

Kynurenic Acid: A Novel Player in Cardioprotection against Myocardial Ischemia/Reperfusion Injuries
 , , ,
, , ,
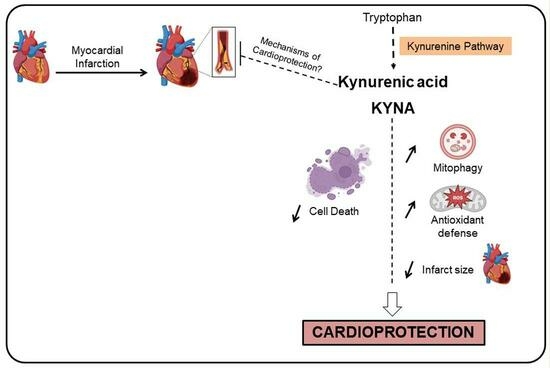
Abstract
:
1. Introduction
2. Results
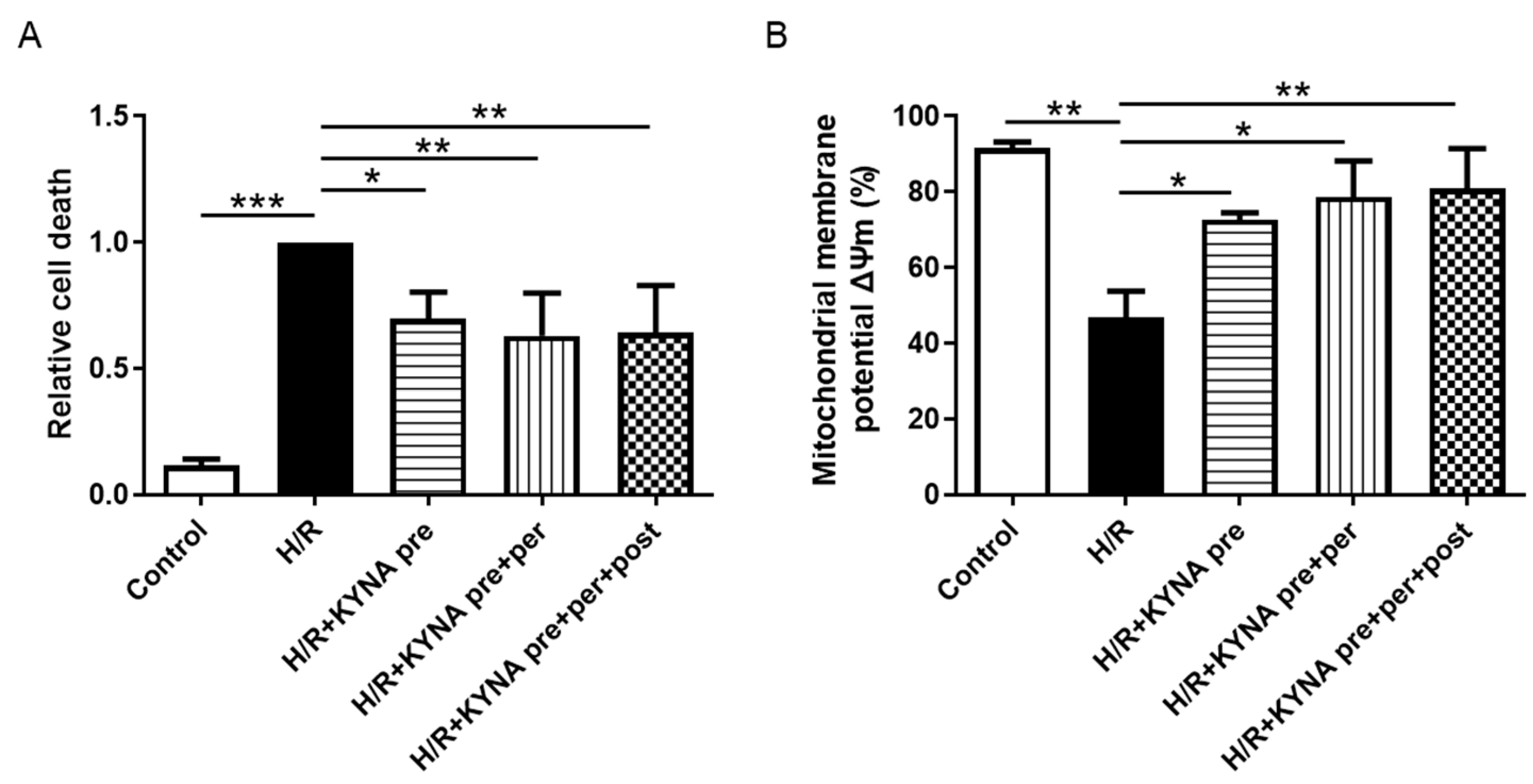
2.1. KYNA Reduced In Vitro Cell Death and Prevented Mitochondrial Membrane Potential Decrease after Hypoxia/Reoxygenation
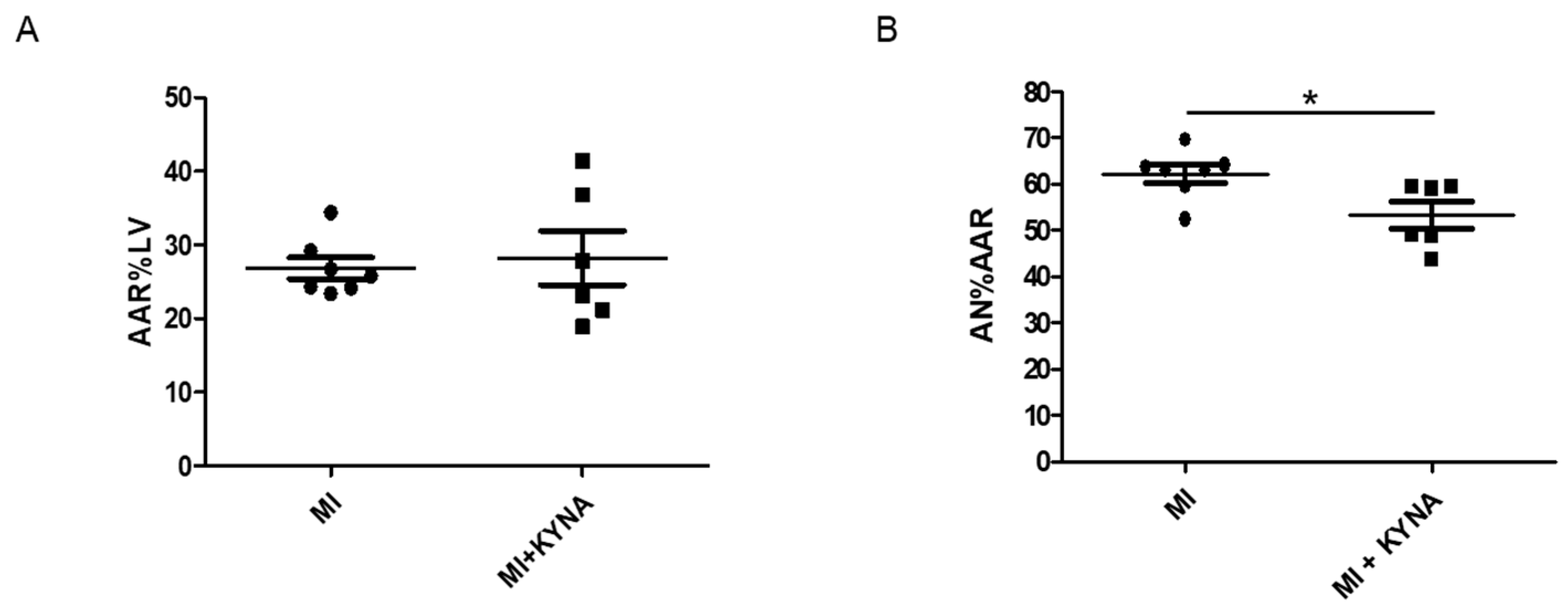
2.2. KYNA Reduced Infarct Size In Vivo
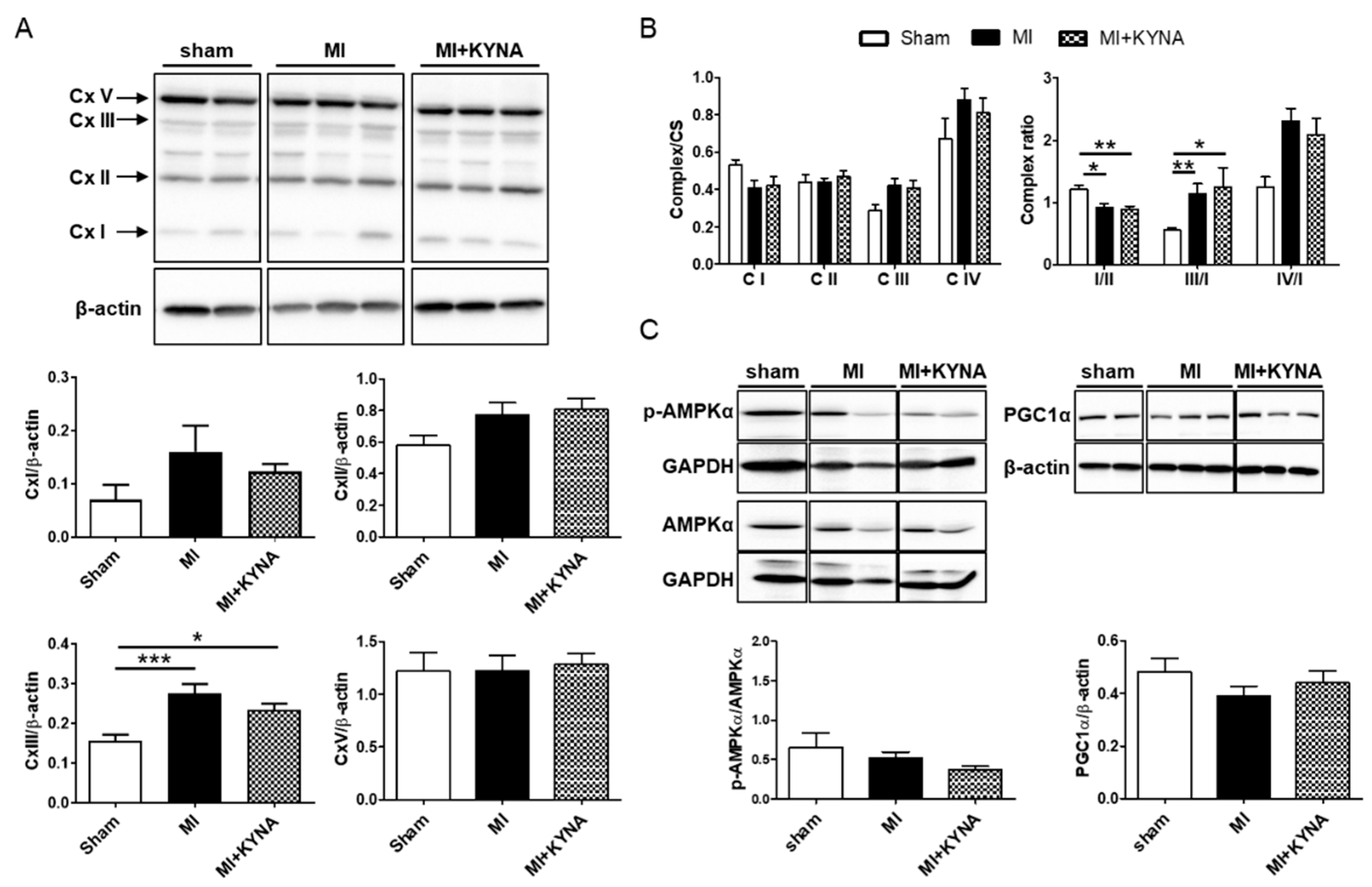
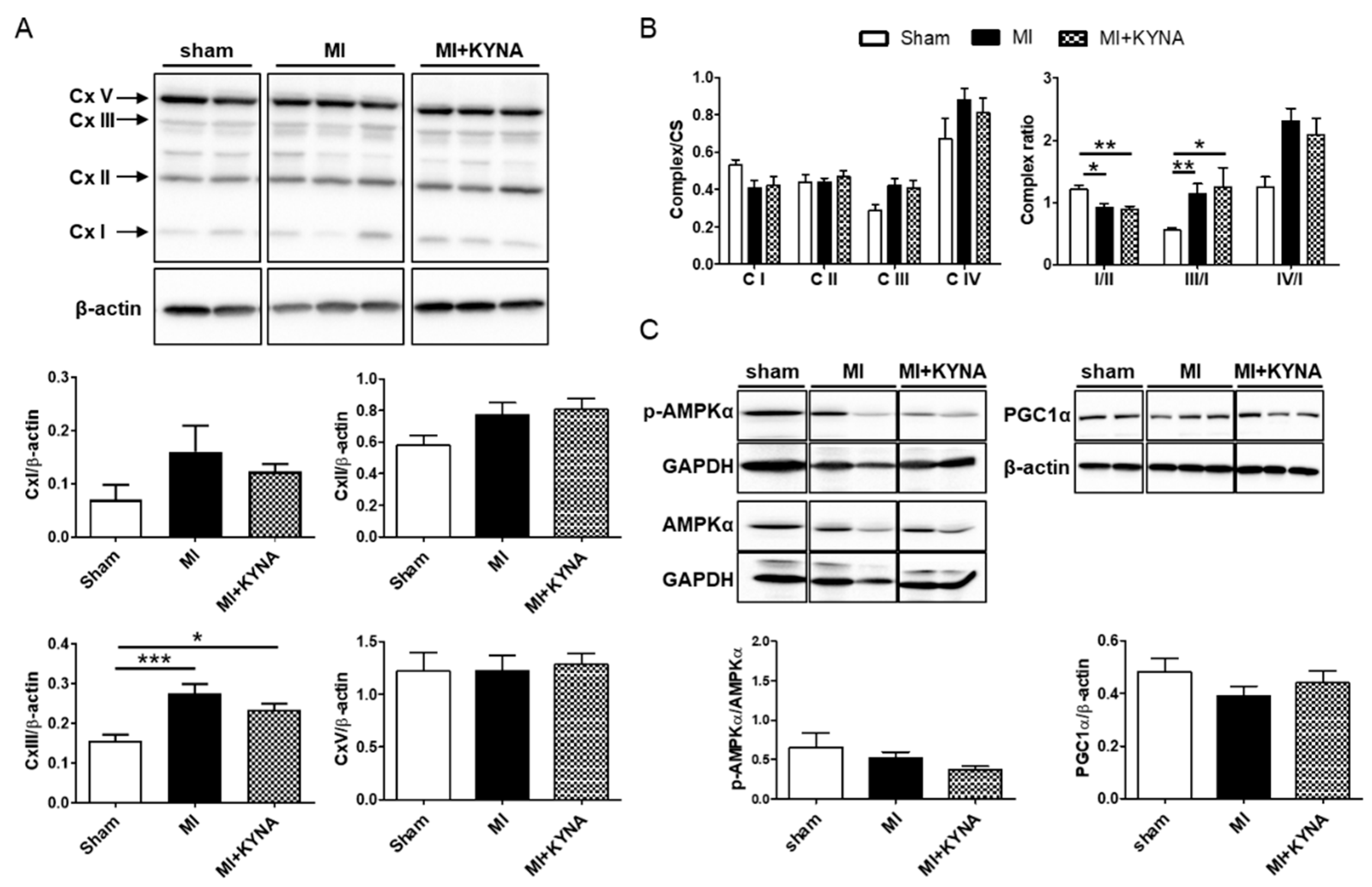
2.3. KYNA Did Not Influence the Mitochondrial Metabolic Function
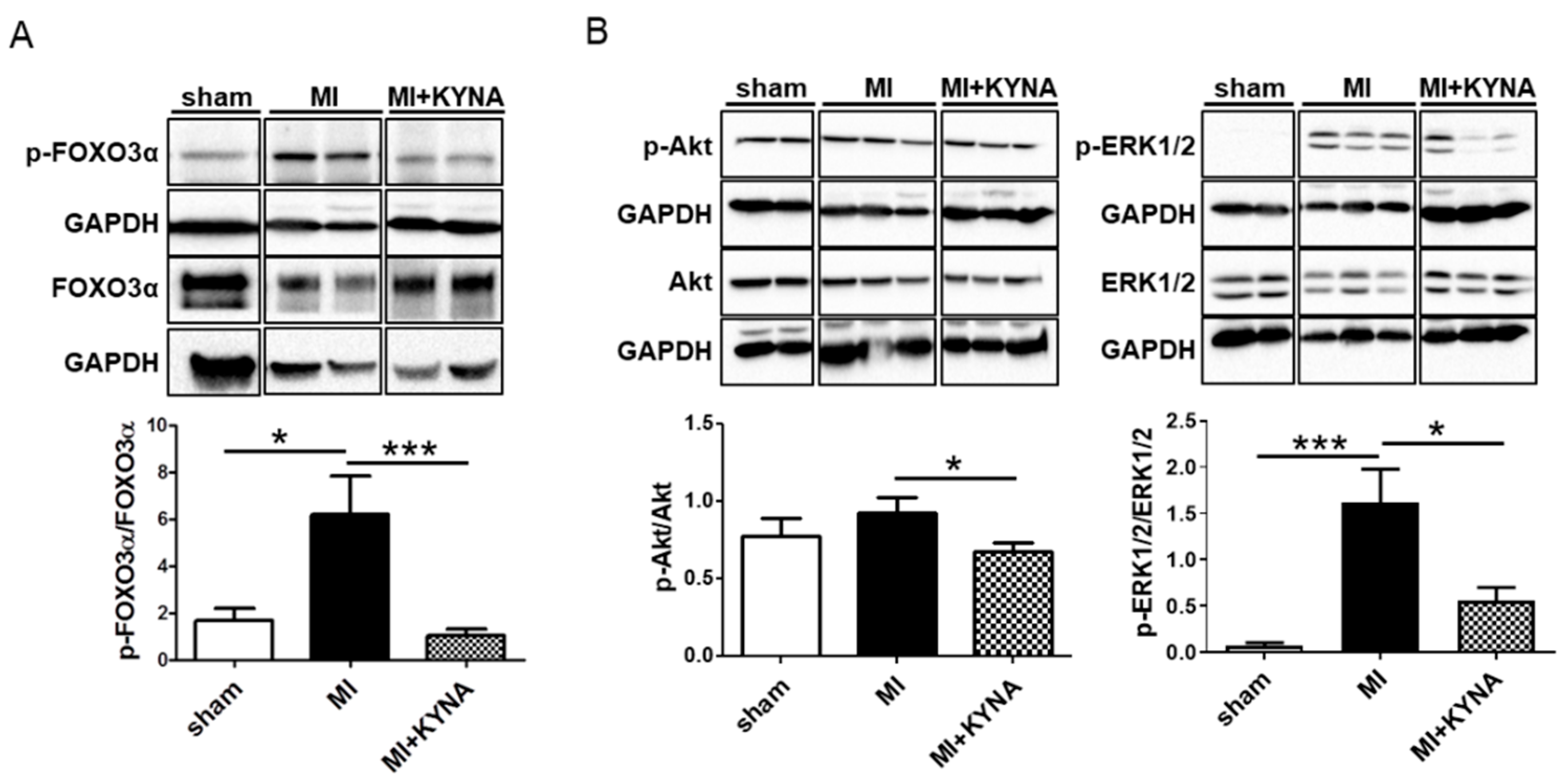
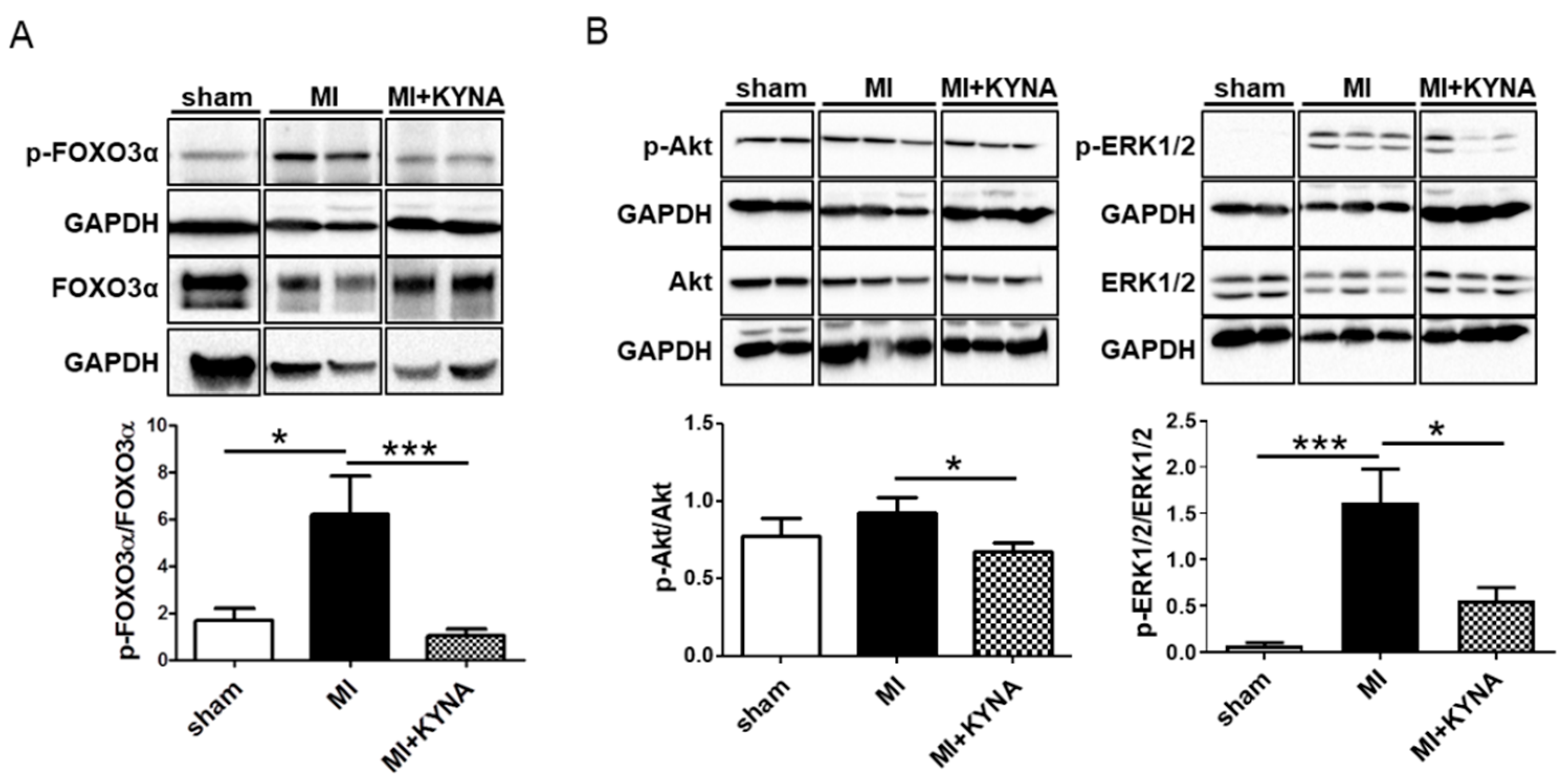
2.4. KYNA Reduced Foxo3α, Akt and ERK1/2 Phosphorylation Levels following Myocardial Ischemia/Reperfusion
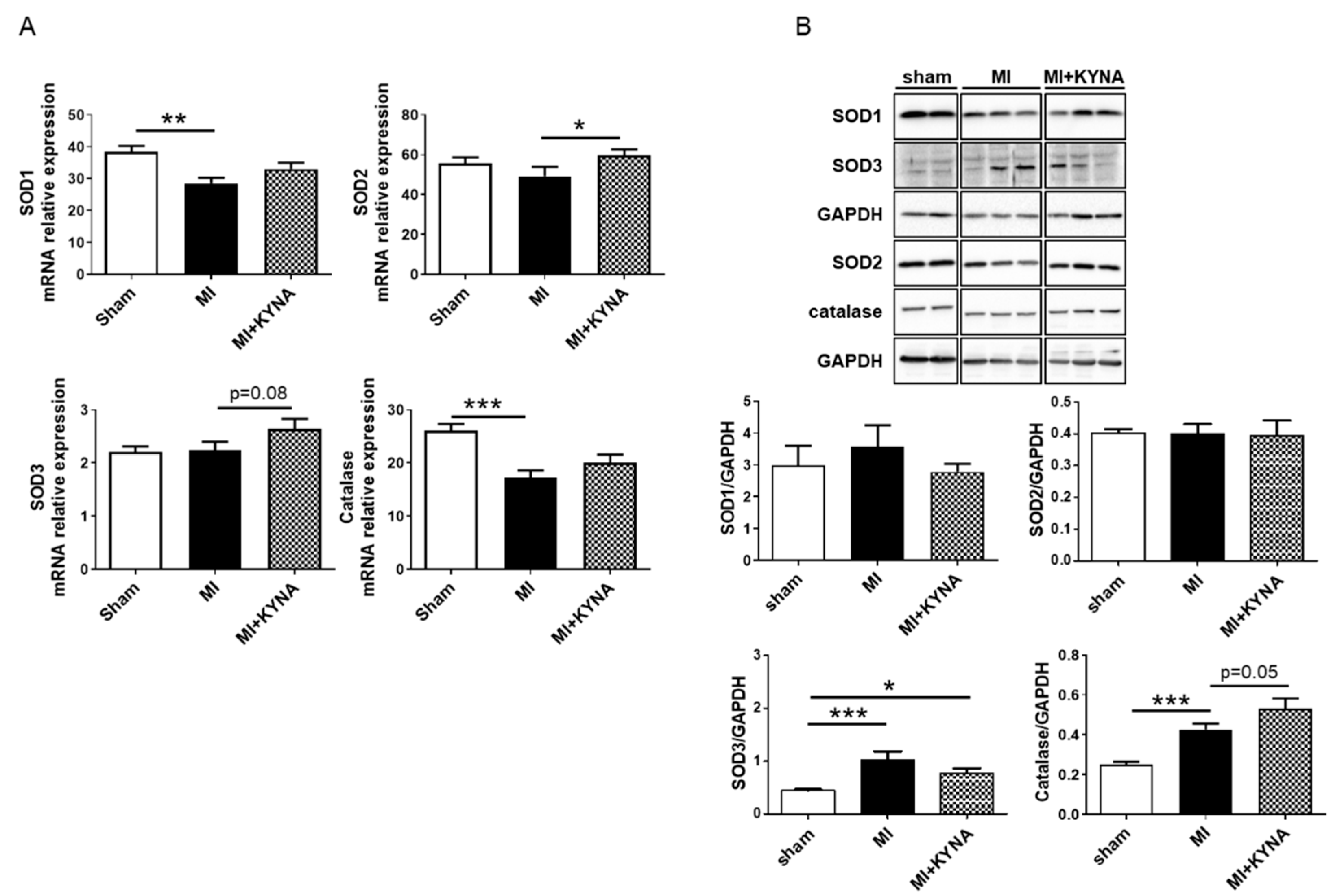
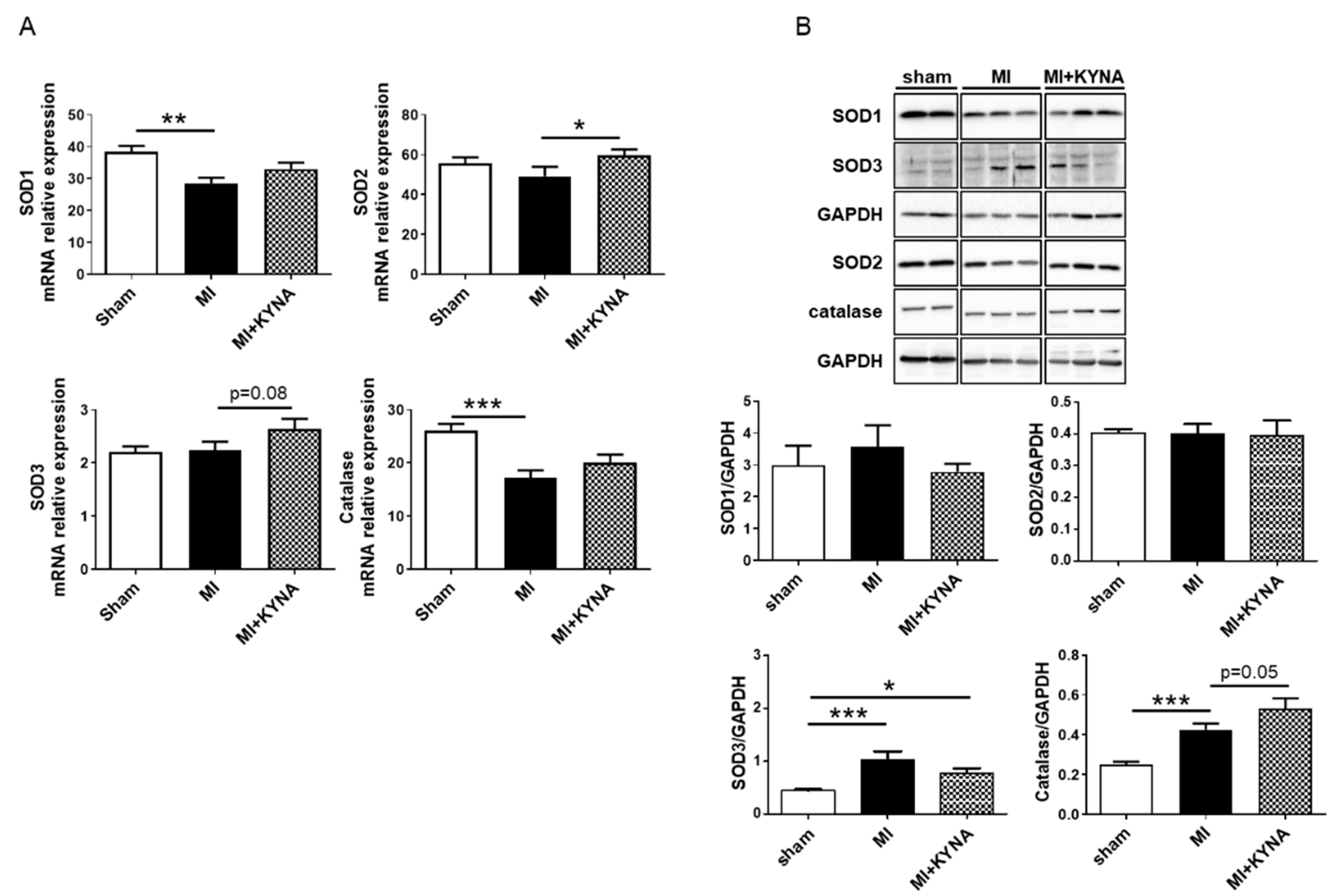
2.5. KYNA Stimulated Antioxidant Defense following Myocardial Ischemia/Reperfusion
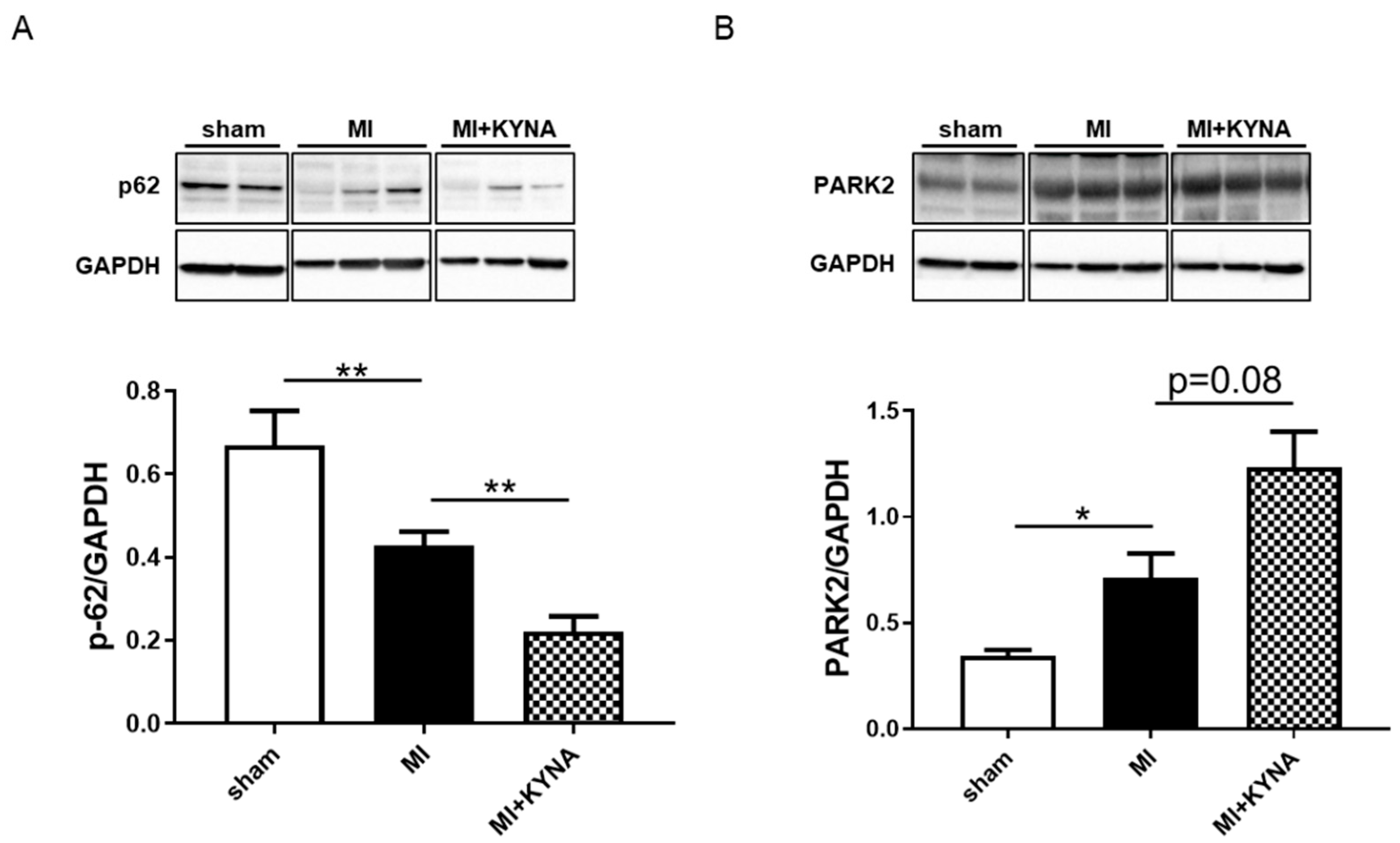
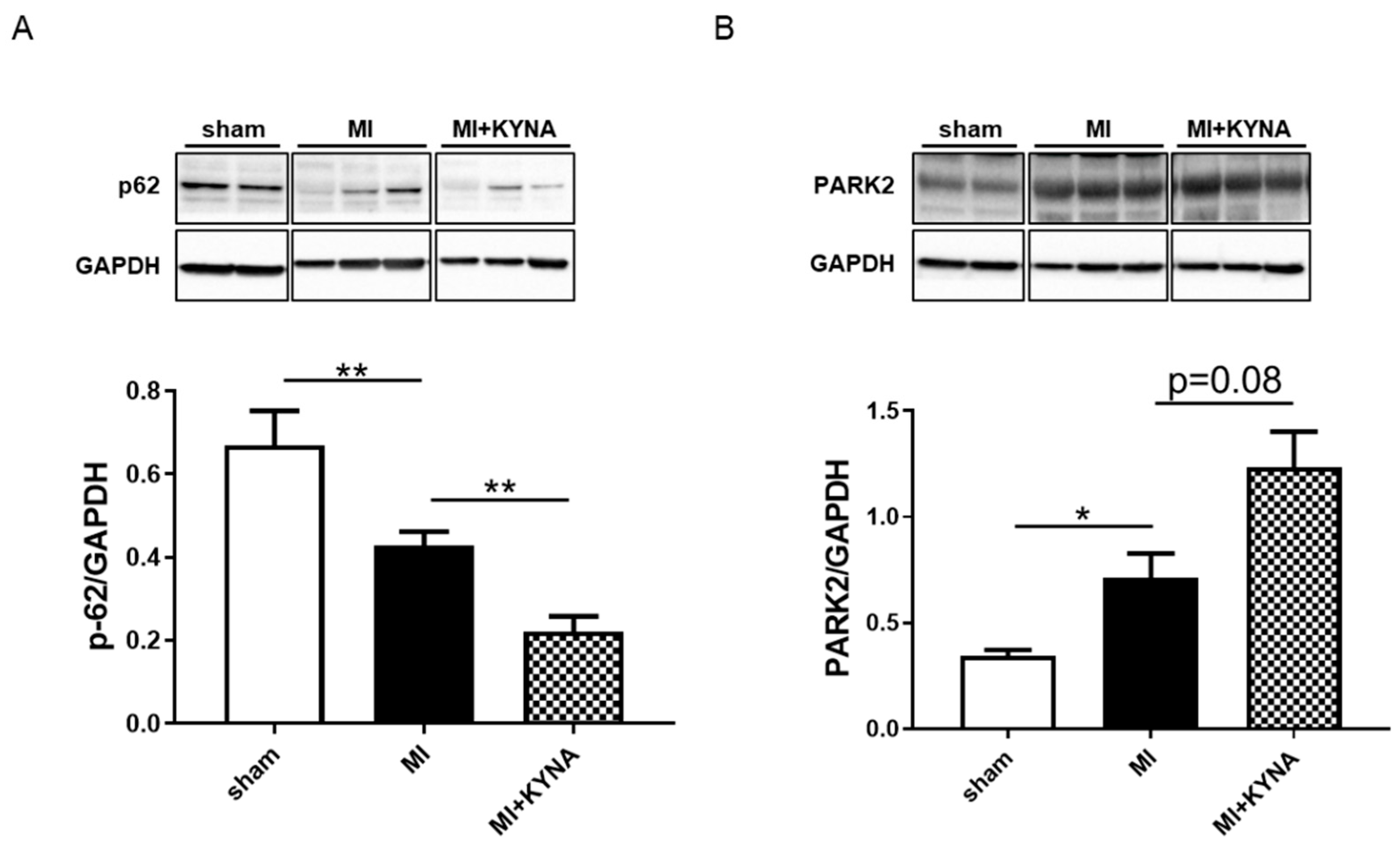
2.6. KYNA Increased Mitophagy Markers following Myocardial Ischemia/Reperfusion
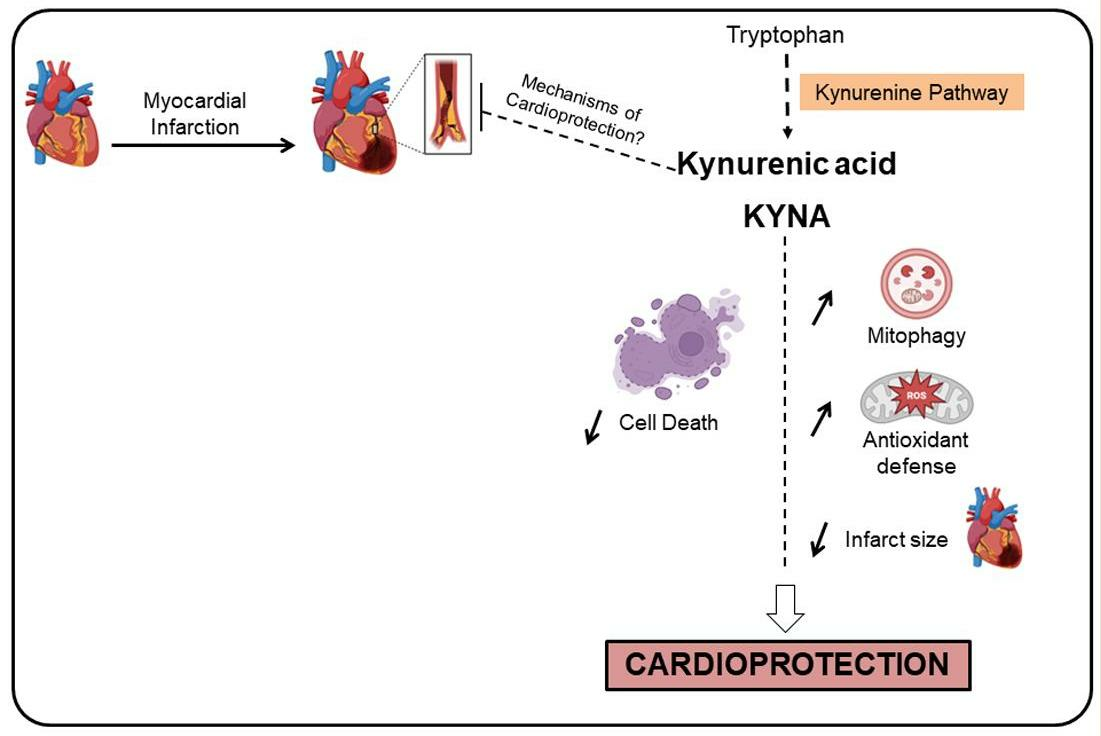
3. Discussion
Limitations
4. Materials and Methods
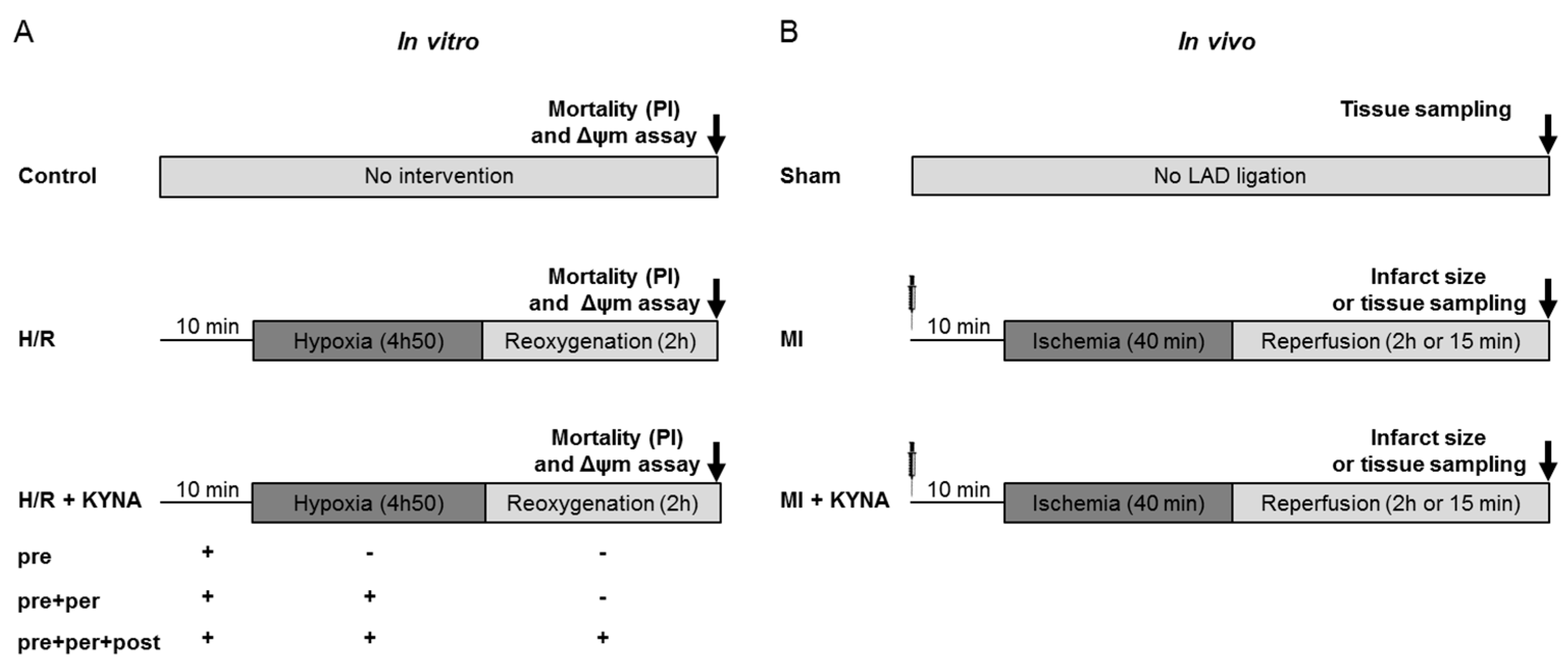
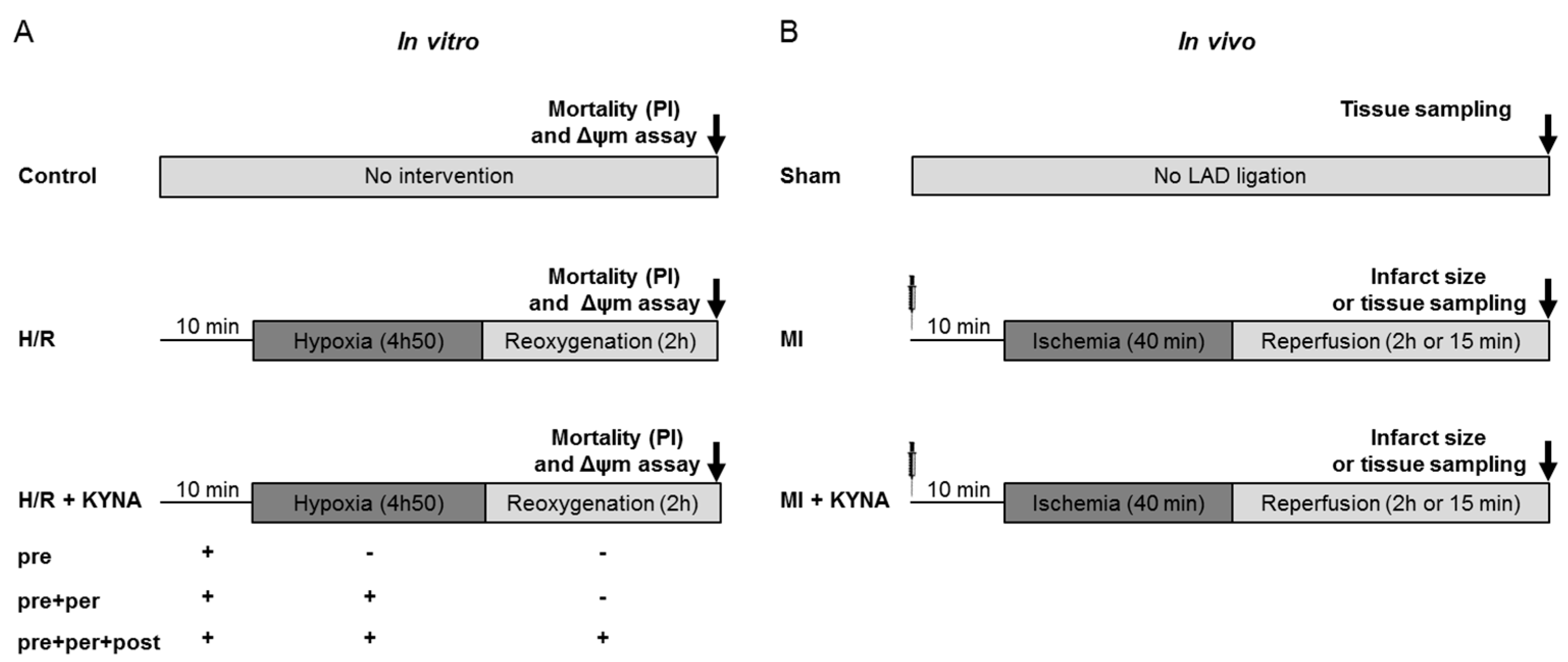
4.1. Study Design and Methodology
4.2. In Vitro H9C2 Hypoxia/Reoxygenation
- –
- Control group: Cells did not undergo any intervention and were kept in normoxic conditions and a culture medium for 7 h.
- –
- Hypoxia/reoxygenation (H/R) group: Cells underwent H/R with the DMSO (vehicle) treatment 10 min before hypoxia and throughout the procedure.
- –
- H/R + KYNA group: Cells underwent H/R with the KYNA treatment (1 µM) either 10 min before hypoxia (pre), pre + during hypoxia (pre + per), or pre + per + during reoxygenation (pre + per + post).
4.3. Cell Death and Mitochondrial Membrane Potential (ΔΨm) Assessment
4.4. Animal Studies
4.5. Myocardial Ischemia/Reperfusion Rat Model
- –
- Sham group: animals undergoing all the surgical procedure except ligature of the coronary artery.
- –
- MI group: animals undergoing myocardial ischemia/reperfusion and injected 10 min before ischemia with NaOH 1 M (vehicle).
- –
4.6. Area at Risk and Infarct Size Determination
4.7. Real-Time Quantitative Reverse Transcription Polymerase Chain Reaction (RT-qPCR)
4.8. Western Blot (WB) Analysis
4.9. Mitochondrial Respiratory Chain Complex Enzymatic Activity Assessment
