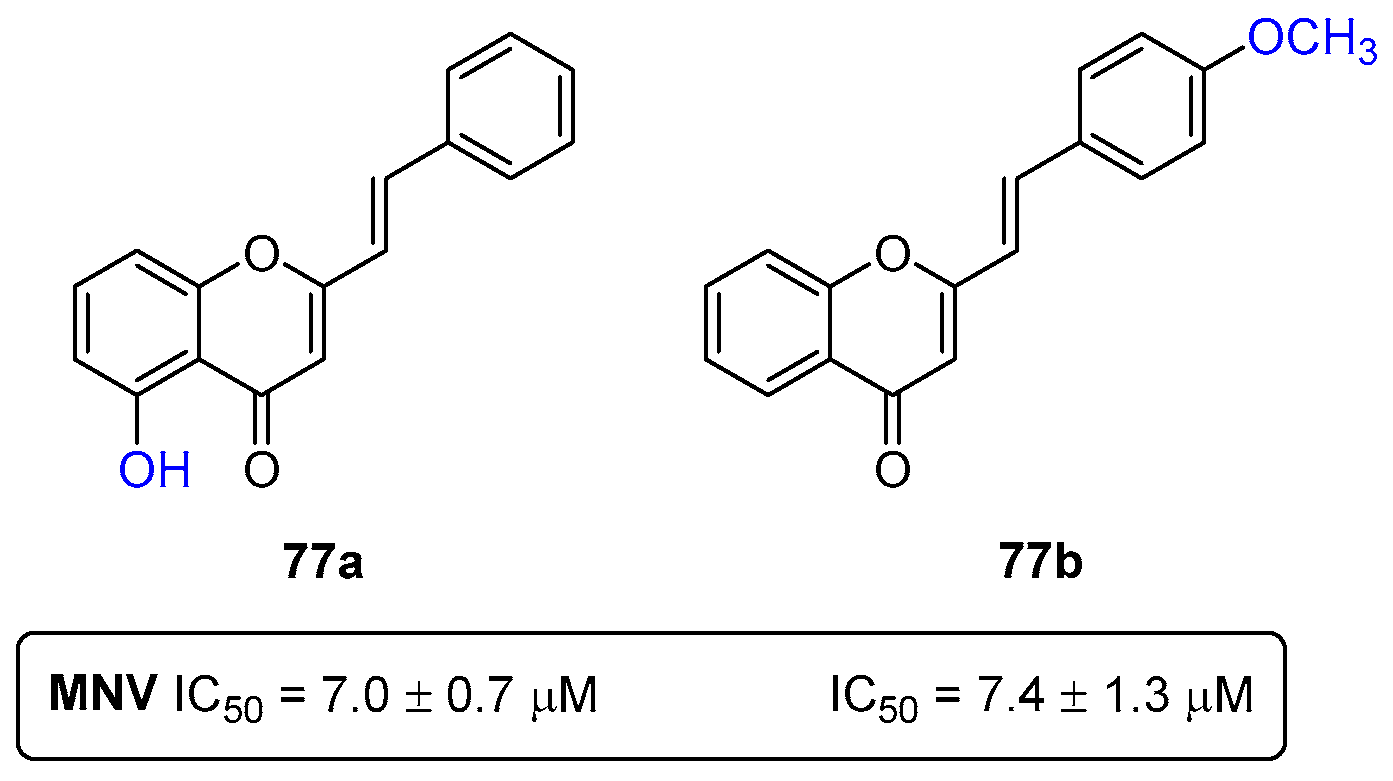
- Rocha-Pereira, J.; Cunha, R.; Pinto, D.C.G.A.; Silva, A.M.S.; Nascimento, M.S.J. (E)-2-Styrylchromones as potential anti-norovirus agents. Bioorganic Med. Chem. 2010, 18, 4195–4201. [Google Scholar] [CrossRef]






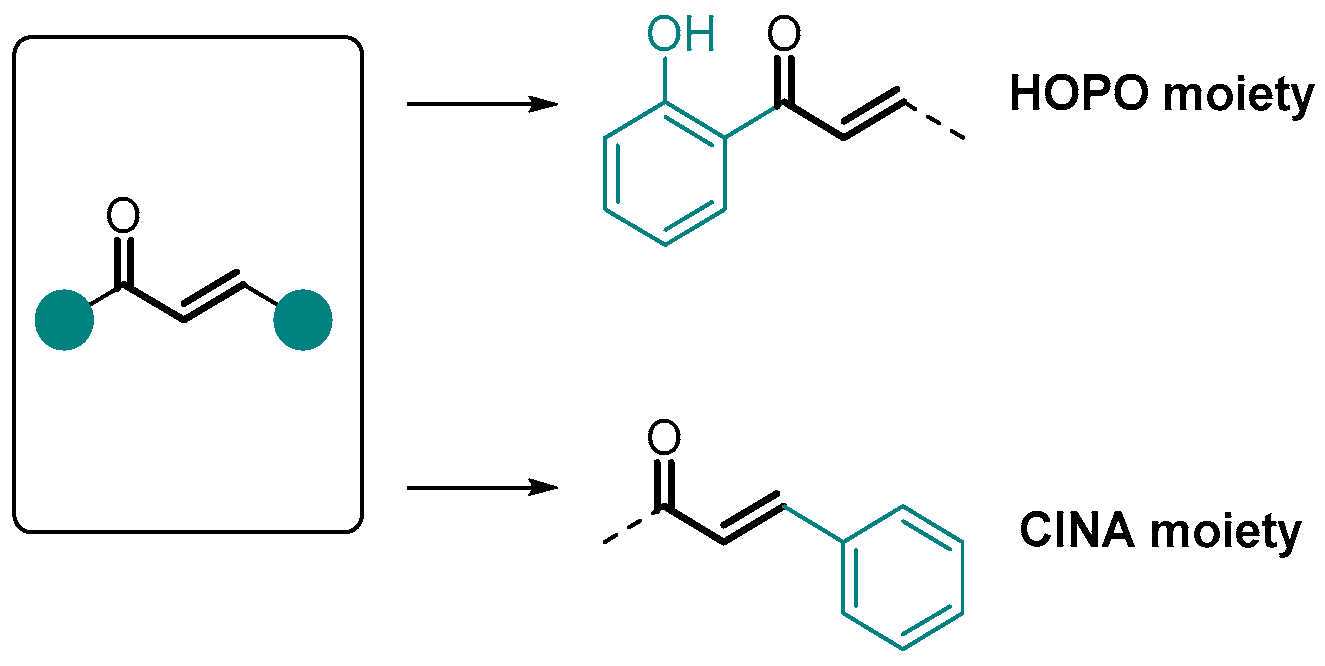

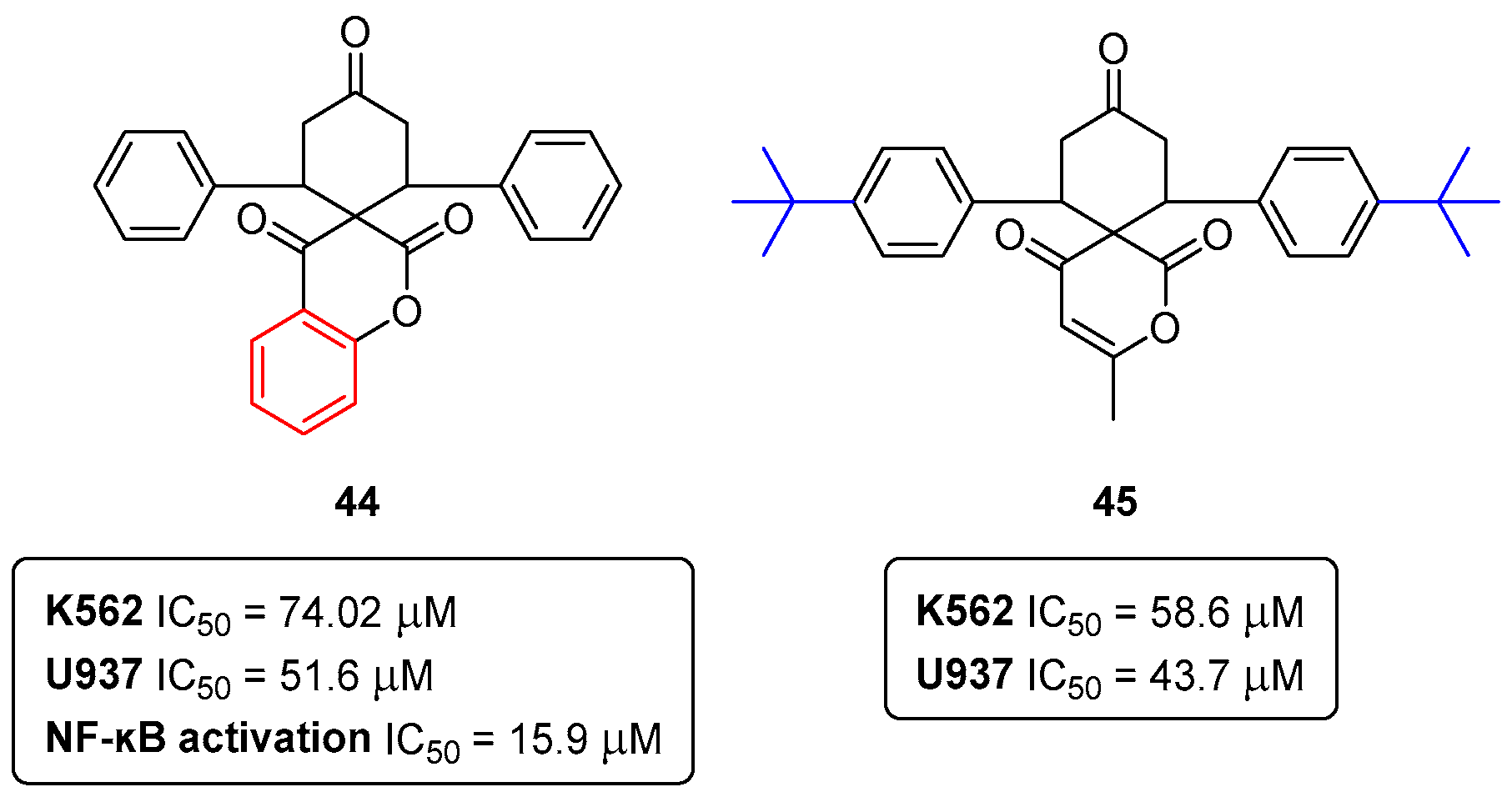
