
Antimicrobial Activity of Rhenium Di- and Tricarbonyl Diimine Complexes: Insights on Membrane-Bound S. aureus Protein Binding
 ,
,  , ,
, , 

Abstract
1. Introduction
2. Results and Discussion
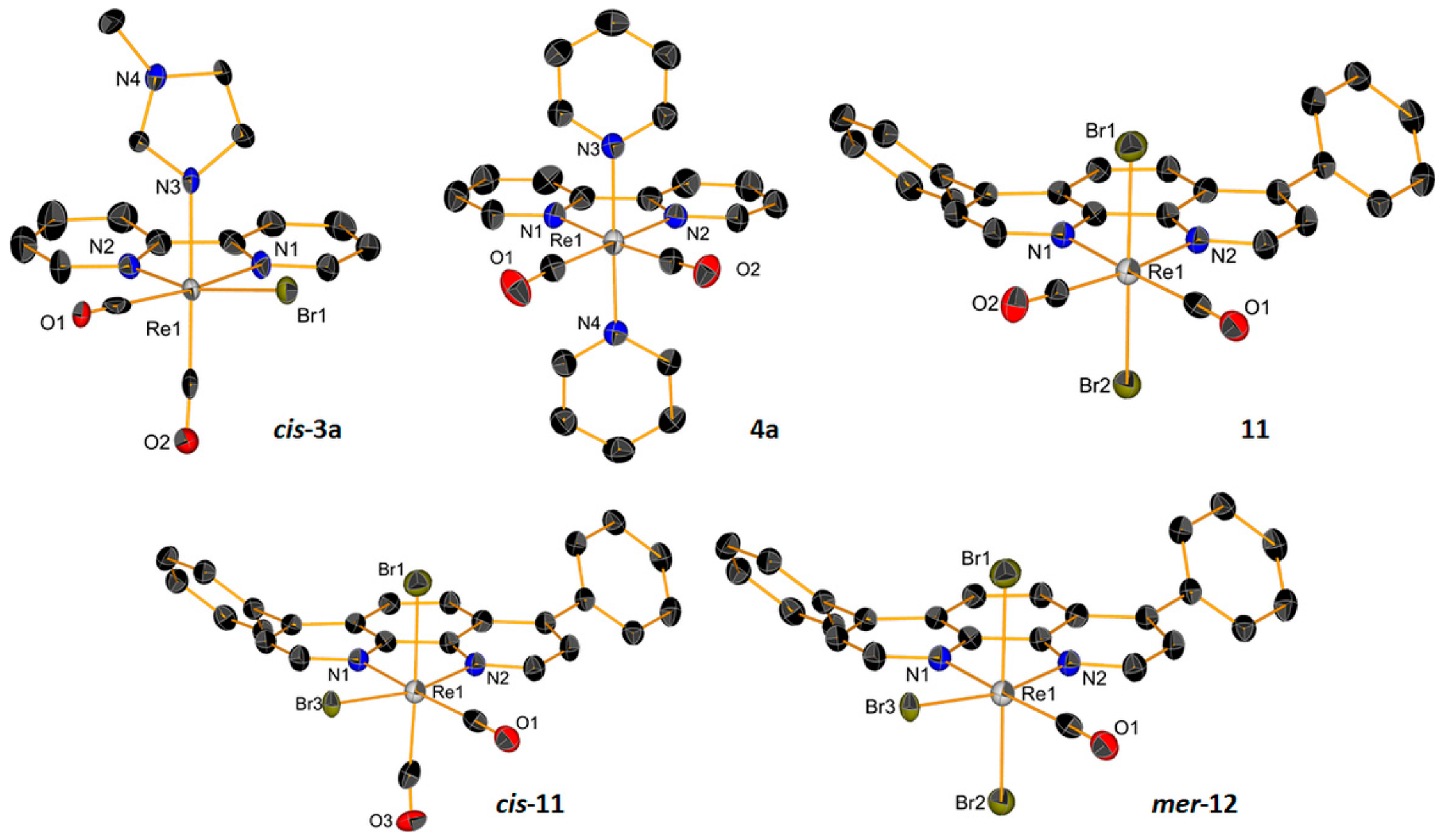
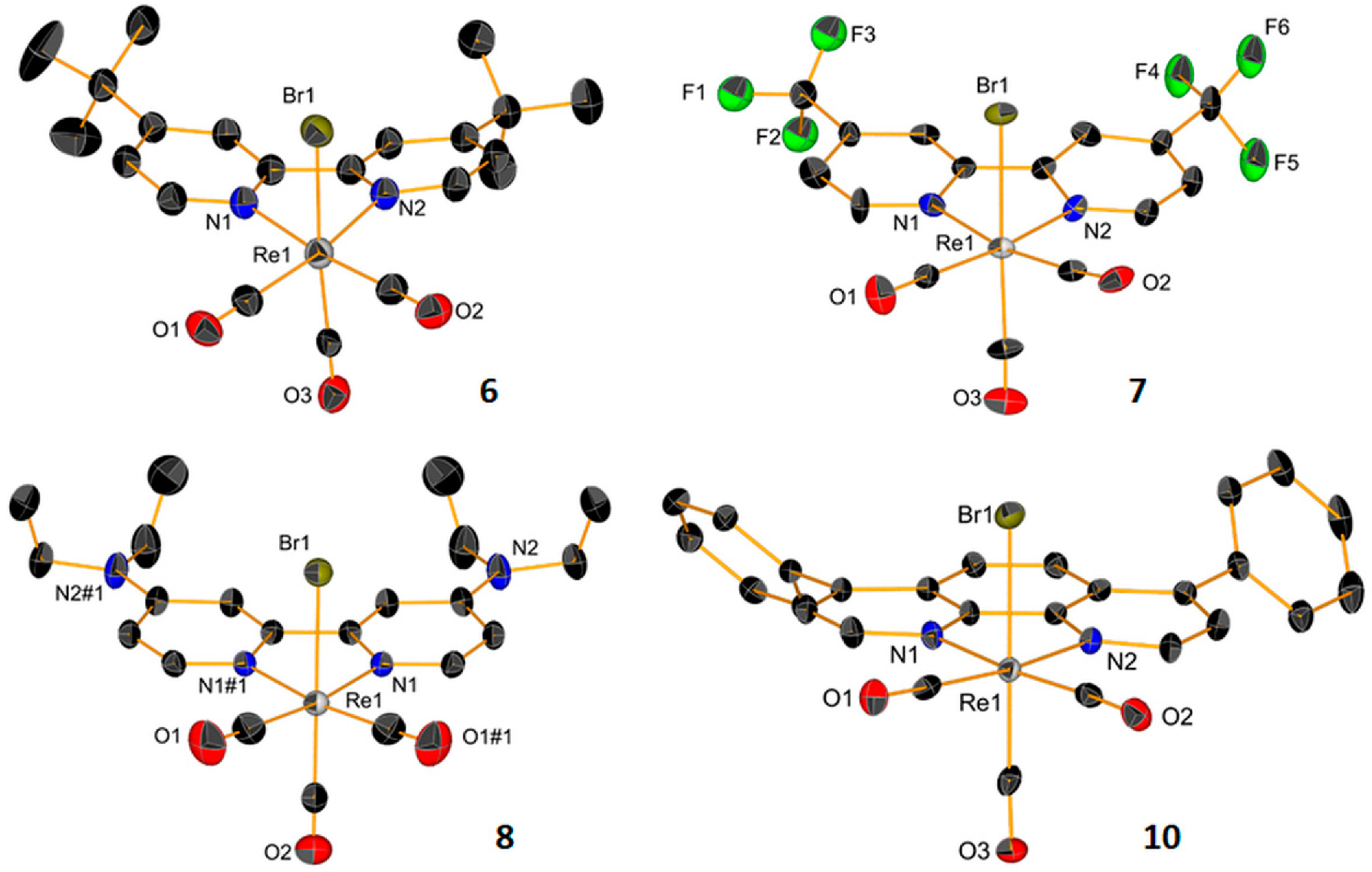
2.1. Synthesis and Characterization of the Metal Complexes
2.2. Antimicrobial Properties of the Complexes
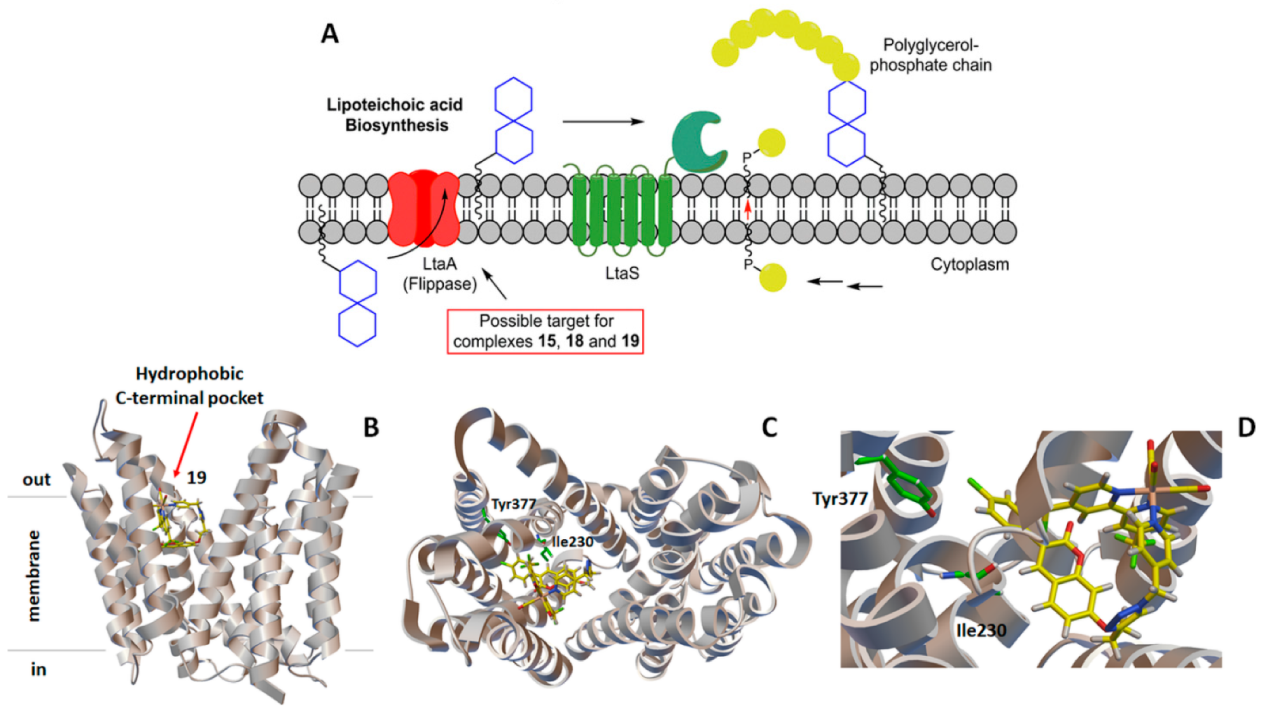
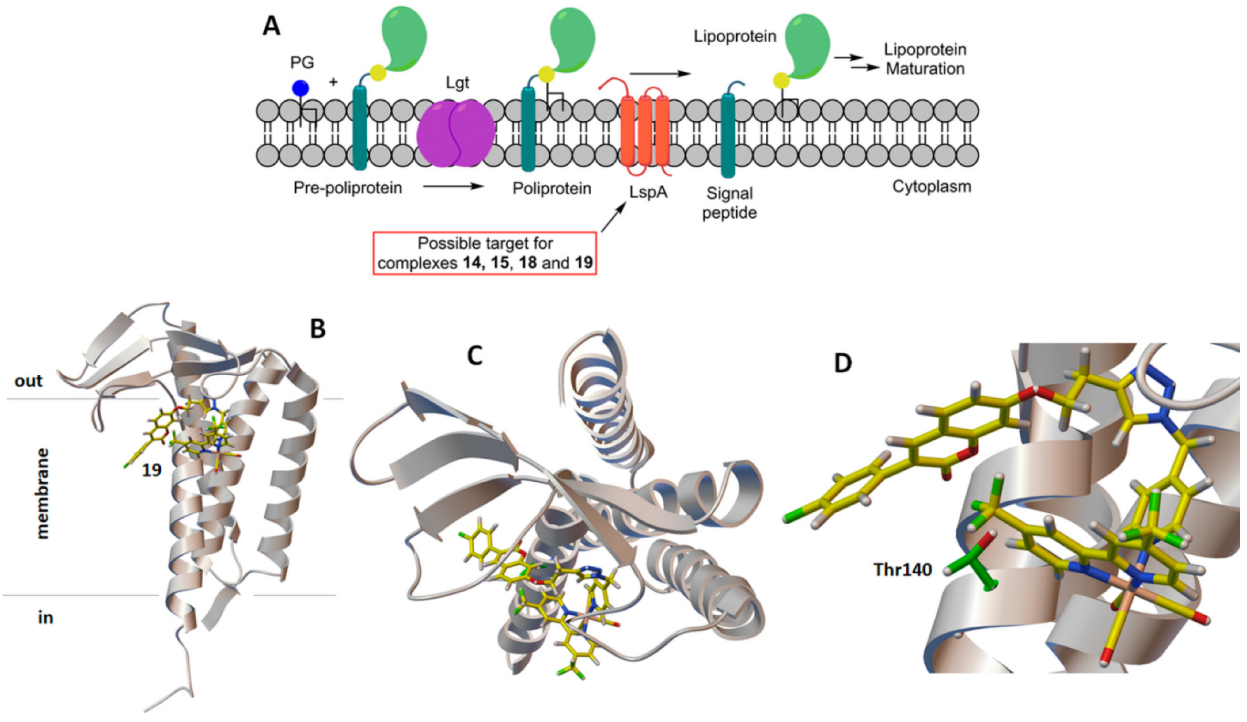
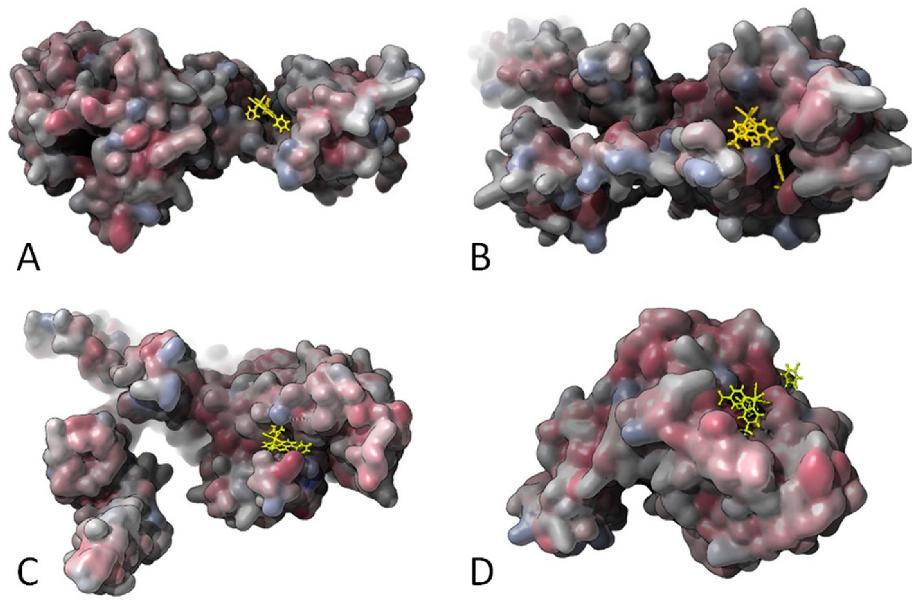
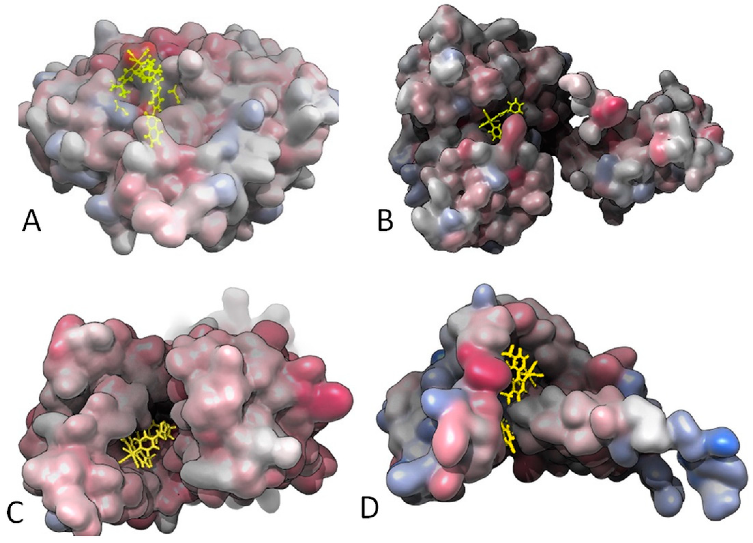
2.3. Molecular Docking Study: Membrane-Bound S. aureus Proteins
- (1)
- With the exception of the cis-[Re(CO)2]n complexes 1b–3b and the fac-[Re(CO)3]+ complexes 6, 7, and 10, none of the inactive rhenium di- or tricarbonyl compounds showed any b.a. for the enzyme evaluated.
- (2)
- The inactive molecules 1b–3b, 6, 7, and 10 showed an affinity for the penicillin-binding protein 4 (PBP4), with b.a. values ranging from −8.9 (1b) to −12.3 (10) kcal/mol.
- (3)
- Compound 10 also showed a strong affinity for lipoteichoic acid flippase (LtaA), with a b.a. of −10.3 kcal/mol.
- (4)
- Amongst the active antimicrobial rhenium complexes (i.e., molecules 13–19, Figure 4), complexes 16 and 17 showed the lowest b.a. values for the selected enzymes. These were higher than those of the inactive compounds but lower than those of the known inhibitors.
- (5)
- With variations within the series, the other active antimicrobial rhenium complexes (13–15 and 18–19) showed good b.a. values for five enzymes. These were the penicillin-binding protein 4 (PBP4, b.a. ranging from −9.1 (13) to −10.7 (19) kcal/mol); type-I signal peptidase (SpsB, all complexes except 14, b.a. ranging from −9.1 (13) to −10.4 (19) kcal/mol); lipoteichoic acid synthase (LtaS, only 15, 18, and 19, b.a. ranging from −9.4 (15) to −10.9 (19) kcal/mol); lipoteichoic acid flippase (LtaA, only 15, 18, and 19, b.a. ranging from −10.4 (15) to −11.3 (19) kcal/mol).; and lipoprotein signal peptidase II (LspA, all complexes except 13, b.a. ranging from −8.7 (15) to −10.6 (18) kcal/mol).