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Montrief, T.; Davis, W.T.; Koyfman, A.; Long, B. Mechanical, Inflammatory, and Embolic Complications of Myocardial Infarction: An Emergency Medicine Review. Am. J. Emerg. Med. 2019, 37, 1175–1183. [Google Scholar] [CrossRef]
- Jennings, R.B.; Sommers, H.M.; Smyth, G.A.; Flack, H.A.; Linn, H. Myocardial Necrosis Induced by Temporary Occlusion of a Coronary Artery in the Dog. Arch. Pathol. 1960, 70, 68–78. [Google Scholar]
- Cung, T.-T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guérin, P.; Elbaz, M.; Delarche, N.; et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N. Engl. J. Med. 2015, 373, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Kharbanda, R.K.; Møller, U.K.; Ramlall, M.; Aarøe, J.; Butler, R.; Bulluck, H.; Clayton, T.; Dana, A.; Dodd, M.; et al. Effect of Remote Ischaemic Conditioning on Clinical Outcomes in Patients with Acute Myocardial Infarction (CONDI-2/ERIC-PPCI): A Single-Blind Randomised Controlled Trial. Lancet 2019, 394, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, D.; Song, P.; Zou, M.-H. Tryptophan-Kynurenine Pathway Is Dysregulated in Inflammation, and Immune Activation. Front. Biosci. 2015, 20, 1116–1143. [Google Scholar] [CrossRef]
- Tanaka, M.; Tóth, F.; Polyák, H.; Szabó, Á.; Mándi, Y.; Vécsei, L. Immune Influencers in Action: Metabolites and Enzymes of the Tryptophan-Kynurenine Metabolic Pathway. Biomedicines 2021, 9, 734. [Google Scholar] [CrossRef]
- Gong, X.; Chang, R.; Zou, J.; Tan, S.; Huang, Z. The Role and Mechanism of Tryptophan—Kynurenine Metabolic Pathway in Depression. Rev. Neurosci. 2023, 34, 313–324. [Google Scholar] [CrossRef]
- Muneer, A. Kynurenine Pathway of Tryptophan Metabolism in Neuropsychiatric Disorders: Pathophysiologic and Therapeutic Considerations. Clin. Psychopharmacol. Neurosci. 2020, 18, 507–526. [Google Scholar] [CrossRef]
- Ciapała, K.; Mika, J.; Rojewska, E. The Kynurenine Pathway as a Potential Target for Neuropathic Pain Therapy Design: From Basic Research to Clinical Perspectives. Int. J. Mol. Sci. 2021, 22, 11055. [Google Scholar] [CrossRef]
- Tsuji, A.; Ikeda, Y.; Yoshikawa, S.; Taniguchi, K.; Sawamura, H.; Morikawa, S.; Nakashima, M.; Asai, T.; Matsuda, S. The Tryptophan and Kynurenine Pathway Involved in the Development of Immune-Related Diseases. Int. J. Mol. Sci. 2023, 24, 5742. [Google Scholar] [CrossRef]
- Boros, F.A.; Vécsei, L. Immunomodulatory Effects of Genetic Alterations Affecting the Kynurenine Pathway. Front. Immunol. 2019, 10, 2570. [Google Scholar] [CrossRef] [PubMed]
- Kozieł, K.; Urbanska, E.M. Kynurenine Pathway in Diabetes Mellitus-Novel Pharmacological Target? Cells 2023, 12, 460. [Google Scholar] [CrossRef] [PubMed]
