
Hybrid Molecules as Potential Drugs for the Treatment of HIV: Design and Applications
 , , ,
, , ,
Abstract
1. Introduction
2. Hybrid Drugs: Concept and Approaches
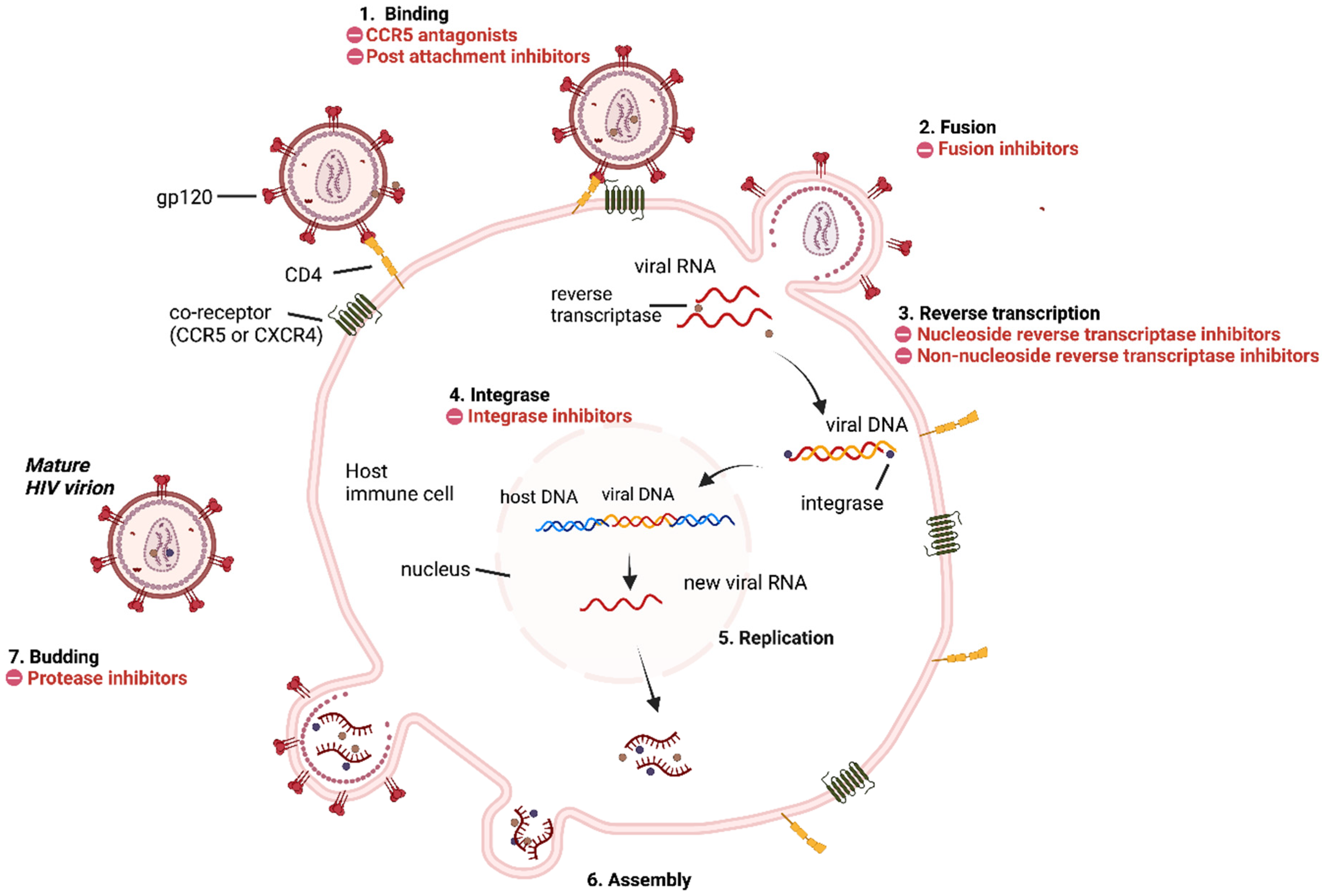
3. HIV Replication Cycle and Inhibition Sites
4. Hybrids Acting on Selected Targets
4.1. RT/PR Inhibition
4.1.1. Cinnamic and Phenylpropionic Amide Derivatives