3. Conclusions
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. Instruments and Analysis
4.3. Synthetic Procedures
4.4. Biological Tests
4.5. In Silico Calculations
4.5.1. Preparation of the Ligand Database and Ligands: Receptors Complexes
4.5.2. Molecular Docking
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Global Shortage of Innovative Antibiotics Fuels Emergence and Spread of Drug-Resistance; WHO: Geneva, Switzerland, 2021; p. 1. [Google Scholar]
- Hu, Q.; Cheng, H.; Yuan, W.; Zeng, F.; Shang, W.; Tang, D.; Xue, W.; Fu, J.; Zhou, R.; Zhu, J.; et al. Panton-Valentine leukocidin (PVL)-positive health care-associated methicillin-resistant Staphylococcus aureus isolates are associated with skin and soft tissue infections and colonized mainly by infective PVL-encoding bacteriophages. J. Clin. Microbiol. 2015, 53, 67–72. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tong, S.Y.C.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G. Staphylococcus aureus Infections: Epidemiology, Pathophysiology, Clinical Manifestations, and Management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [PubMed]
- Laupland, K.B.; Lyytikäinen, O.; Sgaard, M.; Kennedy, K.J.; Knudsen, J.D.; Ostergaard, C.; Galbraith, J.C.; Valiquette, L.; Jacobsson, G.; Collignon, P.; et al. The changing epidemiology of Staphylococcus aureus bloodstream infection: A multinational population-based surveillance study. Clin. Microbiol. Infect. 2013, 19, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Plackett, B. Why big pharma has abandoned antibiotics. Nature 2020, 586, S50–S52. [Google Scholar] [CrossRef]
- Yu, W.; MacKerell, A.D., Jr. Computer-Aided Drug Design Methods. Methods Mol. Biol. 2017, 1520, 85–106. [Google Scholar] [CrossRef] [PubMed]
- Rao, Q.; Shang, W.; Hu, X.; Rao, X. Staphylococcus aureus ST121: A globally disseminated hypervirulent clone. J. Med. Microbiol. 2015, 64, 1462–1473. [Google Scholar] [CrossRef]
- Yu, W.; Lakkaraju, S.K.; Raman, E.P.; Fang, L.; MacKerell, A.D. Pharmacophore Modeling Using Site-Identification by Ligand Competitive Saturation (SILCS) with Multiple Probe Molecules. J. Chem. Inf. Model. 2015, 55, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Nedyalkova, M.; Simeonov, V. Partitioning Pattern of Natural Products Based on Molecular Properties Descriptors Representing Drug-Likeness. Symmetry 2021, 13, 546. [Google Scholar] [CrossRef]
- Barazorda-Ccahuana, H.L.; Nedyalkova, M.; Mas, F.; Madurga, S. Unveiling the Effect of Low pH on the SARS-CoV-2 Main Protease by Molecular Dynamics Simulations. Polymers 2021, 13, 3823. [Google Scholar] [CrossRef] [PubMed]
- Ustach, V.D.; Lakkaraju, S.K.; Jo, S.; Yu, W.; Jiang, W.; MacKerell, A.D. Optimization and Evaluation of Site-Identification by Ligand Competitive Saturation (SILCS) as a Tool for Target-Based Ligand Optimization. J. Chem. Inf. Model. 2019, 59, 3018–3035. [Google Scholar] [CrossRef] [PubMed]
- Gaieb, Z.; Parks, C.D.; Chiu, M.; Yang, H.; Shao, C.; Walters, W.P.; Lambert, M.H.; Nevins, N.; Bembenek, S.D.; Ameriks, M.K.; et al. D3R Grand Challenge 3: Blind prediction of protein–ligand poses and affinity rankings. J. Comput. Aided Mol. Des. 2019, 33, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Śledź, P.; Caflisch, A. Protein structure-based drug design: From docking to molecular dynamics. Curr. Opin. Struct. Biol. 2018, 48, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Cournia, Z.; Allen, B.; Sherman, W. Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations. J. Chem. Inf. Model. 2017, 57, 2911–2937. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.K.; Ahmed, S.F.; Hossain, M.S.; Bhojiya, A.A.; Mathur, A.; Upadhyay, S.K.; Srivastava, A.K.; Vishvakarma, N.K.; Barik, M.; Rahaman, M.M.; et al. Molecular docking and simulation studies of flavonoid compounds against PBP-2a of methicillin-resistant Staphylococcus aureus. J. Biomol. Struct. Dyn. 2021, 1–17. [Google Scholar] [CrossRef]
- Kulanthaivel, L.; Jeyaraman, J.; Biswas, A.; Subbaraj, G.K.; Santhoshkumar, S. Identification of potential inhibitors for Penicillinbinding protein (PBP) from Staphylococcus aureus. Bioinformation 2018, 14, 471–476. [Google Scholar] [CrossRef]
- Chomnawang, M.T.; Surassmo, S.; Wongsariya, K.; Bunyapraphatsara, N. Antibacterial Activity of Thai Medicinal Plants against Methicillin-resistant Staphylococcus aureus. Fitoterapia 2009, 80, 102–104. [Google Scholar] [CrossRef]
- Alhadrami, H.A.; Hamed, A.A.; Hassan, H.M.; Belbahri, L.; Rateb, M.E.; Sayed, A.M. Flavonoids as Potential anti-MRSA Agents through Modulation of PBP2a: A Computational and Experimental Study. Antibiotics 2020, 9, 562. [Google Scholar] [CrossRef]
- Aisha, A.; Zahra, S.; Tahir, I.M.; Hussain, A.; Bano, N.; Roobi, A.; Afsheen, N.; Saleem, Y. Anticancer L-Asparaginase and Phytoactive Compounds From Plant Solanum nigrum Against MDR (Methicillindrug resistant) Staphylococcus aureus and Fungal Isolates. Dose Response 2022, 20, 1–12. [Google Scholar] [CrossRef]
- Liang, M.; Ge, X.; Xua, H.; Ma, K.; Zhang, W.; Zan, Y.; Efferth, T.; Xue, Z.; Hua, X. Phytochemicals with activity against methicillin-resistant Staphylococcus aureus. Phytomed. Int. J. Phytother. Phytopharm. 2022, 100, 154073. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, D.M.; de Oliveira, D.B.C.; Nunes, Y.R.F.; de Almeida Alves, T.M.; Kohlhoff, M.; Andrade, A.A.; Cota, B.B. Natural Occurring Phenolic Derivatives from Mauritia flexuosa (Buriti) Stems and Their Potential Antibacterial Activity against Methicillin-Resistant Staphylococcus aureus (MRSA). Chem. Biodivers. 2022, 19, e202100788. [Google Scholar] [CrossRef] [PubMed]
- WHO. 2020 Antibacterial Agents in Clinical and Preclinical Development: An Overview and Analysis; WHO: Geneva, Switzerland, 2021; pp. 1–76. [Google Scholar]
- Frei, A. Metal Complexes, an Untapped Source of Antibiotic Potential? Antibiotics 2020, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Patra, M.; Gasser, G.; Metzler-Nolte, N. Small organometallic compounds as antibacterial agents. Dalton Trans. 2012, 41, 6350–6358. [Google Scholar] [CrossRef] [PubMed]