- Ala, M.; Fallahpour Khoshdel, M.R.; Mohammad Jafari, R.; Sadrkhanloo, M.; Goudarzi, S.; Asl Soleimani, M.; Dehpour, A.R. Low-Dose Sumatriptan Improves the Outcome of Acute Mesenteric Ischemia in Rats via Downregulating Kynurenine. Pharmacol. Rep. 2023, 75, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Li, G.; Zheng, Q.; Gu, X.; Shi, Q.; Su, Y.; Chu, Q.; Yuan, X.; Bao, Z.; Lu, J.; et al. Tryptophan Metabolism in Health and Disease. Cell Metab. 2023, 35, 1304–1326. [Google Scholar] [CrossRef]
- Song, P.; Ramprasath, T.; Wang, H.; Zou, M.-H. Abnormal Kynurenine Pathway of Tryptophan Catabolism in Cardiovascular Diseases. Cell Mol. Life Sci. 2017, 74, 2899–2916. [Google Scholar] [CrossRef]
- Gáspár, R.; Halmi, D.; Demján, V.; Berkecz, R.; Pipicz, M.; Csont, T. Kynurenine Pathway Metabolites as Potential Clinical Biomarkers in Coronary Artery Disease. Front. Immunol. 2021, 12, 768560. [Google Scholar] [CrossRef]
- Ala, M.; Eftekhar, S.P. The Footprint of Kynurenine Pathway in Cardiovascular Diseases. Int. J. Tryptophan Res. 2022, 15, 11786469221096643. [Google Scholar] [CrossRef]
- Kozhevnikova, M.V.; Krivova, A.V.; Korobkova, E.O.; Ageev, A.A.; Shestakova, K.M.; Moskaleva, N.E.; Appolonova, S.A.; Privalova, E.V.; Belenkov, Y.N. Comparative analysis of tryptophan and downstream metabolites of the kynurenine and serotonin pathways in patients with arterial hypertension and coronary artery disease. Kardiologiia 2022, 62, 40–48. [Google Scholar] [CrossRef]
- Sulo, G.; Vollset, S.E.; Nygård, O.; Midttun, Ø.; Ueland, P.M.; Eussen, S.J.P.M.; Pedersen, E.R.; Tell, G.S. Neopterin and Kynurenine-Tryptophan Ratio as Predictors of Coronary Events in Older Adults, the Hordaland Health Study. Int. J. Cardiol. 2013, 168, 1435–1440. [Google Scholar] [CrossRef]
- Chao de la Barca, J.M.; Bakhta, O.; Kalakech, H.; Simard, G.; Tamareille, S.; Catros, V.; Callebert, J.; Gadras, C.; Tessier, L.; Reynier, P.; et al. Metabolic Signature of Remote Ischemic Preconditioning Involving a Cocktail of Amino Acids and Biogenic Amines. J. Am. Heart Assoc. 2016, 5, e003891. [Google Scholar] [CrossRef]
- Bakhta, O.; Pascaud, A.; Dieu, X.; Beaumont, J.; Kouassi Nzoughet, J.; Kamel, R.; Croyal, M.; Tamareille, S.; Simard, G.; Chao de la Barca, J.M.; et al. Tryptophane-Kynurenine Pathway in the Remote Ischemic Conditioning Mechanism. Basic Res. Cardiol. 2020, 115, 13. [Google Scholar] [CrossRef]