4.1.2. Coumarin Derivatives
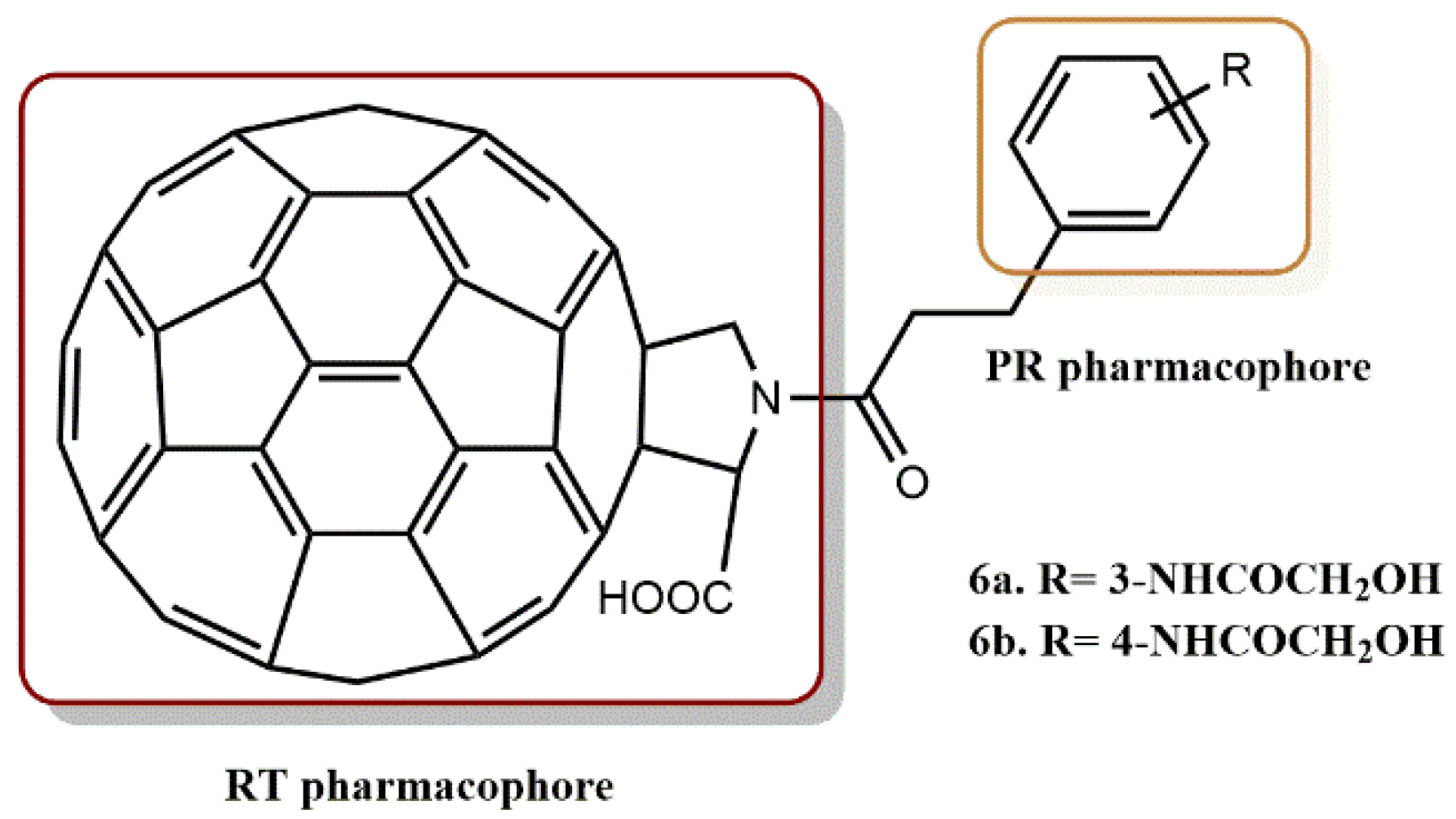
4.1.3. Fullerene Derivatives
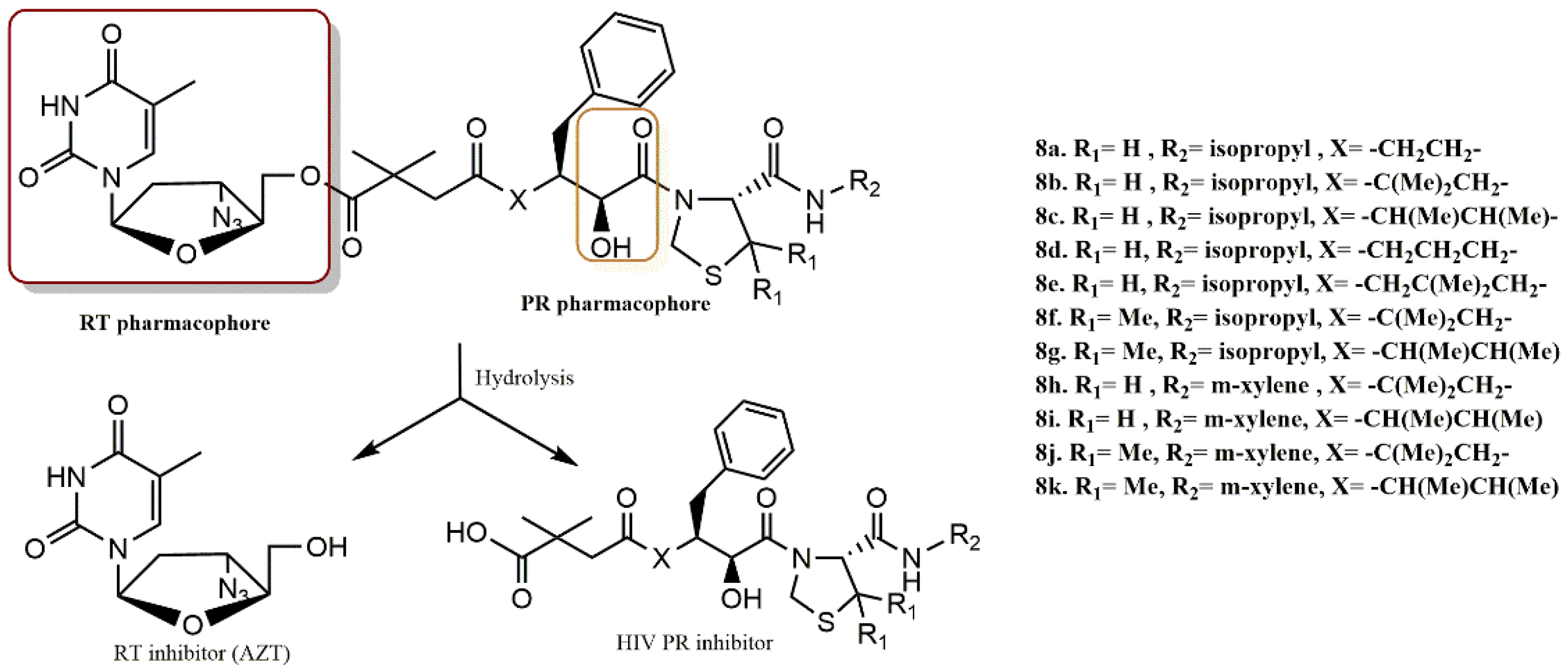
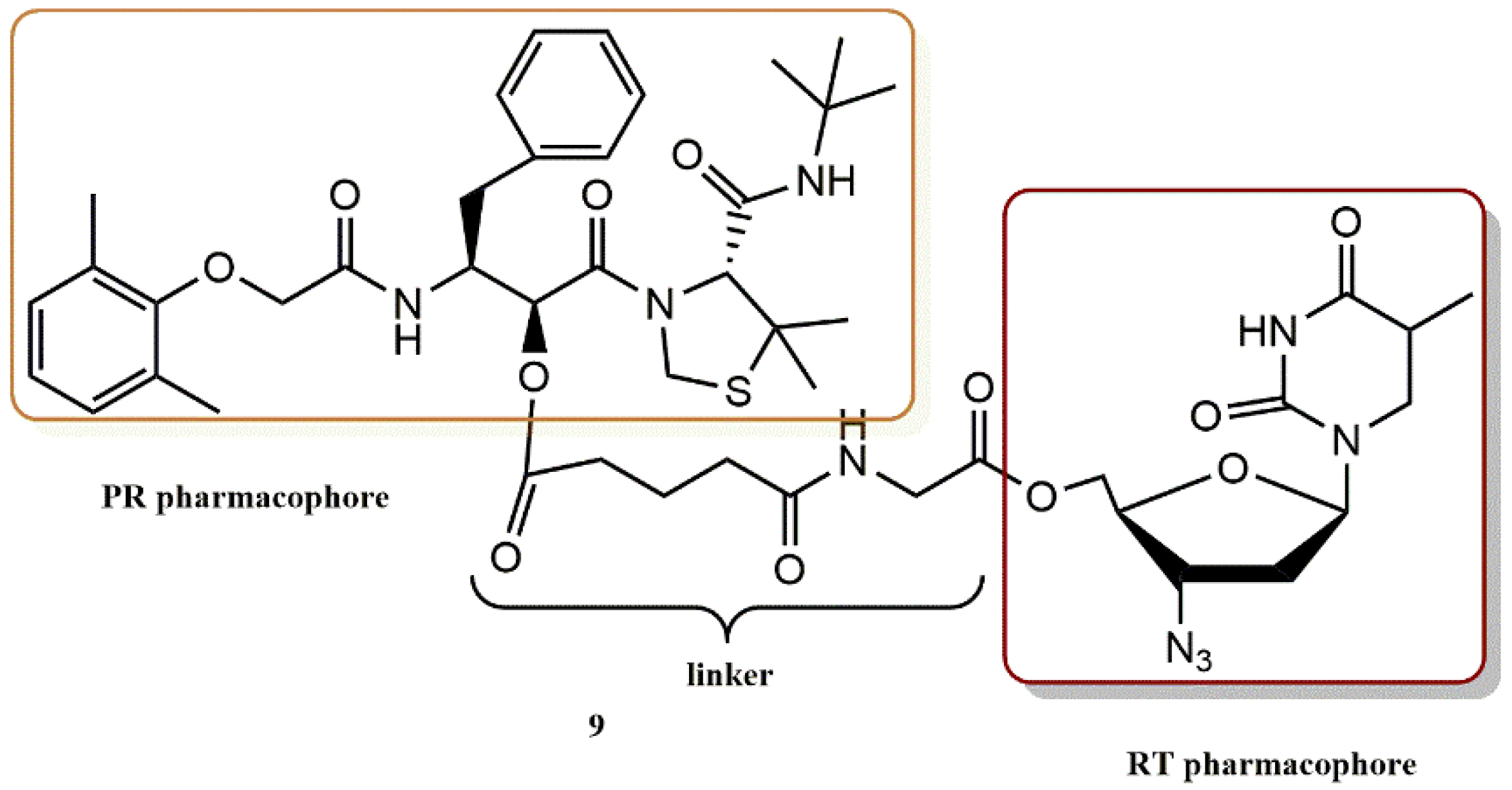
4.1.4. AZT Derivatives
4.2. RT/IN Inhibition
4.2.1. Pyrimidine-2,4-Diones Derivatives
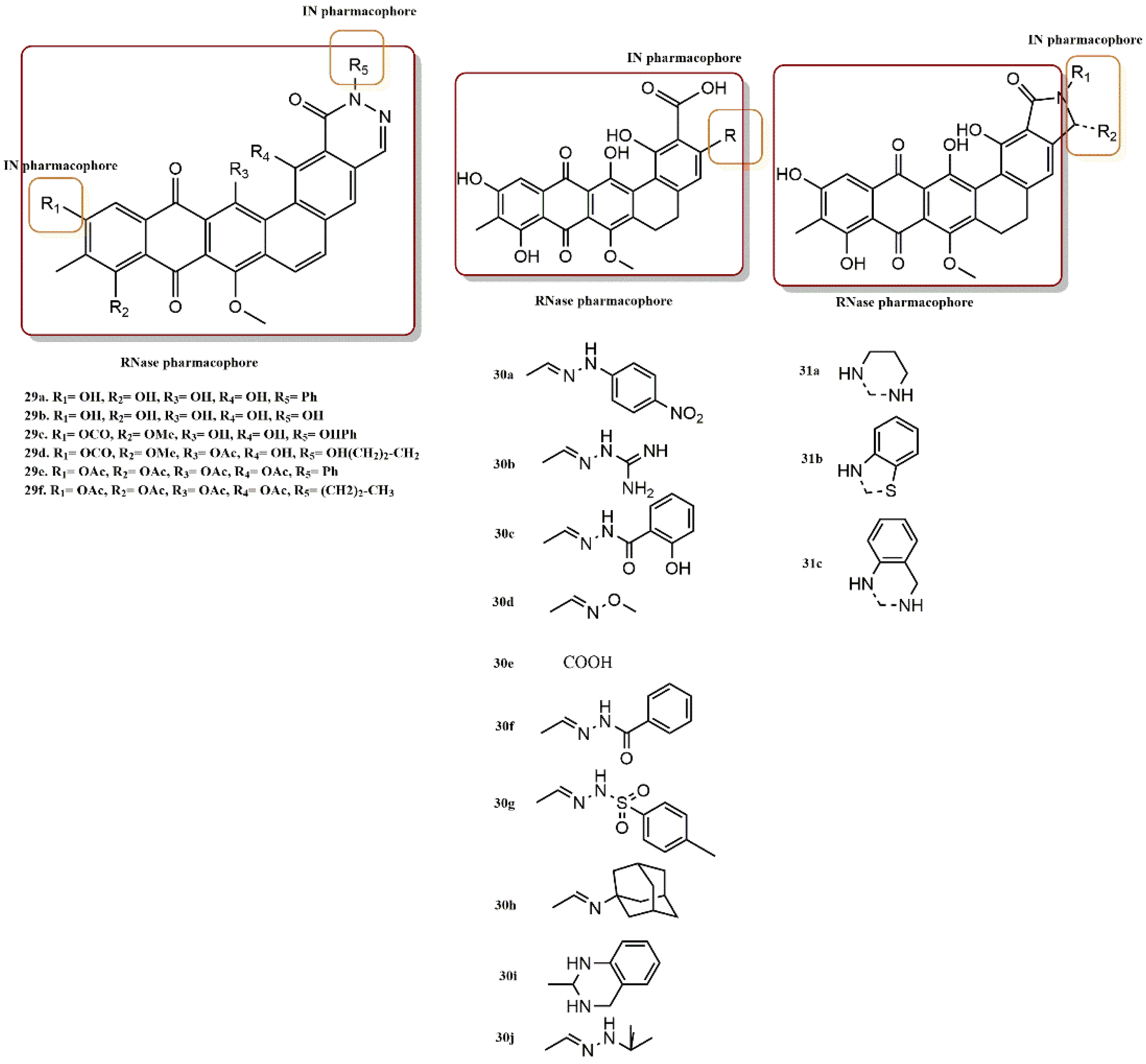
4.2.2. Dihydroxyindole-2-Carboxylic Acid Derivatives
