
The Medicinal Chemistry of Artificial Nucleic Acids and Therapeutic Oligonucleotides


Abstract
1. Introduction
2. The Use of Oligonucleotides in Therapy and Diagnostics and the Mechanisms behind Them
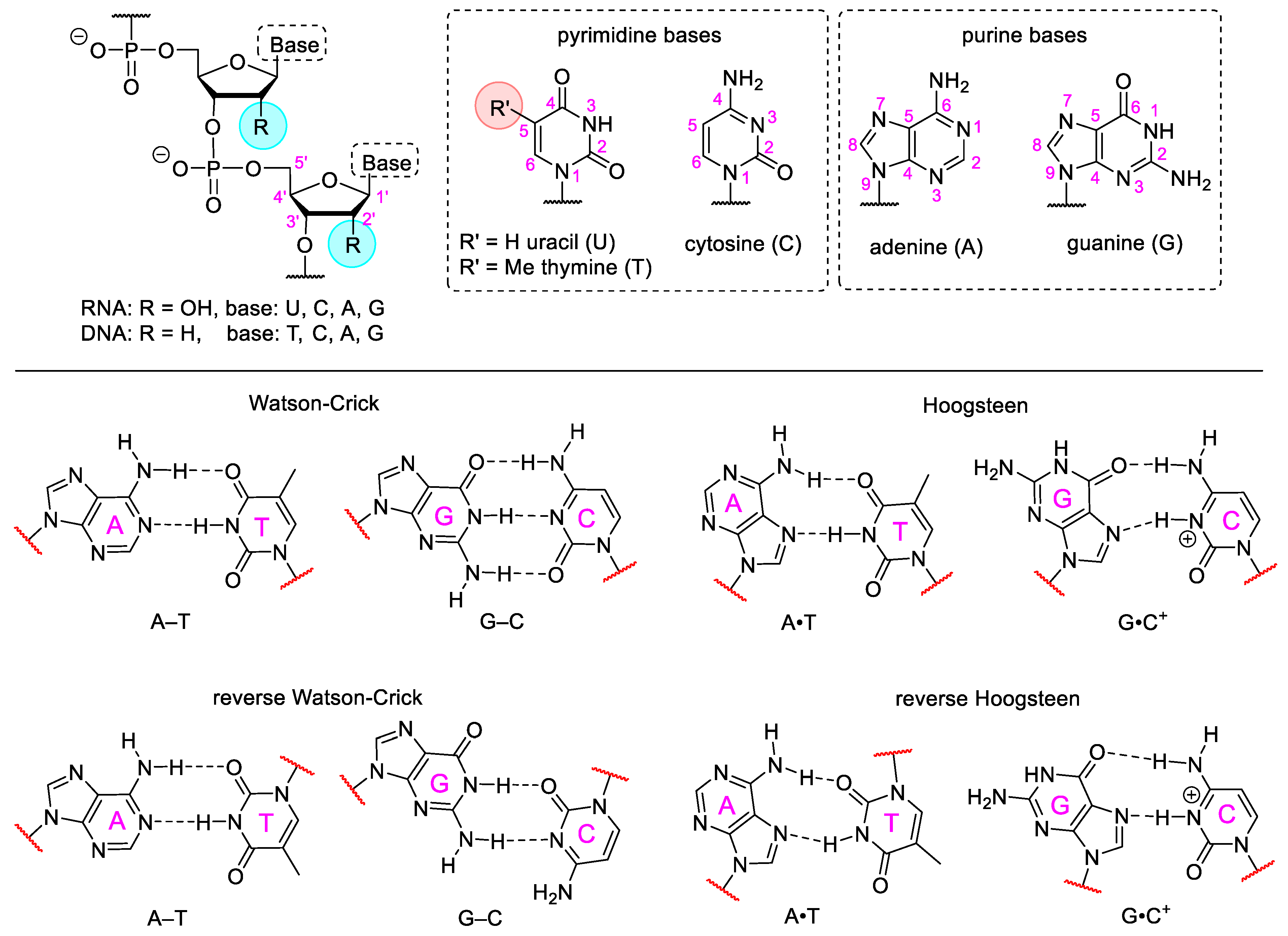
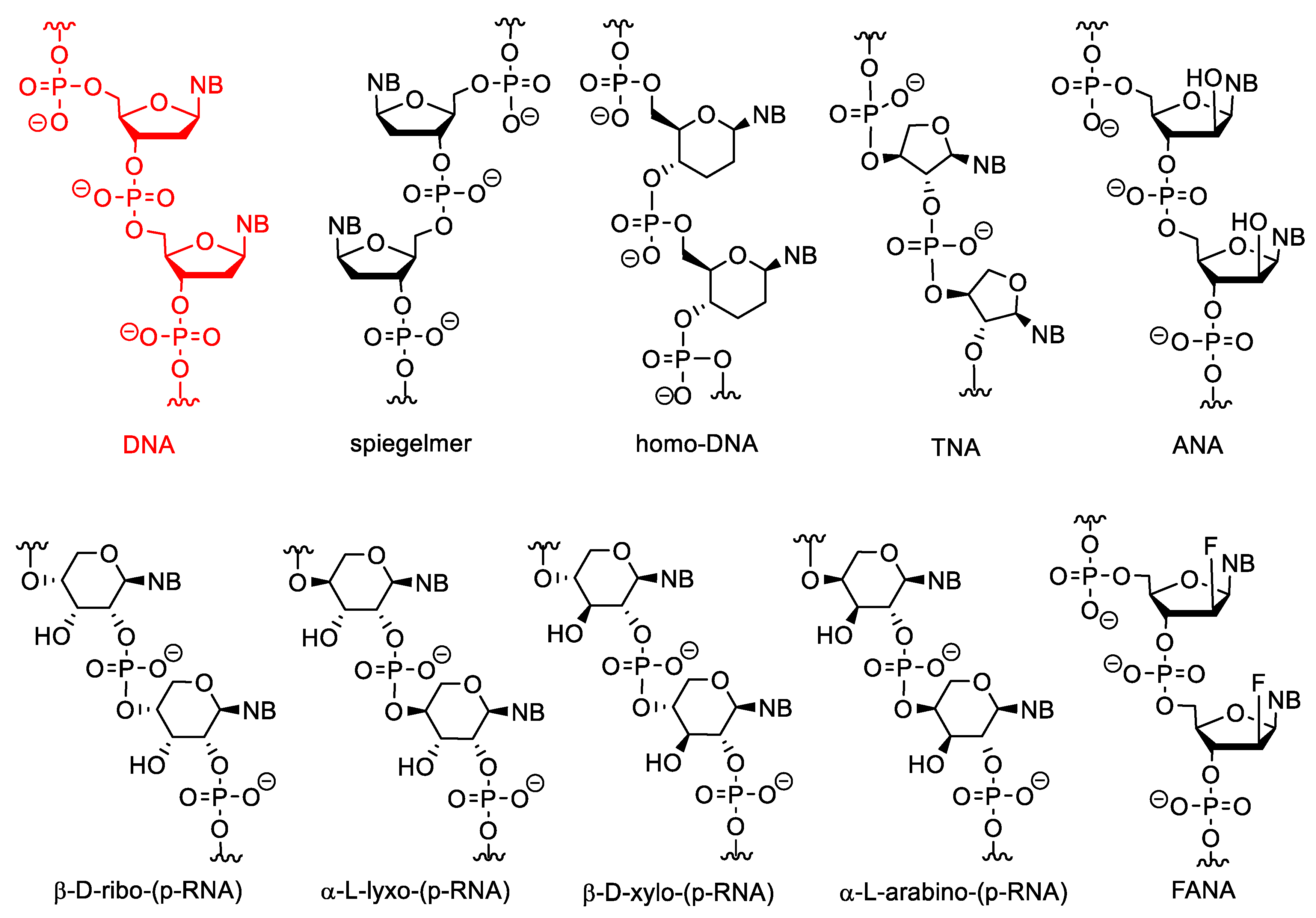
3. Artificial Nucleic Acids
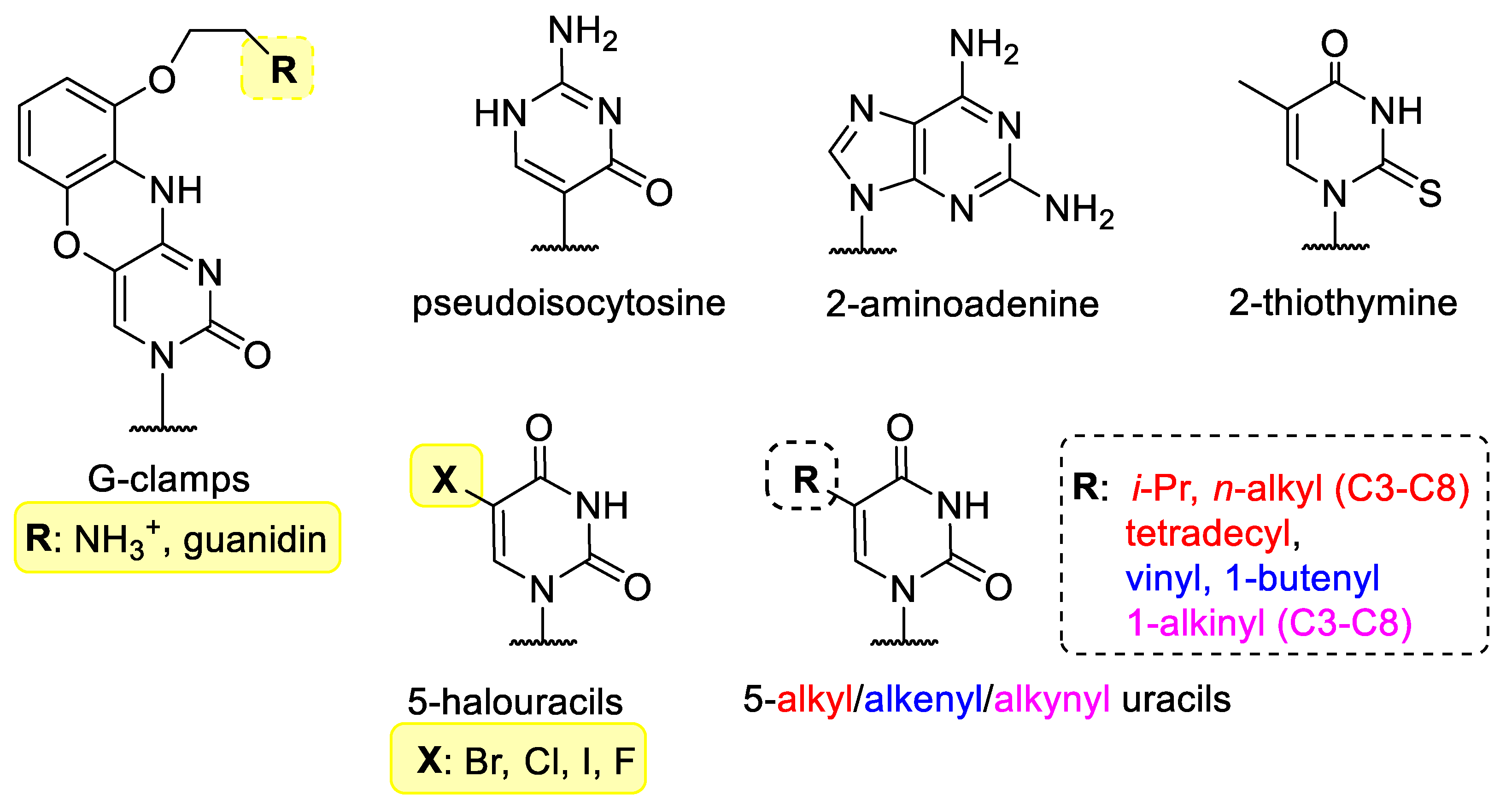
3.1. Base Modified Nucleic Acids
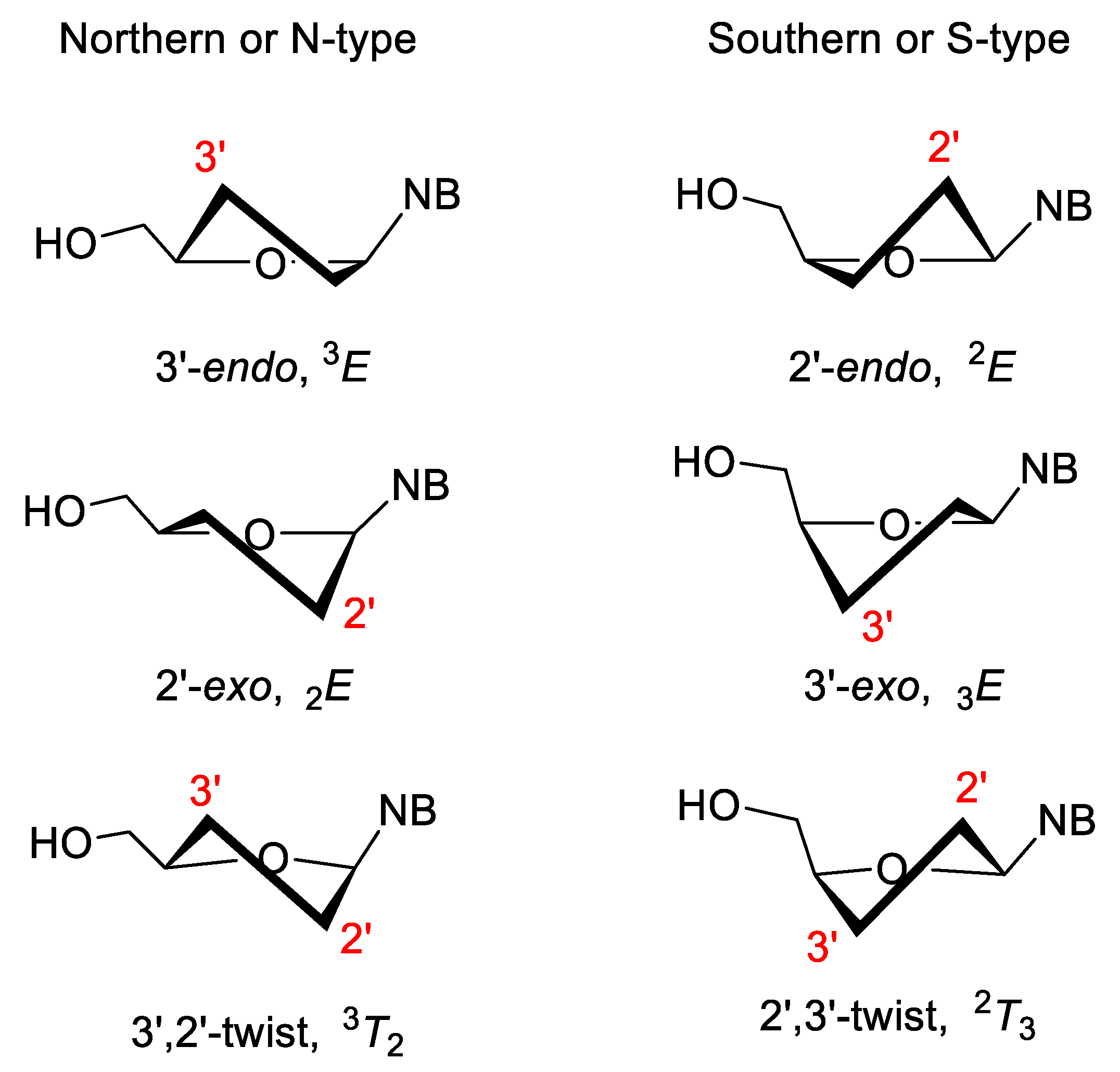
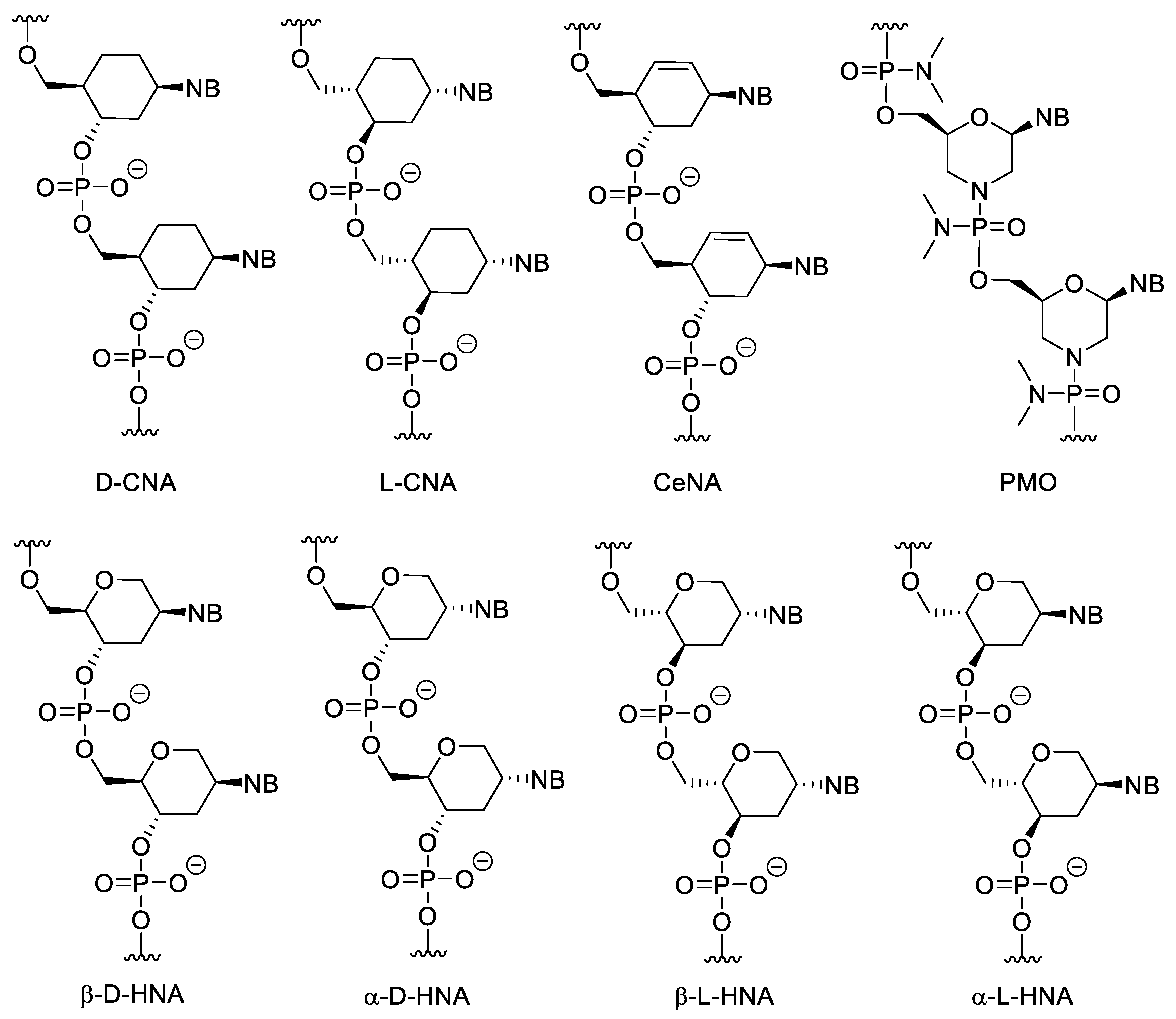
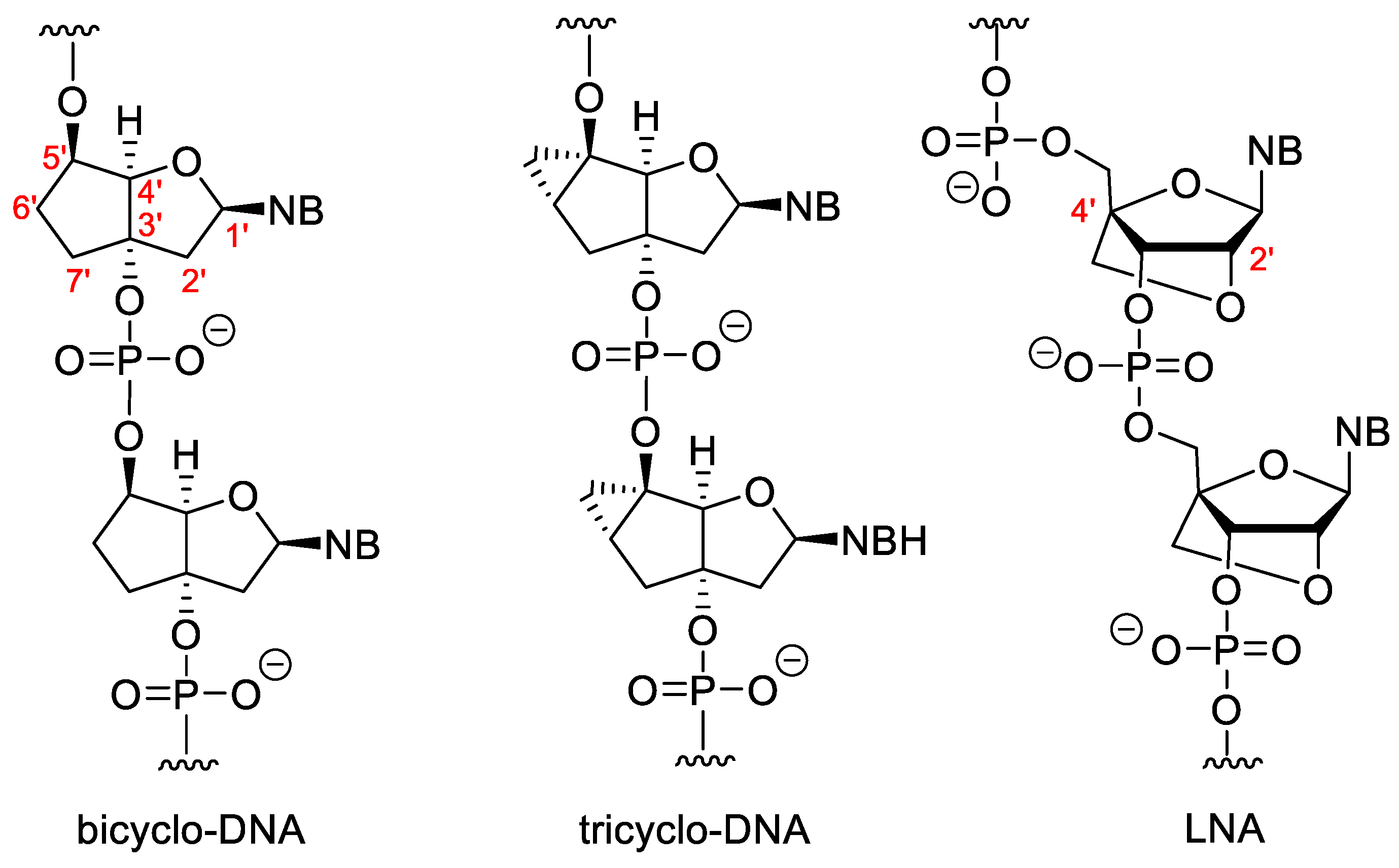
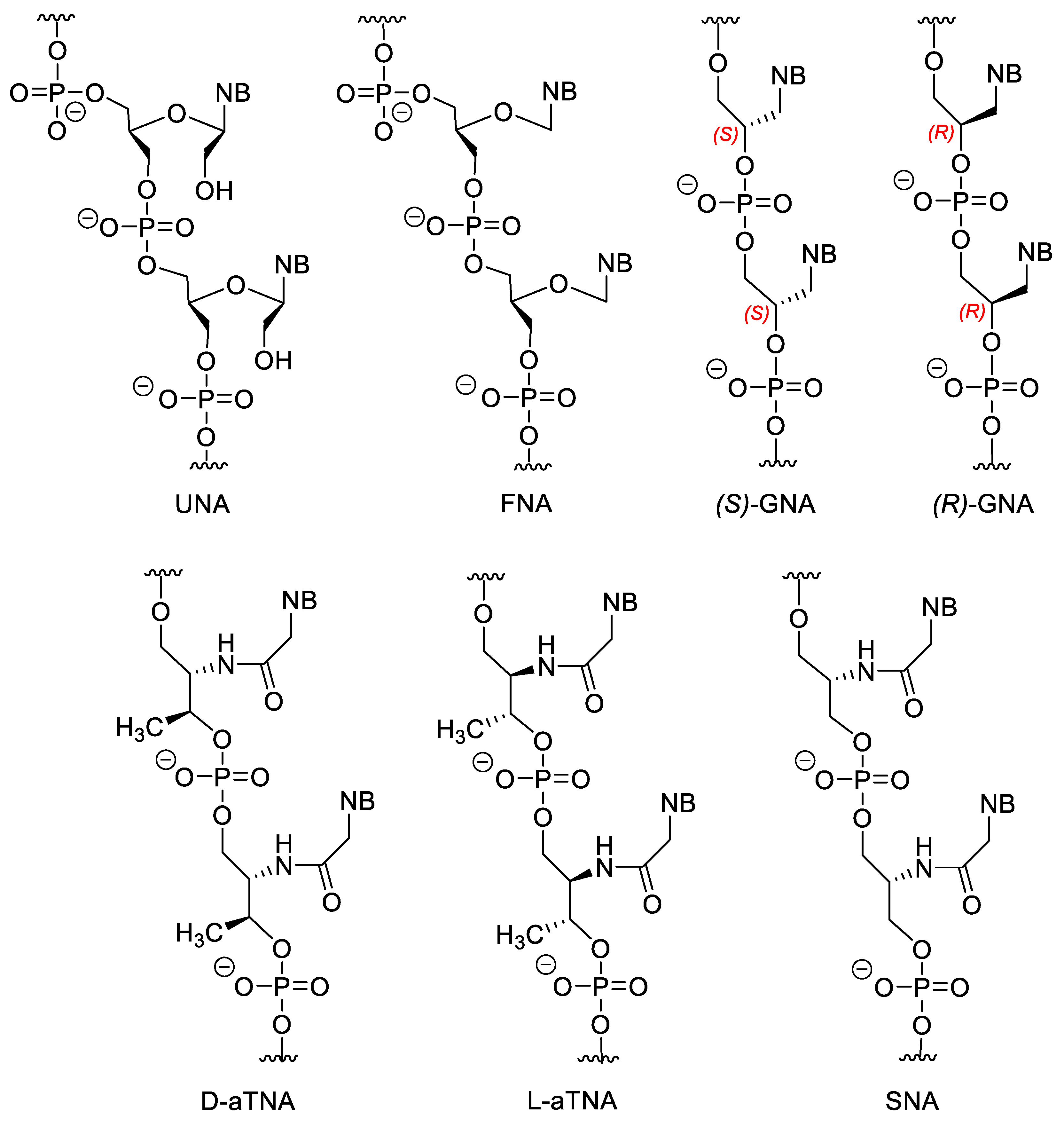
3.2. Sugar-Modified Nucleic Acids
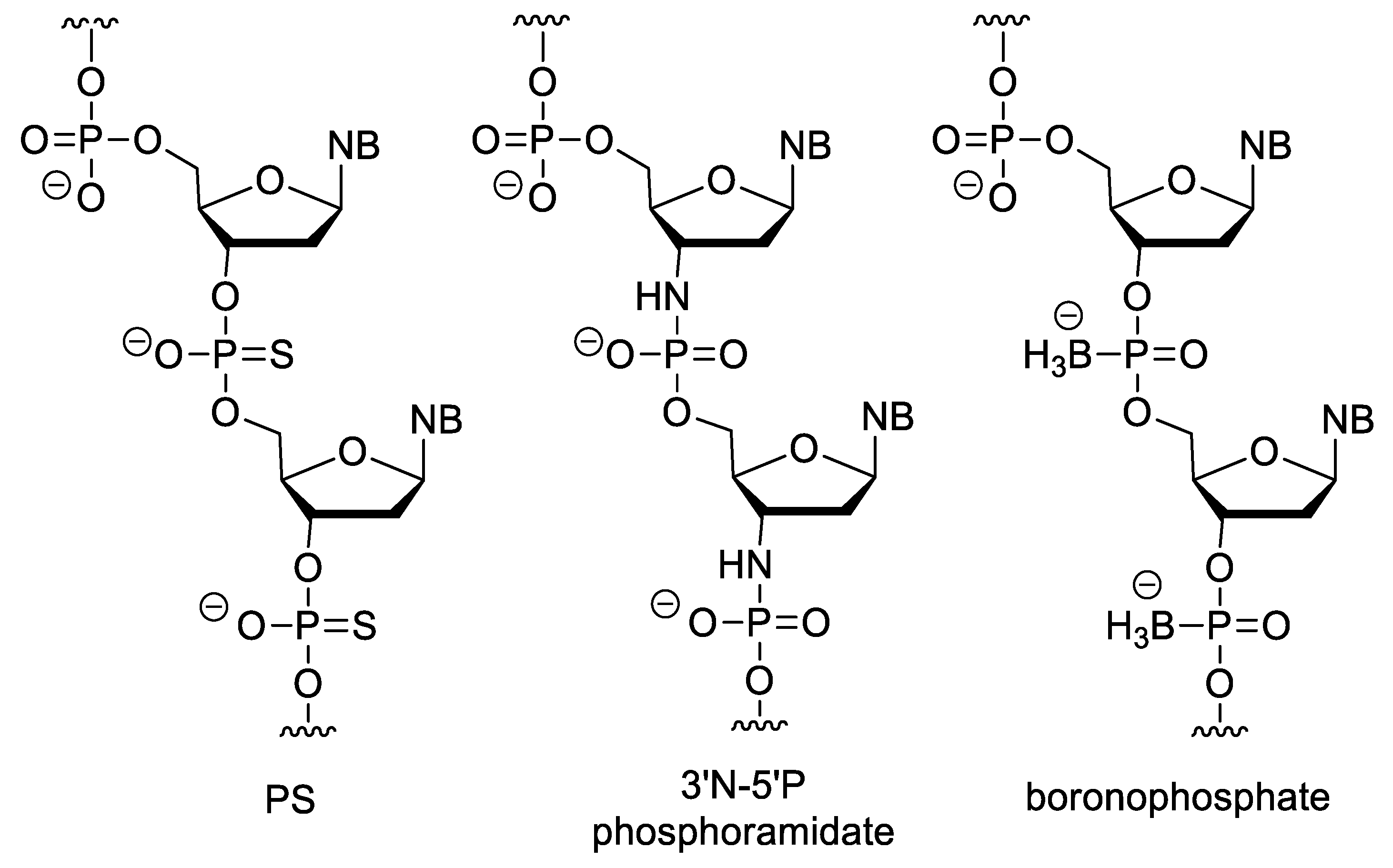
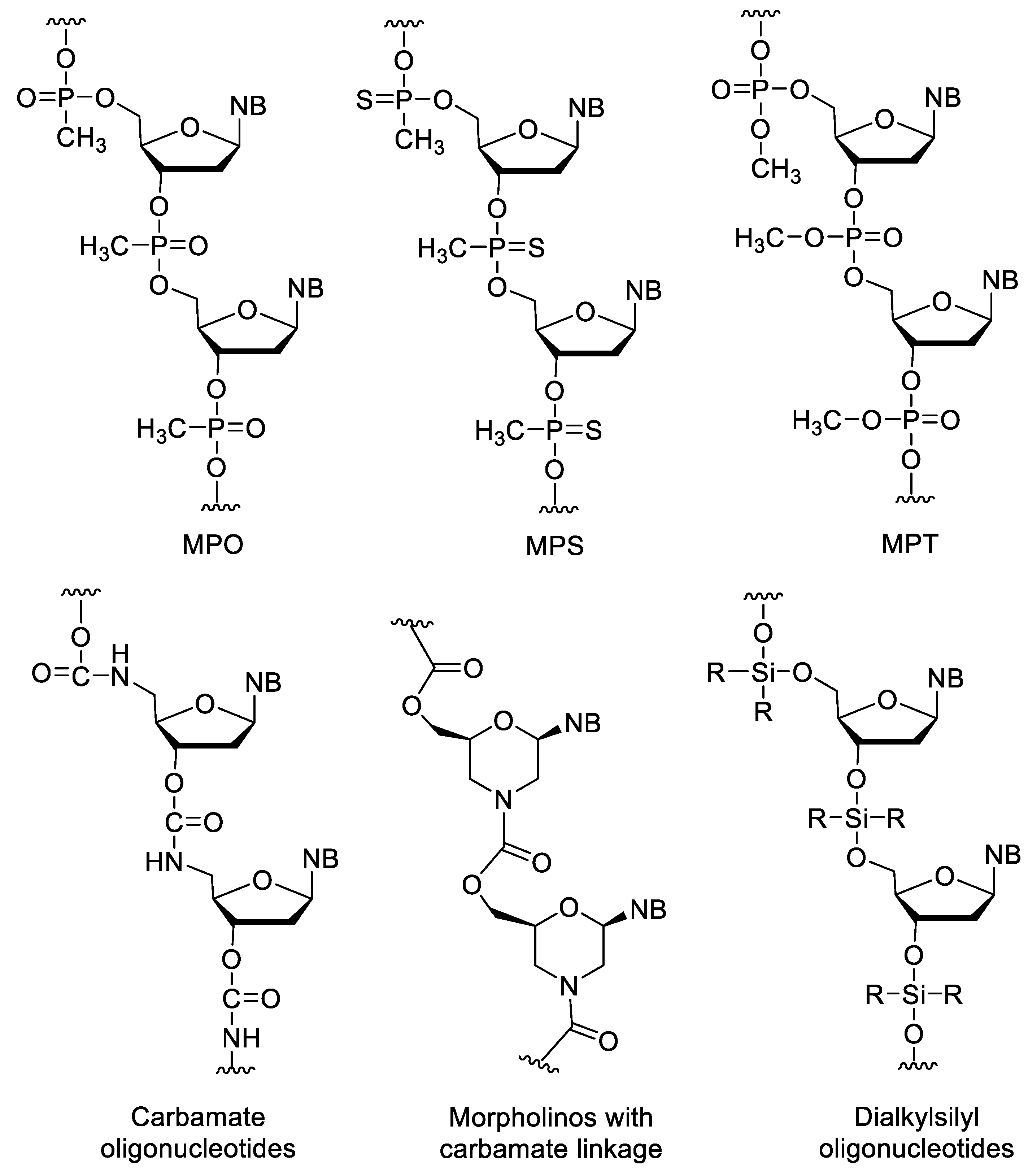
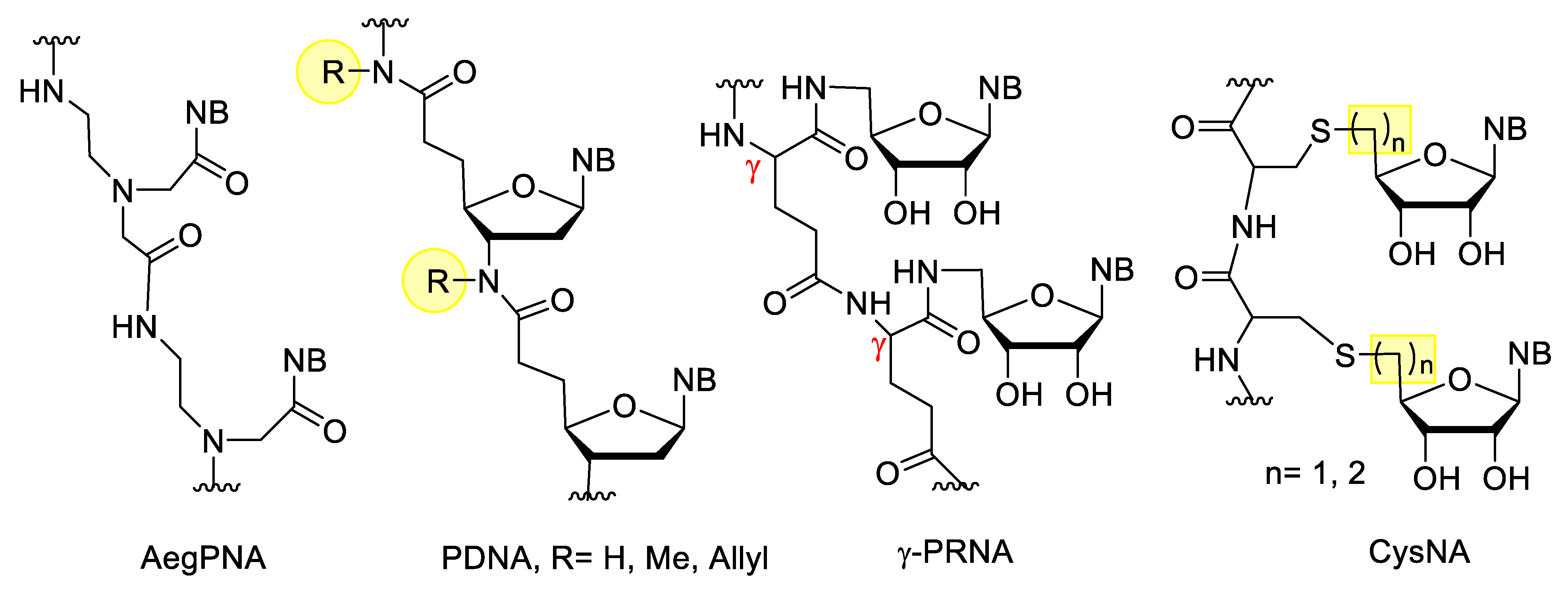
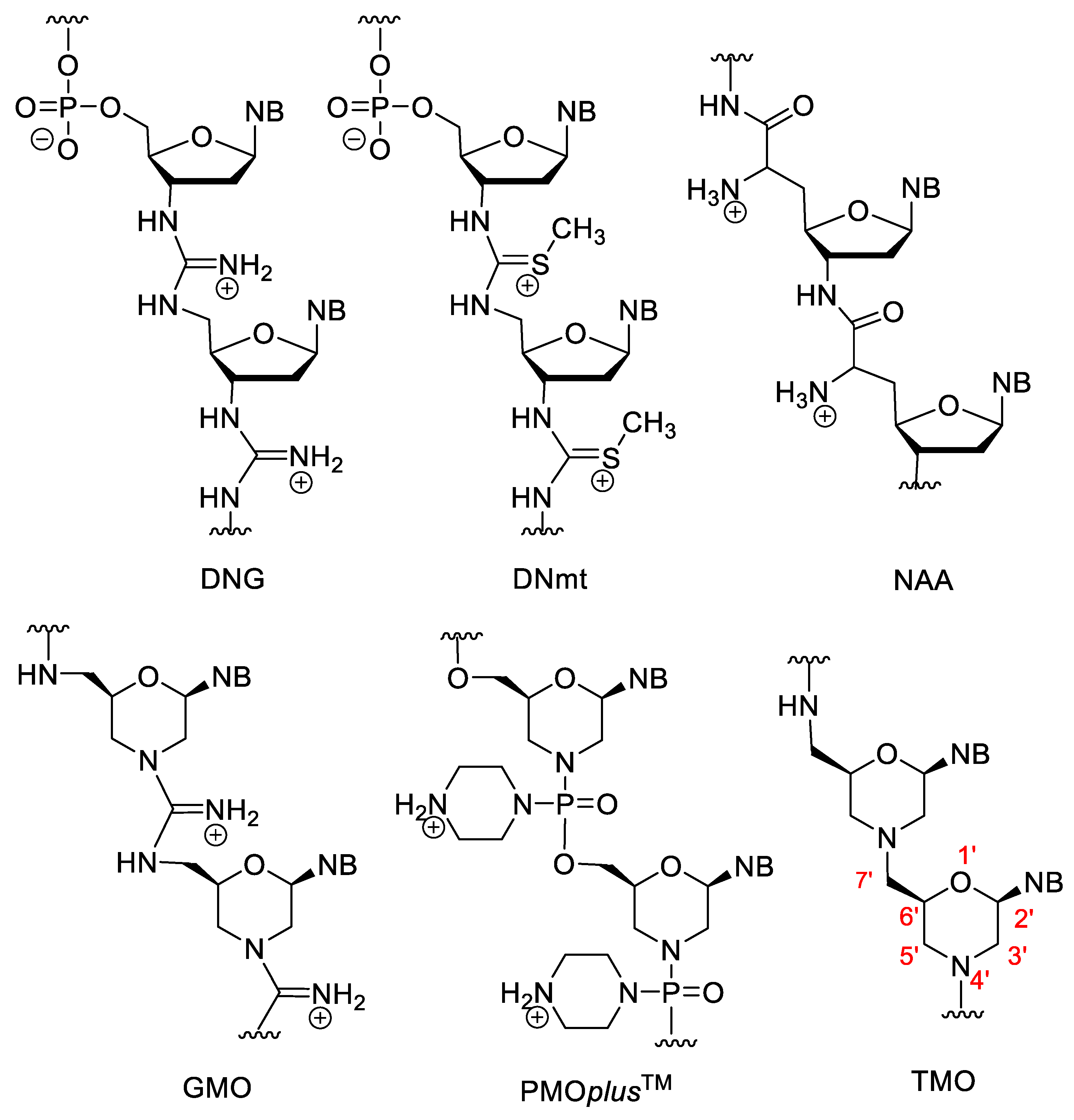
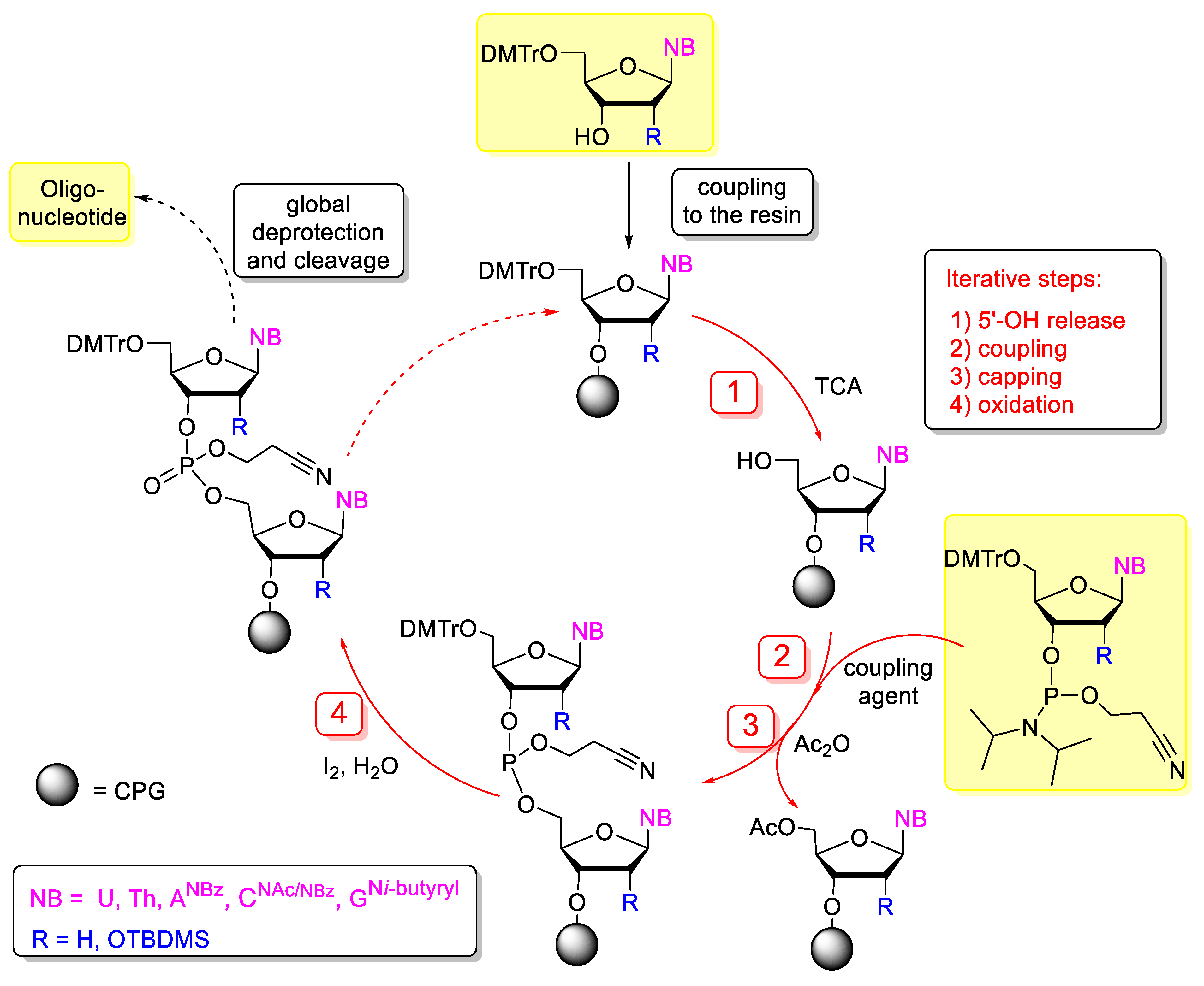
3.3. Nucleotides with Modified Backbones
4. Oligonucleotide Drugs
4.1. Fomivirsen
4.2. Pegaptanib
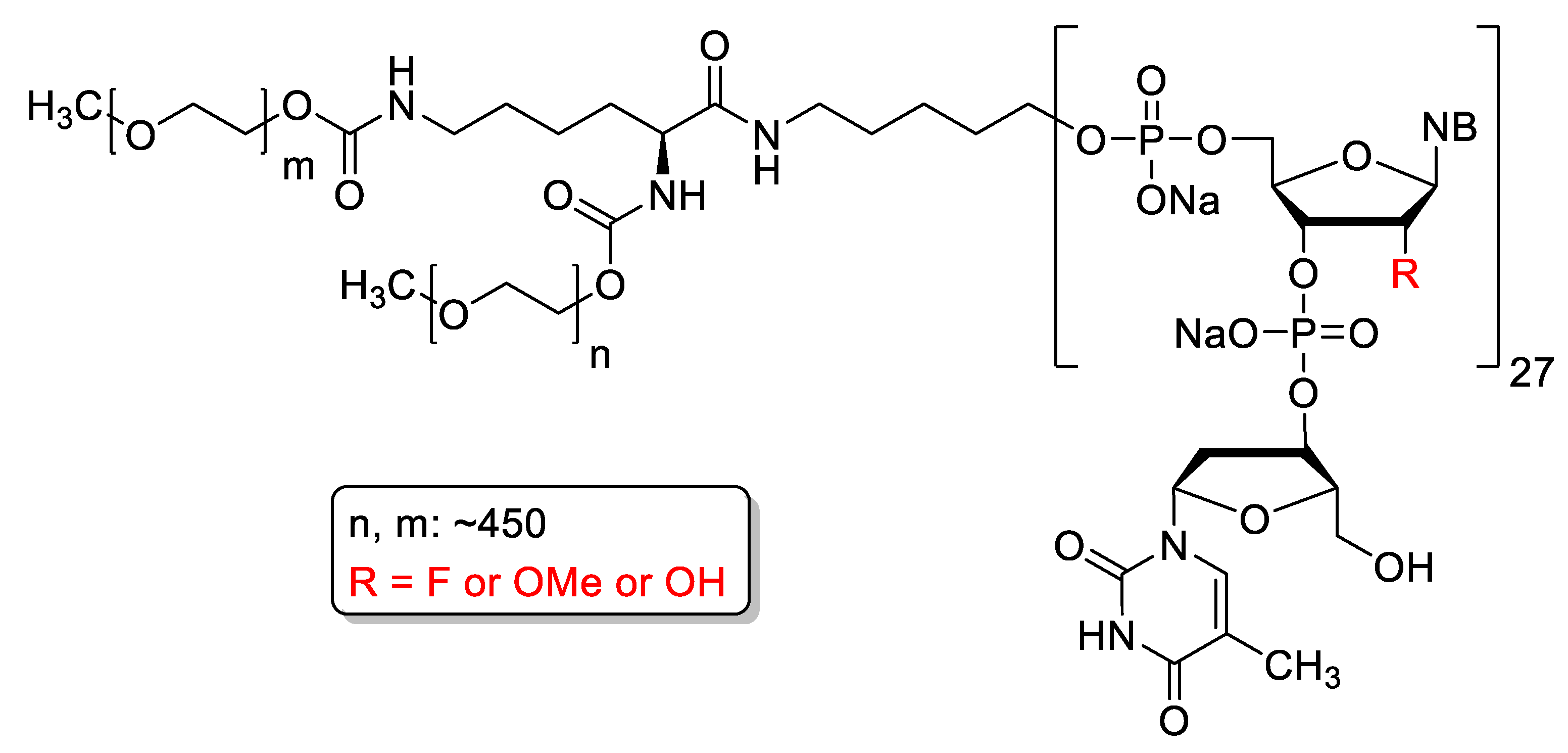
4.3. Mipomersen
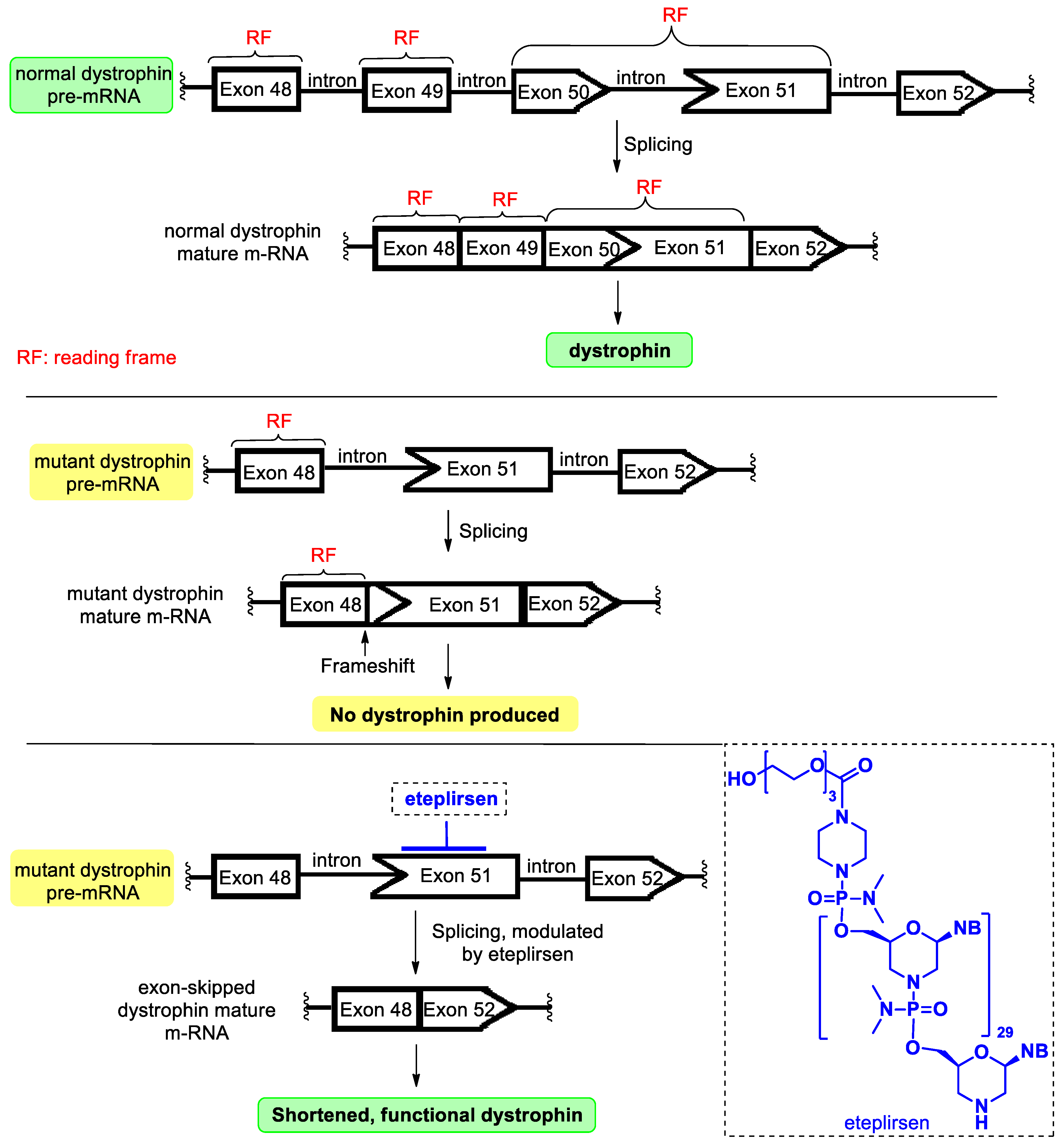
4.4. Eteplirsen
4.5. Defibrotide
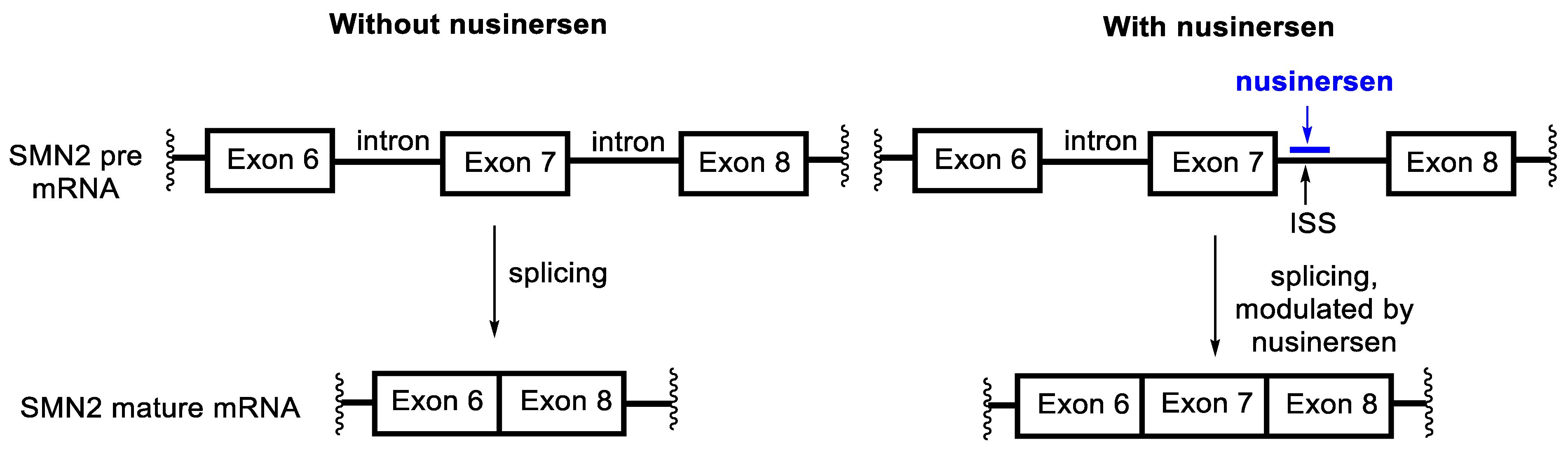
4.6. Nusinersen
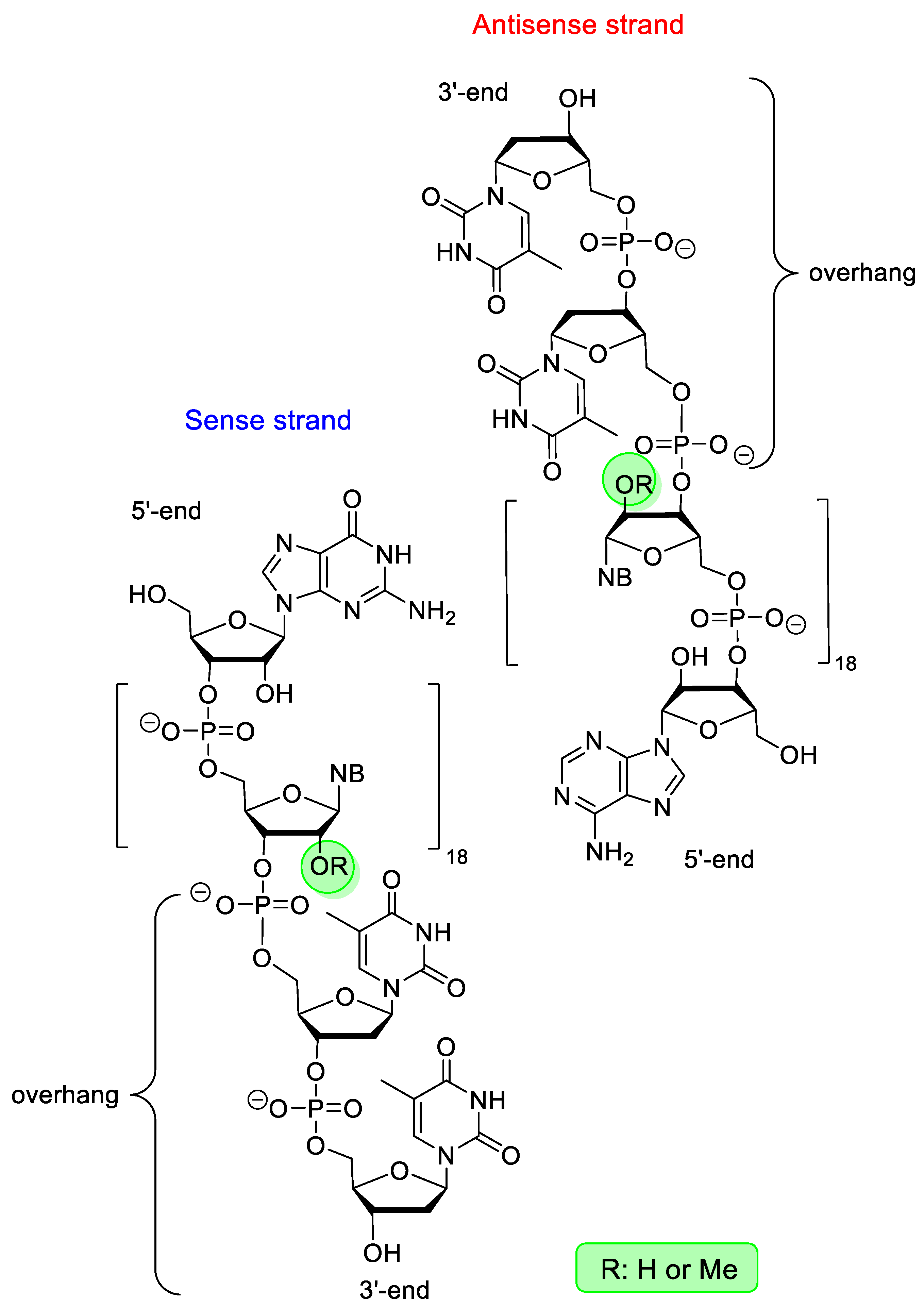
4.7. Patisiran
4.8. Inotersen
4.9. Milasen
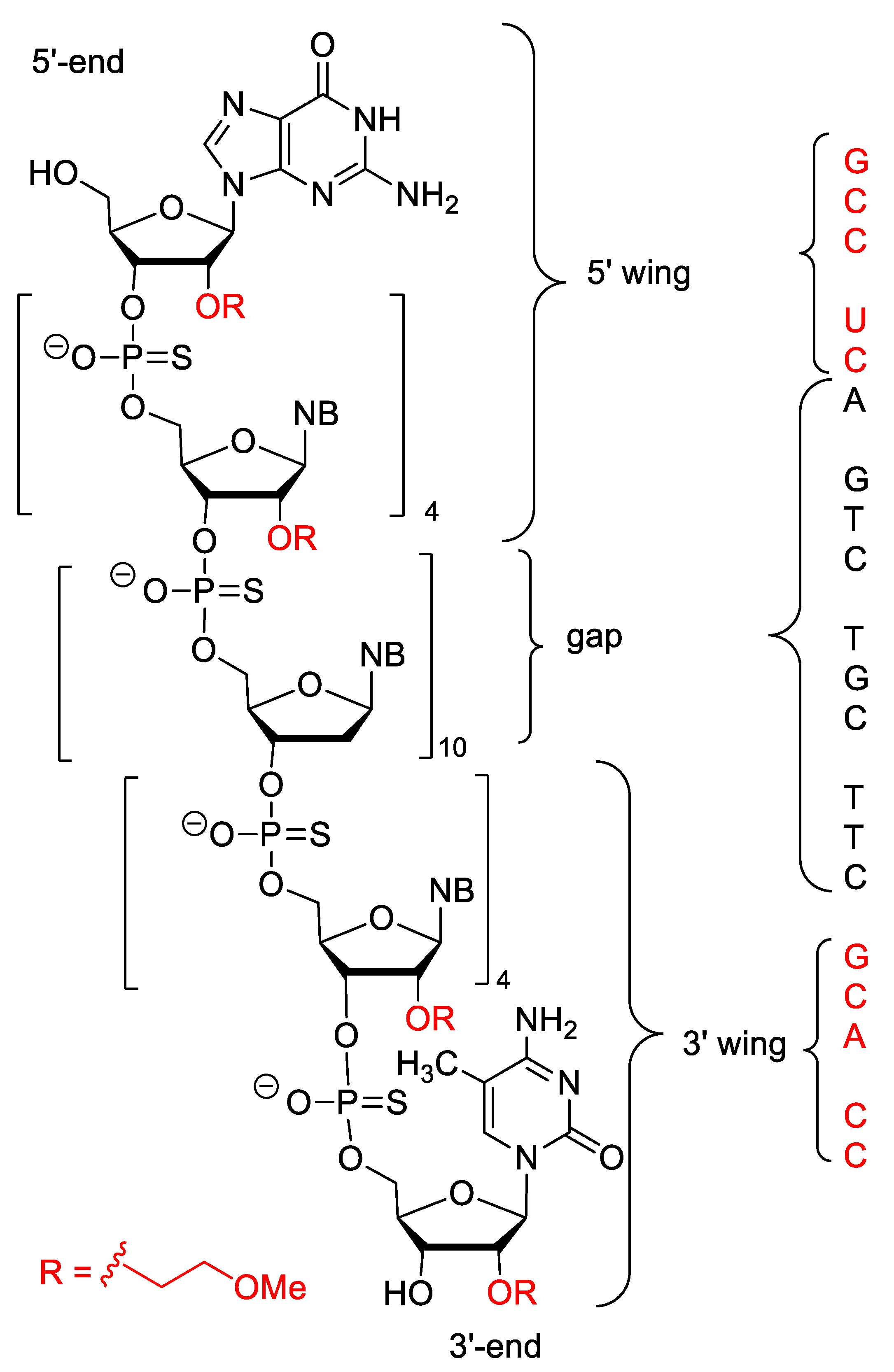
4.10. Volanesorsen
4.11. Givosiran
4.12. Golodirsen
4.13. Viltolarsen
4.14. Inclisiran
4.15. Lumasiran
4.16. Casimersen
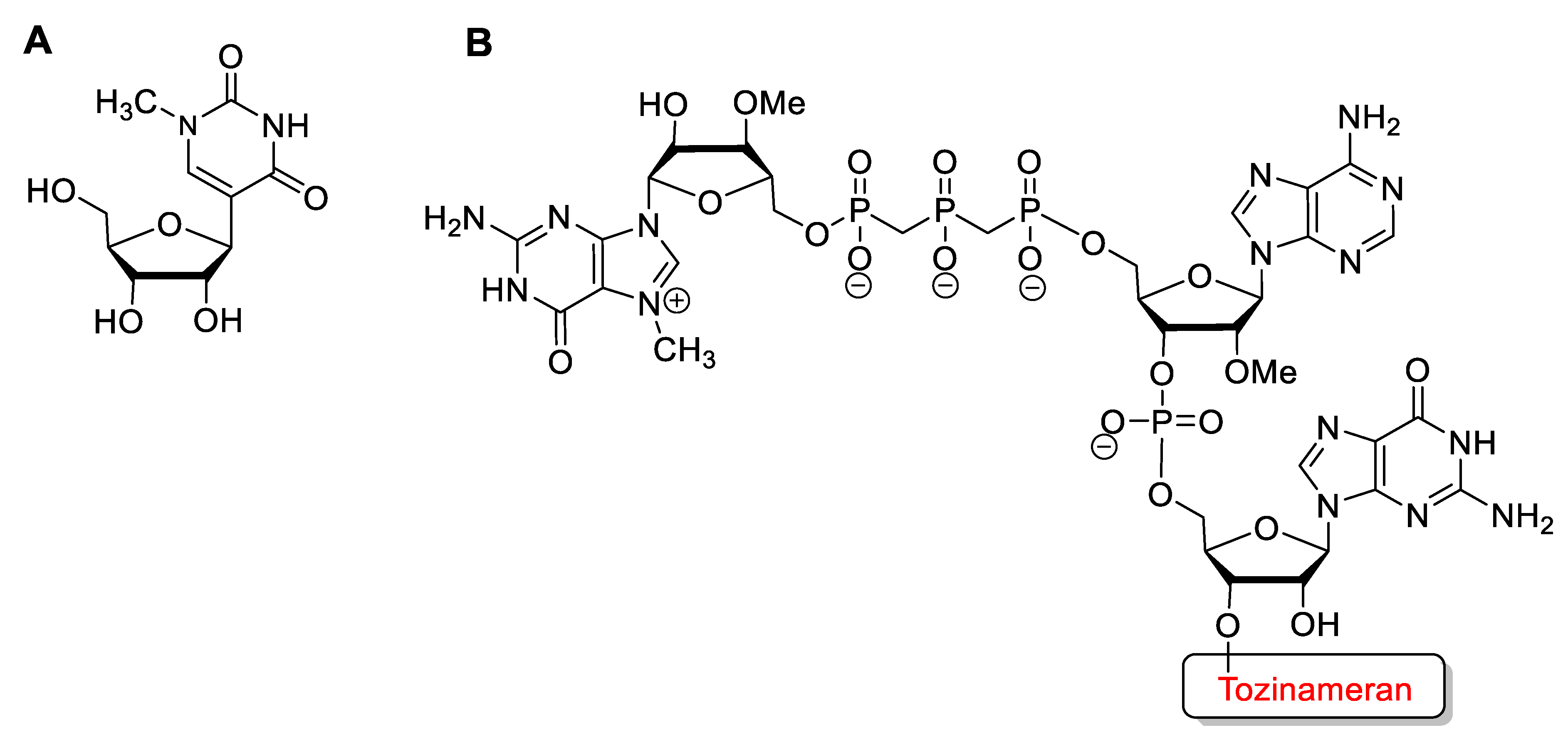
4.17. Tozinameran
4.18. Elasomeran
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Carter, G.T.; McDonald, L.A. Uridyl Peptide Antibiotics: Developments in Biosynthesis and Medicinal Chemistry. In Antimicrobials New and Old Molecules in the Fight against Multi-Resistant Bacteria; Marinelli, F., Genniloud, O., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 177–191. [Google Scholar]
- Suzuki, T. The expanding world of tRNA modifications and their disease relevance. Nat. Rev. 2021, 22, 375–392. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Marcu, D.; Narayanan, P. Modified bases in tRNA: The structures of 5-carbamoylmethyl- and 5-carboxymethyl uridine. Nucleic Acids Res. 1978, 5, 893–903. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rafels-Ybern, Á.; Torres, A.G.; Camacho, N.; Herencia-Ropero, A.; Frigolé, H.R.; Wulff, T.F.; Raboteg, M.; Bordons, A.; Grau-Bove, X.; Ruiz-Trillo, I.; et al. The expansion of Inosine at the wobble position of tRNAs, and its role in the evolution of proteomes. Mol. Biol. Evol. 2019, 36, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Nazar, R.N. Ribosomal RNA Processing and Ribosome Biogenesis in Eukaryotes. Life 2004, 56, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Yu, X.; Hu, S.; Yu, J. A Brief Review on the Mechanisms of miRNA Regulation. Genom. Proteom. Bioinform. 2009, 7, 147–154. [Google Scholar] [CrossRef]
- Hu, B.; Zhong, L.; Weng, Y.; Peng, L.; Huang, Y.; Zhao, Y.; Liang, X. Therapeutic siRNA: State of the art. Signal Transduct. Target. Ther. 2020, 5, 101. [Google Scholar] [CrossRef]
- Kwok, A.; Raulf, N.; Habib, N. Developing small activating RNA as a therapeutic: Current challenges and promises. Ther. Deliv. 2019, 10, 151–164. [Google Scholar] [CrossRef]
- Müller, S.; Appel, B.; Balke, D.; Hieronymus, R.; Nübel, C. Thirty-five years of research into ribozymes and nucleic acid catalysis: Where do we stand today? F1000Research 2016, 5, 1–11. [Google Scholar] [CrossRef]
- Wang, Z.; Mosbaugh, D.W. Uracil-DNA Glycosylase Inhibitor of Bacteriophage PBS2: Cloning and Effects of Expression of the Inhibitor Gene in Escherichia coli. J. Bacteriol. 1988, 170, 1082–1091. [Google Scholar] [CrossRef]