- Kouassi Nzoughet, J.; Bocca, C.; Simard, G.; Prunier-Mirebeau, D.; Chao de la Barca, J.M.; Bonneau, D.; Procaccio, V.; Prunier, F.; Lenaers, G.; Reynier, P. A Nontargeted UHPLC-HRMS Metabolomics Pipeline for Metabolite Identification: Application to Cardiac Remote Ischemic Preconditioning. Anal. Chem. 2017, 89, 2138–2146. [Google Scholar] [CrossRef]
- Wirthgen, E.; Hoeflich, A.; Rebl, A.; Günther, J. Kynurenic Acid: The Janus-Faced Role of an Immunomodulatory Tryptophan Metabolite and Its Link to Pathological Conditions. Front. Immunol. 2017, 8, 1957. [Google Scholar] [CrossRef]
- Lugo-Huitrón, R.; Blanco-Ayala, T.; Ugalde-Muñiz, P.; Carrillo-Mora, P.; Pedraza-Chaverrí, J.; Silva-Adaya, D.; Maldonado, P.D.; Torres, I.; Pinzón, E.; Ortiz-Islas, E.; et al. On the Antioxidant Properties of Kynurenic Acid: Free Radical Scavenging Activity and Inhibition of Oxidative Stress. Neurotoxicol. Teratol. 2011, 33, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Olenchock, B.A.; Moslehi, J.; Baik, A.H.; Davidson, S.M.; Williams, J.; Gibson, W.J.; Chakraborty, A.A.; Pierce, K.A.; Miller, C.M.; Hanse, E.A.; et al. EGLN1 Inhibition and Rerouting of α-Ketoglutarate Suffice for Remote Ischemic Protection. Cell 2016, 164, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Bigelman, E.; Pasmanik-Chor, M.; Dassa, B.; Itkin, M.; Malitsky, S.; Dorot, O.; Pichinuk, E.; Kleinberg, Y.; Keren, G.; Entin-Meer, M. Kynurenic Acid, a Key L-Tryptophan-Derived Metabolite, Protects the Heart from an Ischemic Damage. PLoS ONE 2023, 18, e0275550. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Liu, D.; Zhao, L.; Zhou, D.; Rong, J.; Zhang, L.; Xia, Z. Myocardial Ischemia/Reperfusion Injury: Mechanisms of Injury and Implications for Management (Review). Exp. Ther. Med. 2022, 23, 430. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. Myocardial Ischemia-Reperfusion Injury: A Neglected Therapeutic Target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Andiné, P.; Lehmann, A.; Ellrén, K.; Wennberg, E.; Kjellmer, I.; Nielsen, T.; Hagberg, H. The Excitatory Amino Acid Antagonist Kynurenic Acid Administered after Hypoxic-Ischemia in Neonatal Rats Offers Neuroprotection. Neurosci. Lett. 1988, 90, 208–212. [Google Scholar] [CrossRef]
- Germano, I.M.; Pitts, L.H.; Meldrum, B.S.; Bartkowski, H.M.; Simon, R.P. Kynurenate Inhibition of Cell Excitation Decreases Stroke Size and Deficits. Ann. Neurol. 1987, 22, 730–734. [Google Scholar] [CrossRef]
- Kaelin, W.G.; Ratcliffe, P.J. Oxygen Sensing by Metazoans: The Central Role of the HIF Hydroxylase Pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Tandler, B.; Ye, J.; Slabe, T.J.; Turkaly, J.; Hoppel, C.L. Myocardial Ischemia Decreases Oxidative Phosphorylation through Cytochrome Oxidase in Subsarcolemmal Mitochondria. Am. J. Physiol. 1997, 273, H1544–H1554. [Google Scholar] [CrossRef] [PubMed]