- Sierra, M.A.; Casarrubios, L.; de la Torre, M. Bio-Organometallic Derivatives of Antibacterial Drugs. Chem. Eur. J. 2019, 25, 7232–7242. [Google Scholar] [CrossRef] [PubMed]
- Frei, A.; Zuegg, J.; Elliott, A.G.; Baker, M.; Braese, S.; Brown, C.; Chen, F.; Dowson, C.G.; Dujardin, G.; Jung, N.; et al. Metal complexes as a promising source for new antibiotics. Chem. Sci. 2020, 11, 2627–2639. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Collins, J.G.; Keene, F.R. Ruthenium complexes as antimicrobial agents. Chem. Soc. Rev. 2015, 44, 2529–2542. [Google Scholar] [CrossRef]
- Wenzel, M.; Patra, M.; Senges, C.H.; Ott, I.; Stepanek, J.J.; Pinto, A.; Prochnow, P.; Vuong, C.; Langklotz, S.; Metzler-Nolte, N.; et al. Analysis of the mechanism of action of potent antibacterial hetero-tri-organometallic compounds: A structurally new class of antibiotics. ACS Chem. Biol. 2013, 8, 1442–1450. [Google Scholar] [CrossRef]
- Patra, M.; Wenzel, M.; Prochnow, P.; Pierroz, V.; Gasser, G.; Bandow, J.E.; Metzler-Nolte, N. An organometallic structure-activity relationship study reveals the essential role of a Re(CO)3 moiety in the activity against gram-positive pathogens including MRSA. Chem. Sci. 2015, 6, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, D.; Lorenz, N.; Gothe, Y.; Spies, C.; Geissler, B.; Prochnow, P.; Nuernberger, P.; Bandow, J.E.; Metzler-Nolte, N. Benzannulated Re(I)-NHC complexes: Synthesis, photophysical properties and antimicrobial activity. Dalton Trans. 2017, 46, 15269–15279. [Google Scholar] [CrossRef] [PubMed]
- Frei, A.; Amado, M.; Cooper, M.A.; Blaskovich, M.A.T. Light-activated Rhenium Complexes with Dual Mode of Action against Bacteria. Chem. Eur. J. 2019, 26, 2852–2858. [Google Scholar] [CrossRef]
- Zampakou, M.; Akrivou, M.; Andreadou, E.G.; Raptopoulou, C.P.; Psycharis, V.; Pantazaki, A.A.; Psomas, G. Structure, antimicrobial activity, DNA-and albumin-binding of manganese (II) complexes with the quinolone antimicrobial agents oxolinic acid and enrofloxacin. J. Inorg. Biochem. 2013, 121, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Arthi, P.; Shobana, S.; Srinivasan, P.; Prabhu, D.; Arulvasu, C.; Rahiman, A.K.; Biology, P.B. Dinuclear manganese (II) complexes of hexaazamacrocycles bearing N-benzoylated pendant separated by aromatic spacers: Antibacterial, DNA interaction, cytotoxic and molecular docking studies. J. Photoch. Photobio. B 2015, 153, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Simpson, P.V.; Nagel, C.; Bruhn, H.; Schatzschneider, U. Antibacterial and Antiparasitic Activity of Manganese(I) Tricarbonyl Complexes with Ketoconazole, Miconazole, and Clotrimazole Ligands. Organometallics 2015, 34, 3809–3815. [Google Scholar] [CrossRef]
- Boubakri, L.; Mansour, L.; Harrath, A.; Özdemir, I.; Yaşar, S.; Hamdi, N. N-Heterocyclic carbene-Pd (II)-PPh3 complexes as a new highly efficient catalyst system for the Sonogashira cross-coupling reaction: Synthesis, characterization and biological activities. J. Coord. Chem. 2018, 71, 183–199. [Google Scholar] [CrossRef]
- Kaushal, M.; Lobana, T.S.; Nim, L.; Bala, R.; Arora, D.S.; Garcia-Santos, I.; Duff, C.E.; Jasinski, J.P. Synthesis of 2-acetylpyridine-N-substituted thiosemicarbazonates of copper (II) with high antimicrobial activity against methicillin resistant S. aureus, K. pneumoniae 1 and C. albicans. New J. Chem. 2019, 43, 11727–11742. [Google Scholar] [CrossRef]
- Abu Ali, H.; Omar, S.N.; Darawsheh, M.D.; Fares, H. Synthesis, characterization and antimicrobial activity of zinc (II) ibuprofen complexes with nitrogen-based ligands. J. Coord. Chem. 2016, 69, 1110–1122. [Google Scholar] [CrossRef]
- Kumar, S.V.; Scottwell, S.Ø.; Waugh, E.; McAdam, C.J.; Hanton, L.R.; Brooks, H.J.; Crowley, J.D. Antimicrobial properties of tris (homoleptic) ruthenium (II) 2-Pyridyl-1, 2, 3-triazole “click” complexes against pathogenic bacteria, including methicillin-resistant staphylococcus aureus (MRSA). Inorg. Chem. 2016, 55, 9767–9777. [Google Scholar] [CrossRef] [PubMed]
- van Hilst, Q.V.C.; Vasdev, R.A.S.; Preston, D.; Findlay, J.A.; Scottwell, S.Ø.; Giles, G.I.; Brooks, H.J.L.; Crowley, J.D. Synthesis, Characterisation and Antimicrobial Studies of some 2,6-bis(1,2,3-Triazol-4-yl)Pyridine Ruthenium(II) “Click” Complexes. Asian J. Org. Chem. 2019, 8, 496–505. [Google Scholar] [CrossRef]
- Gichumbi, J.M.; Friedrich, H.B.; Omondi, B.; Naicker, K.; Singh, M.; Chenia, H.Y. Synthesis, characterization, antiproliferative, and antimicrobial activity of osmium (II) half-sandwich complexes. J. Coord. Chem. 2018, 71, 342–354. [Google Scholar] [CrossRef]
- Irgi, E.P.; Geromichalos, G.D.; Balala, S.; Kljun, J.; Kalogiannis, S.; Papadopoulos, A.; Turel, I.; Psomas, G.J.R.A. Cobalt (II) complexes with the quinolone antimicrobial drug oxolinic acid: Structure and biological perspectives. RSC Adv. 2015, 5, 36353–36367. [Google Scholar] [CrossRef]
- Kouris, E.; Kalogiannis, S.; Perdih, F.; Turel, I.; Psomas, G. Cobalt(II) complexes of sparfloxacin: Characterization, structure, antimicrobial activity and interaction with DNA and albumins. J. Inorg. Biochem. 2016, 163, 18–27. [Google Scholar] [CrossRef]
- Fiorini, V.; Zanoni, I.; Zacchini, S.; Costa, A.L.; Hochkoeppler, A.; Zanotti, V.; Ranieri, A.M.; Massi, M.; Stefan, A.; Stagni, S.J.D.T. Methylation of Ir (III)-tetrazolato complexes: An effective route to modulate the emission outputs and to switch to antimicrobial properties. Dalton Trans. 2017, 46, 12328–12338. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Moat, J.; McFeely, D.; Clarkson, G.; Hands-Portman, I.J.; Furner-Pardoe, J.P.; Harrison, F.; Dowson, C.G.; Sadler, P.J. Biguanide Iridium(III) Complexes with Potent Antimicrobial Activity. J. Med. Chem. 2018, 61, 7330–7344. [Google Scholar] [CrossRef] [PubMed]
- Baecker, D.; Sesli, O.; Knabl, L.; Huber, S.; Orth-Holler, D.; Gust, R. Investigating the antibacterial activity of salen/salophene metal complexes: Induction of ferroptosis as part of the mode of action. Eur. J. Med. Chem. 2021, 209. [Google Scholar] [CrossRef]