- Nikolova, E.N.; Kim, E.; Wise, A.A.; O’Brien, P.J.; Andricioaei, I.; Al-Hashimi, H.M. Transient Hoogsteen base pairs in canonical duplex DNA. Nature 2011, 470, 498–504. [Google Scholar] [CrossRef]
- Aishima, J.; Gitti, R.K.; Noah, J.E.; Gan, H.H.; Schlick, T.; Wolberger, C. A Hoogsteen base pair embedded in undistorted B-DNA. Nucleic Acids Res. 2002, 30, 5244–5252. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Giese, T.J.; Lee, T.; York, D.M. Improvement of DNA and RNA Sugar Pucker Profiles from Semiempirical Quantum Methods. J. Chem. Theory Comput. 2014, 10, 1538–1545. [Google Scholar] [CrossRef] [PubMed]
- Maderia, M.; Shenoy, S.; Van, Q.N.; Marquez, V.E.; Barchi, J.J. Biophysical studies of DNA modified with conformationally constrained nucleotides: Comparison of 20-exo (north) and 3 -exo (south) ‘locked’ templates. Nucleic Acid Res. 2007, 35, 1978–1991. [Google Scholar] [CrossRef] [PubMed]
- Evich, M.; Spring-Connel, A.M.; Germann, M.W. Impact of modified ribose sugars on nucleic acid conformation and function. Heterocycl. Commun. 2017, 23, 155–165. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Weidolf, L.; Björkbom, A.; Dahlén, A.; Elebring, M.; Gennemark, P.; Hölttä, M.; Janzén, D.; Li, X.; Andersson, S. Distribution and biotransformation of therapeutic antisense oligonucleotides and conjugates. Drug Discov. Today 2021, 26, 2244–2258. [Google Scholar] [CrossRef]
- Crooke, S.T.; Baker, B.F.; Crooke, R.M.; Liang, X. Antisense technology: An overview and prospectus. Nat. Rev. Drug. Discov. 2021, 20, 427–453. [Google Scholar] [CrossRef]
- Epple, S.; El-Sagheer, A.H.; Brown, T. Artificial nucleic acid backbones and their applications in therapeutics, synthetic biology and biotechnology. Emerg. Top. Life Sci. 2021, 5, 691–697. [Google Scholar] [CrossRef]
- Kumar, P.; Caruthers, M.H. DNA Analogues Modified at the Nonlinking Positions of Phosphorus. Acc. Chem. Res. 2020, 53, 2152–2166. [Google Scholar] [CrossRef]
- McKenzie, L.K.; El-Khoury, R.; Thorpe, J.D.; Damha, M.J.; Hollentein, M. Recent progress in non-native nucleic acid modifications. Chem. Soc. Rev. 2021, 50, 5126–5164. [Google Scholar] [CrossRef]
- Stang, A.; Robers, J.; Schonert, B.; Jöckel, K.H.; Spelsberg, A.; Keil, U.; Cullen, P. The performance of the SARS-CoV-2 RT-PCR test as a tool for detecting SARS-CoV-2 infection in the population. J. Infect. 2021, 83, 237–279. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.R.; Brown, J.M. The polymerase chain reaction: Current and future clinical applications. J. Med. Genet. 1990, 27, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Templeton, M.S. The Polymerase Chain Reaction History Methods and Applications. Diagn. Mol. Pathol. 1992, 1, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Crinelli, R.; Bianchi, M.; Gentilini, L.; Magnani, M. Design and characterization of decoy oligonucleotides containing locked nucleic acids. Nucleic Acids Res. 2002, 30, 2435–2443. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Solis, A.; Han, G.; Gan, L.; Liu, Y.; Markham, J.E.; Cahoon, E.R.; Dunn, T.M.; Cahoon, E.B. Unregulated Sphingolipid Biosynthesis in Gene-Edited Arabidopsis ORM Mutants Results in Nonviable Seeds with Strongly Reduced Oil Content. Plant. Cell. 2020, 32, 2474–2490. [Google Scholar] [CrossRef]
- Crispo, M.; Mulet, A.P.; Tesson, L.; Barrera, N.; Cuadro, F.; dos Santos-Neto, P.C.; Nguyen, T.H.; Crénéguy, A.; Brusselle, L.; Anegón, I.; et al. Efficient Generation of Myostatin Knock-Out Sheep Using CRISPR/Cas9 Technology and Microinjection into Zygotes. PLoS ONE 2015, 10, e0136690. [Google Scholar] [CrossRef] [PubMed]
