

One-Step Pharmaceutical Preparation of PEG-Modified Exosomes Encapsulating Anti-Cancer Drugs by a High-Pressure Homogenization Technique

 , ,
, ,
Abstract
1. Introduction
2. Results
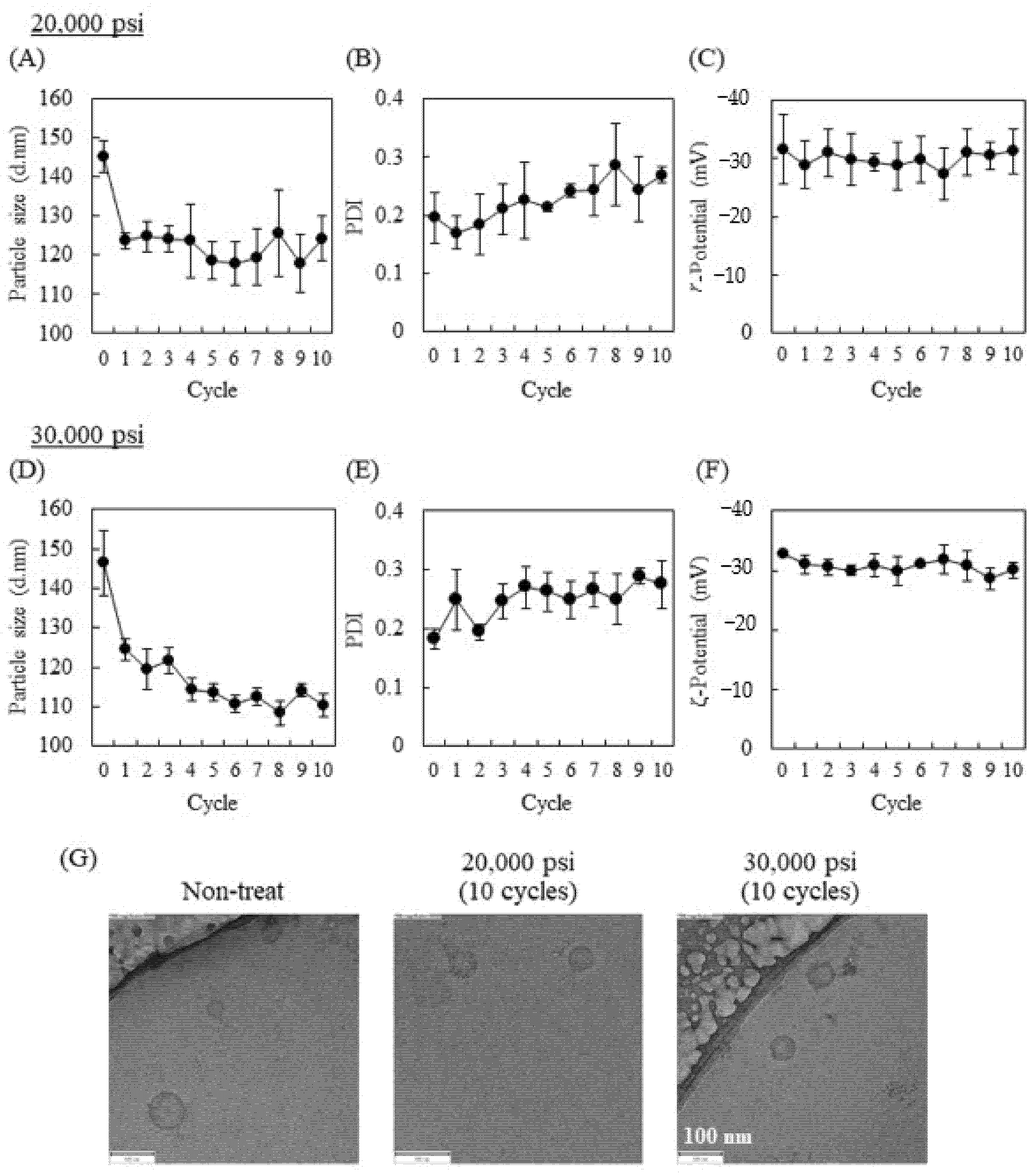
2.1. Influence of High-Pressure Homogenization Using a Microfluidizer on Physicochemical Properties of Exosomes
2.2. Effects of HPH on Expression of Representative Exosomal Marker Protein and Cellular Uptake of RAW-Exos
2.3. PEG Modification onto RAW-Exos by HPH
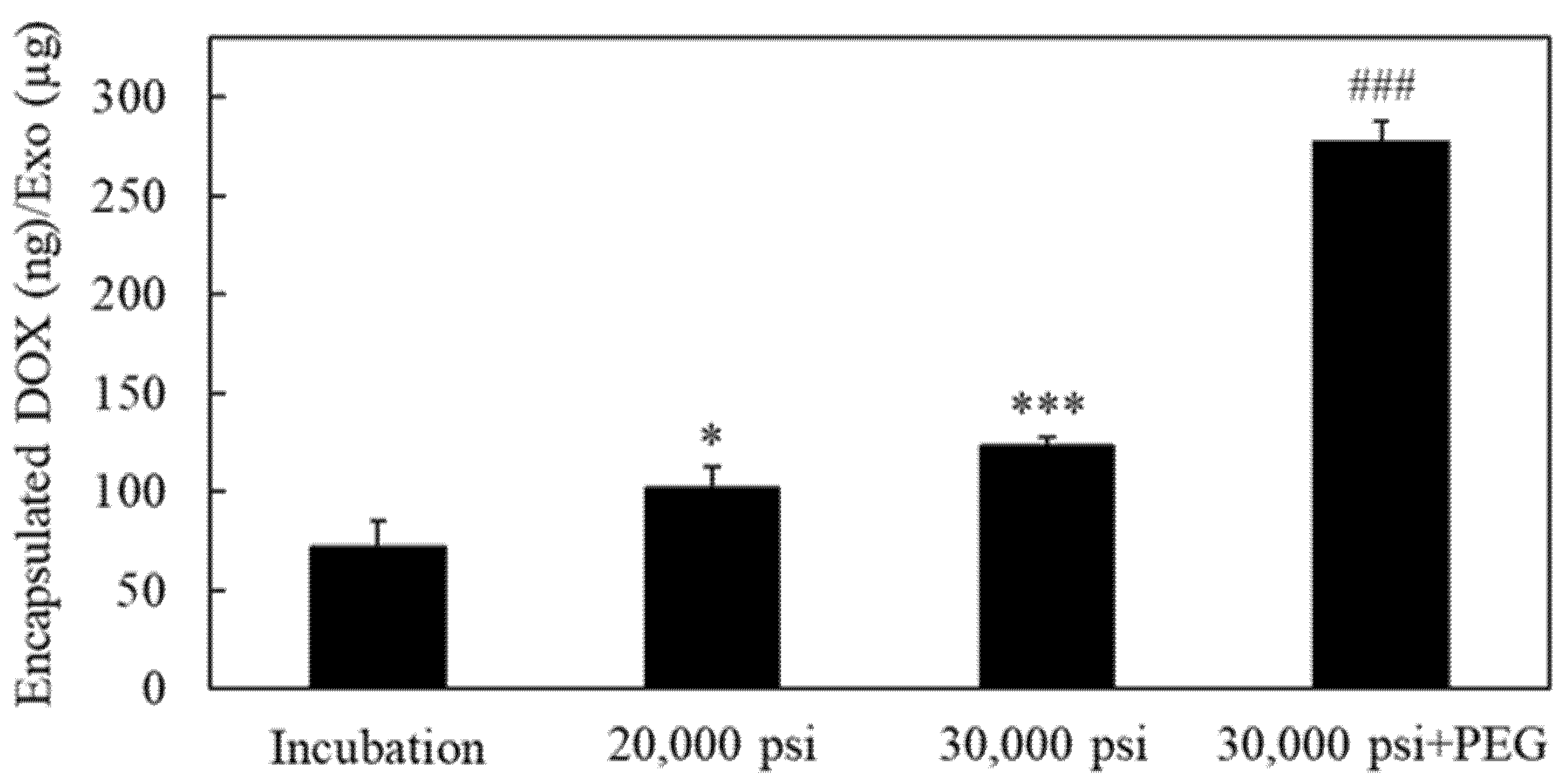
2.4. Encapsulation of the Anti-Cancer Drug Doxorubicin into RAW-Exos by HPH
2.5. Anti-Proliferative Effects of DOX-Encapsulated RAW-Exos on Colon-26 Cells
2.6. Suppression of Tumor Growth by Treatment with DOX-PEG-RAW-Exos
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Isolation of Exosomes
4.3. Measurement of Physicochemical Properties and Protein Concentration of RAW-Exos
4.4. High-Pressure Homogenization (HPH) with a Microfluidizer
4.5. Cryo-Transmission Electron Microscopy
4.6. Western Blotting
4.7. Cellular Uptake of RAW-Exos
4.8. Modification of RAW-Exos with PEG-Lipid
4.9. Measurement of Sample Temperature after HPH
4.10. Doxorubicin Encapsulation into RAW-Exos by HPH
4.11. In Vitro Cytotoxicity Assay
4.12. In Vivo Anti-Cancer Experiments
4.13. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Skotland, T.; Sandvig, K.; Llorente, A. Lipids in exosomes: Current knowledge and the way forward. Prog. Lipid Res. 2017, 66, 30–41. [Google Scholar] [CrossRef]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Rayamajhi, S.; Nguyen, T.D.T.; Marasini, R.; Aryal, S. Macrophage-derived exosome-mimetic hybrid vesicles for tumor targeted drug delivery. Acta Biomater. 2019, 94, 482–494. [Google Scholar] [CrossRef]
- Tominaga, N.; Kosaka, N.; Ono, M.; Katsuda, T.; Yoshioka, Y.; Tamura, K.; Lötvall, J.; Nakagama, H.; Ochiya, T. Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood–brain barrier. Nat. Commun. 2015, 6, 6716. [Google Scholar] [CrossRef]
- Wang, J.; Tang, W.; Yang, M.; Yin, Y.; Li, H.; Hu, F.; Tang, L.; Ma, X.; Zhang, Y.; Wang, Y. Inflammatory tumor microenvironment responsive neutrophil exosomes-based drug delivery system for targeted glioma therapy. Biomaterials 2021, 273, 120784. [Google Scholar] [CrossRef]
- Rankin-Turner, S.; Vader, P.; O’Driscoll, L.; Giebel, B.; Heaney, L.M.; Davies, O.G. A call for the standardised reporting of factors affecting the exogenous loading of extracellular vesicles with therapeutic cargos. Adv. Drug Deliv. Rev. 2021, 173, 479–491. [Google Scholar] [CrossRef]