- Mendes, S.S.; Marques, J.; Mesterházy, E.; Straetener, J.; Arts, M.; Pissarro, T.; Reginold, J.; Berscheid, A.; Bornikoel, J.; Kluj, R.M.; et al. Synergetic Antimicrobial Activity and Mechanism of Clotrimazole-Linked CO-Releasing Molecules. ACS Bio Med. Chem. Au 2022, 2, 419–436. [Google Scholar] [CrossRef]
- Sovari, S.N.; Radakovic, N.; Roch, P.; Crochet, A.; Pavic, A.; Zobi, F. Combatting AMR: A molecular approach to the discovery of potent and non-toxic rhenium complexes active against C. albicans-MRSA co-infection. Eur. J. Med. Chem. 2021, 226, 113858. [Google Scholar] [CrossRef]
- Sovari, S.N.; Vojnovic, S.; Bogojevic, S.S.; Crochet, A.; Pavic, A.; Nikodinovic-Runic, J.; Zobi, F. Design, synthesis and in vivo evaluation of 3-arylcoumarin derivatives of rhenium(I) tricarbonyl complexes as potent antibacterial agents against methicillin-resistant Staphylococcus aureus (MRSA). Eur. J. Med. Chem. 2020, 205, 112533. [Google Scholar] [CrossRef]
- Cooper, S.M.; Siakalli, C.; White, A.J.P.; Frei, A.; Miller, P.W.; Long, N.J. Synthesis and anti-microbial activity of a new series of bis(diphosphine) rhenium(v) dioxo complexes. Dalton Trans. 2022, 51, 12791–12795. [Google Scholar] [CrossRef]
- Delasoie, J.; Schiel, P.; Vojnovic, S.; Nikodinovic-Runic, J.; Zobi, F. Photoactivatable Surface-Functionalized Diatom Microalgae for Colorectal Cancer Targeted Delivery and Enhanced Cytotoxicity of Anticancer Complexes. Pharmaceutics 2020, 12, 480. [Google Scholar] [CrossRef]
- Delasoie, J.; Pavic, A.; Voutier, N.; Vojnovic, S.; Crochet, A.; Nikodinovic-Runic, J.; Zobi, F. Identification of novel potent and non-toxic anticancer, anti-angiogenic and antimetastatic rhenium complexes against colorectal carcinoma. Eur. J. Med. Chem. 2020, 204, 112583. [Google Scholar] [CrossRef] [PubMed]
- Rossier, J.; Hauser, D.; Kottelat, E.; Rothen-Rutishauser, B.; Zobi, F. Organometallic cobalamin anticancer derivatives for targeted prodrug delivery via transcobalamin-mediated uptake. Dalton Trans. 2017, 46, 2159–2164. [Google Scholar] [CrossRef] [PubMed]
- Domenichini, A.; Casari, I.; Simpson, P.V.; Desai, N.M.; Chen, L.; Dustin, C.; Edmands, J.S.; van der Vliet, A.; Mohammadi, M.; Massi, M.; et al. Rhenium N-heterocyclic carbene complexes block growth of aggressive cancers by inhibiting FGFR- and SRC-mediated signalling. J. Exp. Clin. Cancer Res. 2020, 39, 276. [Google Scholar] [CrossRef] [PubMed]
- Collery, P.; Veena, V.; Harikrishnan, A.; Desmaele, D. The rhenium(I)-diselenoether anticancer drug targets ROS, TGF-β1, VEGF-A, and IGF-1 in an in vitro experimental model of triple-negative breast cancers. Investig. New Drugs 2019, 37, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Collery, P.; Santoni, F.; Ciccolini, J.; Tran, T.N.; Mohsen, A.; Desmaele, D. Dose Effect of Rhenium (I)-diselenoether as Anticancer Drug in Resistant Breast Tumor-bearing Mice After Repeated Administrations. Anticancer Res. 2016, 36, 6051–6057. [Google Scholar] [CrossRef][Green Version]
- Collery, P.; Bastian, G.; Santoni, F.; Mohsen, A.; Wei, M.; Collery, T.; Tomas, A.; Desmaele, D.; D’Angelo, J. Uptake and efflux of rhenium in cells exposed to rhenium diseleno-ether and tissue distribution of rhenium and selenium after rhenium diseleno-ether treatment in mice. Anticancer Res. 2014, 34, 1679–1689. [Google Scholar]
- Schindler, K.; Zobi, F. Anticancer and Antibiotic Rhenium Tri- and Dicarbonyl Complexes: Current Research and Future Perspectives. Molecules 2022, 27, 539. [Google Scholar] [CrossRef]
- Nasiri Sovari, S.; Kolly, I.; Schindler, K.; Cortat, Y.; Liu, S.C.; Crochet, A.; Pavic, A.; Zobi, F. Efficient Direct Nitrosylation of alpha-Diimine Rhenium Tricarbonyl Complexes to Structurally Nearly Identical Higher Charge Congeners Activable towards Photo-CO Release. Molecules 2021, 26, 5302. [Google Scholar] [CrossRef]
- Schindler, K.; Crochet, A.; Zobi, F. Aerobically stable and substitutionally labile α-diimine rhenium dicarbonyl complexes. RSC Adv. 2021, 11, 7511–7520. [Google Scholar] [CrossRef]
- Abram, U.; Hübener, R.; Alberto, R.; Schibli, R. Darstellung und Strukturen von (Et4N)2[Re(CO)3(NCS)3] und (Et4N)[Re(CO)2Br4]. Z. Anorg. Allg. Chem. 1996, 622, 813–818. [Google Scholar] [CrossRef]
- Santoro, G.; Beltrami, R.; Kottelat, E.; Blacque, O.; Bogdanova, A.Y.; Zobi, F. N-Nitrosamine-{cis-Re[CO]2}2+ cobalamin conjugates as mixed CO/NO-releasing molecules. Dalton Trans. 2016, 45, 1504–1513. [Google Scholar] [CrossRef] [PubMed]
- Zobi, F.; Blacque, O.; Jacobs, R.A.; Schaub, M.C.; Bogdanova, A.Y. 17 e– rhenium dicarbonyl CO-releasing molecules on a cobalamin scaffold for biological application. Dalton Trans. 2012, 41, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Carolus, H.; Van Dyck, K.; Van Dijck, P. Candida albicans and Staphylococcus Species: A Threatening Twosome. Front. Microbiol. 2019, 10, 2162. [Google Scholar] [CrossRef]
- Mauri, A. alvaDesc: A Tool to Calculate and Analyze Molecular Descriptors and Fingerprints. In Ecotoxicological QSARs; Springer: Berlin/Heidelberg, Germany, 2020; pp. 801–820. [Google Scholar]
- Lovering, A.L.; de Castro, L.H.; Lim, D.; Strynadka, N.C. Structural insight into the transglycosylation step of bacterial cell-wall biosynthesis. Science 2007, 315, 1402–1405. [Google Scholar] [CrossRef] [PubMed]
- Lovering, A.L.; Gretes, M.C.; Safadi, S.S.; Danel, F.; de Castro, L.; Page, M.G.; Strynadka, N.C. Structural insights into the anti-methicillin-resistant Staphylococcus aureus (MRSA) activity of ceftobiprole. J. Biol. Chem. 2012, 287, 32096–32102. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Kawai, F.; Obayashi, E.; Akashi, S.; Roper, D.I.; Tame, J.R.; Park, S.Y. Crystal structures of penicillin-binding protein 3 (PBP3) from methicillin-resistant Staphylococcus aureus in the apo and cefotaxime-bound forms. J. Mol. Biol. 2012, 423, 351–364. [Google Scholar] [CrossRef]
- Alexander, J.A.N.; Chatterjee, S.S.; Hamilton, S.M.; Eltis, L.D.; Chambers, H.F.; Strynadka, N.C.J. Structural and kinetic analyses of penicillin-binding protein 4 (PBP4)-mediated antibiotic resistance in Staphylococcus aureus. J. Biol. Chem. 2018, 293, 19854–19865. [Google Scholar] [CrossRef]