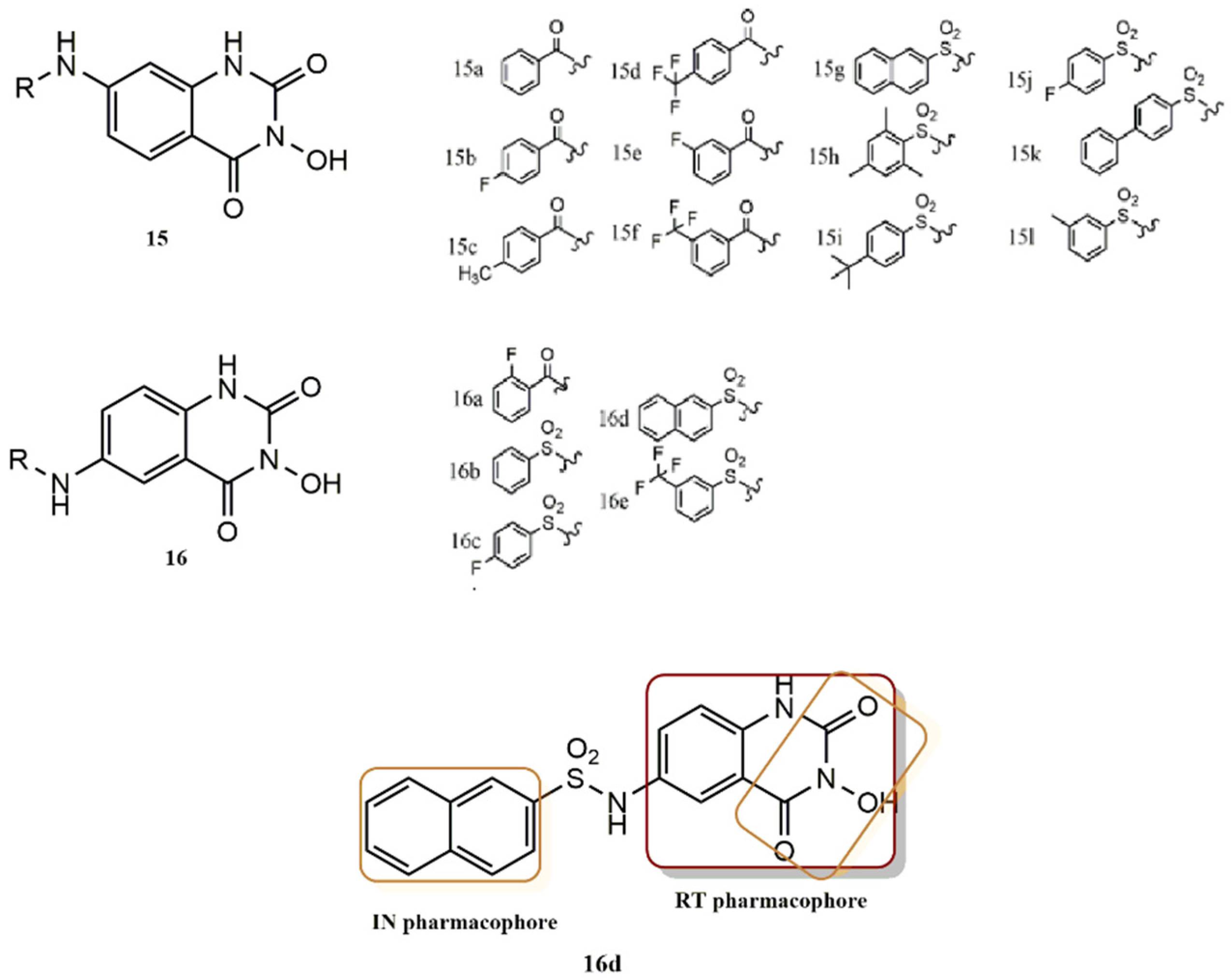
4.2.3. 3-Hydroxyquinazoline-2,4(1H, 3H)-Diones Derivatives
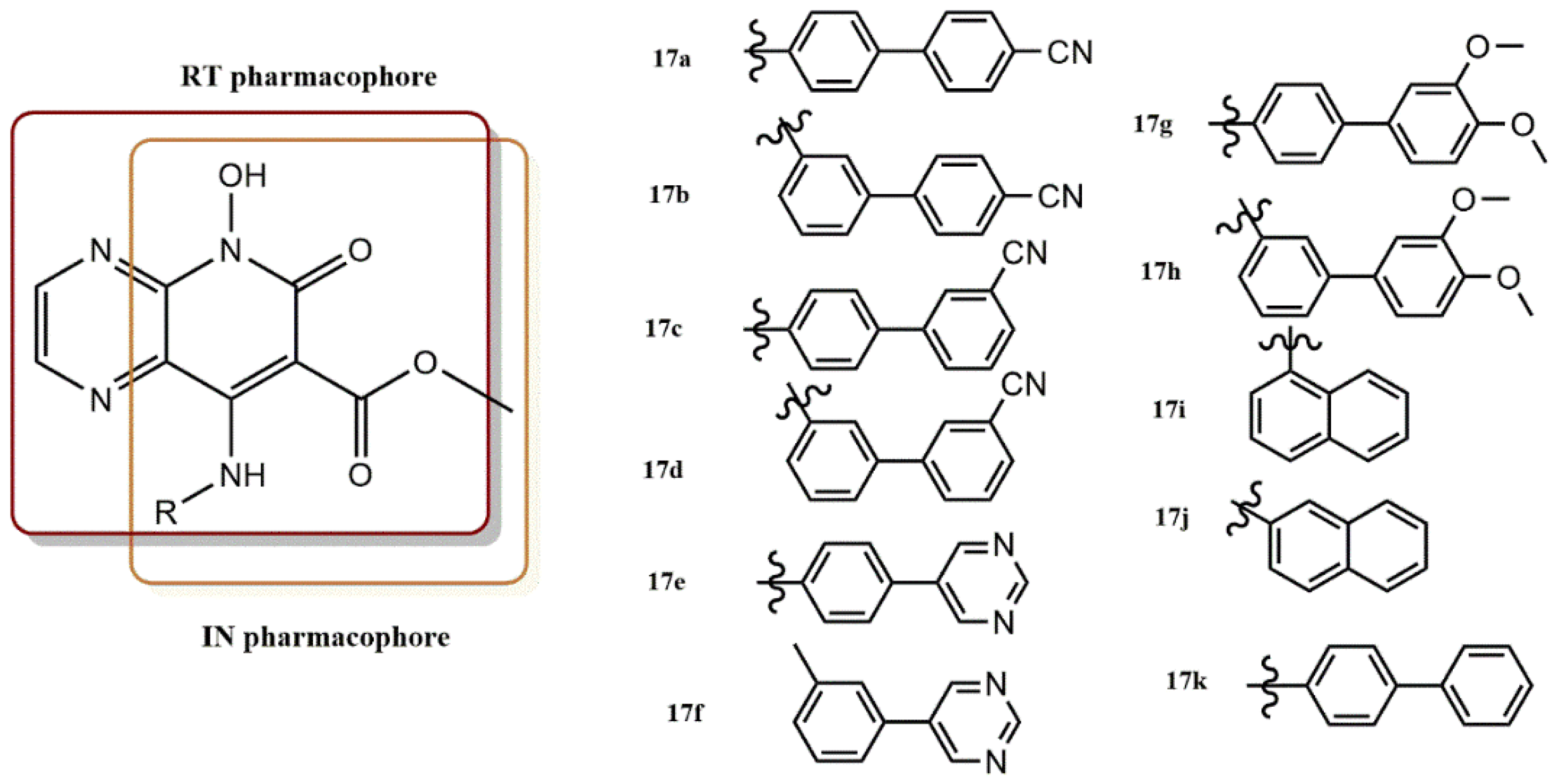
4.2.4. 5-Hydroxypyrido [2,3-b]Pyrazin-6(5H)-One Derivatives
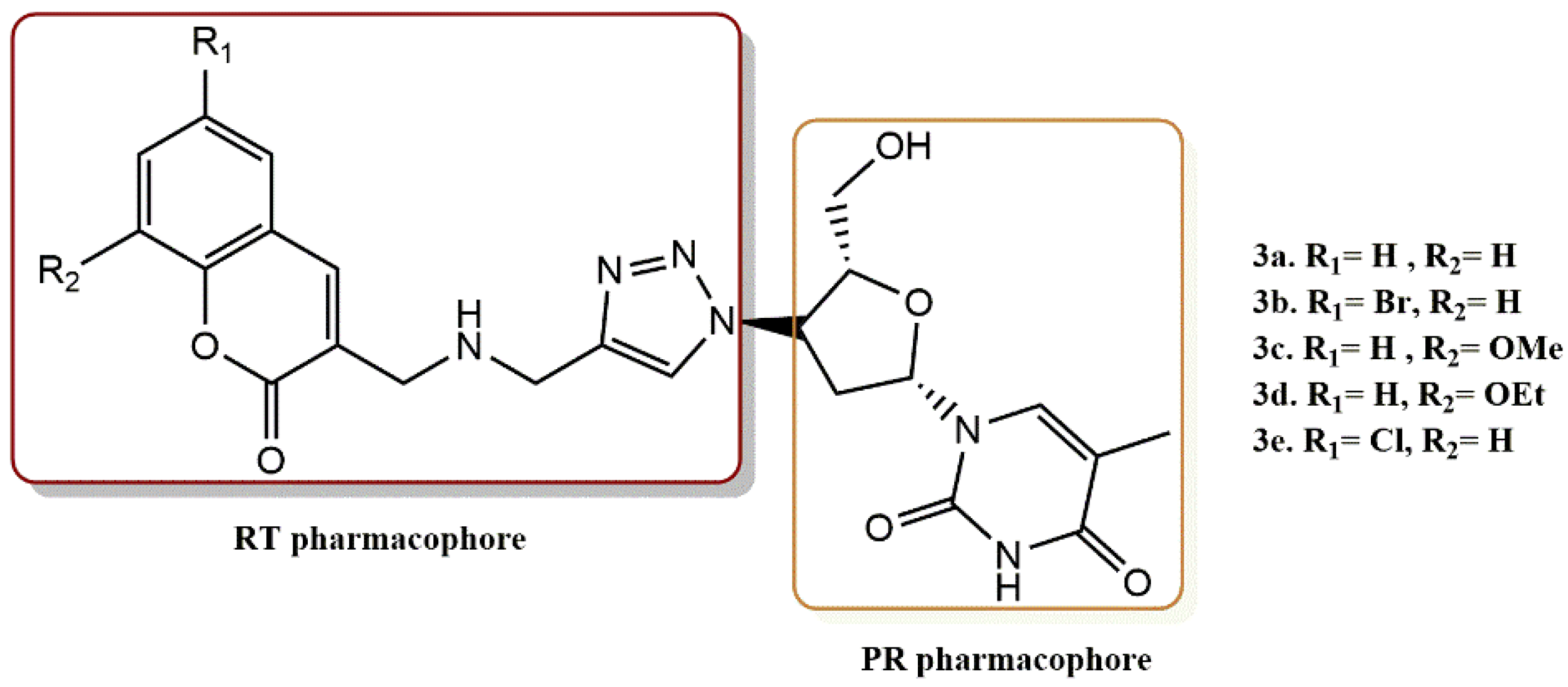
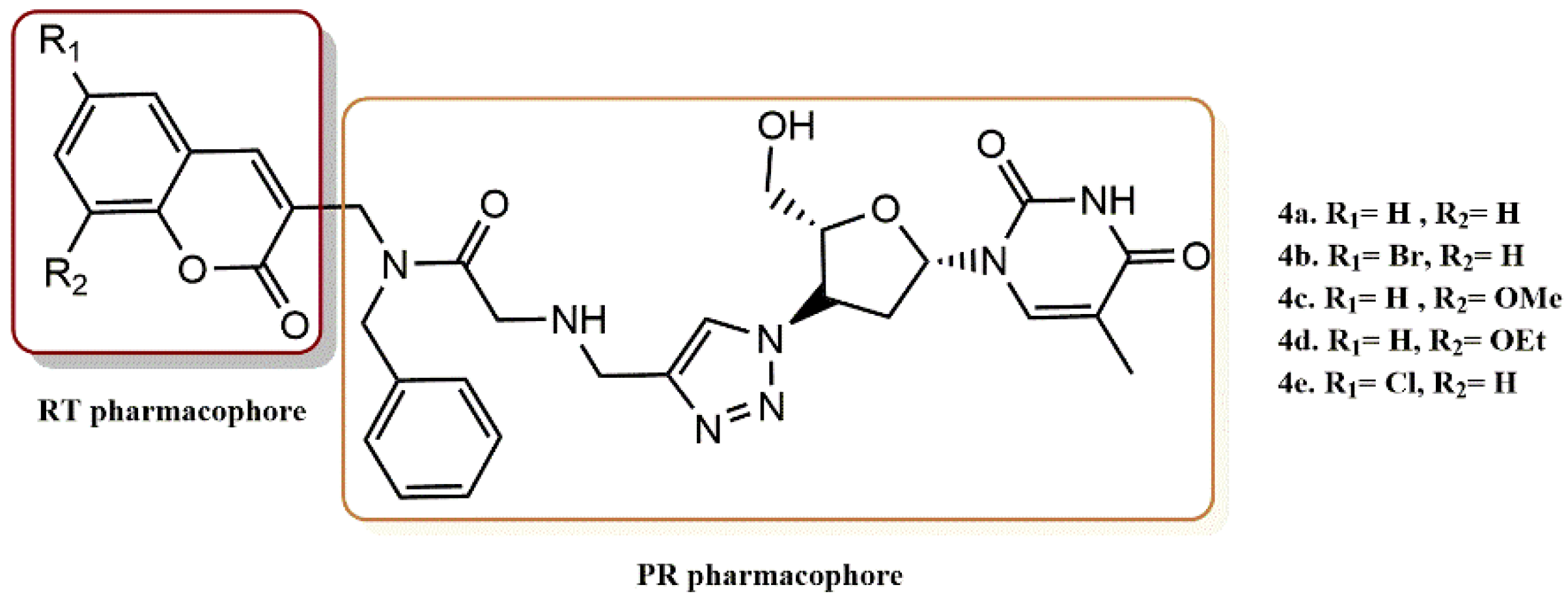
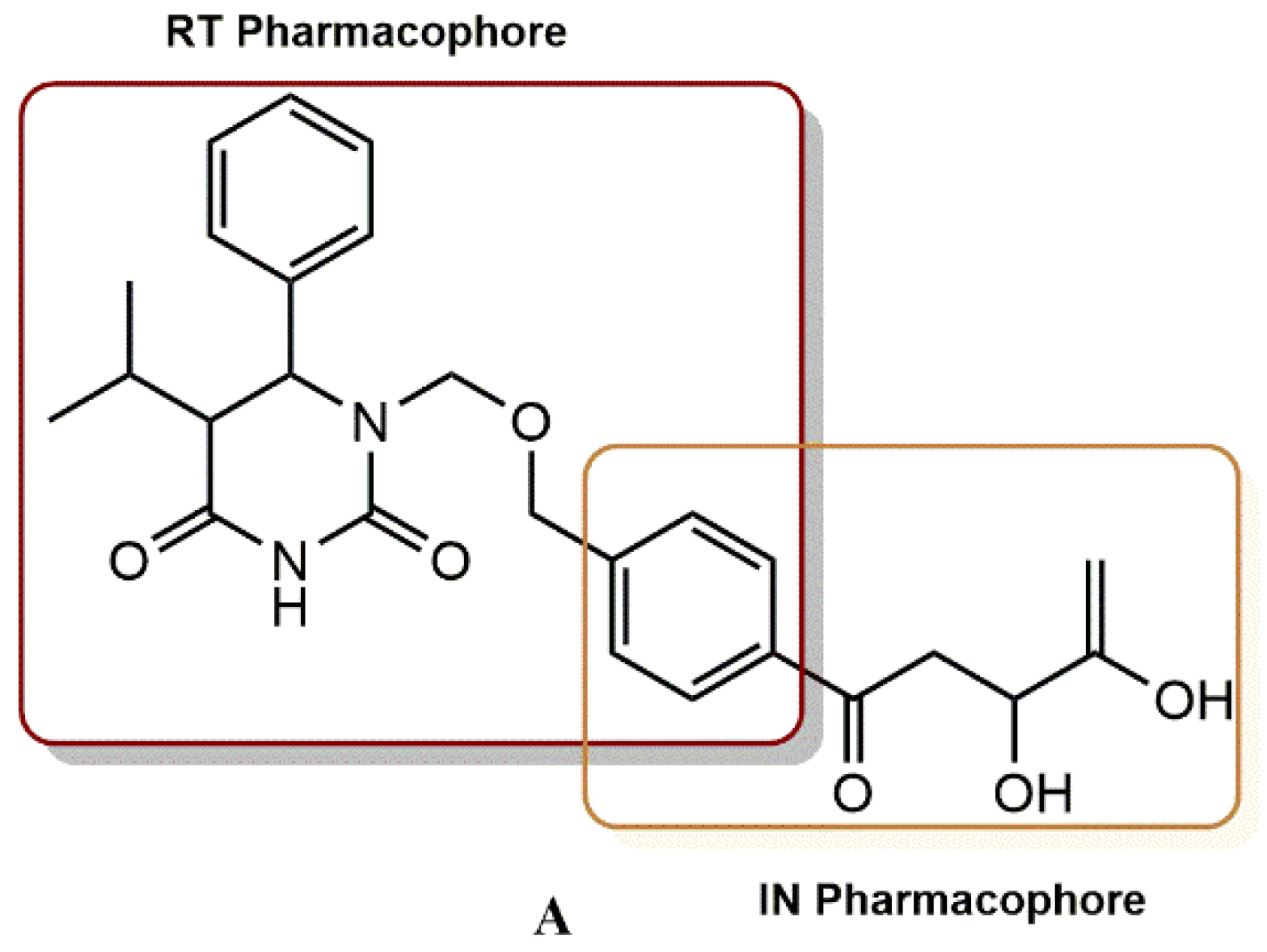
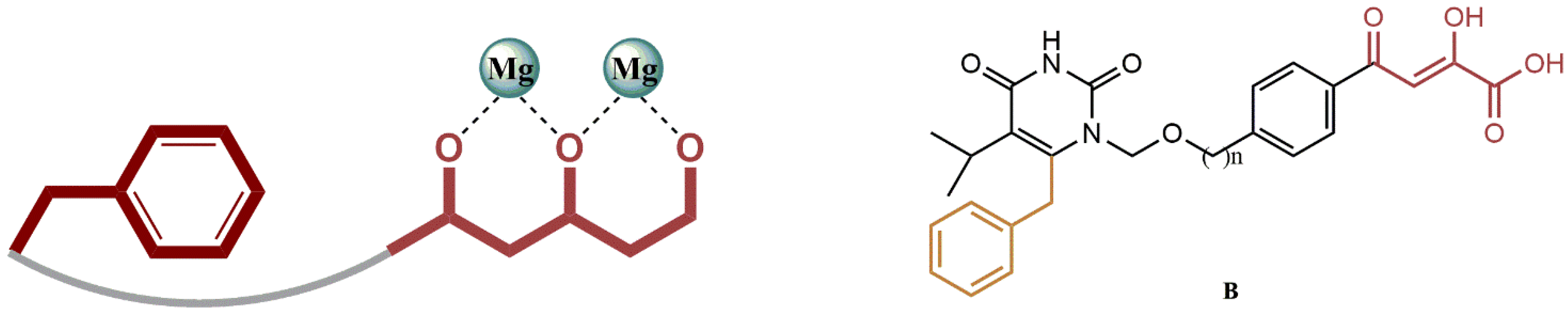