- CarlsonStevermer, J.; Goedland, M.; Steyer, B.; Movaghar, A.; Lou, M.; Kohlenberg, L.; Prestil, R.; Saha, K. High-Content Analysis of CRISPR-Cas9 Gene-Edited Human Embryonic Stem Cells. Stem Cell. Rep. 2016, 6, 109–120. [Google Scholar] [CrossRef]
- Morihiro, K.; Kasahara, Y.; Obika, S. Biological applications of xeno nucleic acids. Mol. Biosyst. 2017, 13, 235–245. [Google Scholar] [CrossRef]
- Ahmad, H.I.; Ahmad, M.J.; Asif, A.R.; Adnan, M.; Iqbal, M.K.; Mehmood, K.; Muhammad, S.A.; Bhuiyan, A.A.; Elokil, A.; Du, X.; et al. A Review of CRISPR-Based Genome Editing: Survival, Evolution and Challenges. Curr. Issues Mol. Biol. 2018, 28, 47–68. [Google Scholar] [CrossRef]
- Ni, X.; Castanares, M.; Mukherjee, A.; Lupold, S.E. Nucleic acid aptamers: Clinical applications and promising new horizons. Curr. Med. Chem. 2011, 18, 4206–4214. [Google Scholar] [CrossRef]
- Kulabhusan, P.K.; Hussain, B.; Yüce, M. Current Perspectives on Aptamers as Diagnostic Tools and Therapeutic Agents. Pharmaceutics 2020, 12, 646. [Google Scholar] [CrossRef] [PubMed]
- Ng, E.W.M.; Shima, D.T.; Calias, P.; Cunningham, E.T.; Guyer, D.R.; Adamis, A.P. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug Discov. 2006, 5, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissma, D. mRNA vaccines-a new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Zamecnik, C.P.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Setten, R.L.; Rossi, J.J.; Han, S. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef]
- Downward, J. RNA Interference. BMJ 2004, 328, 1245–1248. [Google Scholar] [CrossRef]
- Nagel, K.M.; Holstad, S.G.; Isenberg, K.E. Oligonucleotide Pharmacotherapy: An Antigene Strategy. Pharmacotherapy 1993, 13, 177–188. [Google Scholar]
- Praseuth, D.; Guieysse, A.L.; Héléne, C. Review: Triple helix formation and the antigene strategy for sequence-specific control of gene expression. Biochim. Biophys. Acta (BBA)-Gene Struct. Expr. 1999, 1489, 181–206. [Google Scholar] [CrossRef]
- Hobbs, C.A.; Yoon, K. Differential Regulation of Gene Expression in Vivo by Triple Helix-Forming Oligonucleotides as Detected by a Reporter Enzyme. Antisense Res. Dev. 1994, 4, 1–8. [Google Scholar] [CrossRef]
- Crooke, S.T.; Liang, X.; Baker, B.F.; Crooke, R.M. Antisense technology: A review. J. Biol. Chem. 2021, 296, 100416. [Google Scholar] [CrossRef] [PubMed]
- Quemener, A.M.; Bachelot, L.; Forestier, A.; Donnou-Fournet, E.; Gilot, D.; Galibert, M. The powerful world of antisense oligonucleotides: From bench to bedside. Wiley Interdiscip. Rev. RNA 2020, 11, e1594. [Google Scholar] [CrossRef]
- Benett, C.F. Therapeutic Antisense Oligonucleotides Are Coming of Age. Annu. Rev. Med. 2019, 70, 307–321. [Google Scholar] [CrossRef]
- Phylactou, L.A.; Kilpatrick, M.W.; Wood, J.A.M. Ribozymes as therapeutic tools for genetic disease. Hum. Mol. Genet. 1998, 7, 1649–1653. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.B.; Seth, P.P. The Medicinal Chemistry of Therapeutic Oligonucleotides. J. Med. Chem. 2016, 59, 9645–9667. [Google Scholar] [CrossRef] [PubMed]
- Reese, C.B. Oligo- and poly-nucleotides: 50 years of chemical synthesis. Org. Biomol. Chem. 2005, 3, 3851–3868. [Google Scholar] [CrossRef]
- Verma, S.; Eckstein, F. Modified Oligonucleotides: Synthesis and Strategy for Users. Annu. Rev. Biochem. 1998, 67, 99–134. [Google Scholar] [CrossRef]
- Scremin, C.L.; Zhou, L.; Srinivasachar, K.; Beaucage, S.L. Stepwise Regeneration and Recovery of Deoxyribonucleoside Phosphoramidite Monomers during Solid-Phase Oligonucleotide Synthesis. J. Org. Chem. 1994, 59, 1963–1966. [Google Scholar] [CrossRef]