- Lu, D.; Wormann, M.E.; Zhang, X.; Schneewind, O.; Grundling, A.; Freemont, P.S. Structure-based mechanism of lipoteichoic acid synthesis by Staphylococcus aureus LtaS. Proc. Natl. Acad. Sci. USA 2009, 106, 1584–1589. [Google Scholar] [CrossRef]
- Ting, Y.T.; Harris, P.W.; Batot, G.; Brimble, M.A.; Baker, E.N.; Young, P.G. Peptide binding to a bacterial signal peptidase visualized by peptide tethering and carrier-driven crystallization. IUCrJ 2016, 3, 10–19. [Google Scholar] [CrossRef]
- Olatunji, S.; Yu, X.; Bailey, J.; Huang, C.Y.; Zapotoczna, M.; Bowen, K.; Remskar, M.; Muller, R.; Scanlan, E.M.; Geoghegan, J.A.; et al. Structures of lipoprotein signal peptidase II from Staphylococcus aureus complexed with antibiotics globomycin and myxovirescin. Nat. Commun. 2020, 11, 140. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, X.; Lambert, E.; Mas, G.; Hiller, S.; Veening, J.W.; Perez, C. Structure of a proton-dependent lipid transporter involved in lipoteichoic acids biosynthesis. Nat. Struct. Mol. Biol. 2020, 27, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Percy, M.G.; Grundling, A. Lipoteichoic acid synthesis and function in gram-positive bacteria. Annu. Rev. Microbiol. 2014, 68, 81–100. [Google Scholar] [CrossRef]
- Grundling, A.; Schneewind, O. Genes required for glycolipid synthesis and lipoteichoic acid anchoring in Staphylococcus aureus. J. Bacteriol. 2007, 189, 2521–2530. [Google Scholar] [CrossRef] [PubMed]
- Sham, L.T.; Butler, E.K.; Lebar, M.D.; Kahne, D.; Bernhardt, T.G.; Ruiz, N. Bacterial cell wall. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science 2014, 345, 220–222. [Google Scholar] [CrossRef]
- Fishovitz, J.; Hermoso, J.A.; Chang, M.; Mobashery, S. Penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus. IUBMB Life 2014, 66, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.G.; Elli, D.; Kim, H.K.; Hendrickx, A.P.; Sorg, J.A.; Schneewind, O.; Missiakas, D. Small molecule inhibitor of lipoteichoic acid synthesis is an antibiotic for Gram-positive bacteria. Proc. Natl. Acad. Sci. USA 2013, 110, 3531–3536. [Google Scholar] [CrossRef]
- Gründling, A.; Schneewind, O. Synthesis of glycerol phosphate lipoteichoic acid in Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2007, 104, 8478–8483. [Google Scholar] [CrossRef]
- Pasquina, L.W.; Santa Maria, J.P.; Walker, S. Teichoic acid biosynthesis as an antibiotic target. Curr. Opin. Microbiol. 2013, 16, 531–537. [Google Scholar] [CrossRef]
- Kuk, A.C.Y.; Hao, A.; Guan, Z.; Lee, S.-Y. Visualizing conformation transitions of the Lipid II flippase MurJ. Nat. Commun. 2019, 10, 1736. [Google Scholar] [CrossRef]
- Kohga, H.; Mori, T.; Tanaka, Y.; Yoshikaie, K.; Taniguchi, K.; Fujimoto, K.; Fritz, L.; Schneider, T.; Tsukazaki, T. Crystal structure of the lipid flippase MurJ in a “squeezed” form distinct from its inward- and outward-facing forms. Structure 2022. [Google Scholar] [CrossRef]
- Craney, A.; Romesberg, F.E. The inhibition of type I bacterial signal peptidase: Biological consequences and therapeutic potential. Bioorg. Med. Chem. Lett. 2015, 25, 4761–4766. [Google Scholar] [CrossRef] [PubMed]
- Schmaler, M.; Jann, N.J.; Götz, F.; Landmann, R. Staphylococcal lipoproteins and their role in bacterial survival in mice. Int. J. Med. Microbiol. 2010, 300, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Vogeley, L.; Arnaout, T.E.; Bailey, J.; Stansfeld, P.J.; Boland, C.; Caffrey, M. Structural basis of lipoprotein signal peptidase II action and inhibition by the antibiotic globomycin. Science 2016, 351, 876–880. [Google Scholar] [CrossRef]
- Nguyen Minh, T.; Götz, F. Lipoproteins of Gram-Positive Bacteria: Key Players in the Immune Response and Virulence. Microbiol. Mol. Biol. Rev. 2016, 80, 891–903. [Google Scholar] [CrossRef] [PubMed]
- El Arnaout, T.; Soulimane, T. Targeting Lipoprotein Biogenesis: Considerations towards Antimicrobials. Trends Biochem. Sci. 2019, 44, 701–715. [Google Scholar] [CrossRef]
- Jendele, L.; Krivak, R.; Skoda, P.; Novotny, M.; Hoksza, D. PrankWeb: A web server for ligand binding site prediction and visualization. Nucleic Acids Res. 2019, 47, W345–W349. [Google Scholar] [CrossRef]
- Wimley, W.C.; White, S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Mol. Biol. 1996, 3, 842–848. [Google Scholar] [CrossRef]
- Hebditch, M.; Warwicker, J. Web-based display of protein surface and pH-dependent properties for assessing the developability of biotherapeutics. Sci. Rep. 2019, 9, 1969. [Google Scholar] [CrossRef]
- Warwicker, J. Continuum dielectric modelling of the protein-solvent system, and calculation of the long-range electrostatic field of the enzyme phosphoglycerate mutase. J. Theor. Biol. 1986, 121, 199–210. [Google Scholar] [CrossRef]
- Mahesha; Krishnegowda, H.M.; Karthik, C.S.; Kudigana, P.J.; Mallu, P.; Neratur, L.K. μ-phenoxide bridged mixed ligand Cu(II) complex: Synthesis, 3D supramolecular architecture, DFT, energy frameworks and antimicrobial studies. Polyhedron 2020, 185, 114571. [Google Scholar] [CrossRef]
- Prasad, H.S.N.; Ananda, A.P.; Najundaswamy, S.; Nagashree, S.; Mallesha, L.; Dayananda, B.P.; Jayanth, H.S.; Mallu, P. Design, synthesis and molecular docking studies of novel piperazine metal complexes as potential antibacterial candidate against MRSA. J. Mol. Struct. 2021, 1232, 130047. [Google Scholar] [CrossRef]
- Hema, M.K.; Karthik, C.S.; Mahesha; Pampa, K.J.; Mallu, P.; Lokanath, N.K. 4,4,4-trifluoro-1-phenylbutane-1,3-dione metal [Cu(II) and Ni(II)] complexes as an superlative antibacterial agent against MRSA: Synthesis, structural quantum-chemical and molecular docking studies. J. Mol. Struct. 2021, 1243, 130774. [Google Scholar] [CrossRef]
- Burgart, Y.; Shchegolkov, E.; Shchur, I.; Kopchuk, D.; Gerasimova, N.; Borisevich, S.; Evstigneeva, N.; Zyryanov, G.; Savchuk, M.; Ulitko, M.; et al. Promising Antifungal and Antibacterial Agents Based on 5-Aryl-2,2′-bipyridines and Their Heteroligand Salicylate Metal Complexes: Synthesis, Bioevaluation, Molecular Docking. Chemmedchem 2022, 17, e202100577. [Google Scholar] [CrossRef] [PubMed]
