Abstract
Antimicrobial resistance is one of the major human health threats, with significant impacts on the global economy. Antibiotics are becoming increasingly ineffective as drug-resistance spreads, imposing an urgent need for new and innovative antimicrobial agents. Metal complexes are an untapped source of antimicrobial potential. Rhenium complexes, amongst others, are particularly attractive due to their low in vivo toxicity and high antimicrobial activity, but little is known about their targets and mechanism of action. In this study, a series of rhenium di- and tricarbonyl diimine complexes were prepared and evaluated for their antimicrobial potential against eight different microorganisms comprising Gram-negative and -positive bacteria. Our data showed that none of the Re dicarbonyl or neutral tricarbonyl species have either bactericidal or bacteriostatic potential. In order to identify possible targets of the molecules, and thus possibly understand the observed differences in the antimicrobial efficacy of the molecules, we computationally evaluated the binding affinity of active and inactive complexes against structurally characterized membrane-bound S. aureus proteins. The computational analysis indicates two possible major targets for this class of compounds, namely lipoteichoic acids flippase (LtaA) and lipoprotein signal peptidase II (LspA). Our results, consistent with the published in vitro studies, will be useful for the future design of rhenium tricarbonyl diimine-based antibiotics.
Keywords:
rhenium; tricarbonyl; antimicrobial; S. aureus; MRSA; AutoDock; membrane; proteins; LspA; LtaA 1. Introduction
The expansion of resistance to conventional antibiotics has become a notable health threat and necessitated the development of alternative treatment options for battling such a global problem [1]. Amongst the six nosocomial pathogens that exhibit multidrug resistance and virulence, the methicillin-resistant S. aureus (MRSA) is a major cause of community- and hospital-acquired infections worldwide, ranging from superficial skin and soft tissue infections [2] to invasive infections and sepsis [3]. This pathogen represents the most common and the second most common cause of healthcare-associated and bloodstream infections (BSI), as well as the most important cause of BSI death [4]. Since the bacterium is increasingly showing resistance to multiple antibiotics, in 2017 the World Health Organization listed it among the high priority group of human pathogens. Indeed, the same year, the Centers for Disease Control and Prevention (CDC) reported that more than 119,000 people suffered from S. aureus bloodstream infections in the United States, with nearly 20,000 of them (>16%) eventually dying.
What has exacerbated the problem of antimicrobial resistance (AMR) is the fact that fewer new antibiotics are reaching the market, with the last entirely original class of antibiotics discovered in the late 1980s. This is because large pharmaceutical companies have left the market due to a lack of financial incentives [5]. Consequently, in the last few years, academic research groups at universities around the globe have taken up the challenge to prepare and discover new antibiotic drugs that may serve as lead compounds for new structurally viable drugs. In our era, strategies for the discovery and development of new drugs combine computational and experimental approaches. This is true in virtually all medicinal discovery areas, including the design and discovery of molecules as appropriate candidates for treatment of Staphylococcus aureus infection. Computer-aided drug design (CADD) methods are computational approaches to guiding and expediting the experimental findings for new drug design processes [6,7,8,9,10]. CADD can be used in a qualitative and quantitative mode to improve the biovalidity and prediction rates for ligand binding affinity, as well as specificity, in a manner that can lead to the identification of biological targets of known drugs and the design process of new agents in a simpler, more efficient, and less expensive manner. In a modern drug design process, typically hundreds of compounds can be tested in a short time. The existing methodologies such as, e.g., site-identification by ligand competitive saturation (SILCS) [8,11], have become versatile tools for ligand–protein binding prediction. The foundation of the CADD technique is based on molecular docking [12,13,14] and molecular dynamics simulations [15,16].
Within the specific context of this article, CADD has been used, e.g., to evaluate medicinal plant-derived active compounds that could be used as therapeutic alternatives for MRSA infection [17,18,19,20]. The study of receptor–ligand interactions in the framework of molecular docking has drawn attention to the importance of probing the efficiency of these plant-derived antimicrobial agents [17,18] and testing antimicrobial activity using screened lead compounds, focusing on the role of computational screening methods [20,21,22,23] in tackling the problem. However, a major strategy still pursued in the field is that of modifying already approved antibiotics [24]. As may be expected, all of these molecules are purely organic compounds. While some of these new derivatives (some are currently in preclinical or clinical development) will provide feasible short-term solutions, it is probable that the pathogens will rapidly adapt and develop resistance to these molecules as well [24].
As an alternative to organic compounds, there is an increasing awareness in academia of the potential of metal complexes to act as the new class of molecules for this purpose. Indeed, the unique chemistry and larger variety of 3D geometries of metal compounds can address targets and modes of action that are unavailable to organic molecules. Over the last decade, complexes of virtually all the transition metals have been evaluated as antimicrobial agents [25,26,27], with rhenium (Re), among others [28,29], showing promising potential for new antibiotic development [30,31,32,33]. While other transition metal complexes [34,35,36,37,38,39,40,41,42,43,44,45,46,47] act against Gram-negative bacteria, carbonyl rhenium complexes have demonstrated a very potent activity towards Gram-positive pathogens, particularly towards Staphylococcus aureus, involving both the methicillin-resistant (MRSA) and methicillin-sensitive (MSSA) strains [31,32,33,48,49,50,51].
Our group is principally interested in the development of the chemistry of carbonyl rhenium complexes for their use in different medicinal applications, including their evaluation as anticancer [52,53,54] and antibacterial agents [49,50]. Due to their very low in vivo toxicity [55,56,57,58], tricarbonyl complexes of rhenium are particularly attractive. The same type of molecules (i.e., those of the fac-[Re(CO)3]+ core) are also the ones most widely investigated, showing the highest anticancer and antimicrobial effectiveness against S. aureus strains [59]. It is still unclear what molecular features of carbonyl rhenium complexes make them such promising medicinal agents. In a study that we have recently reported [49], we concluded that, unlike anticancer complexes, positively charged rhenium species are most effective against the microbes, and we hypothesized that charged compounds may interact more effectively with phosphatidylglycerol and cardiolipin anionic membrane lipids. Later, however, we found that, by substituting a single neutral carbonyl with a nitrosonium cation, the compounds lose their antimicrobial effect [60]. Thus, in terms of their antibacterial effectiveness, both the required molecular features and mechanism of action of these agents remain largely unknown.
In order to advance knowledge of the issues mentioned above, we evaluated the antimicrobial activity of dicarbonyl rhenium diimine complexes (i.e., of the cis-[Re(CO)2]+/2+ core) and compared it to that of structurally similar fac-[Re(CO)3]+ species. This part of study was performed because (a) no antimicrobial data is available on the carbonyl complexes of the cis-[Re(CO)2]+/2+ core lacking other π-acid ligands, and (b) a comparison of the activity of cis-[Re(CO)2]+/2+ and structurally similar fac-[Re(CO)3]+ species may provide information about the key molecular features required for the design of an effective Re-based antibiotic agent. Furthermore, we computationally evaluated the binding affinity of all the compounds (both active and inactive molecules) against structurally characterized membrane-bound S. aureus proteins. We performed this study principally to (a) identify possible biological targets of the active complexes; (b) possibly understand the underlying reasons for the observed differences in the antimicrobial efficacy of Re complexes; and (c) offer support for the rational design of rhenium complexes based on the computational protocol for CADD.
2. Results and Discussion
2.1. Synthesis and Characterization of the Metal Complexes
Rhenium carbonyl complexes investigated in this study were prepared according to the procedures illustrated in Scheme 1. Tricarbonyl species 6–10 (Figure 1) were obtained in a high yield and purity, according to established routes generally used in the preparation of these compounds. The fac-[Re(CO)3(NN)Br] complexes (6–8, 9a, and 10, where NN = relevant bidentate diimine ligand) may be obtained in one step from [Re(CO)5Br] by boiling this precursor in toluene in the presence of one equivalent of NN. The resulting yellow product, isolated by filtration, is generally of a high purity (>96% by NMR or HPLC) and can be used for further modification by the substitution reaction of the coordinated bromide atom by other monodentate ligands, as in the case of species 9b and 9c. For this reaction, we found that the best conditions consist in the treatment of a fac-[Re(CO)3(NN)Br] complex with trifluoromethanesulfonic acid to produce the intermediate fac-[Re(CO)3(NN)(CF3SO3)] molecule, followed by the addition of L (where L = pyridine: py or N-methyl imidazole: MeIm). The reaction is also high yielding, but the desired fac-[Re(CO)3(NN)L]CF3SO3 salt requires purification on alumina or via HPLC.
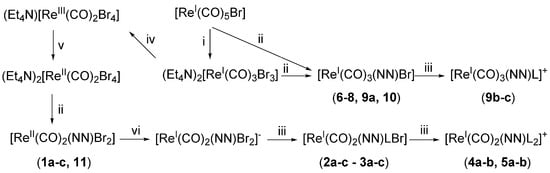
Scheme 1.
Synthetic scheme for the preparation of the complexes investigated in this study. NN = relevant bidentate diamine ligand; L = pyridine (py) or N-methyl imidazole (MeIm). General conditions: i: Et4NBr, diglyme; ii: NN, ethanol/water, toluene, or CH2Cl2; iii: L, methanol or neat L; iv: Br2, CH2Cl2; v: tetrakis(dimethylamino)ethylene, acetonitrile, under N2; vi: tetrakis(dimethylamino)ethylene, CH2Cl2, under N2. For more details, refer to Section 4.
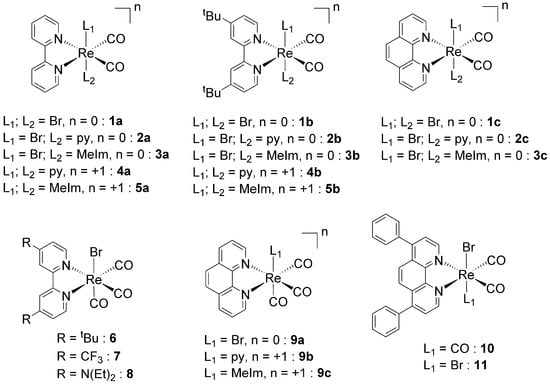
Figure 1.
Structures and codes of the tested Re dicarbonyl (1–5) and tricarbonyl (6–10) complexes.
The preparation of dicarbonyl cis-[Re(CO)2(NN)X2] species (1–5 and 11, where X = Br or L, Figure 1) is more demanding and requires several steps, starting from the common [Re(CO)5Br] precursor. We have recently published the details of this chemistry [61], showing that the synthetic route is favorable if X is a halide or an aromatic heterocycle (or a combination of both). However, yields of cis-[Re(CO)2(NN)X2] species are much lower than the comparable fac-[Re(CO)3(NN)Br] complexes. Briefly, cis-[Re(CO)2(NN)X2] species may be prepared following the sequential two-electron oxidation of fac-[Re(CO)3Br3]2− to cis-[Re(CO)2Br4]− [62], its one-electron reduction to cis-[Re(CO)2Br4]2−, the complexation of NN to cis-[Re(CO)2(NN)Br2], its one-electron reduction to cis-[Re(CO)2(NN)Br2]−, and, finally, the stepwise substitution of Br with L to obtain cis-[Re(CO)2(NN)BrL2] and cis-[Re(CO)2(NN)L2]+. It is interesting to point out here that, contrary to other similar complexes, the presence of NN in the coordination sphere of the 17-electron ReII complexes (1a–c and 11) imparts stability to the molecules, which are stable in solution and do not decompose by releasing CO [63,64].