- Flanagan, W.M.; Wolf, J.J.; Olson, P.; Grant, D.; Lin, K.; Wagner, R.W.; Matteucci, M.D. A cytosine analog that confers enhanced potency to antisense oligonucleotides. Proc. Natl. Acad. Sci. USA 1999, 96, 3513–3518. [Google Scholar] [CrossRef]
- Chenna, V.; Rapireddy, S.; Sahu, B.; Ausin, C.; Pedroso, E.; Ly, D.H. A Simple Cytosine to G-Clamp Nucleobase Substitution Enables Chiral γ-PNAs to Invade Mixed-Sequence Double-Helical B-form DNA. ChemBioChem 2008, 9, 2388–2391. [Google Scholar] [CrossRef]
- Holmes, S.C.; Arzumanov, A.A.; Gait, M.J. Steric inhibition of human immunodeficiency virus type-1 Tat-dependent trans-activation in vitro and in cells by oligonucleotides containing 2′-O-methyl G-clamp ribonucleoside analogues. Nucleic Acids Res. 2003, 31, 2759–2768. [Google Scholar] [CrossRef] [PubMed]
- Wojciechowski, F.; Hudson, R.H.E. A Fmoc/Boc pseudoisocytosine monomer for peptide nucleic acid synthesis. Can. J. Chem. 2008, 86, 1026–1029. [Google Scholar] [CrossRef]
- Neuer, P.; Monaci, P. New Fmoc Pseudoisocytosine Monomer for the Synthesis of Bis-PNA Molecule by Automated Solid-Phase Fmoc Chemistry. Bioconjugate Chem. 2002, 13, 676–678. [Google Scholar] [CrossRef] [PubMed]
- Compagno, D.; Lampe, J.N.; Bourget, C.; Kutyavin, I.V.; Yurchenko, L.; Lukhtanov, E.A.; Gorn, V.V.; Gamper, H.B.; Toulmé, J. Antisense oligonucleotides containing modified bases inhibit in vitro translation of Leishmania amazonensis mRNAs by invading the mini-exon hairpin. J. Biol. Chem. 1999, 274, 8191–8198. [Google Scholar] [CrossRef]
- Kutyavin, I.V.; Rhinehart, R.L.; Lukhtanov, E.A.; Gorn, V.V.; Meyer, R.B.; Gamper, H.B. Oligonucleotides Containing 2-Aminoadenine and 2-Thiothymine Act as Selectively Binding Complementary Agents. Biochemistry 1996, 35, 11170–11176. [Google Scholar] [CrossRef]
- Patil, K.M.; Toh, D.K.; Yuan, Z.; Meng, Z.; Shu, Z.; Zhang, H.; Ong, A.A.L.; Krishna, M.S.; Lu, L.; Lu, Y.; et al. Incorporating uracil and 5-halouracils into short peptide nucleic acids for enhanced recognition of A-U pairs in dsRNAs. Nucleic Acids Res. 2018, 46, 7506–7521. [Google Scholar] [CrossRef]
- Sági, J.; Szemző, A.; Ebinger, K.; Szabolcs, A.; Sági, G.; Ruff, É.; Ötvös, L. Base Modified oligodeoxynucleotides I. Effect of 5-Alkyl, 5-(1-Alkenyl), and 5-(1-Alkinyl) Substitution of the Pirimidines on Duplex Stability and Hydrophobicity. Tetrahedron Lett. 1993, 35, 2191–2194. [Google Scholar] [CrossRef]
- Wlotzka, B.; Leva, S.; Eschgfäller, B.; Burmeister, J.; Kleinjung, F.; Kaduk, C.; Muhn, P.; Hess-Stumpf, H.; Klussmann, S. In vivo properties of an anti-GnRH Spiegelmer: An example of an oligonucleotide based therapeutic substance class. Proc. Natl. Acad. Sci. USA 2002, 13, 8898–8902. [Google Scholar] [CrossRef]
- Vater, A.; Klussmann, S. Turning-mirror image oligonucleotides into drugs: The evolution of Spiegelmer therapeutics. Drug Discov. Today 2015, 20, 147–155. [Google Scholar] [CrossRef]
- Latha, Y.S.; Yathrinda, N. Stereochemical Studies on Nucleic Acid Analogues I. Conformations of α-Nucleosides and α-Nucleotides: Interconversion of Sugar Puckers via O-4′exo. Bipolymers 1992, 32, 249–269. [Google Scholar] [CrossRef]
- Boiziau, C.; Kurfurst, R.; Cazenave, C.; Roig, V.; Thoung, N.T.; Toulmé, J.J. Inhibition of translation initiation by antisense oligonucleotides via an RNase-H independent mechanism. Nucleic Acid Res. 1991, 19, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Frieden, M.; Christensen, S.M.; Mikkelsen, N.D.; Rosenbohm, C.; Thrue, C.A.; Westergaard, M.; Hansen, H.F.; Orum, H.; Koch, T. Expanding the design Horison of antisense oligonucleotides with alpha-L-LNA. Nucleic Acids Res. 2003, 31, 6365–6372. [Google Scholar] [CrossRef] [PubMed]