- Rouslin, W. Mitochondrial Complexes I, II, III, IV, and V in Myocardial Ischemia and Autolysis. Am. J. Physiol. 1983, 244, H743–H748. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Gudz, T.I.; Migita, C.T.; Ikeda-Saito, M.; Hassan, M.O.; Turkaly, P.J.; Hoppel, C.L. Ischemic Injury to Mitochondrial Electron Transport in the Aging Heart: Damage to the Iron-Sulfur Protein Subunit of Electron Transport Complex III. Arch. Biochem. Biophys. 2001, 385, 117–128. [Google Scholar] [CrossRef]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of Reactive Oxygen Species by Mitochondria: Central Role of Complex III. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef]
- Ambrosio, G.; Zweier, J.L.; Duilio, C.; Kuppusamy, P.; Santoro, G.; Elia, P.P.; Tritto, I.; Cirillo, P.; Condorelli, M.; Chiariello, M. Evidence That Mitochondrial Respiration Is a Source of Potentially Toxic Oxygen Free Radicals in Intact Rabbit Hearts Subjected to Ischemia and Reflow. J. Biol. Chem. 1993, 268, 18532–18541. [Google Scholar] [CrossRef]
- Chen, Q.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Reversible Blockade of Electron Transport during Ischemia Protects Mitochondria and Decreases Myocardial Injury Following Reperfusion. J. Pharmacol. Exp. Ther. 2006, 319, 1405–1412. [Google Scholar] [CrossRef]
- Chen, Q.; Younus, M.; Thompson, J.; Hu, Y.; Hollander, J.M.; Lesnefsky, E.J. Intermediary Metabolism and Fatty Acid Oxidation: Novel Targets of Electron Transport Chain-Driven Injury during Ischemia and Reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H787–H795. [Google Scholar] [CrossRef]
- Baran, H.; Staniek, K.; Kepplinger, B.; Gille, L.; Stolze, K.; Nohl, H. Kynurenic Acid Influences the Respiratory Parameters of Rat Heart Mitochondria. Pharmacology 2001, 62, 119–123. [Google Scholar] [CrossRef]
- Baran, H.; Staniek, K.; Bertignol-Spörr, M.; Attam, M.; Kronsteiner, C.; Kepplinger, B. Effects of Various Kynurenine Metabolites on Respiratory Parameters of Rat Brain, Liver and Heart Mitochondria. Int. J. Tryptophan Res. 2016, 9, 17–29. [Google Scholar] [CrossRef]