New complexes were characterized by standard techniques, including X-ray crystallography for the dicarbonyl species 4a and 11 (Figure 2) and tricarbonyl complexes 6–8 and 10 (Figure 3, with relevant bond distances and angles for the solid-state molecular structures in the Supplementary Materials). Within the series of dicarbonyl cis-[Re(CO)2(NN)Br2] species, the preparation of compound 11 (where NN = bathophenanthroline: batho-phen) was particularly challenging. Indeed, the reaction of either cis-[ReIII(CO)2Br4]− or cis-[ReII(CO)2Br4]2− with bathophen leads to a mixture of products which are very difficult to separate. Normally, cis-[Re(CO)2(NN)Br2] complexes are obtained as cis-cis-trans species (with the two Br atoms in the trans position to each other). Only when one of the bromides is substituted for L, the intermediate penta-coordinated complexes undergo Berry pseudorotation, which establishes an equilibrium between the cis-cis-trans and cis-cis-cis isomers [61]. These can be separated by column chromatography and crystallized separately (as in the case of 3a, Figure 2). In the preparation of 11, we found not only that the reaction leads to disproportionation, giving 10, but also that the cis-cis-cis isomer of 11 (cis-11) and the mono carbonyl mer-[Re(CO)(NN)Br3] complex (mer-12, Figure 2) are formed. Complex 11 can be separated from the mixture, but despite our efforts, the other complexes formed could not be eluted separately in our chromatographic purification procedures. We should underline here that we were able to identify the products obtained in the reaction only by co-crystallizing them in a mixture. We also note that, to our knowledge, mer-[Re(CO)(NN)Br3] (mer-12) is a unique example of a diimine rhenium mono carbonyl complex that is structurally characterized.
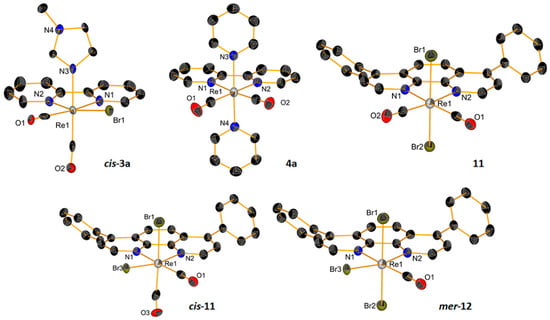
Figure 2.
ORTEP representations of the crystal structures of Re dicarbonyl complexes. Thermal ellipsoids are at 30% probability. Hydrogen atoms are omitted for clarity. Note: the compounds cis-11 and mer-12 co-crystallize in a mixture, where 10 and 11 are also present.
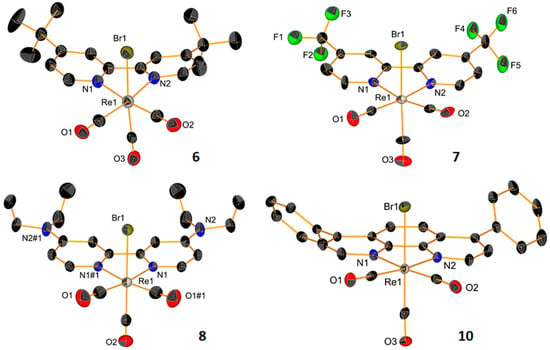
Figure 3.
ORTEP representations of the crystal structures of Re tricarbonyl complexes 6–8 and 10. Thermal ellipsoids are at 30% probability. Hydrogen atoms are omitted for clarity.
2.2. Antimicrobial Properties of the Complexes
The antimicrobial activity of complexes 1–11 (15 neutral, 6 cationic) was determined against eight different microorganisms, including four Gram-negative bacteria (Enterobacter cloacae ATCC 3047, Klebsiella pneumoniae ATCC 13803, Acinetobacter baumannii ATCC 19606, and Pseudomonas aeruginosa PAO1 NCTC10332), two Gram-positive bacteria (methicillin-resistant Staphylococcus aureus MRSA43300 and methicillin-sensitive S. aureus ATCC25923), and two fungi (Candida albicans SC5314 and C. auris, a clinical strain). The species of these two genera are responsible for the majority of hospital-acquired infections and are challenging to treat, especially in the case of their co-infections [65]. The results of our study are given in Table 1. We found that none of the dicarbonyl complexes showed antimicrobial potential. Only compounds 4b, 5b, and 11 were weakly active against the S. aureus strains, but their MIC values (25 and 50 μM, respectively) were much higher than those of the active rhenium complexes 13–19 (Figure 4) [31,32,48,49,50].

Table 1.
Antimicrobial activity, assessed by determining the minimal inhibitory concentrations (MIC, µM, mean of n = 3 independent experiments) of different Re-bearing complexes.
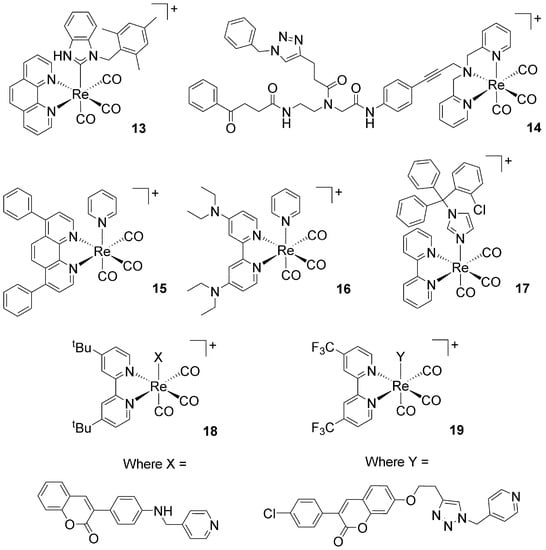
Figure 4.
Structures of previously published active fac-[Re(CO)3]+ complexes. Complex 13 [32]; complex 14 [31]; complexes 15, 16, 18, and 19 [49,50]; complex 17 [48].
2.3. Molecular Docking Study: Membrane-Bound S. aureus Proteins
The results obtained from our in vitro antimicrobial investigation raised a fundamental question, namely: “what sets apart cationic fac-[Re(CO)3]+ complexes from other structurally similar neutral complexes or compounds lacking the tricarbonyl core?” Or, in other words, “why are complexes 13–19 (Figure 4) active antimicrobial agents while other rhenium complexes are not?” Compounds 13–19 are different molecules, but they share some common features (e.g., the same charge, with a lipophilic diimine or polydentate ligand with a pyridine in the coordination sphere). In addition, Table 2 presents the predictability rates of the drug-likeness properties for these rhenium complexes. The descriptor values were retrieved using the AlvaDesc v.2 software (Milano, Italy) [66].
Table 2.
Drug-likeness properties of active antimicrobial rhenium complexes 13–19.
At this early stage of investigation, to aid in our search for an answer to the question, a purely experimental approach focused on, e.g., microbial gene expression analysis and transcriptomic data would be costly and time consuming. We thus decided to adopt an in silico approach in order to guide future synthetic, SAR, and mechanistic studies. There are, fortunately, some experimental facts that helped us to focus our attention on specific enzymes that may be considered as possible targets for one or more of the compounds 13–19. Although mechanistic studies are limited and specific biological targets are still unknown, the effective antimicrobial fac-[Re(CO)3]+ complexes appear to act predominately on the membrane of the bacteria. The complex of Metzler-Nolte and Bandow, i.e., compound 14 in Figure 4, targets the cytoplasmic membrane of Bacillus subtilis, affecting its architecture and disrupting essential cellular processes taking place at the membrane, such as respiration, as well as cell wall biosynthesis and integrity [30]. Similarly, Mendes et al. have shown that the mechanism of action of the fac-[Re(CO)3(bpy)(ctz)]+ complex (17 in Figure 4, where ctz = the drug clotrimazole) involves a sequence of events initiated by membrane insertion, followed by membrane disorganization, the inhibition of peptidoglycan biosynthesis, and the breakdown of the membrane potential [48].
Based on these data, in order to possibly understand the differences in the antimicrobial effects of the previously published active fac-[Re(CO)3]+ complexes (13- 19, Figure 4) and inactive fac-[Re(CO)3]+ and cis-[Re(CO)2]n complexes, we decided to investigate the binding affinity of all the above compounds against membrane-bound S. aureus proteins. The in silico docking studies were also performed in order to gain insights on the possible targets of the molecules by the careful analysis of the data. A PDB search revealed that nine structurally characterized membrane-bound S. aureus MRSA proteins are available on the database. Of these, we selected eight, comprising four penicillin-binding proteins (PBPs) [67,68,69,70] and the following enzymes: lipoteichoic acid synthase [71] (specifically its extracellular catalytic domain, eLtaS), type-I signal peptidase (SpsB) [72], lipoprotein signal peptidase II (LspA) [73], and lipoteichoic acid flippase (LtaA) [74]. The pre-screening of the binding affinities (b.a.) was performed with the AutoDock Vina software [14]. The calculated b.a. were recorded as docking scores in kilocalories per mole (kcal/mol), and the results are given in the Supplementary Materials (Table S1a,b). In the initial screening, the metal complexes were first docked at the known inhibitor-binding site of the specific protein, and the b.a. was compared to that of the same inhibitor. At this stage, only proteins where complexes showed a b.a. of ca. −9.0 kcal/mol that was greater than the corresponding inhibitor’s b.a. (Δ values in Table S1a,b), or a b.a. of ca. −10.0 kcal/mol that was comparable to the corresponding inhibitor’s b.a., were considered as possible targets for the complexes.
Within the abovementioned constrains, in general terms, our initial analysis revealed the following (detailed values are in the Supplementary Materials, Table S1a,b):
- (1)
- With the exception of the cis-[Re(CO)2]n complexes 1b–3b and the fac-[Re(CO)3]+ complexes 6, 7, and 10, none of the inactive rhenium di- or tricarbonyl compounds showed any b.a. for the enzyme evaluated.
- (2)
- The inactive molecules 1b–3b, 6, 7, and 10 showed an affinity for the penicillin-binding protein 4 (PBP4), with b.a. values ranging from −8.9 (1b) to −12.3 (10) kcal/mol.
- (3)
- Compound 10 also showed a strong affinity for lipoteichoic acid flippase (LtaA), with a b.a. of −10.3 kcal/mol.
- (4)
- Amongst the active antimicrobial rhenium complexes (i.e., molecules 13–19, Figure 4), complexes 16 and 17 showed the lowest b.a. values for the selected enzymes. These were higher than those of the inactive compounds but lower than those of the known inhibitors.
- (5)
- With variations within the series, the other active antimicrobial rhenium complexes (13–15 and 18–19) showed good b.a. values for five enzymes. These were the penicillin-binding protein 4 (PBP4, b.a. ranging from −9.1 (13) to −10.7 (19) kcal/mol); type-I signal peptidase (SpsB, all complexes except 14, b.a. ranging from −9.1 (13) to −10.4 (19) kcal/mol); lipoteichoic acid synthase (LtaS, only 15, 18, and 19, b.a. ranging from −9.4 (15) to −10.9 (19) kcal/mol); lipoteichoic acid flippase (LtaA, only 15, 18, and 19, b.a. ranging from −10.4 (15) to −11.3 (19) kcal/mol).; and lipoprotein signal peptidase II (LspA, all complexes except 13, b.a. ranging from −8.7 (15) to −10.6 (18) kcal/mol).