- Eschenmoser, A.; Dobler, M. Warum Pentose und nicht Hexose-Nukleinsäuren? Teil I. Einleitung und Problemstellung, Konformationsanalyse fur Oligonucleotid-Ketten aus 2′, 3′-Dideoxyglucopyranosyl-Bausteinen (‘Homo-DNS’) sowie Betrachtungen zur Konformation von A-und B-DNS. Helv. Chim. Acta 1992, 75, 218–259. [Google Scholar] [CrossRef]
- Bohringer, M.; Roth, H.; Hunziker, J.; Gobe, M.; Krishnan, R.; Giger, A.; Schweizer, B.; Schreiber, J.; Leumann, C.; Eschenmoser, A. Warum Pentose und nicht Hexose-Nukleinsäuren? Teil II. Oligonucleotide aus 2′,3′-Dideoxy-β-d-glucopyranosyl-Bausteinen (‘Homo-DNS’): Herstellung. Helv. Chim. Acta 1992, 75, 1416–1477. [Google Scholar] [CrossRef]
- Hunziker, J.; Roth, H.; Böhringer, M.; Giger, A.; Diederischen, U.; Göbel, M.; Krishnan, R.; Jaun, B.; Leumann, C.; Eschenmoser, A. Warum Pentose und nicht Hexose-Nukleinsäuren? Teil III. Oligo(2′,3′-dideoxy-β-D-glucopyranosyl)nucleotide (‘Homo-DNS’) Paarungseigenschaften. Helv. Chim. Acta 1993, 76, 259–352. [Google Scholar] [CrossRef]
- Egli, M.; Lubini, P.; Pallan, P.S. The long and winding road to the structure of homo-DNA. Chem. Soc. Rev. 2007, 36, 31–45. [Google Scholar] [CrossRef]
- Egli, M.; Pallan, P.S.; Pattanayek, R.; Wilds, C.J.; Lubini, P.; Minsaov, G.; Dobler, M.; Leumann, C.J.; Eschenmoser, A. Crystal Structure of Homo-DNA and Nature’s Choice of Pentose over Hexose int he Genetic System. J. Am. Chem. Soc. 2006, 128, 10847–10856. [Google Scholar] [CrossRef]
- Pitsch, S.; Krishnamurthy, R.; Bolli, M.; Wendehorn, S.; Holzner, A.; Minton, M.; Lesuenr, C.; Schlonvogt, I.; Jaun, B.; Eschenmoser, A. Pyranosyl-RNA (‘p-RNA’): Base-Pairing Selectivity and Potential to Replicate. Helv. Chim. Acta 1995, 78, 1621–1635. [Google Scholar] [CrossRef]
- Beier, M.; Reck, F.; Wagner, T.; Krishnamurty, R.; Eschenmoser, A. Chemical Etiology of Nucleic Acid Structure: Comparing Pentopyranosyl-(2′-4′) Oligonucleotides with RNA. Science 1999, 283, 699–703. [Google Scholar] [CrossRef]
- Reck, F.; Wippo, H.; Kudick, R.; Bolli, M.; Ceulemans, G.; Krishnamurthy, R.; Eschenmoser, A. L-r-Lyxopyranosyl (4′-3′) Oligonucleotides: A Base-Pairing System Containing a Shortened Backbone. Org. Lett. 1999, 1, 1531–1534. [Google Scholar] [CrossRef]
- Jungmann, O.; Wippo, H.; Stanek, M.; Huynh, H.K.; Krishnamurthy, R.; Eschenmoser, A. Promiscuous Watson−Crick Cross-Pairing within the Family of Pentopyranosyl (4′-2′) Oligonucleotides. Org. Lett. 1999, 1, 1527–1530. [Google Scholar] [CrossRef] [PubMed]
- Schöning, K.; Scholz, P.; Guntha, S.; Wu, X.; Krishnamurthy, R.; Eschenmoser, A. Chemical Etiology of Nucleic Acid Structure: The α-Threofuranosyl-(3′-2′) Oligonucleotide System. Science 2000, 290, 1347–1351. [Google Scholar] [CrossRef] [PubMed]
- Noronha, A.M.; Wilds, C.J.; Lok, C.; Viazovkina, K.; Arion, D.; Parniak, M.A.; Damha, M.J. Synthesis and Biophysical Properties of Arabinonucleic Acids (ANA): Circular Dichroic Spectra, Melting Temperatures, and Ribonuclease H Susceptibility of ANA∙RNA Hybrid Duplexes. Biochemistry 2000, 39, 7050–7062. [Google Scholar] [CrossRef] [PubMed]
- Wilds, C.J.; Damha, M.J. 2′-Deoxy-2′-fluoro-β-D-arabinonucleosides and oligonucleotides (2′F-ANA): Synthesis and physicochemical studies. Nucleic Acids Res. 2000, 28, 3625–3635. [Google Scholar] [CrossRef]