- Fu, S.; Wang, Y.; Xia, X.; Zheng, J.C. Exosome engineering: Current progress in cargo loading and targeted delivery. NanoImpact 2020, 20, 100261. [Google Scholar] [CrossRef]
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H.-G. A novel nanoparticle drug delivery system: The anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol. Ther. 2010, 18, 1606–1614. [Google Scholar] [CrossRef]
- Munagala, R.; Aqil, F.; Jeyabalan, J.; Gupta, R.C. Bovine milk-derived exosomes for drug delivery. Cancer Lett. 2016, 371, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Goh, W.J.; Lee, C.K.; Zou, S.; Woon, E.C.; Czarny, B.; Pastorin, G. Doxorubicin-loaded cell-derived nanovesicles: An alternative targeted approach for anti-tumor therapy. Int. J. Nanomed. 2017, 12, 2759. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Mahajan, V.; Deygen, I.; Klyachko, N.L.; Inskoe, E.; Piroyan, A.; Sokolsky, M.; Okolie, O. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomedicine 2016, 12, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef]
- Pascucci, L.; Coccè, V.; Bonomi, A.; Ami, D.; Ceccarelli, P.; Ciusani, E.; Viganò, L.; Locatelli, A.; Sisto, F.; Doglia, S.M. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: A new approach for drug delivery. J. Control. Release 2014, 192, 262–270. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Bai, M.; Wang, J.; Zhu, K.; Liu, R.; Ge, S.; Li, J.; Ning, T.; Deng, T. Exosomes serve as nanoparticles to suppress tumor growth and angiogenesis in gastric cancer by delivering hepatocyte growth factor siRNA. Cancer Sci. 2018, 109, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Zhao, Y.; Banks, W.A.; Bullock, K.M.; Haney, M.; Batrakova, E.; Kabanov, A.V. Macrophage exosomes as natural nanocarriers for protein delivery to inflamed brain. Biomaterials 2017, 142, 1–12. [Google Scholar] [CrossRef]
- Claridge, B.; Lozano, J.; Poh, Q.H.; Greening, D.W. Development of Extracellular Vesicle Therapeutics: Challenges, Considerations, and Opportunities. Front. Cell Dev. Biol. 2021, 9, 734720. [Google Scholar] [CrossRef]
- Romano, E.; Netti, P.A.; Torino, E. A high throughput approach based on dynamic high pressure for the encapsulation of active compounds in exosomes for precision medicine. Int. J. Mol. Sci. 2021, 22, 9896. [Google Scholar] [CrossRef]