Interestingly, PBP4, LtaS, and LtaA are all involved in bacterial wall biosynthesis [70,72,74,75,76,77,78]. PBP4 is a transpeptidase that performs the crosslinking reaction in the synthesis of the peptidoglycan backbone [78]. LtaS catalyzes the polymerization of lipoteichoic acid (LTA) polyglycerol phosphate, a reaction that presumably uses phosphatidylglycerol as a substrate [71]. This enzyme is required for staphylococcal growth and the cell division process [79,80]. LtaA acts upstream of LtaS [81], and it is presumed to catalyze the translocation reaction of anchor lipid-linked disaccharide gemfibrozil-diacylglycerol from the cytoplasmic leaflet of the membrane to the extracellular side of the plasma membrane, where lipoteichoic acids are assembled [74,75,76,77]. A flippase with a similar structure (MurJ) [82,83] is also involved in the translocation of disaccharide-pentapeptide building blocks, attached to a polyisoprene lipid carrier (called lipid II), across the cytoplasmic membrane, where peptidoglycan polymerization (i.e., the polysaccharide matrix that protects bacteria from osmotic lysis) takes place [77]. The remaining two proteins are SpsB and LspA. SpsB is a proteolytic enzyme that plays a crucial role in bacterial viability by processing proteins that are translocated across the membrane [72,84], while LspA is involved in bacterial lipoprotein post-translational processing [85] and is essential for the survival and virulence of Gram-positive bacteria [86,87]. This latter enzyme is considered as one of the major targets for the development of new antibiotics [88]. The calculated binding affinities of active Re complexes for these possible targets (b.a. ranging from ca. −9 to −11 kcal/mol) are fully consistent with the experimental results reported by Wenzel et al. [30] and Mendes et al. [48], in that the inhibition of these proteins leads to membrane disorganization and affects peptidoglycan/wall biosynthesis [70,74,75,76,77,78].
Following this initial screening, the active complexes 13–19 were more comprehensively analyzed for their binding affinities for the selected receptors. Extensive semi-flexible docking was performed, introducing flexibility into the amino acid side chains and complex rotatable bonds of the binding pockets of the receptors. The number of modes was set to 200, and the exhaustiveness was set to 40. Each docked complex was calculated in triplicate mode. The triplication test detects whether there is a variation in the obtained cluster compactness of the poses and changes in the top-ranked compounds from the previous run; thus, one avoids bias in the scoring. If a bias in the scoring is present, the solution for such a case, along with the control experiment, is a reduction in the chemical space search (e.g., narrowing of the search box). The performed protocol provides information as to whether the best selected molecules remain amongst the highest scored compounds in the rank-ordered docking list. After the calibration procedure for the docking, the molecules were virtually screened against the eight target proteins. The localization of the active pocket amino acid residues was predicted according to Jendele et al. [89]. The results are summarized in Table 3, while the detailed ranking of the obtained pockets is in the Supplementary Materials (Table S2).
Table 3.
Predicted binding sites.
Accordingly, the computational results of this library of compounds are shown in Table 4. For the PBP receptors, the docking protocol identified 15 and 19 as having the greatest b.a. for these enzymes, particularly for PBP2a and PBP4 (Table 4). As other non-active rhenium complexes showed a b.a. for PBP4, we posit that this protein is not a probable target for active complexes. Conversely, the b.a. of 15 and 19 for PBP2a is of interest (b.a. values of −9.2 and −9.8 kcal/mol respectively, Table 4). The expression of the penicillin-binding protein 2a (PBP2a) is responsible in methicillin-resistant S. aureus (MRSA) for the high-level resistance of the bacteria to β-lactam antibiotics [78]. PBP2a is a unique transpeptidase, as it is capable of catalyzing cell wall crosslinking despite the presence of β-lactam antibiotics. The inhibition of PBP2a by 15 and 19 may thus possibly additionally account for the strong antimicrobial activity of these complexes against MRSA [49,50]. Computationally, in the case of 19, the stabilization of the protein–drug complex is based on the detected H-bonds between the compound and the surrounding amino acid environment (Ser, Thr, and Gln residues). A detailed distribution of the amino acids of the best complexes is given in the Supplementary Materials (Table S3). In this case, the intramolecular backbone H-bonds stabilize the β-turn structure with the ligand position.
Table 4.
Molecular docking scores and related properties.
For the second group of receptors (namely LtaS, SpsB, LtaA, and LspA), the docking protocol identified complexes 14, 15, 18, and 19 as having a high b.a. for lipoteichoic acid flippase (LtaA, all complexes except 14) and lipoprotein signal peptidase II (LspA, see Table 4). As mentioned above, flippases such as LtaA catalyze the translocation reactions of anchor lipid-linked disaccharide gentiobiosyl-diacylglycerol and lipid II across the cytoplasmic membrane, where essential cell wall polymers (i.e., lipoteichoic acid and peptidoglycan) are assembled (Figure 5) [74,75,76,77,81,82,83]. LspA, on the other hand, is involved in bacterial lipoprotein post-translational processing [85] and is essential for the survival and virulence of Gram-positive bacteria (Figure 6) [86,87]. The possible inhibition of these enzymes by active antibiotic rhenium complexes would disrupt essential cellular processes taking place at the membrane and ultimately lead to cell death. It should be mentioned that our computational analysis did not identify possible targets of complexes 13, 16, and 17. If, for the former complexes, this indicates that the compounds may exert their antibiotic activity against MRSA via mechanisms not involving membrane-bound proteins, for 17, the results appear to support the experimental evidence of Mendes et al. [48]. Indeed, the authors reported that 17 interferes with the cycling of the undecaprenyl precursor in peptidoglycan biosynthesis (“lipid II cycle”), leading to the accumulation of UDP-MurNAc-pentapeptide (i.e., lipid I, the ultimate cytoplasmic peptidoglycan precursor) in the cytoplasm of treated cells. Thus, 17 inhibits the MurG-mediated conversion of lipid I to lipid II [31]. The X-ray structure of MRSA MurG is not available in the PDB database; thus, we could not computationally confirm the experimental data of Mendes et al. [48].
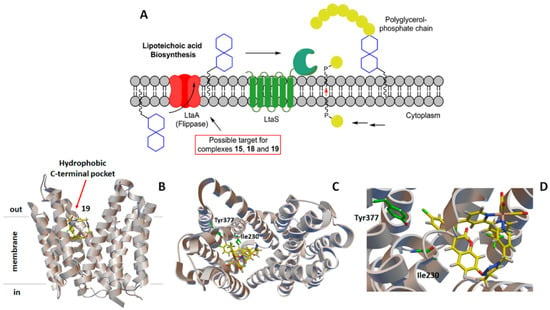
Figure 5.
(A) Schematic diagram of the lipoteichoic acid synthetic machinery in MRSA with the possible target of the active antimicrobial rhenium complexes. For more details about scheme (A), see [75]. Computer-generated lowest energy pose of the selected complex 19 in the hydrophobic C-terminal pocket of lipoteichoic acid flippase (LtaA): (B) side view; (C) top view; (D) detail of the binding region. In (C,D), the two amino acid residues most likely involved in the H-bonding interactions with 19 are shown in green.
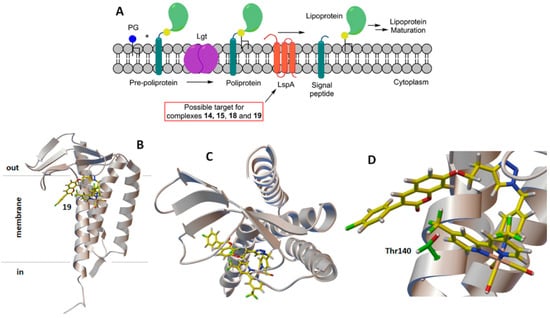
Figure 6.
(A) Schematic diagram of the lipoprotein post-translational processing pathway with the possible target of the active antimicrobial rhenium complexes. For more details about scheme (A), see [73]. Computer-generated lowest energy pose of selected complex 19 in lipoprotein signal peptidase II (LspA): (B) side view; (C) top view; (D) detail of the binding region. In (C,D), the amino acid residue most likely involved in the H-bonding interactions with 19 is shown in green.
Finally, in Figure 7 and Figure 8, the hydrophobic gaussian surface was used for the graphical representation of the binding pockets of the ligands. The hydrophobicity scales of Wimley and White were used for defining the hydrophobicity of the amino acid residues [90]. This prediction assumes that apolar sites are preferentially disposed to the molecular interior, forming a hydrophobic core, whereas polar sites are disposed outside the molecular interior. In the Supplementary Materials (Figure S8), the representation of the protein surface of the non-polar polar ratio (NPP) [91] and the patch analysis of the electrostatic surface potential are depicted [92]. To analyze the effect on the distortion of the receptor and the conformation changes when binding the complex, the results of the Rg for the receptors and the complexes are presented in Table 2. As can be understood from the values, the Rgs of the explored systems did not change significantly for any of the shown complexes. The solvent accessible surface area (SASA) was also assessed for all cases. We did not observe intrinsic flexibility changes of the receptor SASA and system SASA, which can also be seen from the data in the Table 2. We found, in most cases, that the interfaces gain accessibility in order to promote stable interactions. The localization of the complexes preserves the SASA, which is an indication of the protein stability in the presence and absence of the complexes (i.e., ligands). With this property, we have a clearer picture of the existing changes in the protein conformation. The available surface area is maintained before and after the docking, and, as intuitively predicted, the rhenium complexes prefer localizing in the hydrophobic pockets of the possible target enzymes (Figure 7 and Figure 8).
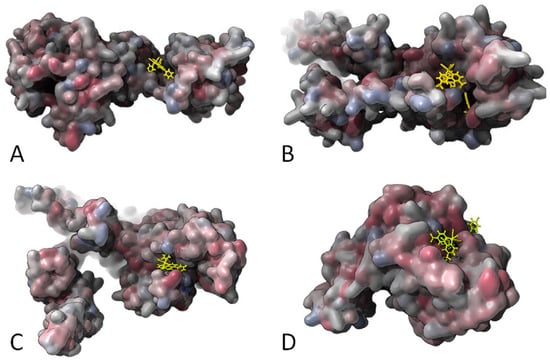
Figure 7.
Gaussian surface representation of the hydrophobicity of (A) 15 and PBP2 (2OLV); (B) 19 and PBP2a (4DKI); (C) 15 and PBP3 (3VSL); and (D) 19 and PBP4 (5TXI). The red-blue color palette changes from hydrophilic blue to hydrophobic red.
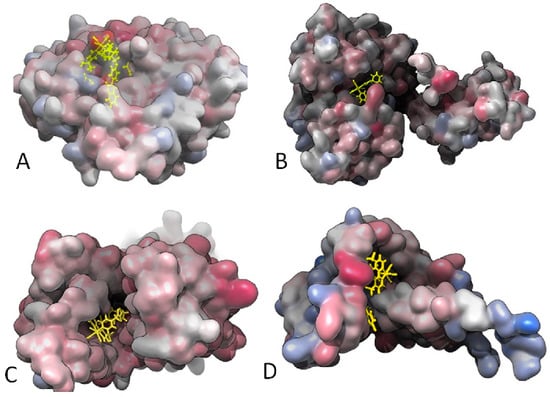
Figure 8.
Gaussian surface representation of the hydrophobicity of (A) 18 and Lipoteichoic acid synthase (LtaS; 2W5Q); (B) 15 and type-I signal peptidase (SpsB; 4WVJ); (C) 19 and Lipoteichoic acid flippase (LtaA), top view (6S7V); and (D) 19 and Lipoprotein signal peptidase II (LspA; 6RYP). The red-blue color palette changes from hydrophilic blue to hydrophobic red.