- Baran, H.; Staniek, K.; Kepplinger, B.; Stur, J.; Draxler, M.; Nohl, H. Kynurenines and the Respiratory Parameters on Rat Heart Mitochondria. Life Sci. 2003, 72, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A Cold-Inducible Coactivator of Nuclear Receptors Linked to Adaptive Thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an Energy Sensing Network That Controls Energy Expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a Nodal Regulator of Mitochondrial Biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-Y.; Hung, M.-C. A New Fork for Clinical Application: Targeting Forkhead Transcription Factors in Cancer. Clin. Cancer Res. 2009, 15, 752–757. [Google Scholar] [CrossRef]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 Blocks the Cardiac Hypertrophic Response by Augmenting Foxo3a-Dependent Antioxidant Defense Mechanisms in Mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef]
- Ronnebaum, S.M.; Patterson, C. The FoxO Family in Cardiac Function and Dysfunction. Annu. Rev. Physiol. 2010, 72, 81–94. [Google Scholar] [CrossRef]
- Wang, X.; Hu, S.; Liu, L. Phosphorylation and Acetylation Modifications of FOXO3a: Independently or Synergistically? Oncol. Lett. 2017, 13, 2867–2872. [Google Scholar] [CrossRef]
- Wang, X.-X.; Wang, X.-L.; Tong, M.; Gan, L.; Chen, H.; Wu, S.; Chen, J.-X.; Li, R.-L.; Wu, Y.; Zhang, H.; et al. SIRT6 Protects Cardiomyocytes against Ischemia/Reperfusion Injury by Augmenting FoxO3α-Dependent Antioxidant Defense Mechanisms. Basic Res. Cardiol. 2016, 111, 13. [Google Scholar] [CrossRef]
- Yu, W.; Gao, B.; Li, N.; Wang, J.; Qiu, C.; Zhang, G.; Liu, M.; Zhang, R.; Li, C.; Ji, G.; et al. Sirt3 Deficiency Exacerbates Diabetic Cardiac Dysfunction: Role of Foxo3A-Parkin-Mediated Mitophagy. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1973–1983. [Google Scholar] [CrossRef]
- Mei, Y.; Zhang, Y.; Yamamoto, K.; Xie, W.; Mak, T.W.; You, H. FOXO3a-Dependent Regulation of Pink1 (Park6) Mediates Survival Signaling in Response to Cytokine Deprivation. Proc. Natl. Acad. Sci. USA 2009, 106, 5153–5158. [Google Scholar] [CrossRef] [PubMed]
- Moyzis, A.G.; Sadoshima, J.; Gustafsson, Å.B. Mending a Broken Heart: The Role of Mitophagy in Cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H183–H192. [Google Scholar] [CrossRef] [PubMed]
- Kubli, D.A.; Zhang, X.; Lee, Y.; Hanna, R.A.; Quinsay, M.N.; Nguyen, C.K.; Jimenez, R.; Petrosyan, S.; Murphy, A.N.; Gustafsson, A.B. Parkin Protein Deficiency Exacerbates Cardiac Injury and Reduces Survival Following Myocardial Infarction. J. Biol. Chem. 2013, 288, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Andres, A.M.; Ratliff, E.P.; Hernandez, G.; Lee, P.; Gottlieb, R.A. Preconditioning Involves Selective Mitophagy Mediated by Parkin and P62/SQSTM1. PLoS ONE 2011, 6, e20975. [Google Scholar] [CrossRef] [PubMed]