- Ekennia, A.C.; Onwudiwe, D.C.; Osowole, A.A.; Okpareke, O.C.; Olubiyi, O.O.; Lane, J.R. Coordination compounds of heterocyclic bases: Synthesis, characterization, computational and biological studies. Res. Chem. Intermed. 2019, 45, 1169–1205. [Google Scholar] [CrossRef]
- Zobi, F.; Kromer, L.; Spingler, B.; Alberto, R. Synthesis and Reactivity of the 17 e− Complex [ReIIBr4(CO)2]2−: A Convenient Entry into Rhenium(II) Chemistry. Inorg. Chem. 2009, 48, 8965–8970. [Google Scholar] [CrossRef]
- Kurz, P.; Probst, B.; Spingler, B.; Alberto, R. Ligand Variations in [ReX(diimine)(CO)3] Complexes: Effects on Photocatalytic CO2 Reduction. Eur. J. Inorg. Chem. 2006, 2006, 2966–2974. [Google Scholar] [CrossRef]
- Murphy, B.L.; Marker, S.C.; Lambert, V.J.; Woods, J.J.; MacMillan, S.N.; Wilson, J.J. Synthesis, characterization, and biological properties of rhenium(I) tricarbonyl complexes bearing nitrogen-donor ligands. J. Organomet. Chem. 2020, 907, 121064. [Google Scholar] [CrossRef]
- Chakraborty, I.; Jimenez, J.; Sameera, W.M.C.; Kato, M.; Mascharak, P.K. Luminescent Re(I) Carbonyl Complexes as Trackable PhotoCORMs for CO delivery to Cellular Targets. Inorg. Chem. 2017, 56, 2863–2873. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. V. Systematic optimization of exchange-correlation functionals. J. Chem. Phys. 1997, 107, 8554–8560. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, W. Empirical correction to density functional theory for van der Waals interactions. J. Chem. Phys. 2002, 116, 515–524. [Google Scholar] [CrossRef]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-adjustedab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Yan, C.; Yuan, R.; Pfalzgraff, W.C.; Nishida, J.; Wang, L.; Markland, T.E.; Fayer, M.D. Unraveling the dynamics and structure of functionalized self-assembled monolayers on gold using 2D IR spectroscopy and MD simulations. Proc. Natl. Acad. Sci. USA 2016, 113, 4929–4934. [Google Scholar] [CrossRef] [PubMed]
- Vasighi, M.; Romanova, J.; Nedyalkova, M. A multilevel approach for screening natural compounds as an antiviral agent for COVID-19. Comput. Biol. Chem. 2022, 98, 107694. [Google Scholar] [CrossRef]










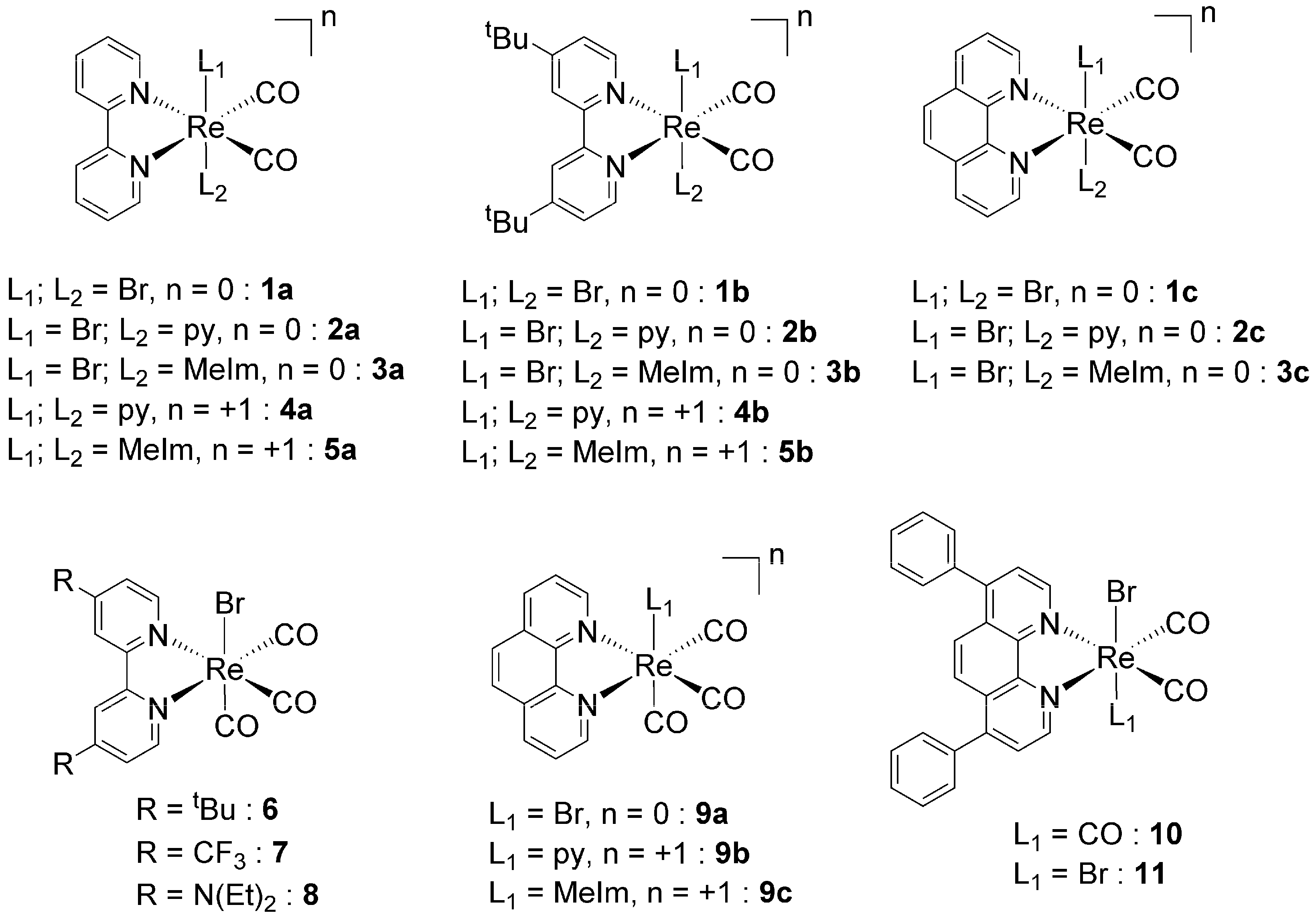{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
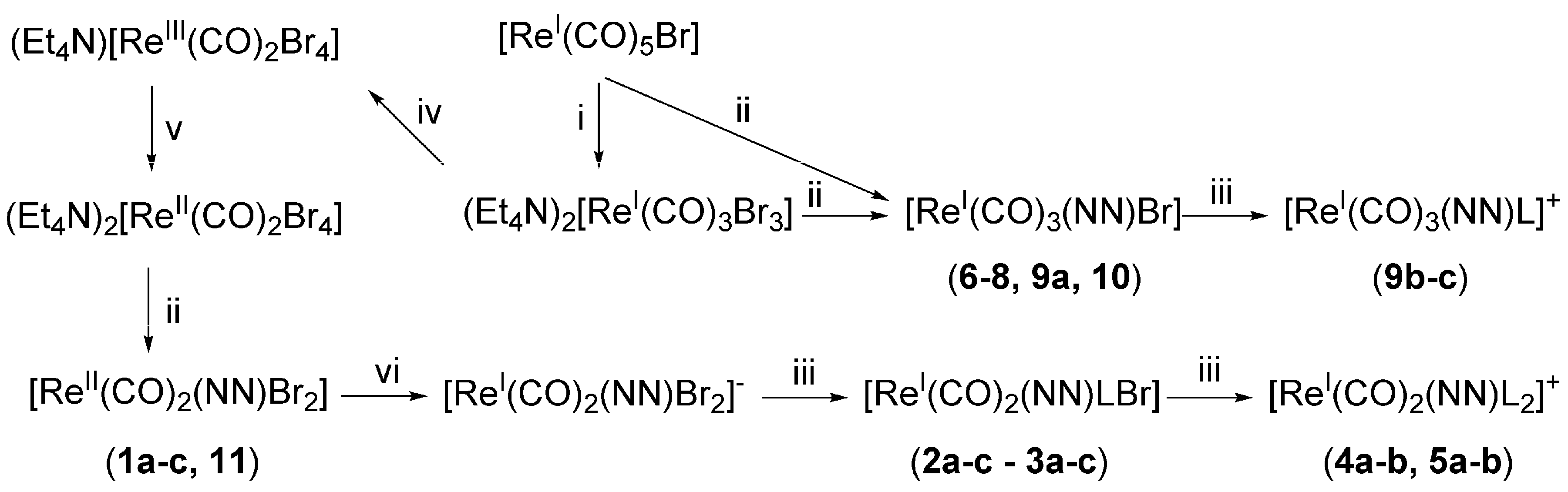{kind=link}
| Compound. | A. baumannii | P. auruginosa | K. pneumoniae | S. aureus MRSA | S. aureus MSSA | E. cloacae | C. albicans | C. auris |
|---|---|---|---|---|---|---|---|---|
| 1a–5a | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 1b–3b | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 4b | >100 | >100 | >100 | 25 | 25 | >100 | >100 | >100 |
| 5b | >100 | >100 | >100 | 50 | 50 | >100 | >100 | >100 |
| 1c–3c | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 6–8, 9a–c, 10 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
| 11 | >100 | >100 | >100 | 50 | 50 | >100 | >100 | >100 |
| 13 | n.a. | n.a. | n.a. | 0.7 | 22.8 | n.a. | n.a. | n.a. |
| 14 | n.a. | n.a. | n.a. | 1.6 | 1.6 | n.a. | n.a. | n.a. |
| 15 | n.a. | n.a. | n.a. | 0.4 | 0.6 | n.a. | 6.2 | 50 |
| 16 | n.a. | n.a. | n.a. | 0.8 | 0.8 | n.a. | 6.2 | 50 |
| 17 | 8 | 32 | 32 | 0.25 | 0.25 | n.a. | n.a. | n.a. |
| 18 | n.a. | n.a. | n.a. | 0.8 | 3.1 | n.a. | 3.1 | 50 |
| 19 | n.a. | n.a. | n.a. | 1.6 | 6.2 | n.a. | 50 | 50 |
| Meropenem | >100 | 1.25 | >100 | 6.25 | >100 | 50 | - | - |
| Vankomycin | >100 | >100 | >100 | 6.25 | 6.25 | - | - | - |
| Fluconazole | - | - | - | - | - | - | 1 | >64 |
| Amphotericin B | - | - | - | - | - | - | 0.3 | 1 |
| Compound | MW | RBN | TPSA(Tot) | HBA | HBD | LOGP99 | BLTF96 | BLTA96 | BLTD48 | ESOL | cRo5 | Ro5 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 13 | 700.827 | 3 | 81.79 | 6 | 1 | 6.8 | −3.09 | −3.22 | −3.23 | −7.19 | 1 | 0 |
| 14 | 1072.267 | 16 | 187.36 | 14 | 2 | 4.8 | 1.13 | 1.49 | 1.73 | −5.67 | 0 | 1 |
| 15 | 681.767 | 3 | 66 | 0 | 6 | 6.7 | −2.99 | −3.11 | −3.11 | −7.02 | 1 | 0 |
| 16 | 647.827 | 7 | 72.48 | 0 | 8 | 4.1 | −2.03 | −2.04 | −1.98 | −4.94 | 1 | 0 |
| 17 | 771.297 | 5 | 70.93 | 8 | 0 | 7.1 | −3.94 | −4.17 | −4.23 | −7.85 | 1 | 0 |
| 18 | 867.067 | 3 | 108.24 | 2 | 1 | 8.5 | −4.13 | −4.38 | −4.45 | −8.74 | 1 | 0 |
| 19 | 1021.367 | 8 | 136.15 | 0 | 12 | 8.0 | −3.84 | −4.06 | −4.11 | −9.29 | 0 | 1 |
| PBD ID | Area (Å2) | Volume (Å3) | Pocket Residues ID/Flexible Chains |
|---|---|---|---|
| 2OLV (PBP2) | 3500.5 | 7715.2 | ALA_112, VAL_367, GLY_339, LYS_127, LYS_135, THR_150, VAL_153, THR_148, GLU_171, LYS_194, PRO_231, ASN_193, GLY_229 |
| 4DKI (PBP2a) | 5537.8 | 9122.9 | THR_398, PRO_401, VAL_443, THR_444, SER_461, TYR_519, GLY_520, THR_582, ALA_601, ARG_612, ASP_638 |
| 3VSL (PBP3) | 9921.9 | 13845.0 | GLY_424, VAL_390, LEU_425, THR_426, MET_453, LEU_518, ASP_519, LYS_618, TYR_636 |
| 5TXI (PBP4) | 4258.4 | 4521.5 | SER_75, ALA_74, THR_77, LYS_78, SER_137, SER_185, SER_262, PHE_241, THR_260, GLY_261, PRO_113, LEU_115, GLU_114 |
| 2W5Q (LtaS) | 132.1 | 103.4 | LEU_254, GLU_255, GLN_297, GLY_298, LYS_299, THR_300, SER_301, HIS_347, PHE_353, TRP_354, ASN_355, LYS_397, HIS_416 |
| 4WVJ (SpsB) | 1922.1 | 3375.8 | TRP_236, GLU_117, GLU_159, TYR_161, ASN_18, ASP_20, LYS_21, LEU_268, SER_343, TRP_346, TYR_347, ARG_350, LYS_48 |
| 6S7V (LtaA) | 1758.2 | 2257.5 | LEU_219, PRO_221, LEU_225, ALA_229, ILE_230, ALA_230, VAL_234 |
| 6RYP (LspA) | 8452.7 | 1485.4 | ALA_103, _367, GLY_339, LYS_127, LYS_135, THR_150, VAL_153, THR_148, GLU_171, LYS_194, PRO_231, ASN_193, GLY_229 |
| Receptor | Drug | Affinity * | H-Bonds | Receptor’s Rgyr (nm) | System’s Rgyr (nm) | Receptor’s SASA (nm2) | Receptor’s Prob. Drugability | Ligand’s SASA (nm2) | System’s SASA (nm2) | Contact Area (nm2) | Detected H-Bonds with AA Residue |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2OLV (PBP2) | 13 | −6.9 | 3 | 3.29 | 3.29 | 295.26 | 0.82 | 7.60 | 295.24 | 3.80 | ASP 156, LYS 194, PRO 231 |