3. Conclusions
In this study, we reported the synthesis, characterization, and antimicrobial effects of a series of rhenium di- and tricarbonyl diimine complexes. Due to the lack of activity of the tested species, in an effort to identify the possible targets of the active complexes (and thus possibly understand the underlying reasons for the observed differences in the antimicrobial efficacy of Re complexes), we computationally evaluated the binding affinity of the active and inactive molecules against structurally characterized membrane-bound S. aureus proteins. Whereas the inactive compounds did not show an affinity for the enzymes, our docking protocol identified two possible major targets for some molecules of this class of compounds, namely lipoteichoic acid flippase (LtaA) and lipoprotein signal peptidase II (LspA). To our knowledge, our study is the first reported attempt to computationally identify MRSA biological targets of antibiotic rhenium complexes [93,94,95,96,97]. Experimental data are required in the future to confirm the in silico results, but our data are in line with the limited mechanistic studies that have previously been published on microbicidal rhenium species. Indeed, if the complexes inhibit the catalytic activity of LtaA and LspA, essential cell wall polymers cannot be assembled, leading to microbial death. We emphasize that LtaA and LspA may be targets for a fraction of known active antimicrobial Re complexes (namely 14, 15, 18, and 19 in this study). The penicillin-binding protein 2a (PBP2a) might also be targeted by 15 and 19, while MurG may be inhibited by 17. We were not able to identify possible targets for compounds 14 and 16; thus, their mechanism of action and targets remain unknown. We also showed that active rhenium complexes tend to localize in the hydrophobic pockets of target enzymes. In terms of the key molecular features common to active rhenium carbonyl complexes, our data support the notion that active diimine species are only cationic complexes of the fac-[Re(CO)3]+ core. If a CO ligand is substituted, leading to dicarbonyl cis-[Re(CO)2]n, regardless of the overall charge of the compounds, the molecules are devoid of any antimicrobial activity. Arguably, the most significant outcome of our study, i.e., the identification of LtaA and LspA as possible targets for this class of antibiotics, is that it offers the scientific community involved in this research support for the rational design of rhenium complexes based on the computational protocol for computer-aided drug design.
4. Materials and Methods
4.1. Reagents and Chemicals
All reagents and solvents were purchased from standard sources and used without further purification. The compound [Re(CO)5Br] was purchased from Sigma-Aldrich. Complexes (Et4N)[Re(CO)2Br4] [62], 1a–2a [60], 1b–5b [61], 1c [98], 9a [99], 9b [100], 9c [101], and 10 [49] were synthesized according to published procedures. Unless otherwise noted, the solvents used in the preparation of all molecules were dry and O2-free.
4.2. Instruments and Analysis
A Bruker Advance III 400 MHz spectrometer was used to record the NMR spectra. The 1H chemical shifts of molecules are reported relative to the residual solvent protons. A Bruker FTMS 4.7-T Apex II spectrometer was used to perform the mass analysis in the positive mode. UV-vis spectra were measured on a Thermo Fisher Scientific Jasco V730 spectrophotometer. A Bruker TENSOR II spectrometer was used to record the IR spectra, with the following parameters: 16 scans for the background, and 32 scans for a sample, with a resolution of 4 cm−1 in the 4000–600 cm−1 region. Single crystals were measured on a Stoe IPDS2 diffractometer (CuKα1 (λ = 1.5406 Å)) equipped with a cryostat from Oxford Cryosystems. The ShelXT structure solution program was used to solve the crystal structures. We used the Intrinsic Phasing refined with the ShelXL refinement package, using least squares minimization [102,103]. Data were deposited in the Cambridge Crystallographic Data Centre. The CCDC numbers are 2184717–2184724. The elemental analysis was performed using a LECO CHNS-932 elemental analyzer.
4.3. Synthetic Procedures
(TDAE)[Re(CO)2(bpy)Br2]2 (1′) was synthesized according to a similar published procedure [61]. Briefly, [Re(CO)2(bpy)Br2] (1a, 63.5 mg, 114.0 µmol) was dissolved in dry CH2Cl2 (DCM) (20 mL) in a glove box. Tetrakis(dimethylamino)ethylene (TDAE, 13.24 µL, 57.0 µmol, 0.5 eq.) was dissolved in dry DCM (1 mL). The latter solution was added dropwise to the solution of 1a. The mixture was stirred under inert conditions for 15 min. The solvent was removed under reduced pressure, giving compound 1′ as a purple solid. Yield: 71.9 mg, 55.0 µmol, 96%. IR (cm−1), νCO: 1861, 1775. UV-vis (DMF), λmax [nm]: 593, 426, 308, 300. The results of the elemental analysis are as follows: calculated for C34H40Br4N8O4Re2: C, 31.01%; H, 3.06%; N, 8.51%; found: C, 30.71%; H, 2.94%; N, 7.89%.
[Re(CO)2(bpy)(MeIm)Br] (3a). Degassed complex 1′ (35.6 mg, 27.0 µmol) was dissolved in dry toluene (20 mL). Anhydrous N-methyl imidazole (MeIm, 4.32 µL, 54 µmol, 2 eq.) was added, and the mixture was stirred at 100 °C for 48 h. The mixture was cooled to room temperature and the brown precipitate was isolated by centrifugation, giving compound 3a. Yield: 15.8 mg, 28.1 µmol, 52%. Single crystals suitable for X-ray diffraction were grown by layering pentane on a CH2Cl2 solution of the compound, giving dark brown crystals. IR (cm−1), νCO: 1877, 1779. UV-vis (DMSO), λmax [nm]: 528, 375, 310, 303. 1H NMR (400MHz, CD2Cl2, ppm) δ: 9.06 (dd, J = 5.5, 1.1 Hz, 2 H), 8.70 (d, J = 8.2 Hz, 2 H), 8.30 (td, J = 7.9, 1.5 Hz, 2 H), 7.69 (ddd, J = 7.6, 5.6, 0.9 Hz, 2 H), 7.28 (s, 1 H), 6.79 (t, J = 1.4 Hz, 1 H), 6.59 (t, J = 1.5 Hz, 1 H), 3.57 (s, 3 H). ESI-MS (MeOH): m/z, 582.9 [M + Na]+. The results of the elemental analysis are as follows: calculated for C16H14BrN4O2Re: C, 34.29%; H, 2.52%; N, 10.00%; found: C, 34.70%; H, 2.40%; N, 9.52%.
[Re(CO)2(bpy)(py)2]PF6 (4a) was synthesized according to a similar published procedure [61]. Briefly, complex 2a (54 mg, 97 µmol) and pyridine (py, 1 mL, ca. 100 eq.) were dissolved in MeOH (20 mL), and the mixture was stirred at 70 °C overnight. The solvent was removed under reduced pressure. The residue was dissolved in water (75 mL), and a solution of KPF6 (36 mg, 194 µmol, 2 eq.) in water (5 mL) was added dropwise to the rhenium. The precipitate was isolated by centrifugation, giving compound 4a as a brown-orange solid. Yield: 45 mg, 64.1 µmol, 66%. Single crystals suitable for X-ray diffraction were grown by the diffusion of pentane into an acetone solution of the compound, giving dark orange crystals. IR (cm−1), νCO: 1901, 1823. UV-vis (DMF), λmax [nm]: 481, 357, 302. 1H NMR (400 MHz, CD2Cl2, ppm) δ: 9.32 (ddd, J = 0.73, 1.56, 5.41 Hz, 2H), 8.36–8.40 (m, 4H), 8.34 (d, J = 8.19 Hz, 2H), 8.18 (dt, J = 1.59, 7.95 Hz, 2H), 7.74 (ddd, J = 1.28, 5.47, 7.67 Hz, 2H), 7.59–7.65 (m, 2H), 7.05–7.11 (m, 4H). 13C NMR (101 MHz, CD2Cl2, ppm) δ: 205.9 (2C), 156.6 (2C), 155.3 (4C), 152.5 (2C), 141.1 (2C), 137.6 (2C), 129.3 (2C), 126.5 (4C), 125.1 (2C). ESI-MS (MeOH): m/z, 556.7 [M]+. The results of the elemental analysis are as follows: calculated for C22H18F6N4O2PRe: C, 37.66%; H, 2.59%; N, 7.99%; found: C, 37.53%; H, 2.72%; N, 7.67%.
[Re(CO)2(bpy)(MeIm)2]PF6 (5a). Compound 1′ (132 mg, 100 µmol) was dissolved in anhydrous MeIm (8 mL) and the mixture was stirred at 110 °C for 60 min. The solvent was removed under reduced pressure and the residue was purified by flash column chromatography (eluent: EtOAc 100%, then DCM/MeOH 100:0, increased to 98:2). The first fraction, compound 3a, was collected with the first gradient (100% EtOAc) as a brown solid (amount: traces). The second fraction was collected with the last gradient as mobile phase. Once dried, the counter ion was exchanged with KPF6 (17.2 mg, 93.4 µmol) in H2O (15 mL). Complex 5a was isolated by centrifugation as a violet solid. Yield: 24 mg, 33.9 µmol, 17%. IR (cm−1), νCO: 1885, 1802. UV-vis (DMF), λmax [nm]: 500, 363, 307, 300. 1H NMR (400 MHz, CD2Cl2, ppm) δ: 9.21–9.26 (m, 2H), 8.32 (dd, J = 0.86, 8.19 Hz, 2H), 8.12 (dt, J = 1.59, 7.95 Hz, 2H), 7.62 (ddd, J = 1.22, 5.44, 7.64 Hz, 2H), 7.29 (s, 2H), 6.63–6.67 (t, 2H), 6.44–6.51 (t, 2H), 3.53 (s, 6H). 13C NMR (101 MHz, CD2Cl2, ppm) δ: 207.6 (2C), 156.7 (2C), 152.5 (2C), 141.2 (2C), 140.4 (2C), 132.0 (2C), 128.6 (2C), 124.5 (2C), 122.3 (2C), 34.7 (2C). ESI-MS (MeOH): m/z, 562.7 [M]+. The results of the elemental analysis are as follows: calculated for C20H20F6N6O2PRe: C, 33.95%; H, 2.85%; N, 11.88%; found: C, 34.29%; H, 2.74%; N, 11.52%.
(TDAE)[Re(CO)2(phen)Br2]2 (1″) was synthesized according to a similar published procedure [61]. Briefly, cis-[Re(CO)2(phen)Br2] (62.8 mg, 107.9 µmol) was dissolved in dry DCM (17 mL) in a glove box. TDAE (12.56 µL, 53.9 µmol, 0.5 eq.) was dissolved in dry DCM (1.5 mL). The latter solution was added dropwise to the rhenium in the glove box, and the mixture was stirred for 15 min at room temperature. The solvent was removed under reduced pressure, giving 1″ as a brown-purple solid. Yield: 67.7 mg, 49.6 µmol, 92%. IR (cm−1), νCO: 1856, 1771.
[Re(CO)2(phen)(py)Br] (2c). Degassed complex 1″ (20 mg, 14.7 µmol) was dissolved in degassed pyridine (2 mL), and the mixture was stirred at 100 °C for 20 min. The reaction mixture was cooled down to room temperature and extracted in DCM (50 mL) with HCL 0.1 M (3 × 50 mL). The organic phase was dried over sodium sulfate, and the solvent was removed under reduced pressure. The crude was purified by flash column chromatography (stationary phase: aluminum oxide, mobile phase: pentane/EtOAc/MeOH 1:2:0, increased to 0:1:0 and finally 0:99:1), giving compound 2c as a brown solid. Yield: 0.9 mg, 1.6 µmol, 5%. IR (cm−1), νCO: 1864, 1778. UV-vis (DMSO), λmax [nm]: 511, 380, 269. 1H NMR (400MHz, CD3CN, ppm) δ: 9.61 (dd, J = 5.1, 1.2 Hz, 2H), 8.84 (dd, J = 8.3, 1.3 Hz, 2H), 8.25–8.29 (m, 2H), 8.17 (s, 2H), 8.11 (dd, J = 8.3, 5.1 Hz, 2H), 7.71–7.78 (m, 1H), 7.19 (s, 2H). The results of the elemental analysis are as follows: calculated for C19H13BrN3O2Re: C, 39.25%; H, 2.25%; N, 7.23%; found: C, 39.42%; H, 2.15%; N, 6.99%.