- Khairnar, S.V.; Pagare, P.; Thakre, A.; Nambiar, A.R.; Junnuthula, V.; Abraham, M.C.; Kolimi, P.; Nyavanandi, D.; Dyawanapelly, S. Review on the Scale-Up Methods for the Preparation of Solid Lipid Nanoparticles. Pharmaceutics 2022, 14, 1886. [Google Scholar] [CrossRef] [PubMed]
- Shegokar, R.; Singh, K.K.; Muller, R.H. Production & stability of stavudine solid lipid nanoparticles—From lab to industrial scale. Int. J. Pharm. 2011, 416, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Lajunen, T.; Hisazumi, K.; Kanazawa, T.; Okada, H.; Seta, Y.; Yliperttula, M.; Urtti, A.; Takashima, Y. Topical drug delivery to retinal pigment epithelium with microfluidizer produced small liposomes. Eur. J. Pharm. Sci. 2014, 62, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, Y.; Iwao, Y.; Noguchi, S.; Itai, S. Effect of alkyl chain length and unsaturation of the phospholipid on the physicochemical properties of lipid nanoparticles. Chem. Pharm. Bull. 2015, 63, 731–736. [Google Scholar] [CrossRef]
- Matsuo, S.; Higashi, K.; Moribe, K.; Kimura, S.I.; Itai, S.; Kondo, H.; Iwao, Y. Combination of Roll Grinding and High-Pressure Homogenization Can Prepare Stable Bicelles for Drug Delivery. Nanomaterials 2018, 8, 998. [Google Scholar] [CrossRef]
- Lai, C.P.; Mardini, O.; Ericsson, M.; Prabhakar, S.; Maguire, C.A.; Chen, J.W.; Tannous, B.A.; Breakefield, X.O. Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano 2014, 8, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, H.; Gu, J.; Zhang, J.; Shi, H.; Qian, H.; Wang, D.; Xu, W.; Pan, J.; Santos, H.A. Engineered Extracellular Vesicles for Cancer Therapy. Adv. Mater. 2021, 33, e2005709. [Google Scholar] [CrossRef]
- Matsumoto, A.; Takahashi, Y.; Nishikawa, M.; Sano, K.; Morishita, M.; Charoenviriyakul, C.; Saji, H.; Takakura, Y. Role of Phosphatidylserine-Derived Negative Surface Charges in the Recognition and Uptake of Intravenously Injected B16BL6-Derived Exosomes by Macrophages. J. Pharm. Sci. 2017, 106, 168–175. [Google Scholar] [CrossRef]
- Matsumoto, A.; Takahashi, Y.; Ogata, K.; Kitamura, S.; Nakagawa, N.; Yamamoto, A.; Ishihama, Y.; Takakura, Y. Phosphatidylserine-deficient small extracellular vesicle is a major somatic cell-derived sEV subpopulation in blood. iScience 2021, 24, 102839. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update post COVID-19 vaccines. Bioeng. Transl. Med. 2021, 6, e10246. [Google Scholar] [CrossRef]
- Fukuta, T.; Oku, N.; Kogure, K. Application and Utility of Liposomal Neuroprotective Agents and Biomimetic Nanoparticles for the Treatment of Ischemic Stroke. Pharmaceutics 2022, 14, 361. [Google Scholar] [CrossRef]