| 14 | −7.4 | 3 | 3.29 | 3.29 | 295.26 | 0.82 | 12.14 | 294.91 | 6.24 | ASP 156, LYS 194, PRO 231 | |
| 15 | −8.1 | 2 | 3.29 | 3.28 | 295.26 | 0.82 | 7.87 | 294.87 | 4.13 | ASP 89 | |
| 16 | −5.7 | 4 | 3.29 | 3.28 | 295.26 | 0.82 | 8.05 | 295.15 | 4.08 | THR 87, GLN 92, HIS 94, GLU 95 | |
| 17 | −7.8 | 1 | 3.29 | 3.32 | 295.26 | 0.82 | 7.90 | 299.57 | 3.13 | ASP 156 | |
| 18 | −7.9 | 1 | 3.29 | 3.29 | 295.26 | 0.82 | 10.77 | 295.17 | 5.43 | PRO 72 | |
| 19 | −7.2 | 1 | 3.29 | 3.30 | 295.26 | 0.82 | 9.35 | 295.14 | 4.73 | ASN 237 | |
| 4DKI * (PBP2a) | 13 | −6.7 | 1 | 3.66 | 3.66 | 317.73 | 0.76 | 7.09 | 316.08 | 4.10 | THR 398 |
| 14 | −7.2 | 3 | 3.66 | 3.65 | 317.73 | 0.76 | 11.21 | 316.08 | 6.43 | THR 398, GLY 520 | |
| 15 | −9.2 | 1 | 3.66 | 3.66 | 317.73 | 0.76 | 7.55 | 315.86 | 4.71 | LYS 394 | |
| 16 | −5.9 | 4 | 3.66 | 3.66 | 317.73 | 0.76 | 7.87 | 316.27 | 4.66 | THR 600, LEU 603, MER 605 | |
| 17 | −6.7 | 4 | 3.66 | 3.67 | 317.73 | 0.76 | 7.7 | 317.2 | 3.9 | ASP 516, GLN 521, MET | |
| 18 | −8.5 | 1 | 3.66 | 3.66 | 317.73 | 0.76 | 10.75 | 316.08 | 6.94 | SER 400 | |
| 19 | −9.8 | 4 | 3.66 | 3.67 | 317.73 | 0.76 | 11.57 | 315.25 | 7.02 | SER 403, GLN 521, THR 600, SER 400 | |
| 3VSL (PBP3) | 13 | −7.0 | 3 | 3.11 | 3.11 | 301.97 | 0.81 | 7.25 | 300.48 | 4.37 | TYR 525, GLU 623, GLN 626 |
| 14 | −7.0 | 3 | 3.11 | 3.11 | 301.97 | 0.81 | 12.00 | 299.12 | 7.42 | ||
| 15 | −8.6 | 3 | 3.11 | 3.11 | 301.97 | 0.81 | 7.80 | 300.81 | 4.48 | TYR 525, ASP 519, GLU 623 | |
| 16 | −5.3 | 1 | 3.11 | 3.11 | 301.97 | 0.81 | 8.01 | 301.13 | 4.42 | GLN 626 | |
| 17 | −6.9 | 0 | 3.11 | 3.11 | 301.97 | 0.81 | 7.69 | 300.78 | 4.44 | - | |
| 18 | −7.6 | 2 | 3.11 | 3.11 | 301.97 | 0.81 | 11.22 | 302.30 | 5.44 | GLU 623 | |
| 19 | −6.7 | 3 | 3.11 | 3.11 | 301.97 | 0.81 | 11.88 | 301.39 | 6.23 | GLU 623 | |
| 5TXI * (PBP4) | 13 | −6.3 | 0 | 2.16 | 2.17 | 151.84 | 0.8 | 7.7212 | 155.88 | 1.83 | - |
| 14 | −9.1 | 5 | 2.16 | 2.16 | 151.84 | 0.8 | 11.821 | 150.85 | 6.40 | GLU 114, SER 262, TYR 268, TYR 291, GLU 297 | |
| 15 | −7.0 | 0 | 2.16 | 2.17 | 151.84 | 0.8 | 7.951 | 156.74 | 1.5 | - | |
| 16 | −5.6 | 0 | 2.16 | 2.16 | 151.84 | 0.8 | 7.9606 | 150.89 | 4.45 | - | |
| 17 | −7.1 | 2 | 2.16 | 2.17 | 151.84 | 0.8 | 7.6 | 155.2 | 2.6 | THR 240, GLY 247 | |
| 18 | −8.1 | 3 | 2.16 | 151.84 | 0.8 | 10.5309 | 151.17 | 5.60 | GLU 114, SER 262, TYR 268, TYR 291 | ||
| 19 | −10.02 | 3 | 2.16 | 2.16 | 151.84 | 0.8 | 12.6803 | 151.04 | 6.73 | SER 116 | |
| 2W5Q (LtaS) | 13 | −6 | 1 | 2.07 | 2.06 | 177.84 | 0.81 | 7.25 | 178.03 | 3.53 | ASP 502 |
| 14 | −6.2 | 0 | 2.07 | 2.07 | 177.84 | 0.81 | 10.73 | 177.85 | 5.36 | - | |
| 15 | −7.8 | 1 | 2.07 | 2.07 | 177.84 | 0.81 | 7.99 | 178.05 | 3.89 | ASP 366 | |
| 16 | −5.7 | 0 | 2.07 | 2.06 | 177.84 | 0.81 | 7.94 | 176.11 | 4.83 | - | |
| 17 | −7.5 | 1 | 2.07 | 2.07 | 177.84 | 0.81 | 7.72 | 184 | 0.7 | ASP 521 | |
| 18 | −8.9 | 2 | 2.07 | 2.06 | 177.84 | 0.81 | 10.98 | 176.25 | 6.28 | GLY 296, GLY 478 | |
| 19 | −7.5 | 0 | 2.07 | 2.0697 | 177.84 | 0.81 | 9.2 | 177.15 | 4.98 | - | |
| 4WV J* (SpsB) | 13 | −7.3 | 2 | 2.77 | 2.75 | 239.62 | 0.82 | 7.26 | 238.84 | 4.02 | SER 343 |
| 14 | −8.3 | 2 | 2.77 | 2.75 | 239.62 | 0.82 | 12.90 | 237.89 | 7.31 | TYR 182, ALA 330 | |
| 15 | −9.5 | 2 | 2.77 | 2.76 | 239.62 | 0.82 | 7.76 | 238.10 | 4.64 | ASP 20 | |
| 16 | −6.1 | 0 | 2.77 | 2.75 | 239.62 | 0.82 | 8.09 | 237.67 | 5.02 | - | |
| 17 | −7.1 | 0 | 2.77 | 2.76 | 239.62 | 0.82 | 7.70 | 238.36 | 4.1 | - | |
| 18 | −7.5 | 2 | 2.77 | 2.75 | 239.62 | 0.82 | 8.89 | 238.49 | 5.01 | GLU 51, PRO 340 | |
| 19 | −8.9 | 2 | 2.77 | 2.74 | 239.62 | 0.82 | 10.14 | 238.36 | 5.70 | GLU 50, VAL 378 | |
| 6S7V * (LtaA) | 13 | −8.3 | 1 | 2.13 | 2.12 | 192.79 | 0.81 | 7.55 | 190.60 | 4.87 | GLY 259 |
| 14 | −8.6 | 1 | 2.13 | 2.12 | 192.79 | 0.81 | 11.06 | 187.8256 | 8.01 | ILE 256 | |
| 15 | −10.0 | 1 | 2.13 | 2.12 | 192.79 | 0.81 | 7.91 | 190.39 | 5.15 | TYR 377 | |
| 16 | −6.2 | 0 | 2.13 | 2.12 | 192.79 | 0.81 | 7.84 | 190.33 | 5.15 | - | |
| 17 | −8.0 | 0 | 2.13 | 2.12 | 192.79 | 0.81 | 7.8 | 189.5 | 3.8 | - | |
| 18 | −9.7 | 0 | 2.13 | 2.12 | 192.79 | 0.81 | 11.06 | 189.55 | 7.15 | - | |
| 19 | −10.2 | 2 | 2.13 | 2.12 | 192.79 | 0.81 | 9.71 | 189.48 | 6.51 | ILE 230, TYR 377 | |
| 6RYP * (LspA) | 13 | −7.4 | 1 | 1.86 | 1.84 | 108.41 | 0.82 | 7.31 | 107.33 | 4.19 | GLY 54 |
| 14 | −10.0 | 2 | 1.86 | 1.83 | 108.41 | 0.82 | 12.55 | 105.94 | 7.51 | ASP 136 | |
| 15 | −10.6 | 0 | 1.86 | 1.84 | 108.41 | 0.82 | 7.91 | 106.88 | 4.71 | - | |
| 16 | −7 | 0 | 1.86 | 1.84 | 108.41 | 0.82 | 7.82 | 105.99 | 5.15 | - | |
| 17 | −8.1 | 2 | 1.86 | 1.85 | 108.41 | 0.82 | 7.5 | 107.3 | 4.05 | ILE 120, THR 140 | |
| 18 | −9.2 | 2 | 1.86 | 1.83 | 108.41 | 0.82 | 9.76 | 106.50 | 5.83 | GLY 54, THR 140 | |
| 19 | −11.5 | 1 | 1.86 | 1.83 | 108.41 | 0.82 | 10.58 | 106.47 | 6.26 | THR 140 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schindler, K.; Cortat, Y.; Nedyalkova, M.; Crochet, A.; Lattuada, M.; Pavic, A.; Zobi, F. Antimicrobial Activity of Rhenium Di- and Tricarbonyl Diimine Complexes: Insights on Membrane-Bound S. aureus Protein Binding. Pharmaceuticals 2022, 15, 1107. https://doi.org/10.3390/ph15091107
Schindler K, Cortat Y, Nedyalkova M, Crochet A, Lattuada M, Pavic A, Zobi F. Antimicrobial Activity of Rhenium Di- and Tricarbonyl Diimine Complexes: Insights on Membrane-Bound S. aureus Protein Binding. Pharmaceuticals. 2022; 15(9):1107. https://doi.org/10.3390/ph15091107
Chicago/Turabian StyleSchindler, Kevin, Youri Cortat, Miroslava Nedyalkova, Aurelien Crochet, Marco Lattuada, Aleksandar Pavic, and Fabio Zobi. 2022. "Antimicrobial Activity of Rhenium Di- and Tricarbonyl Diimine Complexes: Insights on Membrane-Bound S. aureus Protein Binding" Pharmaceuticals 15, no. 9: 1107. https://doi.org/10.3390/ph15091107
APA StyleSchindler, K., Cortat, Y., Nedyalkova, M., Crochet, A., Lattuada, M., Pavic, A., & Zobi, F. (2022). Antimicrobial Activity of Rhenium Di- and Tricarbonyl Diimine Complexes: Insights on Membrane-Bound S. aureus Protein Binding. Pharmaceuticals, 15(9), 1107. https://doi.org/10.3390/ph15091107