[Re(CO)2(phen)(MeIm)Br] (3c). Degassed complex 1″ (17 mg, 12.5 µmol) was dissolved in anhydrous MeIm (2 mL) and the mixture was stirred at 110 °C for 20 min. The reaction mixture was cooled down to room temperature and extracted in DCM (50 mL) with HCl 0.1 M (3 × 50 mL). The organic phase was dried over sodium sulfate, and the solvent was removed under reduced pressure. The crude was purified by flash column chromatography (stationary phase: aluminum oxide, mobile phase: pentane/EtOAc 1:1, increased to 0:1), giving compound 3c as a violet solid. Yield: 1.8 mg, 3.1 µmol, 12%. IR (cm−1), νCO: 1876, 1773. UV-vis (DMSO), λmax [nm]: 528, 275. 1H NMR (400MHz, (CD3)2SO, ppm) δ: 9.46–9.50 (m, 2H), 8.73–8.78 (m, 2H), 8.21 (s, 2H), 8.01–8.07 (m, 2H), 7.64–7.67 (m, 1H), 6.81–6.84 (m, 1H), 6.46–6.49 (m, 1H), 3.41 (s, 3H). The results of the elemental analysis are as follows: calculated for C18H14BrN4O2Re: C, 36.99%; H, 2.41%; N, 9.59%; found: C, 37.38%; H, 2.73%; N, 10.05%.
The following general procedure was applied for the synthesis of complexes 6–8 [50]. To a solution of [Re(CO)5Br] (1.0 equiv.) in hot toluene, the appropriate bipyridine (bpy) ligand (1.0 equiv.) was added, and the mixture was refluxed for 7–9 h. After the solution had cooled to the room temperature, the reaction mixture was filtered and washed with cold toluene (2×), yielding fac-[Re(CO)3(bpy)Br] as a bright fluorescent yellow powder. The solid was then dried in vacuo for 24h. The complexes were found to be pure (≥ 96%) by NMR and HPLC.
fac-[Re(CO)3(tBu-bpy)Br] (6), where tBu-bpy is 4,4′-di-tert-butyl-2,2′-bipyridine. Pale yellow solid, yield 92%. IR (solid, cm−1); νCO: 2016, 1912, 1889, 1869. Uv-vis (DMF), λmax [nm]: 368, 292. 1H-NMR (400 MHz, CD3CN, ppm): 8.96 (d, J = 5.99 Hz, 2H) 8.10 (d, J = 1.71 Hz, 2H) 7.51 (dd, J = 5.87, 1.96 Hz, 2H) 1.45 (s, 18H). ESI+-MS (MeOH): m/z, 576.9 [Re(CO)3(C18H24N2)(H2O)]+, [M-Br + H2O]+. Single crystals suitable for X-ray diffraction were grown by the diffusion of pentane into a DCM solution of the compound, giving yellow needles. The results of the elemental analysis are as follows: calculated for C21H24BrN2O3Re: C, 40.78%; H, 3.91%; N, 4.53%; found: C, 40.43%; H, 4.08%; N, 4.44%.
fac-[Re(CO)3(CF3-bpy)Br] (7), where CF3-bpy is 4,4′-bis(trifluoromethyl)-2,2′-bipyridine. Orange solid, yield 87%. IR (solid, cm−1); νCO: 2015, 1932, 1897. UV-vis (DMF), λmax [nm]: 417, 304. 1H-NMR (400 MHz, CD3CN, ppm): 9.33 (d, J = 5.75 Hz, 2H) 8.46 (s, 2H) 7.84 (dd, J = 5.75, 1.22 Hz, 2H). ESI+-MS (MeOH): m/z, 580.7 [Re(CO)3(C12H6F6N2)(H2O)]+, [M-Br + H2O]+. Single crystals suitable for X-ray diffraction were grown by the diffusion of hexane into a DCM solution of the compound, giving orange needles. The results of the elemental analysis are as follows: calculated for C15H6BrF6N2O3Re: C, 28.05%; H, 0.94%; N, 4.36 %; found: C, 27.59%; H, 1.12%; N, 4.23%.
fac-[Re(CO)3((Et)2N-bpy)Br] (8), where (Et)2N-bpy is N4,N4,N4′,N4′-tetraethyl-[2,2′-bipyridine]-4,4′-diimine. Pale yellow solid, yield 92%. IR (solid, cm−1); νCO: 2008, 1886, 1866. UV-vis (DMF), λmax [nm]: 367, 373. 1H-NMR (400 MHz, CD3CN, ppm): 8.49 (d, J = 6.60 Hz, 2H) 7.04 (d, J = 2.81 Hz, 2H) 6.54 (dd, J = 6.72, 2.69 Hz,2H) 3.49 (q, J = 7.21 Hz, 8H) 1.28 (t, J = 7.21 Hz, 12 H). ESI+-MS (MeOH): m/z, 568.9 [Re(CO)3(C18H26N4)]+, [M-Br]+. Single crystals suitable for X-ray diffraction were grown by the diffusion of pentane into a DCM solution of the compound, giving yellow needles. The results of the elemental analysis are as follows: calculated for C21H26BrN4O3Re: C, 38.89%; H, 4.04%; N, 8.64%; found: C, 38.45%; H, 4.17%; N, 8.47%.
cis-[Re(CO)2(batho-phen)Br2] (11). Degassed (Et4N)[Re(CO)2Br4] (500 mg, 722 µmol) and batho-phen (240 mg, 722 µmol) were dissolved in dry DCM (80 mL). The mixture was stirred under inert conditions at room temperature for 72 h. The solvent was removed under reduced pressure, and the crude was purified by flash column chromatography on silica (Eluent: DCM / Pentane 1:9), giving complex 11 as an orange-red solid. Yield: 82 mg, 112 µmol, 15%. Single crystals suitable for X-ray diffraction were grown by the slow evaporation of DCM solution of the compound, giving dark brown needles. IR (cm−1), νCO: 1999, 1849. UV-Vis (DMF), λmax [nm]: 429, 288. The results of the elemental analysis are as follows: calculated for C26H16Br2N2O2Re: C, 42.52%; H, 2.20%; N, 3.81%; found: C, 42.00%; H, 2.42%; N, 3.84%.
4.4. Biological Tests
The culture conditions and in vitro antimicrobial activity determination were performed exactly as previously reported [60]. We thank Dr Aleksandra Barac (University Clinical Center of Serbia) and Prof. Cornelia Lass-Floerl (University of Innsbruck) for kindly providing the clinical C. auris strain 7. All other microbial strains (Enterobacter cloacae ATCC 3047, Klebsiella pneumoniae ATCC 13803, Acinetobacter baumannii ATCC 19606, Pseudomonas aeruginosa PAO1 NCTC10332, Staphylococcus aureus MRSA43300 (methicillin-resistant) and S. aureus ATCC25923 (methicillin-sensitive), and Candida albicans SC5314) were obtained from the American Type Culture Collection (ATCC) and the National Collection of Type Cultures (NCTC).
4.5. In Silico Calculations
4.5.1. Preparation of the Ligand Database and Ligands: Receptors Complexes
The docking calculations were performed with AutoDock Vina version 1.2.0 (The Scripps Research Institute, La Jolla, San Diego, CA, USA) [14] and AutoDock4 version 4.2.6 (AD4, The Scripps Research Institute, La Jolla, San Diego, CA, USA) [104]. The receptor/protein PDBQT files were prepared, and the grid box size was determined using the AutoDock Tools version 1.5.7 (ADT, Scripps Research Institute, La Jolla, San Diego, CA, USA) [104]. Biovia Discovery Studio Visualizer 2021, version 21.1.0.20298 (Dassault Systèmes, San Diego, California, CA, USA), was used to visualize the receptor and ligand interactions. Figures were prepared with the ADT software. The structures of complexes 5–11 and 15 were obtained by the determined X-ray structures. Chemical structures as .CIF files were converted to .MOL2 files using the Mercury (Build RC1) software, version 3.7 (CCDC 2001–2015). All complexes (ligands) were optimized with the hybrid meta-GGA functional wB97XD [105,106,107,108,109], designed to account for dispersion, which was used in combination with the standard SDD basis sets [110]. The optimized structures were subject to frequency analysis to verify that they represented minima on the potential energy surface. All calculations were performed with Gaussian 09 software (version 5.0.9, Carnegie Mellon University, Gaussian, Inc., Wallingford, CT, USA).
The ADT software was then used to investigate the complexes’ structures in terms of their combinations with nonpolar hydrogens, additions of Gasteiger changes, and rotatable bonds. The rhenium atom is not parametrized in AD4 and AutoDock Vina; thus, AutoDock Vina calculations were performed using Mn instead of Re. The resulting binding poses of the Mn complexes were then cross-checked with the corresponding Re complexes using AD4, where the following line was added to the AD4 atom parameters file: “atom_par Re 2.95 0.066 12.000 -0.00110 0.0 0.0 0 -1 -1 1 # Non-H-bonding”. The binding poses of the Mn and Re complexes were found to be the same. Moreover, due to the fact that ADT failed to assign a Gasteiger change to the metal ion, a charge of 0.320 (to either Mn or Re) was assigned to the atom by editing the corresponding .PDBQT file [111].
The crystal structure of S. aureus proteins were obtained from the RCSB protein data bank (http://www.rcsb.org). Only the structures of membrane protein annotation (PDBTM, MemProtMD, OPM or mpstruc) were considered and selected. All water molecules were removed, and the required files for AutoDock Vina and AD4 were prepared by assigning hydrogens and Kollman charges to the protein structures, and finally converting them from the .PDB file format to .PDBQT file format.
4.5.2. Molecular Docking
The docking calculations were conducted using the AutoDock Vina software (https://vina.scripps.edu/) with adapted parameters for the rhenium complexes. The extended version of the Vina code was used via the integrated platform SAMSON [https://www.samson-connect.net] as a SAMSON extension [112]. This provides additional functionality when preparing receptors and ligands, docking libraries, and analyzing and exporting docking results. Both the number of flexible side chains and the size of the search domain were different for all the cases because of the receptor’s conformation (i.e., the chain orientation and position of residues). On average, there were about 30 flexible side chains with unlocked rotatable bonds. The search space was defined by a docking box wrapper of the space around the receptors. The scaling of the box depended on the defined pocket score. The number of modes was set to 200 with an energy range = 3 kcal/mol (default value). The energy range is the maximum energy difference between the best binding mode and the unfavorable one displayed (kcal/mol). Energy (affinity) values that differed by more than 3 kcal/mol from the best mode were not saved among the results. In the configuration file, the parameter called “exhaustiveness” was set to 40. This parameter controls how comprehensive the search space is. In AutoDock Vina, the electrostatic interactions were handled with the hydrophobic and the hydrogen bonding terms. The post-docking analysis of the favorable ligand–receptor complexes was performed via the Protein–Ligand Interaction Analyzer Extension in SAMSON [112]. With the help of the Protein–Ligand Interaction Analyzer, it was possible to calculate the radius of gyration, hydrogen bonds, residues surrounding the ligand, and solvent-accessible surface area (SASA) of the receptor and ligand and of the form complexes. The multistep validation protocol was considered in this study, and the ability of the combined methodology was examined independently through initial screening and extensive semi-flexible docking.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ph15091107/s1, Figures S1–S8: 1H-NMR spectra of compounds; Figure S9: IR spectra (solid state) of compounds; Figure S10: UV-vis spectra (in DMF) of compounds; Figure S11: Visualization of the surface protein surface polarity: (A) non-polar to polar SASA color-coded from low NPP ratio (purple) to high NPP ratio (green); and (B) color-coded from negative charge (red) to positive charge (blue). Regions of high hydrophobicity are colored green, low hydrophobicity are colored purple; Figure S12a: Binding orientation of the compounds with hydrogen–acceptor and hydrogen–donor distances: A. 15 and PBP2: 2OLV; B. 19 and PBP2a: 4DKI; C. 15 and PBP3: 3VSL; D. 19 and PBP4: 5TXI; Figure S12b: Binding orientation of the compounds with hydrogen–acceptor and hydrogen–donor distances: A. 18 and lipoteichoic acid synthase (LtaS): 2W5Q; B. 15 and type-I signal peptidase (SpsB): 4WVJ; C. 19 and lipoteichoic acid flippase (LtaA): 6S7V; D. 19 and lipoprotein signal peptidase II (LspA): 6RYP; Table S1a: In silico pre-screening of binding affinities (b.a.; docking scores, kcal/mol) of rhenium complexes against structurally characterized membrane-bound S. aureus proteins: penicillin-binding proteins (PBPs); Table S1b: In silico pre-screening of binding affinities (b.a.; docking scores, kcal/mol) of non-toxic complexes against other structurally characterized membrane-bound S. aureus proteins; Table S2: Pockets prediction: mapping the ranking with residue environment distribution; Table S3: Percentage distribution of the surrounding residue types for the two groups of proteins; Table S4a–S4i: Selected bond lengths of the complexes; Table S4a’–S4i’: Selected bond angles of the complexes.