- Maurinsh, Y.; Rosemeyer, H.; Esnouf, R.; Medvedovici, A.; Wang, J.; Ceulemans, G.; Lescrinier, E.; Hendrix, C.; Busson, R.; Sandra, P.; et al. Synthesis and Pairing Properties of Oligonucleotides Containing 3-Hydroxy-4-hydroxymethyl-1-cyclohexanyl Nucleosides. Chem. Eur. J. 1999, 5, 2139–2150. [Google Scholar] [CrossRef]
- Wang, J.; Verbeure, B.; Luyten, I.; Lescrinier, E.; Froeyen, M.; Hendrix, C.; Rosemeyer, H.; Seela, F.; Aerschot, A.; Herdewijn, P. Cyclohexene Nucleic Acids (CeNA): Serum Stable Oligonucleotides that Activate RNase H and Increase Duplex Stability with Complementary RNA. J. Am. Chem. Soc. 2000, 122, 8595–8602. [Google Scholar] [CrossRef]
- Verbeure, B.; Lescrinier, E.; Wang, J.; Herdewijn, P. RNAse H mediated cleavage of RNA by cyclohexene nucleic acid (CeNA). Nucleic Acid Res. 2001, 29, 4941–4947. [Google Scholar] [CrossRef]
- D’Alonzo, D.; Froeyen, M.; Schepers, G.; Fabio, G.D.; Aerschot, A.; Hardewijn, P.; Palumbo, G.; Guaragna, A. 1′, 5′-Anhydro-L-ribo-hexitol Adenine Nucleic Acids (α-L-HNA-A): Synthesis and Chiral Selection Properties in the Mirror Image World. J. Org. Chem. 2015, 80, 5014–5022. [Google Scholar] [CrossRef]
- Bouvere, B.D.; Kerremans, L.; Hendrix, C.; Winter, H.D.; Schepers, G.; Aerschot, A.; Herdewijn, P. Hexitol Nucleic Acids (HNA): Synthesis and Properties. Nucleosides Nucleotides 1997, 16, 973–976. [Google Scholar] [CrossRef]
- Kang, H.; Fischer, M.H.; Xu, D.; Miyamoto, Y.J.; Marchand, A.; Aerschot, A.; Herdewijn, P.; Juliano, R.L. Inhibition of MDR1 gene expression by chimeric HNA antisense oligonucleotides. Nucleic Acid. Res. 2004, 32, 4411–4419. [Google Scholar] [CrossRef]
- Fisher, M.; Abramov, M.; Aerschot, A.; Rozenski, J.; Dixit, V.; Juliano, R.L.; Herdewijn, P. Biological effects of hexitol and altritol-modified siRNAs targeting B-Raf. Eur. J. Pharmacol. 2009, 606, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Summerton, J.; Weller, D. Morpholino Antisense Oligomers: Design, Preparation, and Properties. Antisense Nucleic Acid Drug Dev. 1997, 7, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Zhang, Y. Antisense Phosphorodiamidate Morpholino Oligomers as Novel Antiviral Compounds. Front. Microbiol. 2018, 9, 750. [Google Scholar] [CrossRef] [PubMed]
- Corey, D.R.; Abrams, J.M. Morpholino antisense oligonucleotides: Tools for investigating vertebrate development. Genome Biol. 2001, 2, 1015.1. [Google Scholar] [CrossRef]
- Nasevicius, A.; Ekker, S.C. Effective targeted gene ‘knockdown’ in zebrafish. Nat. Genet. 2000, 26, 216–220. [Google Scholar] [CrossRef]
- Heasman, J.; Kofron, M.; Wylie, C. β-Catenin Signaling Activity Dissected in the Early Xenopus Embryo: A Novel Antisense Approach. Dev. Biol. 2000, 222, 124–134. [Google Scholar] [CrossRef]
- Howard, E.W.; Newman, L.A.; Oleksyn, D.W.; Angerer, R.C.; Angerer, L.M. SpKrl: A direct target of β-catenin regulation required for endoderm differentiation in sea urchin embryos. Development 2001, 128, 365–375. [Google Scholar] [CrossRef]
- Bolli, M.; Litten, J.C.; Schütz, R.; Leumann, C.J. Bicyclo-DNA: A Hoogsteen-selective pairing system. Chem. Biol. 1996, 3, 197–206. [Google Scholar] [CrossRef]
- Tarköy, M.; Bolli, M.; Leumann, C. Nucleic-Acid Analogues with Restricted Conformational Flexibility in the Sugar-Phosphate Backbone (‘Bicyclo-DNA’) (Part 3) Synthesis, Pairing Properties, and Calorimetric Determination of Duplex and Triplex Stability of Decanucleotides from [(3′S,5′R)-2′-Deoxy-3′,5′-ethano-β-D-ribofuranosyl]adenine and -thymine. Helv. Chim. Acta 1994, 77, 716–744. [Google Scholar]
- Steffens, R.; Leumann, C.J. Tricyclo-DNA: A Phosphodiester-Backbone Based DNA Analog Exhibiting Strong Complementary Base-Pairing Properties. J. Am. Chem. Soc. 1997, 119, 11548–11549. [Google Scholar] [CrossRef]