- Emam, S.E.; Abu Lila, A.S.; Elsadek, N.E.; Ando, H.; Shimizu, T.; Okuhira, K.; Ishima, Y.; Mahdy, M.A.; Ghazy, F.S.; Ishida, T. Cancer cell-type tropism is one of crucial determinants for the efficient systemic delivery of cancer cell-derived exosomes to tumor tissues. Eur. J. Pharm. Biopharm. 2019, 145, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Munye, M.M.; Tagalakis, A.D.; Manunta, M.D.; Hart, S.L. The role of the helper lipid on the DNA transfection efficiency of lipopolyplex formulations. Sci. Rep. 2014, 4, 7107. [Google Scholar] [CrossRef]
- Kuntsche, J.; Horst, J.C.; Bunjes, H. Cryogenic transmission electron microscopy (cryo-TEM) for studying the morphology of colloidal drug delivery systems. Int. J. Pharm. 2011, 417, 120–137. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, H.; Park, K.; Shin, S. Rapid and efficient isolation of exosomes by clustering and scattering. J. Clin. Med. 2020, 9, 650. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, H.; Akita, H.; Kogure, K.; Oishi, M.; Nagasaki, Y.; Kihira, Y.; Ueno, M.; Kobayashi, H.; Kikuchi, H.; Harashima, H. Development of a novel systemic gene delivery system for cancer therapy with a tumor-specific cleavable PEG-lipid. Gene Ther. 2007, 14, 68–77. [Google Scholar] [CrossRef]
- Gong, C.; Tian, J.; Wang, Z.; Gao, Y.; Wu, X.; Ding, X.; Qiang, L.; Li, G.; Han, Z.; Yuan, Y.; et al. Functional exosome-mediated co-delivery of doxorubicin and hydrophobically modified microRNA 159 for triple-negative breast cancer therapy. J. Nanobiotechnol. 2019, 17, 93. [Google Scholar] [CrossRef]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef]
- Maritim, S.; Boulas, P.; Lin, Y. Comprehensive analysis of liposome formulation parameters and their influence on encapsulation, stability and drug release in glibenclamide liposomes. Int. J. Pharm. 2021, 592, 120051. [Google Scholar] [CrossRef] [PubMed]
- Hald Albertsen, C.; Kulkarni, J.A.; Witzigmann, D.; Lind, M.; Petersson, K.; Simonsen, J.B. The role of lipid components in lipid nanoparticles for vaccines and gene therapy. Adv. Drug Deliv. Rev. 2022, 188, 114416. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, R.; Lu, X.; Lu, W.; Zhang, C.; Liang, W. Pegylated phospholipids-based self-assembly with water-soluble drugs. Pharm. Res. 2010, 27, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Ogawara, K.; Un, K.; Tanaka, K.; Higaki, K.; Kimura, T. In vivo anti-tumor effect of PEG liposomal doxorubicin (DOX) in DOX-resistant tumor-bearing mice: Involvement of cytotoxic effect on vascular endothelial cells. J. Control. Release 2009, 133, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, R.; Ishima, Y.; Ikeda, M.; Kragh-Hansen, U.; Fang, J.; Nakamura, H.; Chuang, V.T.G.; Tanaka, R.; Maeda, H.; Kodama, A.; et al. S-Nitrosated human serum albumin dimer as novel nano-EPR enhancer applied to macromolecular anti-tumor drugs such as micelles and liposomes. J. Control. Release 2015, 217, 1–9. [Google Scholar] [CrossRef]
- Ishii, T.; Asai, T.; Oyama, D.; Fukuta, T.; Yasuda, N.; Shimizu, K.; Minamino, T.; Oku, N. Amelioration of cerebral ischemia–reperfusion injury based on liposomal drug delivery system with asialo-erythropoietin. J. Control. Release 2012, 160, 81–87. [Google Scholar] [CrossRef]
- Shimizu, K.; Takeuchi, Y.; Otsuka, K.; Mori, T.; Narita, Y.; Takasugi, S.; Magata, Y.; Matsumura, Y.; Oku, N. Development of tissue factor-targeted liposomes for effective drug delivery to stroma-rich tumors. J. Control. Release 2020, 323, 519–529. [Google Scholar] [CrossRef]
- Skotland, T.; Hessvik, N.P.; Sandvig, K.; Llorente, A. Exosomal lipid composition and the role of ether lipids and phosphoinositides in exosome biology. J. Lipid Res. 2019, 60, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Simbari, F.; McCaskill, J.; Coakley, G.; Millar, M.; Maizels, R.M.; Fabrias, G.; Casas, J.; Buck, A.H. Plasmalogen enrichment in exosomes secreted by a nematode parasite versus those derived from its mouse host: Implications for exosome stability and biology. J. Extracell. Vesicles 2016, 5, 30741. [Google Scholar] [CrossRef]
- Suga, K.; Matsui, D.; Watanabe, N.; Okamoto, Y.; Umakoshi, H. Insight into the Exosomal Membrane: From Viewpoints of Membrane Fluidity and Polarity. Langmuir 2021, 37, 11195–11202. [Google Scholar] [CrossRef]
- McElhaney, R.N.; De Gier, J.; Van der Neut-Kok, E. The effect of alterations in fatty acid composition and cholesterol content on the nonelectrolyte permeability of Acholeplasma laidlawii B cells and derived liposomes. Biochim. Biophys. Acta 1973, 298, 500–512. [Google Scholar] [CrossRef]
- Moribe, K.; Maruyama, K.; Iwatsuru, M. Encapsulation characteristics of nystatin in liposomes: Effects of cholesterol and polyethylene glycol derivatives. Int. J. Pharm. 1999, 188, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Fukuta, T.; Nishikawa, A.; Kogure, K. Low level electricity increases the secretion of extracellular vesicles from cultured cells. Biochem. Biophys. Rep. 2020, 21, 100713. [Google Scholar] [CrossRef] [PubMed]