Author Contributions
Conceptualization, F.Z. and M.N.; methodology, M.N. and F.Z.; software, M.N., A.C., M.L. and F.Z.; validation, M.N., A.C., A.P. and F.Z.; formal analysis, K.S., Y.C., M.N., A.C., A.P. and F.Z.; investigation, K.S., Y.C., M.N., A.C., A.P. and F.Z.; resources, M.L., A.P. and F.Z.; data curation, K.S., Y.C., M.N., A.C., A.P. and F.Z.; writing—original draft preparation, K.S., Y.C., M.N., A.P. and F.Z.; writing—review and editing, all authors; visualization, M.N., A.C., A.P. and F.Z.; supervision, M.L., A.P. and F.Z.; project administration, A.P. and F.Z.; funding acquisition, F.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Swiss National Science Foundation (project# 200021_196967 and NCCR Bioinspired Materials) and the Ministry of Education, Science and Technological Development of the Republic of Serbia (project no. 451-03-9/2021-14/200042, 2021).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Financial support from the Fonds de Recherche du Centenaire de l’Université de Fribourg (project no. 818) and National Competence Centre for Research (NCCR), “Bioinspired Materials”, is gratefully acknowledged. A.P. has received the financial support from Ministry of Education, Science and Technological Development of the Republic of Serbia (Project No 451-03-68/2022-14/200042).
Conflicts of Interest
The authors declare no conflict of interest.
References
- WHO. Global Shortage of Innovative Antibiotics Fuels Emergence and Spread of Drug-Resistance; WHO: Geneva, Switzerland, 2021; p. 1. [Google Scholar]
- Hu, Q.; Cheng, H.; Yuan, W.; Zeng, F.; Shang, W.; Tang, D.; Xue, W.; Fu, J.; Zhou, R.; Zhu, J.; et al. Panton-Valentine leukocidin (PVL)-positive health care-associated methicillin-resistant Staphylococcus aureus isolates are associated with skin and soft tissue infections and colonized mainly by infective PVL-encoding bacteriophages. J. Clin. Microbiol. 2015, 53, 67–72. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tong, S.Y.C.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G. Staphylococcus aureus Infections: Epidemiology, Pathophysiology, Clinical Manifestations, and Management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [PubMed]
- Laupland, K.B.; Lyytikäinen, O.; Sgaard, M.; Kennedy, K.J.; Knudsen, J.D.; Ostergaard, C.; Galbraith, J.C.; Valiquette, L.; Jacobsson, G.; Collignon, P.; et al. The changing epidemiology of Staphylococcus aureus bloodstream infection: A multinational population-based surveillance study. Clin. Microbiol. Infect. 2013, 19, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Plackett, B. Why big pharma has abandoned antibiotics. Nature 2020, 586, S50–S52. [Google Scholar] [CrossRef]
- Yu, W.; MacKerell, A.D., Jr. Computer-Aided Drug Design Methods. Methods Mol. Biol. 2017, 1520, 85–106. [Google Scholar] [CrossRef] [PubMed]
- Rao, Q.; Shang, W.; Hu, X.; Rao, X. Staphylococcus aureus ST121: A globally disseminated hypervirulent clone. J. Med. Microbiol. 2015, 64, 1462–1473. [Google Scholar] [CrossRef]
- Yu, W.; Lakkaraju, S.K.; Raman, E.P.; Fang, L.; MacKerell, A.D. Pharmacophore Modeling Using Site-Identification by Ligand Competitive Saturation (SILCS) with Multiple Probe Molecules. J. Chem. Inf. Model. 2015, 55, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Nedyalkova, M.; Simeonov, V. Partitioning Pattern of Natural Products Based on Molecular Properties Descriptors Representing Drug-Likeness. Symmetry 2021, 13, 546. [Google Scholar] [CrossRef]
- Barazorda-Ccahuana, H.L.; Nedyalkova, M.; Mas, F.; Madurga, S. Unveiling the Effect of Low pH on the SARS-CoV-2 Main Protease by Molecular Dynamics Simulations. Polymers 2021, 13, 3823. [Google Scholar] [CrossRef] [PubMed]
- Ustach, V.D.; Lakkaraju, S.K.; Jo, S.; Yu, W.; Jiang, W.; MacKerell, A.D. Optimization and Evaluation of Site-Identification by Ligand Competitive Saturation (SILCS) as a Tool for Target-Based Ligand Optimization. J. Chem. Inf. Model. 2019, 59, 3018–3035. [Google Scholar] [CrossRef] [PubMed]
- Gaieb, Z.; Parks, C.D.; Chiu, M.; Yang, H.; Shao, C.; Walters, W.P.; Lambert, M.H.; Nevins, N.; Bembenek, S.D.; Ameriks, M.K.; et al. D3R Grand Challenge 3: Blind prediction of protein–ligand poses and affinity rankings. J. Comput. Aided Mol. Des. 2019, 33, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Śledź, P.; Caflisch, A. Protein structure-based drug design: From docking to molecular dynamics. Curr. Opin. Struct. Biol. 2018, 48, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Cournia, Z.; Allen, B.; Sherman, W. Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations. J. Chem. Inf. Model. 2017, 57, 2911–2937. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.K.; Ahmed, S.F.; Hossain, M.S.; Bhojiya, A.A.; Mathur, A.; Upadhyay, S.K.; Srivastava, A.K.; Vishvakarma, N.K.; Barik, M.; Rahaman, M.M.; et al. Molecular docking and simulation studies of flavonoid compounds against PBP-2a of methicillin-resistant Staphylococcus aureus. J. Biomol. Struct. Dyn. 2021, 1–17. [Google Scholar] [CrossRef]
- Kulanthaivel, L.; Jeyaraman, J.; Biswas, A.; Subbaraj, G.K.; Santhoshkumar, S. Identification of potential inhibitors for Penicillinbinding protein (PBP) from Staphylococcus aureus. Bioinformation 2018, 14, 471–476. [Google Scholar] [CrossRef]
- Chomnawang, M.T.; Surassmo, S.; Wongsariya, K.; Bunyapraphatsara, N. Antibacterial Activity of Thai Medicinal Plants against Methicillin-resistant Staphylococcus aureus. Fitoterapia 2009, 80, 102–104. [Google Scholar] [CrossRef]
- Alhadrami, H.A.; Hamed, A.A.; Hassan, H.M.; Belbahri, L.; Rateb, M.E.; Sayed, A.M. Flavonoids as Potential anti-MRSA Agents through Modulation of PBP2a: A Computational and Experimental Study. Antibiotics 2020, 9, 562. [Google Scholar] [CrossRef]
- Aisha, A.; Zahra, S.; Tahir, I.M.; Hussain, A.; Bano, N.; Roobi, A.; Afsheen, N.; Saleem, Y. Anticancer L-Asparaginase and Phytoactive Compounds From Plant Solanum nigrum Against MDR (Methicillindrug resistant) Staphylococcus aureus and Fungal Isolates. Dose Response 2022, 20, 1–12. [Google Scholar] [CrossRef]
- Liang, M.; Ge, X.; Xua, H.; Ma, K.; Zhang, W.; Zan, Y.; Efferth, T.; Xue, Z.; Hua, X. Phytochemicals with activity against methicillin-resistant Staphylococcus aureus. Phytomed. Int. J. Phytother. Phytopharm. 2022, 100, 154073. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, D.M.; de Oliveira, D.B.C.; Nunes, Y.R.F.; de Almeida Alves, T.M.; Kohlhoff, M.; Andrade, A.A.; Cota, B.B. Natural Occurring Phenolic Derivatives from Mauritia flexuosa (Buriti) Stems and Their Potential Antibacterial Activity against Methicillin-Resistant Staphylococcus aureus (MRSA). Chem. Biodivers. 2022, 19, e202100788. [Google Scholar] [CrossRef] [PubMed]
- WHO. 2020 Antibacterial Agents in Clinical and Preclinical Development: An Overview and Analysis; WHO: Geneva, Switzerland, 2021; pp. 1–76. [Google Scholar]
- Frei, A. Metal Complexes, an Untapped Source of Antibiotic Potential? Antibiotics 2020, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Patra, M.; Gasser, G.; Metzler-Nolte, N. Small organometallic compounds as antibacterial agents. Dalton Trans. 2012, 41, 6350–6358. [Google Scholar] [CrossRef] [PubMed]
- Sierra, M.A.; Casarrubios, L.; de la Torre, M. Bio-Organometallic Derivatives of Antibacterial Drugs. Chem. Eur. J. 2019, 25, 7232–7242. [Google Scholar] [CrossRef] [PubMed]
- Frei, A.; Zuegg, J.; Elliott, A.G.; Baker, M.; Braese, S.; Brown, C.; Chen, F.; Dowson, C.G.; Dujardin, G.; Jung, N.; et al. Metal complexes as a promising source for new antibiotics. Chem. Sci. 2020, 11, 2627–2639. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Collins, J.G.; Keene, F.R. Ruthenium complexes as antimicrobial agents. Chem. Soc. Rev. 2015, 44, 2529–2542. [Google Scholar] [CrossRef]
- Wenzel, M.; Patra, M.; Senges, C.H.; Ott, I.; Stepanek, J.J.; Pinto, A.; Prochnow, P.; Vuong, C.; Langklotz, S.; Metzler-Nolte, N.; et al. Analysis of the mechanism of action of potent antibacterial hetero-tri-organometallic compounds: A structurally new class of antibiotics. ACS Chem. Biol. 2013, 8, 1442–1450. [Google Scholar] [CrossRef]
- Patra, M.; Wenzel, M.; Prochnow, P.; Pierroz, V.; Gasser, G.; Bandow, J.E.; Metzler-Nolte, N. An organometallic structure-activity relationship study reveals the essential role of a Re(CO)3 moiety in the activity against gram-positive pathogens including MRSA. Chem. Sci. 2015, 6, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, D.; Lorenz, N.; Gothe, Y.; Spies, C.; Geissler, B.; Prochnow, P.; Nuernberger, P.; Bandow, J.E.; Metzler-Nolte, N. Benzannulated Re(I)-NHC complexes: Synthesis, photophysical properties and antimicrobial activity. Dalton Trans. 2017, 46, 15269–15279. [Google Scholar] [CrossRef] [PubMed]
- Frei, A.; Amado, M.; Cooper, M.A.; Blaskovich, M.A.T. Light-activated Rhenium Complexes with Dual Mode of Action against Bacteria. Chem. Eur. J. 2019, 26, 2852–2858. [Google Scholar] [CrossRef]