- Renneberg, D.; Bouliong, E.; Reber, U.; Schümperli, D.; Leumann, C.J. Antisense properties of tricyclo-DNA. Nucleic Acid Res. 2002, 30, 2751–2757. [Google Scholar] [CrossRef] [PubMed]
- Aupy, P.; Echevarría, L.; Relizani, K.; Goyenvalle, A. The Use of Tricyclo-DNA Oligomers for the Treatment of Genetic Disorders. Biomedicines 2018, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Renneberg, D.; Leumann, C.J. Watson-Crick Base-Pairing Properties of Tricyclo-DNA. J. Am. Chem. Soc. 2002, 124, 5993–6002. [Google Scholar] [CrossRef] [PubMed]
- Koshkin, A.A.; Singh, S.K.; Nielsen, P.; Rajwanshi, V.K.; Kumar, R.; Meldgaard, M.; Olsen, C.E.; Wengel, J. LNA (Locked Nucleic Acids): Synthesis of the Adenine, Cytosine, Guanine, 5-Methylcytosine, Thymine and Uracil Bicyclonucleoside Monomers, Oligomerisation, and Unprecedented Nucleic Acid Recognition. Tetrahedron 1998, 54, 3607–3630. [Google Scholar] [CrossRef]
- Kaur, H.; Babu, B.R.; Maiti, S. Perspectives on Chemistry and Therapeutic Applications of Locked Nucleic Acid (LNA). Chem. Rev. 2007, 107, 4672–4697. [Google Scholar] [CrossRef]
- Langkjaer, N.; Pasternak, A.; Wengel, J. UNA (unlocked nucleic acid): A flexible RNA mimic that allows engineering of nucleic acid duplex stability. Bioorg. Med. Chem. 2009, 17, 5420–5425. [Google Scholar] [CrossRef]
- Campbell, M.A.; Wengel, J. Locked vs. unlocked nucleic acids (LNA vs. UNA): Contrasting structures work towards common therapeutic goals. Chem. Soc. Rev. 2011, 40, 5680–5689. [Google Scholar] [CrossRef]
- Merle, Y.; Bonneil, E.; Merle, L.; Sági, J.; Szemző, A. Acyclic oligonucleotide analogues. Int. J. Biol. Macromol. 1995, 17, 239–246. [Google Scholar] [CrossRef]
- Heuberger, B.D.; Switzer, C. A Pre-RNA Candidate Revisited: Both Enantiomers of Flexible Nucleoside Triphosphates are DNA Polymerase Substrates. J. Am. Chem. Soc. 2008, 130, 412–413. [Google Scholar] [CrossRef]
- Joyce, G.F.; Schwartz, A.W.; Miller, S.L.; Orgel, L.E. The case for an ancestral genetic system involving simple analogues of the nucleotides. Proc. Natl. Acad. Sci. USA 1987, 84, 4398–4402. [Google Scholar] [CrossRef]
- Schlegel, M.K.; Peritz, A.E.; Kittigowittana, K.; Zhang, L.; Meggers, E. Duplex Formation of the Simplified Nucleic Acid GNA. ChemBioChem 2007, 8, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, M.K.; Xie, X.; Zhang, L.; Meggers, E. Insight into the High Duplex Stability of the Simplified Nucleic Acid GNA. Angew. Chem. Int. Ed. 2009, 48, 960–963. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, H.; Toda, T.; Murayama, K.; Liang, X.; Kashida, H. Unexpectedly Stable Artificial Duplex from Flexible Acyclic Threoninol. J. Am. Chem. Soc. 2010, 132, 14702–14703. [Google Scholar] [CrossRef] [PubMed]
- Murayama, K.; Kashida, H.; Asanuma, H. Acyclic L-threoninol nucleic acid (L-aTNA) with suitable structural rigidity cross-pairs with DNA and RNA. Chem. Commun. 2015, 51, 6500–6503. [Google Scholar] [CrossRef] [PubMed]
- Kashida, H.; Murayama, K.; Asanuma, H. Acyclic artificial nucleic acids with phosphodiester bonds exhibit unique functions. Polym. J. 2016, 48, 781–786. [Google Scholar] [CrossRef]
- Kashida, H.; Murayama, K.; Toda, T.; Asanuma, H. Control of the Chirality and Helicity of Oligomers of Serinol Nucleic Acid (SNA) by Sequence Design. Angew. Chem. Int. Ed. 2011, 50, 1285–1288. [Google Scholar] [CrossRef]
- Freier, S.M.; Altmann, K. The ups and downs of nucleic acid duplex stability: Structure–stability studies on chemically-modified DNA:RNA duplexes. Nucleic Acid Res. 1997, 25, 4429–4443. [Google Scholar] [CrossRef]