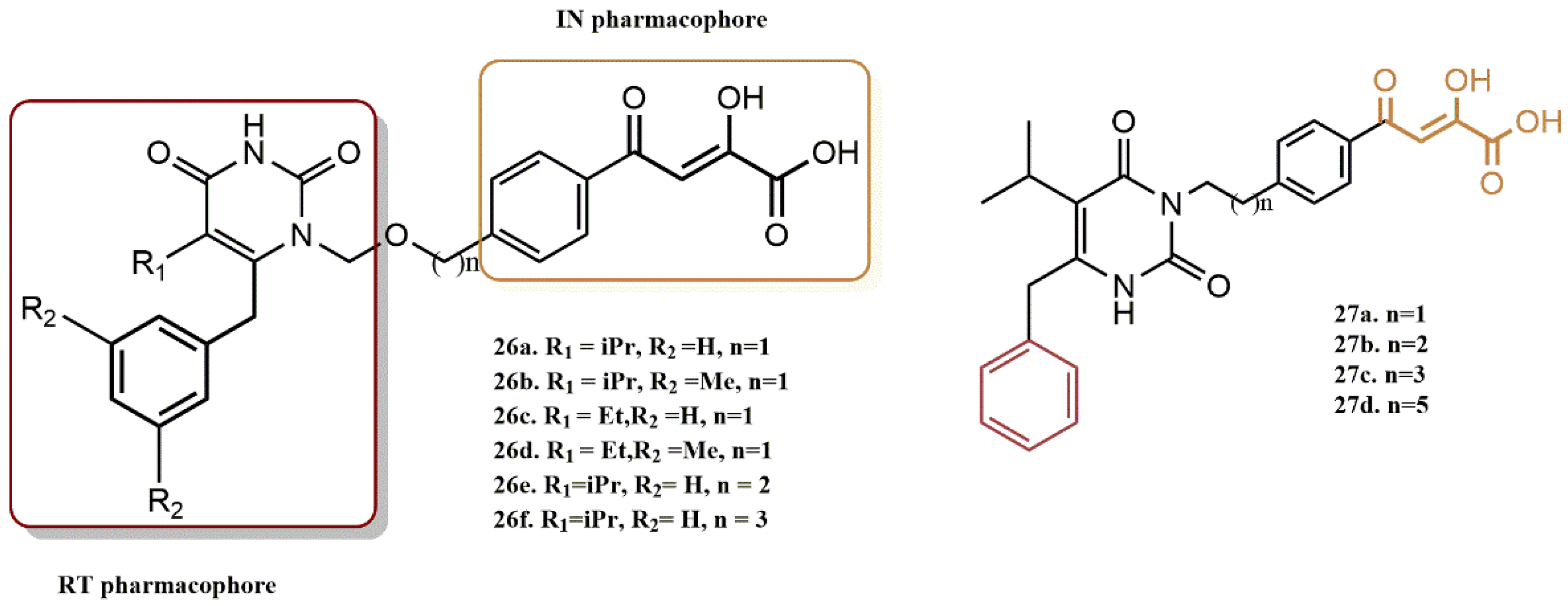
4.2.5. 3-Hydroxy-3-Phenylpropanoate Ester–AZT Derivatives
- (i)
- The dual inhibition RT/IN of these compounds generally followed this order: 3-hydroxy-3-phenylpropanoate ester-AZT conjugates 18a-d > O-benzylated 3-hydroxy-3-phenylpropanoate ester-AZT conjugates 19a-d cinnamate ester-AZT conjugates 20a-e.