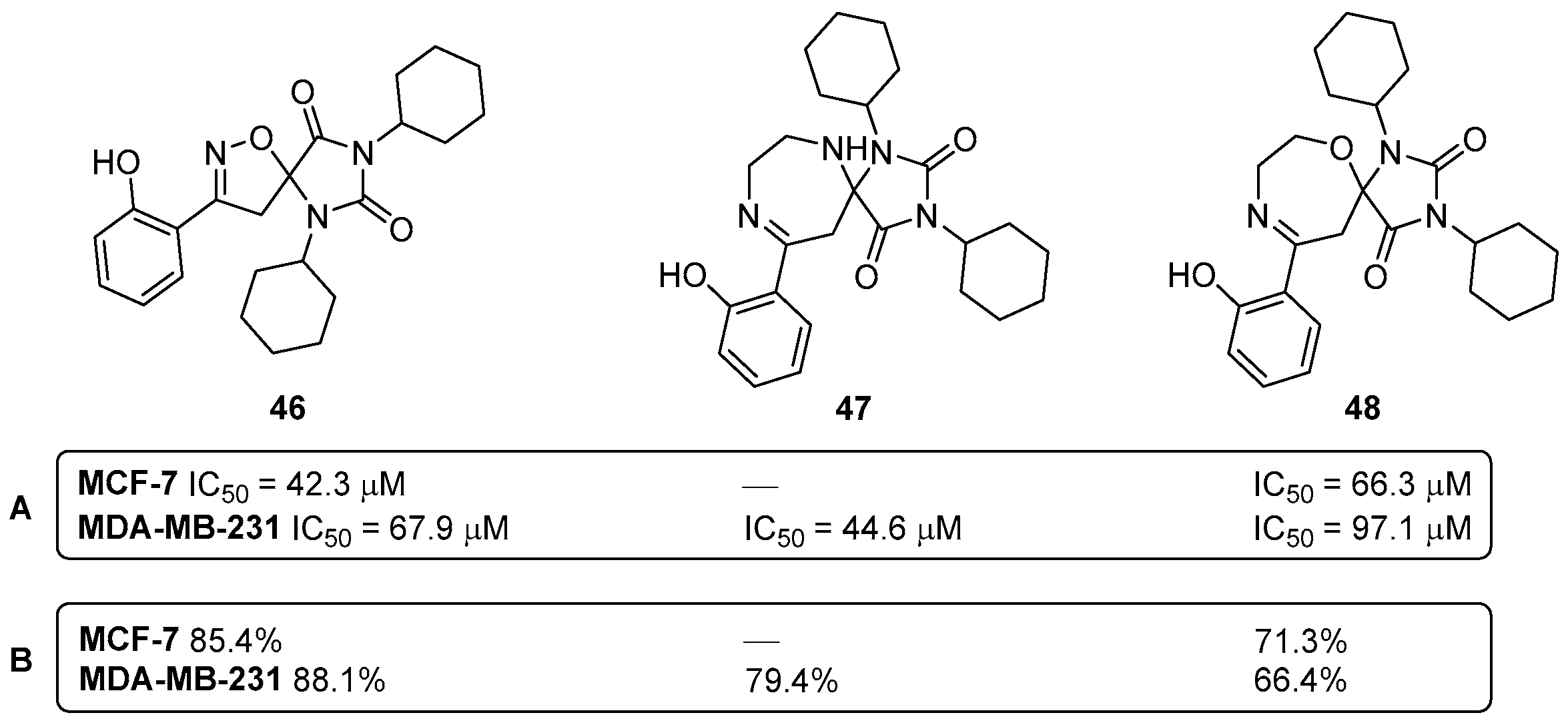
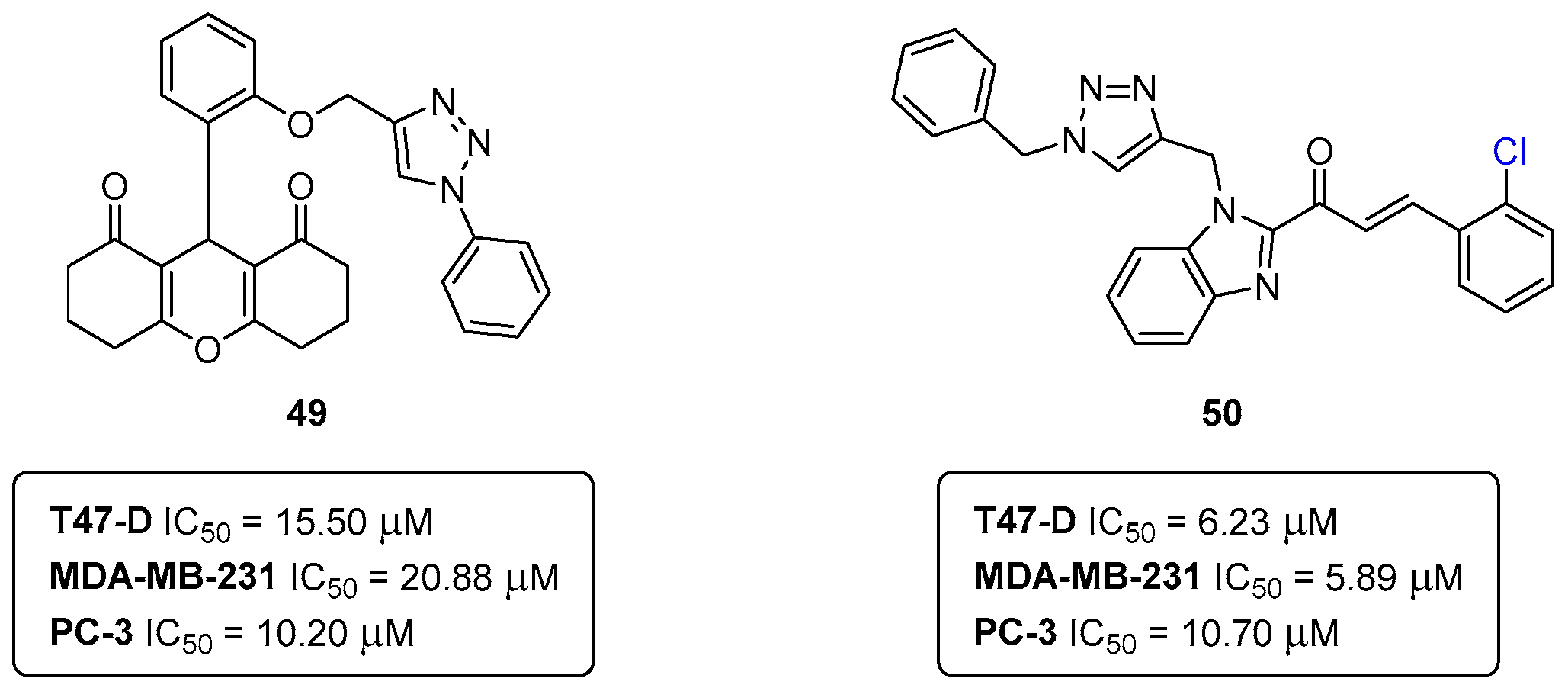







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sousa, J.L.C.; Albuquerque, H.M.T.; Silva, A.M.S. Drug Discovery Based on Oxygen and Nitrogen (Non-)Heterocyclic Compounds Developed @LAQV–REQUIMTE/Aveiro. Pharmaceuticals 2023, 16, 1668. https://doi.org/10.3390/ph16121668
Sousa JLC, Albuquerque HMT, Silva AMS. Drug Discovery Based on Oxygen and Nitrogen (Non-)Heterocyclic Compounds Developed @LAQV–REQUIMTE/Aveiro. Pharmaceuticals. 2023; 16(12):1668. https://doi.org/10.3390/ph16121668
Chicago/Turabian StyleSousa, Joana L. C., Hélio M. T. Albuquerque, and Artur M. S. Silva. 2023. "Drug Discovery Based on Oxygen and Nitrogen (Non-)Heterocyclic Compounds Developed @LAQV–REQUIMTE/Aveiro" Pharmaceuticals 16, no. 12: 1668. https://doi.org/10.3390/ph16121668
APA StyleSousa, J. L. C., Albuquerque, H. M. T., & Silva, A. M. S. (2023). Drug Discovery Based on Oxygen and Nitrogen (Non-)Heterocyclic Compounds Developed @LAQV–REQUIMTE/Aveiro. Pharmaceuticals, 16(12), 1668. https://doi.org/10.3390/ph16121668