- Zampakou, M.; Akrivou, M.; Andreadou, E.G.; Raptopoulou, C.P.; Psycharis, V.; Pantazaki, A.A.; Psomas, G. Structure, antimicrobial activity, DNA-and albumin-binding of manganese (II) complexes with the quinolone antimicrobial agents oxolinic acid and enrofloxacin. J. Inorg. Biochem. 2013, 121, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Arthi, P.; Shobana, S.; Srinivasan, P.; Prabhu, D.; Arulvasu, C.; Rahiman, A.K.; Biology, P.B. Dinuclear manganese (II) complexes of hexaazamacrocycles bearing N-benzoylated pendant separated by aromatic spacers: Antibacterial, DNA interaction, cytotoxic and molecular docking studies. J. Photoch. Photobio. B 2015, 153, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Simpson, P.V.; Nagel, C.; Bruhn, H.; Schatzschneider, U. Antibacterial and Antiparasitic Activity of Manganese(I) Tricarbonyl Complexes with Ketoconazole, Miconazole, and Clotrimazole Ligands. Organometallics 2015, 34, 3809–3815. [Google Scholar] [CrossRef]
- Boubakri, L.; Mansour, L.; Harrath, A.; Özdemir, I.; Yaşar, S.; Hamdi, N. N-Heterocyclic carbene-Pd (II)-PPh3 complexes as a new highly efficient catalyst system for the Sonogashira cross-coupling reaction: Synthesis, characterization and biological activities. J. Coord. Chem. 2018, 71, 183–199. [Google Scholar] [CrossRef]
- Kaushal, M.; Lobana, T.S.; Nim, L.; Bala, R.; Arora, D.S.; Garcia-Santos, I.; Duff, C.E.; Jasinski, J.P. Synthesis of 2-acetylpyridine-N-substituted thiosemicarbazonates of copper (II) with high antimicrobial activity against methicillin resistant S. aureus, K. pneumoniae 1 and C. albicans. New J. Chem. 2019, 43, 11727–11742. [Google Scholar] [CrossRef]
- Abu Ali, H.; Omar, S.N.; Darawsheh, M.D.; Fares, H. Synthesis, characterization and antimicrobial activity of zinc (II) ibuprofen complexes with nitrogen-based ligands. J. Coord. Chem. 2016, 69, 1110–1122. [Google Scholar] [CrossRef]
- Kumar, S.V.; Scottwell, S.Ø.; Waugh, E.; McAdam, C.J.; Hanton, L.R.; Brooks, H.J.; Crowley, J.D. Antimicrobial properties of tris (homoleptic) ruthenium (II) 2-Pyridyl-1, 2, 3-triazole “click” complexes against pathogenic bacteria, including methicillin-resistant staphylococcus aureus (MRSA). Inorg. Chem. 2016, 55, 9767–9777. [Google Scholar] [CrossRef] [PubMed]
- van Hilst, Q.V.C.; Vasdev, R.A.S.; Preston, D.; Findlay, J.A.; Scottwell, S.Ø.; Giles, G.I.; Brooks, H.J.L.; Crowley, J.D. Synthesis, Characterisation and Antimicrobial Studies of some 2,6-bis(1,2,3-Triazol-4-yl)Pyridine Ruthenium(II) “Click” Complexes. Asian J. Org. Chem. 2019, 8, 496–505. [Google Scholar] [CrossRef]
- Gichumbi, J.M.; Friedrich, H.B.; Omondi, B.; Naicker, K.; Singh, M.; Chenia, H.Y. Synthesis, characterization, antiproliferative, and antimicrobial activity of osmium (II) half-sandwich complexes. J. Coord. Chem. 2018, 71, 342–354. [Google Scholar] [CrossRef]
- Irgi, E.P.; Geromichalos, G.D.; Balala, S.; Kljun, J.; Kalogiannis, S.; Papadopoulos, A.; Turel, I.; Psomas, G.J.R.A. Cobalt (II) complexes with the quinolone antimicrobial drug oxolinic acid: Structure and biological perspectives. RSC Adv. 2015, 5, 36353–36367. [Google Scholar] [CrossRef]
- Kouris, E.; Kalogiannis, S.; Perdih, F.; Turel, I.; Psomas, G. Cobalt(II) complexes of sparfloxacin: Characterization, structure, antimicrobial activity and interaction with DNA and albumins. J. Inorg. Biochem. 2016, 163, 18–27. [Google Scholar] [CrossRef]
- Fiorini, V.; Zanoni, I.; Zacchini, S.; Costa, A.L.; Hochkoeppler, A.; Zanotti, V.; Ranieri, A.M.; Massi, M.; Stefan, A.; Stagni, S.J.D.T. Methylation of Ir (III)-tetrazolato complexes: An effective route to modulate the emission outputs and to switch to antimicrobial properties. Dalton Trans. 2017, 46, 12328–12338. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Moat, J.; McFeely, D.; Clarkson, G.; Hands-Portman, I.J.; Furner-Pardoe, J.P.; Harrison, F.; Dowson, C.G.; Sadler, P.J. Biguanide Iridium(III) Complexes with Potent Antimicrobial Activity. J. Med. Chem. 2018, 61, 7330–7344. [Google Scholar] [CrossRef] [PubMed]
- Baecker, D.; Sesli, O.; Knabl, L.; Huber, S.; Orth-Holler, D.; Gust, R. Investigating the antibacterial activity of salen/salophene metal complexes: Induction of ferroptosis as part of the mode of action. Eur. J. Med. Chem. 2021, 209. [Google Scholar] [CrossRef]
- Mendes, S.S.; Marques, J.; Mesterházy, E.; Straetener, J.; Arts, M.; Pissarro, T.; Reginold, J.; Berscheid, A.; Bornikoel, J.; Kluj, R.M.; et al. Synergetic Antimicrobial Activity and Mechanism of Clotrimazole-Linked CO-Releasing Molecules. ACS Bio Med. Chem. Au 2022, 2, 419–436. [Google Scholar] [CrossRef]
- Sovari, S.N.; Radakovic, N.; Roch, P.; Crochet, A.; Pavic, A.; Zobi, F. Combatting AMR: A molecular approach to the discovery of potent and non-toxic rhenium complexes active against C. albicans-MRSA co-infection. Eur. J. Med. Chem. 2021, 226, 113858. [Google Scholar] [CrossRef]
- Sovari, S.N.; Vojnovic, S.; Bogojevic, S.S.; Crochet, A.; Pavic, A.; Nikodinovic-Runic, J.; Zobi, F. Design, synthesis and in vivo evaluation of 3-arylcoumarin derivatives of rhenium(I) tricarbonyl complexes as potent antibacterial agents against methicillin-resistant Staphylococcus aureus (MRSA). Eur. J. Med. Chem. 2020, 205, 112533. [Google Scholar] [CrossRef]
- Cooper, S.M.; Siakalli, C.; White, A.J.P.; Frei, A.; Miller, P.W.; Long, N.J. Synthesis and anti-microbial activity of a new series of bis(diphosphine) rhenium(v) dioxo complexes. Dalton Trans. 2022, 51, 12791–12795. [Google Scholar] [CrossRef]
- Delasoie, J.; Schiel, P.; Vojnovic, S.; Nikodinovic-Runic, J.; Zobi, F. Photoactivatable Surface-Functionalized Diatom Microalgae for Colorectal Cancer Targeted Delivery and Enhanced Cytotoxicity of Anticancer Complexes. Pharmaceutics 2020, 12, 480. [Google Scholar] [CrossRef]
- Delasoie, J.; Pavic, A.; Voutier, N.; Vojnovic, S.; Crochet, A.; Nikodinovic-Runic, J.; Zobi, F. Identification of novel potent and non-toxic anticancer, anti-angiogenic and antimetastatic rhenium complexes against colorectal carcinoma. Eur. J. Med. Chem. 2020, 204, 112583. [Google Scholar] [CrossRef] [PubMed]
- Rossier, J.; Hauser, D.; Kottelat, E.; Rothen-Rutishauser, B.; Zobi, F. Organometallic cobalamin anticancer derivatives for targeted prodrug delivery via transcobalamin-mediated uptake. Dalton Trans. 2017, 46, 2159–2164. [Google Scholar] [CrossRef] [PubMed]
- Domenichini, A.; Casari, I.; Simpson, P.V.; Desai, N.M.; Chen, L.; Dustin, C.; Edmands, J.S.; van der Vliet, A.; Mohammadi, M.; Massi, M.; et al. Rhenium N-heterocyclic carbene complexes block growth of aggressive cancers by inhibiting FGFR- and SRC-mediated signalling. J. Exp. Clin. Cancer Res. 2020, 39, 276. [Google Scholar] [CrossRef] [PubMed]
- Collery, P.; Veena, V.; Harikrishnan, A.; Desmaele, D. The rhenium(I)-diselenoether anticancer drug targets ROS, TGF-β1, VEGF-A, and IGF-1 in an in vitro experimental model of triple-negative breast cancers. Investig. New Drugs 2019, 37, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Collery, P.; Santoni, F.; Ciccolini, J.; Tran, T.N.; Mohsen, A.; Desmaele, D. Dose Effect of Rhenium (I)-diselenoether as Anticancer Drug in Resistant Breast Tumor-bearing Mice After Repeated Administrations. Anticancer Res. 2016, 36, 6051–6057. [Google Scholar] [CrossRef][Green Version]
- Collery, P.; Bastian, G.; Santoni, F.; Mohsen, A.; Wei, M.; Collery, T.; Tomas, A.; Desmaele, D.; D’Angelo, J. Uptake and efflux of rhenium in cells exposed to rhenium diseleno-ether and tissue distribution of rhenium and selenium after rhenium diseleno-ether treatment in mice. Anticancer Res. 2014, 34, 1679–1689. [Google Scholar]
- Schindler, K.; Zobi, F. Anticancer and Antibiotic Rhenium Tri- and Dicarbonyl Complexes: Current Research and Future Perspectives. Molecules 2022, 27, 539. [Google Scholar] [CrossRef]
- Nasiri Sovari, S.; Kolly, I.; Schindler, K.; Cortat, Y.; Liu, S.C.; Crochet, A.; Pavic, A.; Zobi, F. Efficient Direct Nitrosylation of alpha-Diimine Rhenium Tricarbonyl Complexes to Structurally Nearly Identical Higher Charge Congeners Activable towards Photo-CO Release. Molecules 2021, 26, 5302. [Google Scholar] [CrossRef]
- Schindler, K.; Crochet, A.; Zobi, F. Aerobically stable and substitutionally labile α-diimine rhenium dicarbonyl complexes. RSC Adv. 2021, 11, 7511–7520. [Google Scholar] [CrossRef]
- Abram, U.; Hübener, R.; Alberto, R.; Schibli, R. Darstellung und Strukturen von (Et4N)2[Re(CO)3(NCS)3] und (Et4N)[Re(CO)2Br4]. Z. Anorg. Allg. Chem. 1996, 622, 813–818. [Google Scholar] [CrossRef]
- Santoro, G.; Beltrami, R.; Kottelat, E.; Blacque, O.; Bogdanova, A.Y.; Zobi, F. N-Nitrosamine-{cis-Re[CO]2}2+ cobalamin conjugates as mixed CO/NO-releasing molecules. Dalton Trans. 2016, 45, 1504–1513. [Google Scholar] [CrossRef] [PubMed]
- Zobi, F.; Blacque, O.; Jacobs, R.A.; Schaub, M.C.; Bogdanova, A.Y. 17 e– rhenium dicarbonyl CO-releasing molecules on a cobalamin scaffold for biological application. Dalton Trans. 2012, 41, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Carolus, H.; Van Dyck, K.; Van Dijck, P. Candida albicans and Staphylococcus Species: A Threatening Twosome. Front. Microbiol. 2019, 10, 2162. [Google Scholar] [CrossRef]
- Mauri, A. alvaDesc: A Tool to Calculate and Analyze Molecular Descriptors and Fingerprints. In Ecotoxicological QSARs; Springer: Berlin/Heidelberg, Germany, 2020; pp. 801–820. [Google Scholar]
- Lovering, A.L.; de Castro, L.H.; Lim, D.; Strynadka, N.C. Structural insight into the transglycosylation step of bacterial cell-wall biosynthesis. Science 2007, 315, 1402–1405. [Google Scholar] [CrossRef] [PubMed]