- (ii)
- In most cases, the addition of the AZT moiety appeared to increase the percentage of IN viability, suggesting that lengthening the linker between the RT and IN active moieties may be beneficial.
- (iii)
- The 5′-bromo-3-hydroxyl analogs 18b and 19b had concomitant promising potential against the enzymes RT and IN.
- (iv)
- Most ester-AZT conjugates exhibited little or no cell toxicity.
- (v)
- Among the sensitized derivatives, compounds (18a, 18b, 18c and 20a) showed similar or greater inhibitory activity than their propargylated precursors with low cell toxicity (P85% cell viability at 20 µM).
4.2.6. Chromenone Derivatives
4.2.7. Diketo Acid Derivatives
4.2.8. 1-[(2-Hydroxyethoxy)methyl]-6-(Phenylthio)thymine (HEPT) Derivatives
4.2.9. Pyrimidine and Quinolone Derivatives
4.2.10. Madurahydroxylactone Derivatives
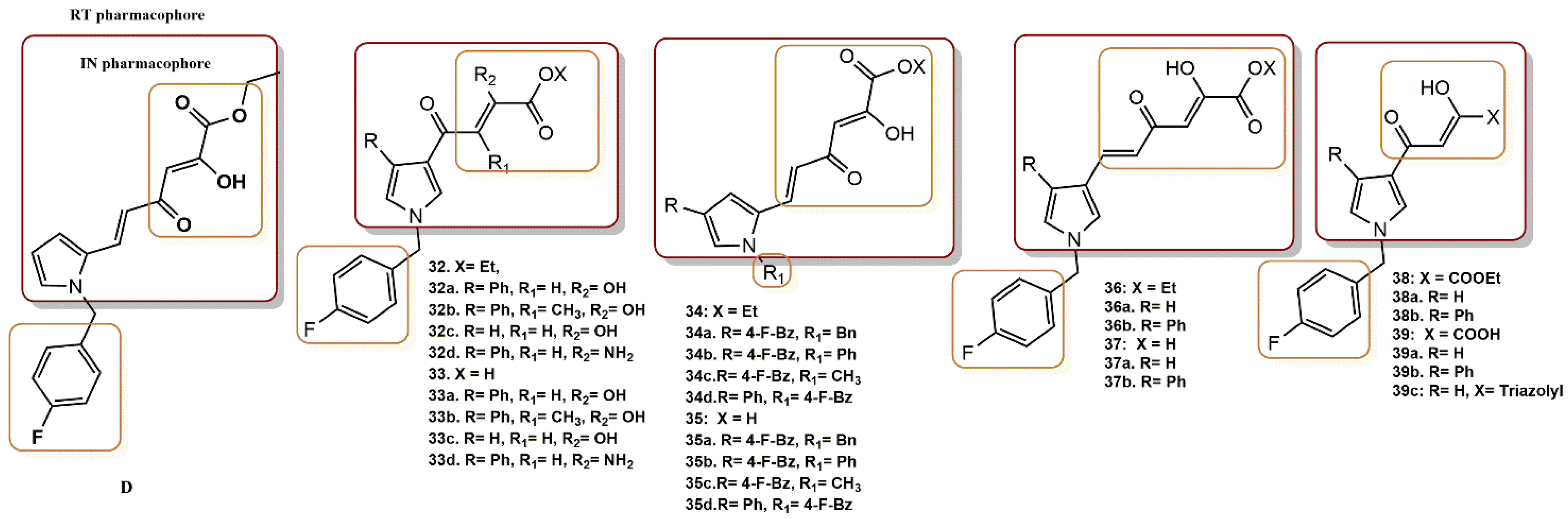
4.2.11. Pyrrolyl Diketo Acid Derivatives
4.3. RT/RT Inhibition
4.3.1. Diarylpyrimidine–Quinolone Hybrids Derivatives
4.3.2. (3Z)-3-(2-[4-(aryl)-1,3-Thiazol-2-yl]ydrazine-1-Ylidene)-2,3-Dihydro-1H-Indol-2-One Derivatives
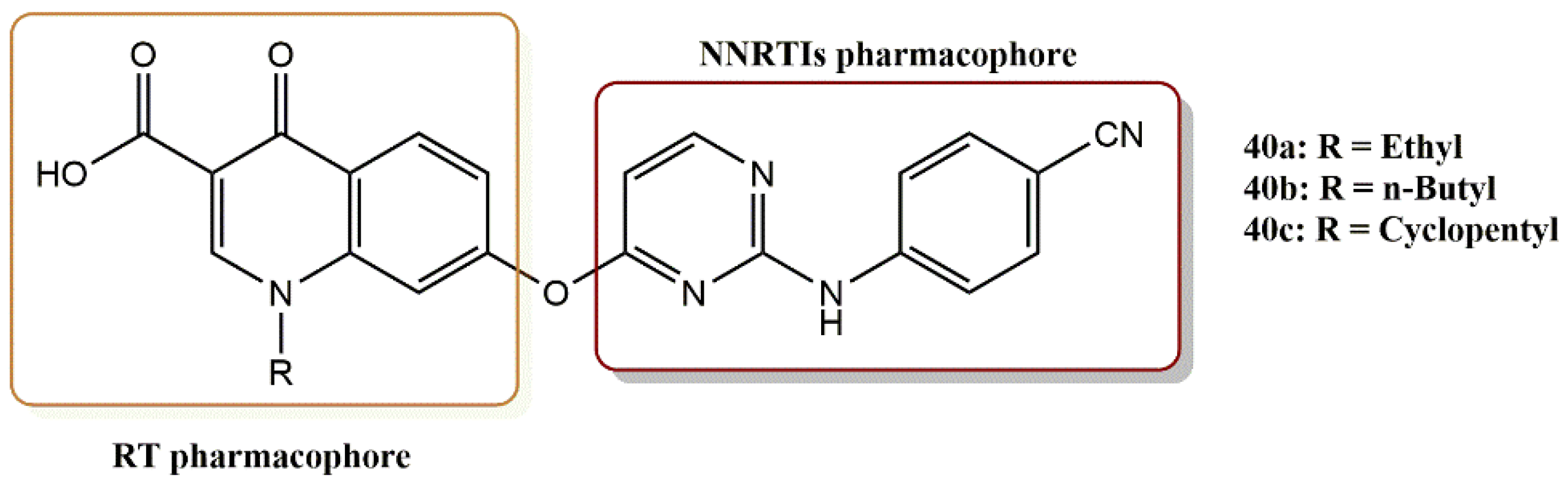
4.3.3. Diarylpyrimidines Derivatives
4.3.4. Dihydropyrimidinone-Isatin Derivatives
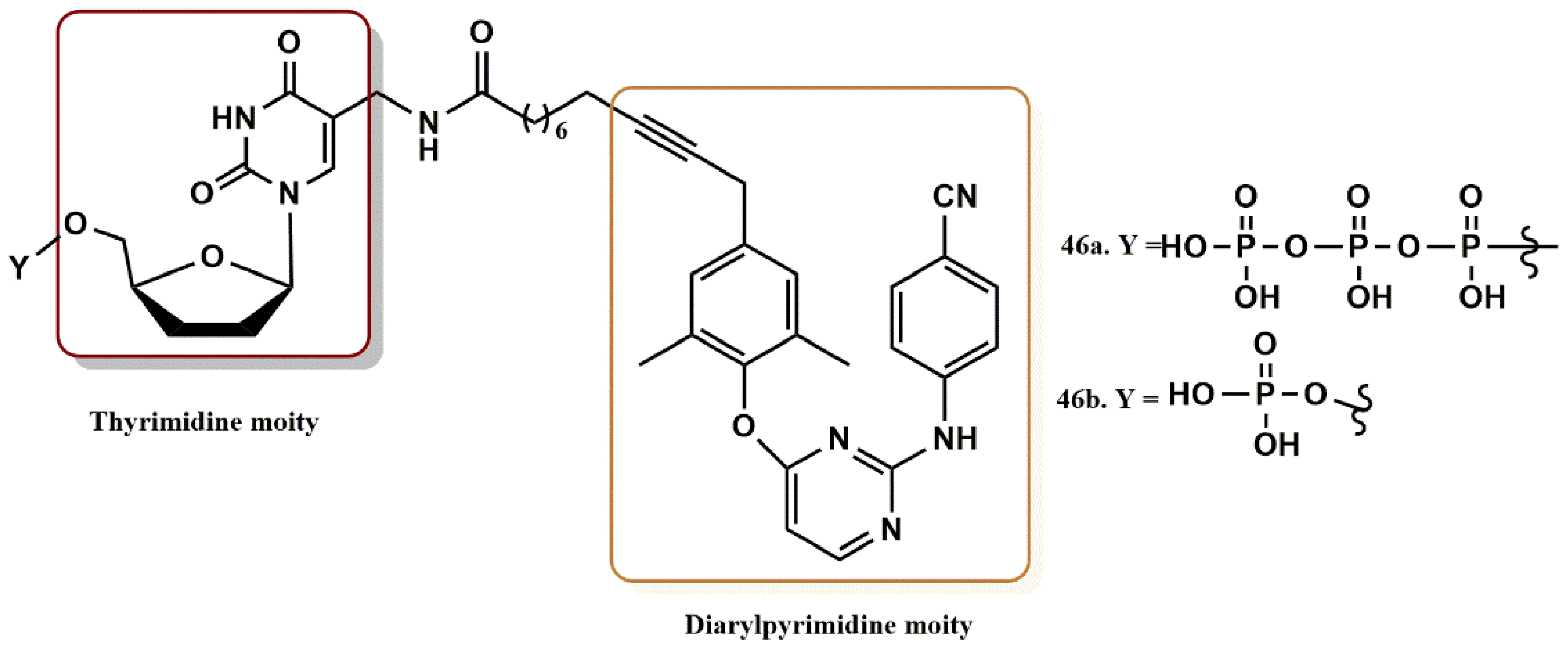
4.3.5. THY-ALK-TMC Derivatives
4.3.6. Hydrazone Derivatives
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bowers, M. HIV nucleoside reverse transcriptase inhibitors. Eur. J. Med. Chem. 2022, 240, 114554. [Google Scholar] [CrossRef]
- Machado, L.D.A.; Gomes, M.F.D.C.; Guimarães, A.C.R. Raltegravir-Induced Adaptations of the HIV-1 Integrase: Analysis of Structure, Variability, and Mutation Co-occurrence. Front. Microbiol. 2019, 10, 1981. [Google Scholar] [CrossRef] [PubMed]
- Hdoufane, I.; Bjij, I.; Soliman, M.; Tadjer, A.; Villemin, D.; Bogdanov, J.; Cherqaoui, D. In Silico SAR Studies of HIV-1 Inhibitors. Pharmaceuticals 2018, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- UNAIDS. Global HIV & AIDS statistics—Fact Sheet. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 25 July 2022).