- Ronkainen, V.-P.; Tuomainen, T.; Huusko, J.; Laidinen, S.; Malinen, M.; Palvimo, J.J.; Ylä-Herttuala, S.; Vuolteenaho, O.; Tavi, P. Hypoxia-Inducible Factor 1-Induced G Protein-Coupled Receptor 35 Expression Is an Early Marker of Progressive Cardiac Remodelling. Cardiovasc. Res. 2014, 101, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, A.E.; Lappin, J.E.; Taylor, D.L.; Nicklin, S.A.; Milligan, G. GPR35 as a Novel Therapeutic Target. Front. Endocrinol. 2011, 2, 68. [Google Scholar] [CrossRef]
- Wyant, G.A.; Yu, W.; Doulamis, I.P.; Nomoto, R.S.; Saeed, M.Y.; Duignan, T.; McCully, J.D.; Kaelin, W.G. Mitochondrial Remodeling and Ischemic Protection by G Protein-Coupled Receptor 35 Agonists. Science 2022, 377, 621–629. [Google Scholar] [CrossRef]
- Oka, S.; Ota, R.; Shima, M.; Yamashita, A.; Sugiura, T. GPR35 Is a Novel Lysophosphatidic Acid Receptor. Biochem. Biophys. Res. Commun. 2010, 395, 232–237. [Google Scholar] [CrossRef]
- Deng, H.; Hu, H.; Fang, Y. Tyrphostin Analogs Are GPR35 Agonists. FEBS Lett. 2011, 585, 1957–1962. [Google Scholar] [CrossRef]
- Zhao, P.; Sharir, H.; Kapur, A.; Cowan, A.; Geller, E.B.; Adler, M.W.; Seltzman, H.H.; Reggio, P.H.; Heynen-Genel, S.; Sauer, M.; et al. Targeting of the Orphan Receptor GPR35 by Pamoic Acid: A Potent Activator of Extracellular Signal-Regulated Kinase and β-Arrestin2 with Antinociceptive Activity. Mol. Pharmacol. 2010, 78, 560–568. [Google Scholar] [CrossRef]
- Bochaton, T.; Crola-Da-Silva, C.; Pillot, B.; Villedieu, C.; Ferreras, L.; Alam, M.R.; Thibault, H.; Strina, M.; Gharib, A.; Ovize, M.; et al. Inhibition of Myocardial Reperfusion Injury by Ischemic Postconditioning Requires Sirtuin 3-Mediated Deacetylation of Cyclophilin D. J. Mol. Cell Cardiol. 2015, 84, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Leib, S.L.; Kim, Y.S.; Ferriero, D.M.; Täuber, M.G. Neuroprotective Effect of Excitatory Amino Acid Antagonist Kynurenic Acid in Experimental Bacterial Meningitis. J. Infect. Dis. 1996, 173, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Tamareille, S.; Mateus, V.; Ghaboura, N.; Jeanneteau, J.; Croué, A.; Henrion, D.; Furber, A.; Prunier, F. RISK and SAFE Signaling Pathway Interactions in Remote Limb Ischemic Perconditioning in Combination with Local Ischemic Postconditioning. Basic Res. Cardiol. 2011, 106, 1329–1339. [Google Scholar] [CrossRef]