- Lovering, A.L.; Gretes, M.C.; Safadi, S.S.; Danel, F.; de Castro, L.; Page, M.G.; Strynadka, N.C. Structural insights into the anti-methicillin-resistant Staphylococcus aureus (MRSA) activity of ceftobiprole. J. Biol. Chem. 2012, 287, 32096–32102. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Kawai, F.; Obayashi, E.; Akashi, S.; Roper, D.I.; Tame, J.R.; Park, S.Y. Crystal structures of penicillin-binding protein 3 (PBP3) from methicillin-resistant Staphylococcus aureus in the apo and cefotaxime-bound forms. J. Mol. Biol. 2012, 423, 351–364. [Google Scholar] [CrossRef]
- Alexander, J.A.N.; Chatterjee, S.S.; Hamilton, S.M.; Eltis, L.D.; Chambers, H.F.; Strynadka, N.C.J. Structural and kinetic analyses of penicillin-binding protein 4 (PBP4)-mediated antibiotic resistance in Staphylococcus aureus. J. Biol. Chem. 2018, 293, 19854–19865. [Google Scholar] [CrossRef]
- Lu, D.; Wormann, M.E.; Zhang, X.; Schneewind, O.; Grundling, A.; Freemont, P.S. Structure-based mechanism of lipoteichoic acid synthesis by Staphylococcus aureus LtaS. Proc. Natl. Acad. Sci. USA 2009, 106, 1584–1589. [Google Scholar] [CrossRef]
- Ting, Y.T.; Harris, P.W.; Batot, G.; Brimble, M.A.; Baker, E.N.; Young, P.G. Peptide binding to a bacterial signal peptidase visualized by peptide tethering and carrier-driven crystallization. IUCrJ 2016, 3, 10–19. [Google Scholar] [CrossRef]
- Olatunji, S.; Yu, X.; Bailey, J.; Huang, C.Y.; Zapotoczna, M.; Bowen, K.; Remskar, M.; Muller, R.; Scanlan, E.M.; Geoghegan, J.A.; et al. Structures of lipoprotein signal peptidase II from Staphylococcus aureus complexed with antibiotics globomycin and myxovirescin. Nat. Commun. 2020, 11, 140. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, X.; Lambert, E.; Mas, G.; Hiller, S.; Veening, J.W.; Perez, C. Structure of a proton-dependent lipid transporter involved in lipoteichoic acids biosynthesis. Nat. Struct. Mol. Biol. 2020, 27, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Percy, M.G.; Grundling, A. Lipoteichoic acid synthesis and function in gram-positive bacteria. Annu. Rev. Microbiol. 2014, 68, 81–100. [Google Scholar] [CrossRef]
- Grundling, A.; Schneewind, O. Genes required for glycolipid synthesis and lipoteichoic acid anchoring in Staphylococcus aureus. J. Bacteriol. 2007, 189, 2521–2530. [Google Scholar] [CrossRef] [PubMed]
- Sham, L.T.; Butler, E.K.; Lebar, M.D.; Kahne, D.; Bernhardt, T.G.; Ruiz, N. Bacterial cell wall. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science 2014, 345, 220–222. [Google Scholar] [CrossRef]
- Fishovitz, J.; Hermoso, J.A.; Chang, M.; Mobashery, S. Penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus. IUBMB Life 2014, 66, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.G.; Elli, D.; Kim, H.K.; Hendrickx, A.P.; Sorg, J.A.; Schneewind, O.; Missiakas, D. Small molecule inhibitor of lipoteichoic acid synthesis is an antibiotic for Gram-positive bacteria. Proc. Natl. Acad. Sci. USA 2013, 110, 3531–3536. [Google Scholar] [CrossRef]
- Gründling, A.; Schneewind, O. Synthesis of glycerol phosphate lipoteichoic acid in Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2007, 104, 8478–8483. [Google Scholar] [CrossRef]
- Pasquina, L.W.; Santa Maria, J.P.; Walker, S. Teichoic acid biosynthesis as an antibiotic target. Curr. Opin. Microbiol. 2013, 16, 531–537. [Google Scholar] [CrossRef]
- Kuk, A.C.Y.; Hao, A.; Guan, Z.; Lee, S.-Y. Visualizing conformation transitions of the Lipid II flippase MurJ. Nat. Commun. 2019, 10, 1736. [Google Scholar] [CrossRef]
- Kohga, H.; Mori, T.; Tanaka, Y.; Yoshikaie, K.; Taniguchi, K.; Fujimoto, K.; Fritz, L.; Schneider, T.; Tsukazaki, T. Crystal structure of the lipid flippase MurJ in a “squeezed” form distinct from its inward- and outward-facing forms. Structure 2022. [Google Scholar] [CrossRef]
- Craney, A.; Romesberg, F.E. The inhibition of type I bacterial signal peptidase: Biological consequences and therapeutic potential. Bioorg. Med. Chem. Lett. 2015, 25, 4761–4766. [Google Scholar] [CrossRef] [PubMed]
- Schmaler, M.; Jann, N.J.; Götz, F.; Landmann, R. Staphylococcal lipoproteins and their role in bacterial survival in mice. Int. J. Med. Microbiol. 2010, 300, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Vogeley, L.; Arnaout, T.E.; Bailey, J.; Stansfeld, P.J.; Boland, C.; Caffrey, M. Structural basis of lipoprotein signal peptidase II action and inhibition by the antibiotic globomycin. Science 2016, 351, 876–880. [Google Scholar] [CrossRef]
- Nguyen Minh, T.; Götz, F. Lipoproteins of Gram-Positive Bacteria: Key Players in the Immune Response and Virulence. Microbiol. Mol. Biol. Rev. 2016, 80, 891–903. [Google Scholar] [CrossRef] [PubMed]
- El Arnaout, T.; Soulimane, T. Targeting Lipoprotein Biogenesis: Considerations towards Antimicrobials. Trends Biochem. Sci. 2019, 44, 701–715. [Google Scholar] [CrossRef]
- Jendele, L.; Krivak, R.; Skoda, P.; Novotny, M.; Hoksza, D. PrankWeb: A web server for ligand binding site prediction and visualization. Nucleic Acids Res. 2019, 47, W345–W349. [Google Scholar] [CrossRef]
- Wimley, W.C.; White, S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Mol. Biol. 1996, 3, 842–848. [Google Scholar] [CrossRef]
- Hebditch, M.; Warwicker, J. Web-based display of protein surface and pH-dependent properties for assessing the developability of biotherapeutics. Sci. Rep. 2019, 9, 1969. [Google Scholar] [CrossRef]
- Warwicker, J. Continuum dielectric modelling of the protein-solvent system, and calculation of the long-range electrostatic field of the enzyme phosphoglycerate mutase. J. Theor. Biol. 1986, 121, 199–210. [Google Scholar] [CrossRef]
- Mahesha; Krishnegowda, H.M.; Karthik, C.S.; Kudigana, P.J.; Mallu, P.; Neratur, L.K. μ-phenoxide bridged mixed ligand Cu(II) complex: Synthesis, 3D supramolecular architecture, DFT, energy frameworks and antimicrobial studies. Polyhedron 2020, 185, 114571. [Google Scholar] [CrossRef]
- Prasad, H.S.N.; Ananda, A.P.; Najundaswamy, S.; Nagashree, S.; Mallesha, L.; Dayananda, B.P.; Jayanth, H.S.; Mallu, P. Design, synthesis and molecular docking studies of novel piperazine metal complexes as potential antibacterial candidate against MRSA. J. Mol. Struct. 2021, 1232, 130047. [Google Scholar] [CrossRef]
- Hema, M.K.; Karthik, C.S.; Mahesha; Pampa, K.J.; Mallu, P.; Lokanath, N.K. 4,4,4-trifluoro-1-phenylbutane-1,3-dione metal [Cu(II) and Ni(II)] complexes as an superlative antibacterial agent against MRSA: Synthesis, structural quantum-chemical and molecular docking studies. J. Mol. Struct. 2021, 1243, 130774. [Google Scholar] [CrossRef]
- Burgart, Y.; Shchegolkov, E.; Shchur, I.; Kopchuk, D.; Gerasimova, N.; Borisevich, S.; Evstigneeva, N.; Zyryanov, G.; Savchuk, M.; Ulitko, M.; et al. Promising Antifungal and Antibacterial Agents Based on 5-Aryl-2,2′-bipyridines and Their Heteroligand Salicylate Metal Complexes: Synthesis, Bioevaluation, Molecular Docking. Chemmedchem 2022, 17, e202100577. [Google Scholar] [CrossRef] [PubMed]
- Ekennia, A.C.; Onwudiwe, D.C.; Osowole, A.A.; Okpareke, O.C.; Olubiyi, O.O.; Lane, J.R. Coordination compounds of heterocyclic bases: Synthesis, characterization, computational and biological studies. Res. Chem. Intermed. 2019, 45, 1169–1205. [Google Scholar] [CrossRef]
- Zobi, F.; Kromer, L.; Spingler, B.; Alberto, R. Synthesis and Reactivity of the 17 e− Complex [ReIIBr4(CO)2]2−: A Convenient Entry into Rhenium(II) Chemistry. Inorg. Chem. 2009, 48, 8965–8970. [Google Scholar] [CrossRef]
- Kurz, P.; Probst, B.; Spingler, B.; Alberto, R. Ligand Variations in [ReX(diimine)(CO)3] Complexes: Effects on Photocatalytic CO2 Reduction. Eur. J. Inorg. Chem. 2006, 2006, 2966–2974. [Google Scholar] [CrossRef]
- Murphy, B.L.; Marker, S.C.; Lambert, V.J.; Woods, J.J.; MacMillan, S.N.; Wilson, J.J. Synthesis, characterization, and biological properties of rhenium(I) tricarbonyl complexes bearing nitrogen-donor ligands. J. Organomet. Chem. 2020, 907, 121064. [Google Scholar] [CrossRef]
- Chakraborty, I.; Jimenez, J.; Sameera, W.M.C.; Kato, M.; Mascharak, P.K. Luminescent Re(I) Carbonyl Complexes as Trackable PhotoCORMs for CO delivery to Cellular Targets. Inorg. Chem. 2017, 56, 2863–2873. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. V. Systematic optimization of exchange-correlation functionals. J. Chem. Phys. 1997, 107, 8554–8560. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, W. Empirical correction to density functional theory for van der Waals interactions. J. Chem. Phys. 2002, 116, 515–524. [Google Scholar] [CrossRef]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-adjustedab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Yan, C.; Yuan, R.; Pfalzgraff, W.C.; Nishida, J.; Wang, L.; Markland, T.E.; Fayer, M.D. Unraveling the dynamics and structure of functionalized self-assembled monolayers on gold using 2D IR spectroscopy and MD simulations. Proc. Natl. Acad. Sci. USA 2016, 113, 4929–4934. [Google Scholar] [CrossRef] [PubMed]
- Vasighi, M.; Romanova, J.; Nedyalkova, M. A multilevel approach for screening natural compounds as an antiviral agent for COVID-19. Comput. Biol. Chem. 2022, 98, 107694. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).