- Tseng, A.; Seet, J.; Phillips, E.J. The evolution of three decades of antiretroviral therapy: Challenges, triumphs and the promise of the future. Br. J. Clin. Pharmacol. 2015, 79, 182–194. [Google Scholar] [CrossRef]
- Brady, M.T.; Oleske, J.M.; Williams, P.L.; Elgie, C.; Mofenson, L.M.; Dankner, W.M.; Van Dyke, R.B.; Pediatric AIDS Clinical Trials Group 219/219C Team. Declines in Mortality Rates and Changes in Causes of Death in HIV-1-Infected Children During the HAART Era. J. Acquir. Immune Defic. Syndr. 1999, 53, 86. [Google Scholar] [CrossRef]
- Teeraananchai, S.; Kerr, S.; Amin, J.; Ruxrungtham, K.; Law, M.G. Life expectancy of HIV-positive people after starting combination antiretroviral therapy: A meta-analysis. HIV Med. 2017, 18, 256–266. [Google Scholar] [CrossRef]
- Bianco, M.D.C.A.D.; Marinho, D.I.L.F.; Hoelz, L.V.B.; Bastos, M.M.; Boechat, N. Pyrroles as Privileged Scaffolds in the Search for New Potential HIV Inhibitors. Pharmaceuticals 2021, 14, 893. [Google Scholar] [CrossRef]
- Szumilak, M.; Wiktorowska-Owczarek, A.; Stanczak, A. Hybrid Drugs—A Strategy for Overcoming Anticancer Drug Resistance? Molecules 2021, 26, 2601. [Google Scholar] [CrossRef]
- Meunier, B. Hybrid Molecules with a Dual Mode of Action: Dream or Reality? Accounts Chem. Res. 2008, 41, 69–77. [Google Scholar] [CrossRef]
- Kerru, N.; Singh, P.; Koorbanally, N.; Raj, R.; Kumar, V. Recent advances (2015–2016) in anticancer hybrids. Eur. J. Med. Chem. 2017, 142, 179–212. [Google Scholar] [CrossRef]
- Agarwal, D.; Gupta, R.D.; Awasthi, S.K. Are Antimalarial Hybrid Molecules a Close Reality or a Distant Dream? Antimicrob. Agents Chemother. 2017, 61, e00249-17. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.; Sharma, M.; Chauhan, P. Trioxaquines: Hybrid molecules for the treatment of malaria. Drug News Perspect. 2010, 23, 632–646. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.J.; Bell, A. Hybrid Drugs for Malaria. Curr. Pharm. Des. 2009, 15, 2970–2985. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Weinstein, A.; Wang, Z.; Cheng, Y.; Kingsley, L.; Levine, A.; Martin, E.; Munro, C.; Ragin, A.B.; Rubin, L.H.; et al. Legacy effect on neuropsychological function in HIV-infected men on combination antiretroviral therapy. AIDS 2022, 36, 19–27. [Google Scholar] [CrossRef]
- Kangueane, P. HIV-1 GP160 (GP120/GP40) Trimer ENV Spike Protein. In Bioinformation Discovery; Springer: Cham, Switzerland, 2018; pp. 173–181. [Google Scholar] [CrossRef]
- Zhan, P.; Pannecouque, C.; De Clercq, E.; Liu, X. Anti-HIV Drug Discovery and Development: Current Innovations and Future Trends. J. Med. Chem. 2016, 59, 2849–2878. [Google Scholar] [CrossRef]
- Mirza, M.U.; Saadabadi, A.; Vanmeert, M.; Salo-Ahen, O.M.H.; Abdullah, I.; Claes, S.; De Jonghe, S.; Schols, D.; Ahmad, S.; Froeyen, M. Discovery of HIV entry inhibitors via a hybrid CXCR4 and CCR5 receptor pharmacophore-based virtual screening approach. Eur. J. Pharm. Sci. 2020, 155, 105537. [Google Scholar] [CrossRef]
- Hdoufane, I.; Stoycheva, J.; Tadjer, A.; Villemin, D.; Najdoska-Bogdanov, M.; Bogdanov, J.; Cherqaoui, D. QSAR and molecular docking studies of indole-based analogs as HIV-1 attachment inhibitors. J. Mol. Struct. 2019, 1193, 429–443. [Google Scholar] [CrossRef]
- Sillapachaiyaporn, C.; Rangsinth, P.; Nilkhet, S.; Moungkote, N.; Chuchawankul, S. HIV-1 Protease and Reverse Transcriptase Inhibitory Activities of Curcuma aeruginosa Roxb. Rhizome Extracts and the Phytochemical Profile Analysis: In Vitro and In Silico Screening. Pharmaceuticals 2021, 14, 1115. [Google Scholar] [CrossRef]
- Moelling, K.; Broecker, F.; Kerrigan, J.E. RNase H: Specificity, Mechanisms of Action, and Antiviral Target. Methods Mol. Biol. 2014, 1087, 71–84. [Google Scholar] [CrossRef]
- Vergni, D.; Santoni, D.; Bouba, Y.; Lemme, S.; Fabeni, L.; Carioti, L.; Bertoli, A.; Gennari, W.; Forbici, F.; Perno, C.F.; et al. Evaluation of HIV-1 integrase variability by combining computational and probabilistic approaches. Infect. Genet. Evol. 2022, 101, 105294. [Google Scholar] [CrossRef]
- Esposito, D.; Craigie, R. HIV Integrase Structure and Function. Adv. Virus Res. 1999, 52, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Sundquist, W.I.; Kräusslich, H.G. HIV-1 Assembly, Budding, and Maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef] [PubMed]
- Gulnik, S.; Erickson, J.W.; Xie, D. HIV protease: Enzyme function and drug resistance. Vitam. Horm. 2000, 58, 213–256. [Google Scholar] [CrossRef] [PubMed]
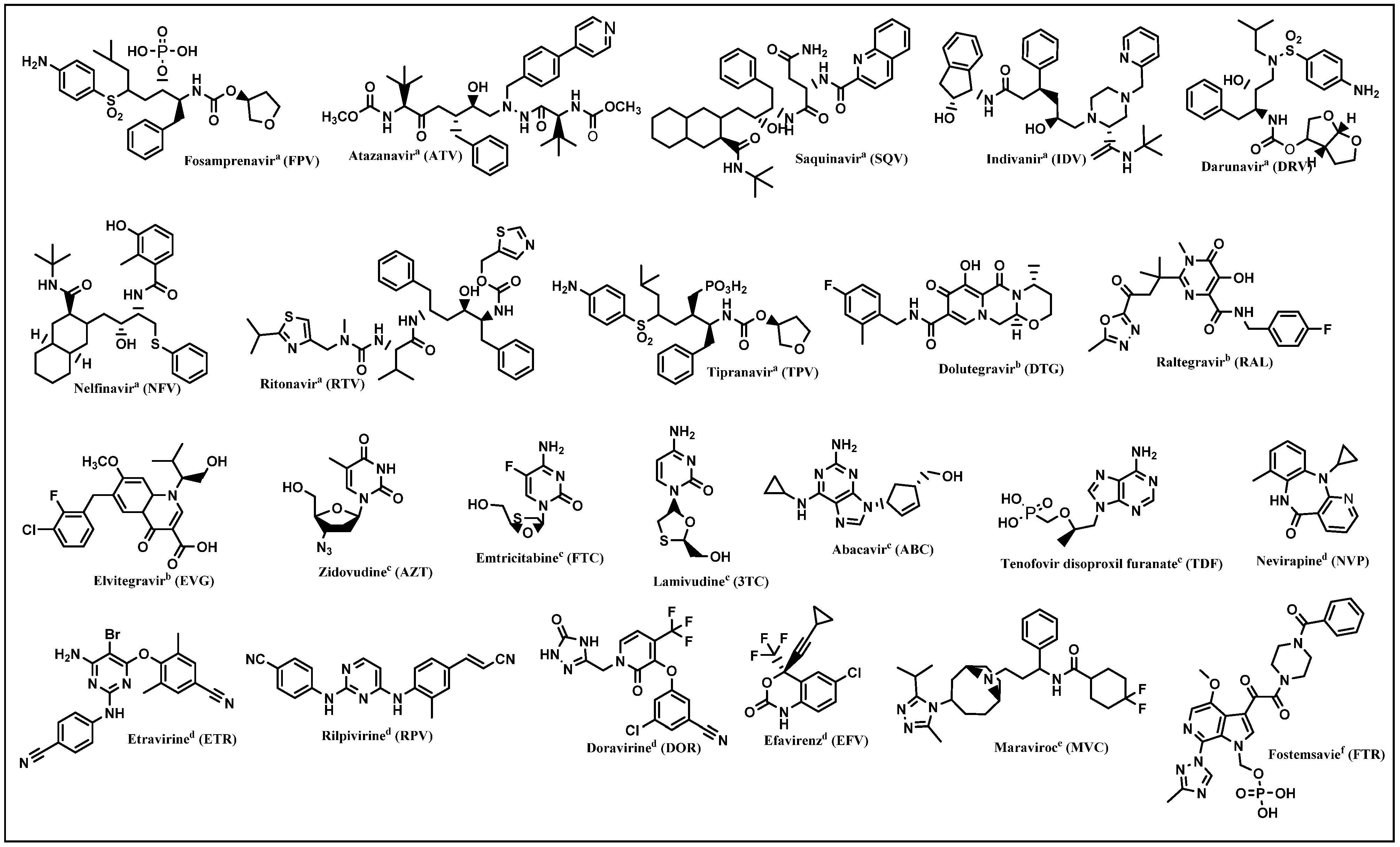
- NIH. FDA-Approved HIV Medicines. Available online: https://hivinfo.nih.gov/understanding-hiv/fact-sheets/fda-approved-hiv-medicines (accessed on 25 July 2022).