- Fukuta, T.; Asai, T.; Kiyokawa, Y.; Nakada, T.; Bessyo-Hirashima, K.; Fukaya, N.; Hyodo, K.; Takase, K.; Kikuchi, H.; Oku, N. Targeted delivery of anticancer drugs to tumor vessels by use of liposomes modified with a peptide identified by phage biopanning with human endothelial progenitor cells. Int. J. Pharm. 2017, 524, 364–372. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Particle Size (d.nm) | Polydispersity Index (PDI) | ζ-Potential (mV) |
|---|---|---|---|
| RAW-Exos | 150.1 ± 4.8 | 0.25 ± 0.03 | −30.0 ± 2.8 |
| PEG-RAW-Exos (Incubation) | 125.6 ± 12.2 | 0.20 ± 0.05 | −29.4 ± 5.9 |
| PEG-RAW-Exos (HPH) | 113.2 ± 27.6 | 0.29 ± 0.04 | −17.4 ± 1.6 |
| Process Pressure (psi) | Before (°C) | After (°C) | Degree of Temperature Change (°C) |
|---|---|---|---|
| 20,000 | 9.6 ± 1.5 | 33.4 ± 0.4 | 24.4 ± 1.8 |
| 30,000 | 12.0 ± 2.2 | 44.7 ± 0.9 | 32.7 ± 2.8 *** |
| Sample | Particle Size (d.nm) | Polydispersity Index (PDI) | ζ-Potential (mV) |
|---|---|---|---|
| DOX-RAW-Exos (20,000 psi) | 1222.7 ± 187.0 | 0.38 ± 0.15 | −19.8 ± 7.2 |
| DOX-RAW-Exos (30,000 psi) | 887.2 ± 445.6 | 0.49 ± 0.16 | −21.3 ± 3.8 |
| DOX-PEG-RAW-Exos | 110.8 ± 12.4 | 0.35 ± 0.04 | −27.5 ± 1.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukuta, T.; Ikeda-Imafuku, M.; Kodama, S.; Kuse, J.; Matsui, K.; Iwao, Y. One-Step Pharmaceutical Preparation of PEG-Modified Exosomes Encapsulating Anti-Cancer Drugs by a High-Pressure Homogenization Technique. Pharmaceuticals 2023, 16, 108. https://doi.org/10.3390/ph16010108
Fukuta T, Ikeda-Imafuku M, Kodama S, Kuse J, Matsui K, Iwao Y. One-Step Pharmaceutical Preparation of PEG-Modified Exosomes Encapsulating Anti-Cancer Drugs by a High-Pressure Homogenization Technique. Pharmaceuticals. 2023; 16(1):108. https://doi.org/10.3390/ph16010108
Chicago/Turabian StyleFukuta, Tatsuya, Mayumi Ikeda-Imafuku, Satoshi Kodama, Junko Kuse, Ko Matsui, and Yasunori Iwao. 2023. "One-Step Pharmaceutical Preparation of PEG-Modified Exosomes Encapsulating Anti-Cancer Drugs by a High-Pressure Homogenization Technique" Pharmaceuticals 16, no. 1: 108. https://doi.org/10.3390/ph16010108
APA StyleFukuta, T., Ikeda-Imafuku, M., Kodama, S., Kuse, J., Matsui, K., & Iwao, Y. (2023). One-Step Pharmaceutical Preparation of PEG-Modified Exosomes Encapsulating Anti-Cancer Drugs by a High-Pressure Homogenization Technique. Pharmaceuticals, 16(1), 108. https://doi.org/10.3390/ph16010108