- Bakhta, O.; Blanchard, S.; Guihot, A.-L.; Tamareille, S.; Mirebeau-Prunier, D.; Jeannin, P.; Prunier, F. Cardioprotective Role of Colchicine Against Inflammatory Injury in a Rat Model of Acute Myocardial Infarction. J. Cardiovasc. Pharmacol. Ther. 2018, 23, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Malgat, M.; Letellier, T.; Durrieu, G.; Mazat, J.-P. Enzymatic and Polarographic Measurements of the Respiratory Chain Complexes. In Mitochondrial Diseases; Lestienne, P., Ed.; Springer: Berlin/Heidelberg, Germany, 1999; pp. 357–377. ISBN 978-3-642-64166-4. [Google Scholar]
- Rustin, P.; Chretien, D.; Bourgeron, T.; Gérard, B.; Rötig, A.; Saudubray, J.M.; Munnich, A. Biochemical and Molecular Investigations in Respiratory Chain Deficiencies. Clin. Chim. Acta 1994, 228, 35–51. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | NCBI Genbank | Forward Sequence | Reverse Sequence |
|---|---|---|---|
| cat | NM_012520.2 | 5′-ttgccaaccacctgaaagat-3′ | 5′-agggtggacgtcagtgaaat-3′ |
| gusb | NM_017015.2 | 5′-ctctggtggccttacctgat-3′ | 5′-cagactcaggtgttgtcatcg-3′ |
| hprt | NM_012583.2 | 5′-gaccggttctgtcatgtcg-3′ | 5′-acctggttcatcatcactaatcac-3′ |
| sod1 | NM_017050.1 | 5′-ggtccagcggatgaagag-3′ | 5′-ggacacattggccacacc-3′ |
| sod2 | NM_017051.2 | 5′-attgccgcctgctctaatc-3′ | 5′-gatagtaagcgtgctcccaca-3′ |
| sod3 | NM_012880.1 | 5′-cttgggagagcttgtcaggt-3′ | 5′-caccagtagcaggttgcaga-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamel, R.; Baetz, D.; Gueguen, N.; Lebeau, L.; Barbelivien, A.; Guihot, A.-L.; Allawa, L.; Gallet, J.; Beaumont, J.; Ovize, M.; et al. Kynurenic Acid: A Novel Player in Cardioprotection against Myocardial Ischemia/Reperfusion Injuries. Pharmaceuticals 2023, 16, 1381. https://doi.org/10.3390/ph16101381
Kamel R, Baetz D, Gueguen N, Lebeau L, Barbelivien A, Guihot A-L, Allawa L, Gallet J, Beaumont J, Ovize M, et al. Kynurenic Acid: A Novel Player in Cardioprotection against Myocardial Ischemia/Reperfusion Injuries. Pharmaceuticals. 2023; 16(10):1381. https://doi.org/10.3390/ph16101381
Chicago/Turabian StyleKamel, Rima, Delphine Baetz, Naïg Gueguen, Lucie Lebeau, Agnès Barbelivien, Anne-Laure Guihot, Louwana Allawa, Jean Gallet, Justine Beaumont, Michel Ovize, and et al. 2023. "Kynurenic Acid: A Novel Player in Cardioprotection against Myocardial Ischemia/Reperfusion Injuries" Pharmaceuticals 16, no. 10: 1381. https://doi.org/10.3390/ph16101381
APA StyleKamel, R., Baetz, D., Gueguen, N., Lebeau, L., Barbelivien, A., Guihot, A.-L., Allawa, L., Gallet, J., Beaumont, J., Ovize, M., Henrion, D., Reynier, P., Mirebeau-Prunier, D., Prunier, F., & Tamareille, S. (2023). Kynurenic Acid: A Novel Player in Cardioprotection against Myocardial Ischemia/Reperfusion Injuries. Pharmaceuticals, 16(10), 1381. https://doi.org/10.3390/ph16101381