- Muregi, F.W.; Ishih, A. Next-generation antimalarial drugs: Hybrid molecules as a new strategy in drug design. Drug Dev. Res. 2010, 71, 20–32. [Google Scholar] [CrossRef]
- Zhu, M.; Shan, Q.; Ma, L.; Wen, J.; Dong, B.; Zhang, G.; Wang, M.; Wang, J.; Zhou, J.; Cen, S.; et al. Design and biological evaluation of cinnamic and phenylpropionic amide derivatives as novel dual inhibitors of HIV-1 protease and reverse transcriptase. Eur. J. Med. Chem. 2021, 220, 113498. [Google Scholar] [CrossRef]
- Olomola, T.O.; Klein, R.; Lobb, K.A.; Sayed, Y.; Kaye, P.T. Towards the synthesis of coumarin derivatives as potential dual-action HIV-1 protease and reverse transcriptase inhibitors. Tetrahedron Lett. 2010, 51, 6325–6328. [Google Scholar] [CrossRef]
- Olomola, T.O.; Klein, R.; Mautsa, N.; Sayed, Y.; Kaye, P.T. Synthesis and evaluation of coumarin derivatives as potential dual-action HIV-1 protease and reverse transcriptase inhibitors. Bioorg. Med. Chem. 2013, 21, 1964–1971. [Google Scholar] [CrossRef]
- Zhu, M.; Ma, L.; Wen, J.; Dong, B.; Wang, Y.; Wang, Z.; Zhou, J.; Zhang, G.; Wang, J.; Guo, Y.; et al. Rational design and Structure−Activity relationship of coumarin derivatives effective on HIV-1 protease and partially on HIV-1 reverse transcriptase. Eur. J. Med. Chem. 2020, 186, 111900. [Google Scholar] [CrossRef]
- Lee, V.S.; Nimmanpipug, P.; Aruksakunwong, O.; Promsri, S.; Sompornpisut, P.; Hannongbua, S. Structural analysis of lead fullerene-based inhibitor bound to human immunodeficiency virus type 1 protease in solution from molecular dynamics simulations. J. Mol. Graph. Model. 2007, 26, 558–570. [Google Scholar] [CrossRef]
- Durdagi, S.; Mavromoustakos, T.; Chronakis, N.; Papadopoulos, M.G. Computational design of novel fullerene analogues as potential HIV-1 PR inhibitors: Analysis of the binding interactions between fullerene inhibitors and HIV-1 PR residues using 3D QSAR, molecular docking and molecular dynamics simulations. Bioorg. Med. Chem. 2008, 16, 9957–9974. [Google Scholar] [CrossRef]
- Ibrahim, M.; Saleh, N.A.; Hameed, A.J.; Elshemey, W.M.; Elsayed, A.A. Structural and electronic properties of new fullerene derivatives and their possible application as HIV-1 protease inhibitors. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2010, 75, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Saleh, N.A. The QSAR and docking calculations of fullerene derivatives as HIV-1 protease inhibitors. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 136, 1523–1529. [Google Scholar] [CrossRef]
- Yasuno, T.; Ohe, T.; Kataoka, H.; Hashimoto, K.; Ishikawa, Y.; Furukawa, K.; Tateishi, Y.; Kobayashi, T.; Takahashi, K.; Nakamura, S.; et al. Fullerene derivatives as dual inhibitors of HIV-1 reverse transcriptase and protease. Bioorg. Med. Chem. Lett. 2021, 31, 127675. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Yasuno, T.; Takahashi, K.; Nakamura, S.; Mashino, T.; Ohe, T. Novel pyridinium-type fullerene derivatives as multitargeting inhibitors of HIV-1 reverse transcriptase, HIV-1 protease, and HCV NS5B polymerase. Bioorg. Med. Chem. Lett. 2021, 49, 128267. [Google Scholar] [CrossRef]
- Matsumoto, H.; Matsuda, T.; Nakata, S.; Mitoguchi, T.; Kimura, T.; Hayashi, Y.; Kiso, Y. Synthesis and biological evaluation of prodrug-type anti-HIV agents: Ester conjugates of carboxylic acid-containing dipeptide HIV protease inhibitors and a reverse transcriptase inhibitor. Bioorg. Med. Chem. 2001, 9, 417–430. [Google Scholar] [CrossRef]
- Matsumoto, H.; Kimura, T.; Hamawaki, T.; Kumagai, A.; Goto, T.; Sano, K.; Hayashi, Y.; Kiso, Y. Design, synthesis, and biological evaluation of anti-HIV double-drugs: Conjugates of HIV protease inhibitors with a reverse transcriptase inhibitor through spontaneously cleavable linkers. Bioorg.Med. Chem. 2001, 9, 1589–1600. [Google Scholar] [CrossRef]
- Tang, J.; Maddali, K.; Dreis, C.D.; Sham, Y.Y.; Vince, R.; Pommier, Y.; Wang, Z. 6-Benzoyl-3-hydroxypyrimidine-2,4-diones as dual inhibitors of HIV reverse transcriptase and integrase. Bioorg. Med. Chem. Lett. 2011, 21, 2400–2402. [Google Scholar] [CrossRef]
- Kankanala, J.; Kirby, K.A.; Huber, A.D.; Casey, M.C.; Wilson, D.J.; Sarafianos, S.G.; Wang, Z. Design, synthesis and biological evaluations of N-Hydroxy thienopyrimidine-2,4-diones as inhibitors of HIV reverse transcriptase-associated RNase H. Eur. J. Med. Chem. 2017, 141, 149–161. [Google Scholar] [CrossRef]
- Tang, J.; Maddali, K.; Dreis, C.D.; Sham, Y.Y.; Vince, R.; Pommier, Y.; Wang, Z. N-3 Hydroxylation of Pyrimidine-2,4-diones Yields Dual Inhibitors of HIV Reverse Transcriptase and Integrase. ACS Med. Chem. Lett. 2011, 2, 63–67. [Google Scholar] [CrossRef]
- Esposito, F.; Sechi, M.; Pala, N.; Sanna, A.; Koneru, P.C.; Kvaratskhelia, M.; Naesens, L.; Corona, A.; Grandi, N.; di Santo, R.; et al. Discovery of dihydroxyindole-2-carboxylic acid derivatives as dual allosteric HIV-1 Integrase and Reverse Transcriptase associated Ribonuclease H inhibitors. Antivir. Res. 2020, 174, 104671. [Google Scholar] [CrossRef]
- Gao, P.; Cheng, X.; Sun, L.; Song, S.; Álvarez, M.; Luczkowiak, J.; Pannecouque, C.; De Clercq, E.; Menéndez-Arias, L.; Zhan, P.; et al. Design, synthesis and biological evaluation of 3-hydroxyquinazoline-2,4(1H,3H)-diones as dual inhibitors of HIV-1 reverse transcriptase-associated RNase H and integrase. Bioorg. Med. Chem. 2019, 27, 3836–3845. [Google Scholar] [CrossRef]
- Sun, L.; Gao, P.; Dong, G.; Zhang, X.; Cheng, X.; Ding, X.; Wang, X.; Daelemans, D.; De Clercq, E.; Pannecouque, C.; et al. 5-Hydroxypyrido [2,3-b]pyrazin-6(5H)-one derivatives as novel dual inhibitors of HIV-1 reverse transcriptase-associated ribonuclease H and integrase. Eur. J. Med. Chem. 2018, 155, 714–724. [Google Scholar] [CrossRef]
- Billamboz, M.; Bailly, F.; Lion, C.; Calmels, C.; Andréola, M.L.; Witvrouw, M.; Christ, F.; Debyser, Z.; De Luca, L.; Chimirri, A.; et al. 2-Hydroxyisoquinoline-1,3(2H,4H)-diones as inhibitors of HIV-1 integrase and reverse transcriptase RNase H domain: Influence of the alkylation of position 4. Eur. J. Med. Chem. 2010, 46, 535–546. [Google Scholar] [CrossRef]
- Manyeruke, M.H.; Olomola, T.; Majumder, S.; Abrahams, S.; Isaacs, M.; Mautsa, N.; Mosebi, S.; Mnkandhla, D.; Hewer, R.; Hoppe, H.; et al. Synthesis and evaluation of 3-hydroxy-3-phenylpropanoate ester–AZT conjugates as potential dual-action HIV-1 Integrase and Reverse Transcriptase inhibitors. Bioorg. Med. Chem. 2015, 23, 7521–7528. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef]
- Esposito, F.; Ambrosio, F.A.; Maleddu, R.; Costa, G.; Rocca, R.; Maccioni, E.; Catalano, R.; Romeo, I.; Eleftheriou, P.; Karia, D.C.; et al. Chromenone derivatives as a versatile scaffold with dual mode of inhibition of HIV-1 reverse transcriptase-associated Ribonuclease H function and integrase activity. Eur. J. Med. Chem. 2019, 182, 111617. [Google Scholar] [CrossRef]
- Wang, Z.; Vince, R. Design and synthesis of dual inhibitors of HIV reverse transcriptase and integrase: Introducing a diketoacid functionality into delavirdine. Bioorg. Med. Chem. 2008, 16, 3587–3595. [Google Scholar] [CrossRef]
- Wang, Z.; Tang, J.; Salomon, C.E.; Dreis, C.D.; Vince, R. Pharmacophore and structure–activity relationships of integrase inhibition within a dual inhibitor scaffold of HIV reverse transcriptase and integrase. Bioorg. Med. Chem. 2010, 18, 4202–4211. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. The Physicochemical Challenges of Designing Multiple Ligands. J. Med. Chem. 2006, 49, 4961–4970. [Google Scholar] [CrossRef]
- Wang, Z.; Vince, R. Synthesis of pyrimidine and quinolone conjugates as a scaffold for dual inhibitors of HIV reverse transcriptase and integrase. Bioorg. Med. Chem. Lett. 2008, 18, 1293–1296. [Google Scholar] [CrossRef]
- Marchand, C.; Beutler, J.A.; Wamiru, A.; Budihas, S.; Möllmann, U.; Heinisch, L.; Mellors, J.W.; Le Grice, S.F.; Pommier, Y. Madurahydroxylactone Derivatives as Dual Inhibitors of Human Immunodeficiency Virus Type 1 Integrase and RNase H. Antimicrob. Agents Chemother. 2008, 52, 361–364. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cuzzucoli Crucitti, G.; Métifiot, M.; Pescatori, L.; Messore, A.; Madia, V.N.; Pupo, G.; Saccoliti, F.; Scipione, L.; Tortorella, S.; Esposito, F.; et al. Structure–Activity Relationship of Pyrrolyl Diketo Acid Derivatives as Dual Inhibitors of HIV-1 Integrase and Reverse Transcriptase Ribonuclease H Domain. J. Med. Chem. 2015, 58, 1915–1928. [Google Scholar] [CrossRef] [PubMed]
- Costi, R.; Métifiot, M.; Esposito, F.; Crucitti, G.C.; Pescatori, L.; Messore, A.; Scipione, L.; Tortorella, S.; Zinzula, L.; Novellino, E.; et al. 6-(1-Benzyl-1H-pyrrol-2-yl)-2,4-dioxo-5-hexenoic Acids as Dual Inhibitors of Recombinant HIV-1 Integrase and Ribonuclease H, Synthesized by a Parallel Synthesis Approach. J. Med. Chem. 2013, 56, 8588–8598. [Google Scholar] [CrossRef] [PubMed]
- Costi, R.; Métifiot, M.; Chung, S.; Cuzzucoli Crucitti, G.; Maddali, K.; Pescatori, L.; Messore, A.; Madia, V.N.; Pupo, G.; Scipione, L.; et al. Basic Quinolinonyl Diketo Acid Derivatives as Inhibitors of HIV Integrase and their Activity against RNase H Function of Reverse Transcriptase. J. Med. Chem. 2014, 57, 3223–3234. [Google Scholar] [CrossRef]
- Shaw-Reid, C.A.; Munshi, V.; Graham, P.; Wolfe, A.; Witmer, M.; Danzeisen, R.; Olsen, D.B.; Carroll, S.S.; Embrey, M.; Wai, J.S.; et al. Inhibition of HIV-1 Ribonuclease H by a Novel Diketo Acid, 4-[5-(Benzoylamino)thien-2-yl]-2,4-dioxobutanoic Acid. J. Biol. Chem. 2003, 278, 2777–2780. [Google Scholar] [CrossRef]
- Mao, T.Q.; He, Q.Q.; Wan, Z.Y.; Chen, W.X.; Chen, F.E.; Tang, G.F.; De Clercq, E.; Daelemans, D.; Pannecouque, C. Anti-HIV diarylpyrimidine–quinolone hybrids and their mode of action. Bioorg. Med. Chem. 2015, 23, 3860–3868. [Google Scholar] [CrossRef]
- Meleddu, R.; Distinto, S.; Corona, A.; Bianco, G.; Cannas, V.; Esposito, F.; Artese, A.; Alcaro, S.; Matyus, P.; Bogdan, D.; et al. (3Z)-3-(2-[4-(aryl)-1,3-thiazol-2-yl]hydrazin-1-ylidene)-2,3-dihydro-1H-indol-2-one derivatives as dual inhibitors of HIV-1 reverse transcriptase. Eur. J. Med. Chem. 2015, 93, 452–460. [Google Scholar] [CrossRef]
- Feng, D.; Zuo, X.; Jing, L.; Chen, C.-H.; Olotu, F.A.; Lin, H.; Soliman, M.; De Clercq, E.; Pannecouque, C.; Lee, K.-H.; et al. Design, synthesis, and evaluation of “dual-site”-binding diarylpyrimidines targeting both NNIBP and the NNRTI adjacent site of the HIV-1 reverse transcriptase. Eur. J. Med. Chem. 2020, 211, 113063. [Google Scholar] [CrossRef]
- Devale, T.L.; Parikh, J.; Miniyar, P.; Sharma, P.; Shrivastava, B.; Murumkar, P. Dihydropyrimidinone-isatin hybrids as novel non-nucleoside HIV-1 reverse transcriptase inhibitors. Bioorg. Chem. 2017, 70, 256–266. [Google Scholar] [CrossRef]
- Iyidogan, P.; Sullivan, T.J.; Chordia, M.D.; Frey, K.M.; Anderson, K.S. Design, Synthesis, and Antiviral Evaluation of Chimeric Inhibitors of HIV Reverse Transcriptase. ACS Med. Chem. Lett. 2013, 4, 1183–1188. [Google Scholar] [CrossRef][Green Version]
- Distinto, S.; Esposito, F.; Kirchmair, J.; Cardia, M.C.; Gaspari, M.; Maccioni, E.; Alcaro, S.; Markt, P.; Wolber, G.; Zinzula, L.; et al. Identification of HIV-1 reverse transcriptase dual inhibitors by a combined shape-, 2D-fingerprint- and pharmacophore-based virtual screening approach. Eur. J. Med. Chem. 2012, 50, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Su, H.P.; Yan, Y.; Prasad, G.S.; Smith, R.F.; Daniels, C.L.; Abeywickrema, P.D.; Reid, J.C.; Loughran, H.M.; Kornienko, M.; Sharma, S.; et al. Structural Basis for the Inhibition of RNase H Activity of HIV-1 Reverse Transcriptase by RNase H Active Site-Directed Inhibitors. J. Virol. 2010, 84, 7625–7633. [Google Scholar] [CrossRef] [PubMed]
- Himmel, D.M.; Sarafianos, S.G.; Dharmasena, S.; Hossain, M.M.; McCoy-Simandle, K.; Ilina, T.; Clark, A.D., Jr.; Knight, J.L.; Julias, J.G.; Clark, P.K.; et al. HIV-1 Reverse Transcriptase Structure with RNase H Inhibitor Dihydroxy Benzoyl Naphthyl Hydrazone Bound at a Novel Site. ACS Chem. Biol. 2006, 1, 702–712. [Google Scholar] [CrossRef] [PubMed]


































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trade Name | Generic Name (Dosage) | Target |
|---|---|---|
| Combivir | Lamivudine (150 mg) and Zidovudine (300 mg) | RT, RT |
| Trizivir | Abacavir (300 mg), Zidovudine (300 mg), and Lamivudine (150 mg) | RT, RT, RT |
| Kaletra | Lopinavir (80 mg) and Ritonavir a (20 mg) | CYP3A b, PR |
| Epzicom | Abacavir (600 mg) and Lamivudine (300 mg) | RT, RT |
| Truvada | Emtricitabine (200 mg) and Tenofovir disoproxil fumarate (245 mg) | RT, RT |
| Atripla | Efavirenz (600 mg), Emtricitabine (200 mg), and Tenofovir disoproxil fumarate (245 mg) | RT, RT, RT |
| Complera | Emtricitabine (200 mg), Tenofovir disoproxil fumarate (300 mg), and Rilpivirine (25 mg) | RT, RT, RT |
| Stribild | Elvitegravir (150 mg), Cobicistat a (150 mg), Tenofovir disoproxil fumarate (245 mg), and Emtricitabine (200 mg) | IN, CYP3A b, RT, RT |
| Triumeq | Abacavir (600 mg), Lamivudine (300 mg), and Dolutegravir (50 mg) | RT, RT, IN |
| Evotaz | Atazanavir (300 mg) and Cobicistat a (150 mg) | PR, CYP3A b |
| Prezcobix | Darunavir (800 mg) and Cobicistat a (150 mg) | PR, CYP3A b |
| Genvoya | Elvitegravir (150 mg), Cobicistat a (150 mg), Tenofovir alafenamide (100 mg), and Emtricitabine (200 mg) | IN, CYP3A b, RT, RT |
| Odefsey | Tenofovir alafenamide (25 mg), and Emtricitabine (200 mg), Rilpivirine (25 mg) | RT, RT |
| Descovy | Emtricitabine (200 mg) and Tenofovir alafenamide (10 mg) | RT, RT |
| Juluca | Dolutegravir (50 mg) and Rilpivirine (25 mg) | IN, RT |
| Symfi | Efavirenz (600 mg), Lamivudine (300 mg), and Tenofovir disoproxil fumarate (300 mg) | RT, RT, RT |
| Symfi Lo | Efavirenz (400 mg), Tenofovir disoproxil fumarate (300 mg), and Lamivudine (300 mg) | RT, RT, RT |
| Cimduo | Tenofovir disoproxil fumarate (300 mg) and Lamivudine (300 mg) | RT, RT |
| Delstrigo | Tenofovir disoproxil fumarate (245 mg), Doravirine (100 mg), and Lamivudine (300 mg) | RT, PR, RT |
| Biktarvy | Bictegravir (50 mg), Tenofovir alafenamide (25 mg), and Emtricitabine (200 mg) | IN, RT, RT |
| Symtuza | Darunavir (800 mg), Emtricitabine (200 mg), Cobicistat a (150 mg), and Tenofovir alafenamide (10 mg) | PR, RT, CYP3A b, RT |
| Dovato | Dolutegravir (50 mg) and Lamivudine (300 mg) | IN, RT |
| Cabenuva | Cabotegravir (200 mg) and Rilpivirine (300 mg) | IN, RT |
| Triumeq PD | Abacavir (600 mg), Lamivudine (300 mg), and Dolutegravir (50 mg) | RT, RT, IN |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liman, W.; Ait Lahcen, N.; Oubahmane, M.; Hdoufane, I.; Cherqaoui, D.; Daoud, R.; El Allali, A. Hybrid Molecules as Potential Drugs for the Treatment of HIV: Design and Applications. Pharmaceuticals 2022, 15, 1092. https://doi.org/10.3390/ph15091092
Liman W, Ait Lahcen N, Oubahmane M, Hdoufane I, Cherqaoui D, Daoud R, El Allali A. Hybrid Molecules as Potential Drugs for the Treatment of HIV: Design and Applications. Pharmaceuticals. 2022; 15(9):1092. https://doi.org/10.3390/ph15091092
Chicago/Turabian StyleLiman, Wissal, Nouhaila Ait Lahcen, Mehdi Oubahmane, Ismail Hdoufane, Driss Cherqaoui, Rachid Daoud, and Achraf El Allali. 2022. "Hybrid Molecules as Potential Drugs for the Treatment of HIV: Design and Applications" Pharmaceuticals 15, no. 9: 1092. https://doi.org/10.3390/ph15091092
APA StyleLiman, W., Ait Lahcen, N., Oubahmane, M., Hdoufane, I., Cherqaoui, D., Daoud, R., & El Allali, A. (2022). Hybrid Molecules as Potential Drugs for the Treatment of HIV: Design and Applications. Pharmaceuticals, 15(9), 1092. https://doi.org/10.3390/ph15091092