- Baker, B.F.; Lot, S.S.; Condon, T.P.; Cheng-Flournoy, S.; Lesnik, E.A.; Sasmor, H.M.; Bennett, C.F. 2*-O-(2-Methoxy)ethyl-modified Anti-intercellular Adhesion Molecule 1 (ICAM-1) Oligonucleotides Selectively Increase the ICAM-1 mRNA Level and Inhibit Formation of the ICAM-1 Translation Initiation Complex in Human Umbilical Vein Endothelial Cells. J. Biol. Chem. 1997, 272, 11994–12000. [Google Scholar] [CrossRef]
- Lind, K.E.; Mohan, V.; Manoharan, M.; Ferguson, D.M. Structural characteristics of 2′-O-(2-methoxyethyl)-modified nucleic acids from molecular dynamics simulations. Nucleic Acid Res. 1998, 26, 3694–3699. [Google Scholar] [CrossRef]
- Sheng, L.; Rigo, F.; Bennett, C.F.; Krainer, A.R.; Hua, Y. Comparison of the efficacy of MOE and PMO modifications of systemic antisense oligonucleotides in a severe SMA mouse model. Nucleic Acid Res. 2020, 48, 2853–2865. [Google Scholar] [CrossRef]
- Srinivasan, S.K.; Iversen, P. Review of In Vivo Pharmacokinetics and Toxicology of Phosphorothioate Oligonucleotides. J. Clin. Lab. Anal. 1995, 9, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Eckstein, F. Phosphorothioates, Essential Components of Therapeutic Oligonucleotides. Nucleic Acid Ther. 2014, 24, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.A. A review of issues in the pharmacokinetics and toxicology of phosphorothioate antisense oligonucleotides. Biochim. Biophys. Acta 1999, 1489, 69–84. [Google Scholar] [CrossRef]
- Flierl, U.; Nero, T.L.; Lim, B.; Arthur, J.F.; Yao, Y.; Jung, S.M.; Gitz, E.; Pollitt, A.Y.; Zaldivia, M.T.K.; Jandrot-Perrus, M.; et al. Phosphorothioate backbone modifications of nucleotide-based drugs are potent platelet activators. J. Exp. Med. 2015, 212, 129–137. [Google Scholar] [CrossRef]
- Hartmann, G.; Krug, A.; Waller-Fontaine, K.; Endres, S. Oligodeoxynucleotides Enhance Lipopolysaccharide- Stimulated Synthesis of Tumor Necrosis Factor: Dependence on Phosphorothioate Modification and Reversal by Heparin. Mol. Med. 1996, 2, 429–438. [Google Scholar] [CrossRef]
- Jahrsdörfer, B.; Jox, R.; Mühlenhoff, L.; Tschoep, K.; Krug, A.; Rothenfusser, S.; Meinhardt, G.; Emmerich, B.; Endres, S.; Hartmann, G. Modulation of malignant B cell activation and apoptosis by bcl-2 antisense ODN and immunostimulatory CpG ODN. J. Leukoc. Biol. 2002, 72, 83–92. [Google Scholar]
- Karlin, S.; Ladunga, I.; Blaisdell, B.E. Heterogeneity of genomes: Measures and values. Proc. Natl. Acad. Sci. USA 1994, 91, 12837–12841. [Google Scholar] [CrossRef]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial RNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef]
- Crooke, S.T.; Vickers, T.A.; Liang, X. Phosphorothioate modified oligonucleotide–protein interactions. Nucleic Acid Res. 2020, 48, 5235–5253. [Google Scholar] [CrossRef]
- Gryaznov, S.; Chen, J. ligodeoxyribonucleotide N3‘4P5’ Phosphoramidates: Synthesis and Hybridization Properties. J. Am. Chem. Soc. 1994, 116, 3143–3144. [Google Scholar] [CrossRef]
- Micklefield, J. Backbone Modification of Nucleic Acids: Synthesis, Structure and Therapeutic Applications. Curr. Med. Chem. 2001, 8, 1157–1179. [Google Scholar] [CrossRef] [PubMed]
- Schultz, R.G.; Gryaznov, S.M. Oligo-2′-fluoro-2′-deoxynucleotide N3′→P5′ phosphoramidates: Synthesis and properties. Nucleic Acid Res. 1996, 24, 2966–2973. [Google Scholar] [CrossRef] [PubMed]
- Tereshko, V.; Gryaznov, S.; Egli, M. Consequences of Replacing the DNA 3′-Oxygen by an Amino Group: High-Resolution Crystal Structure of a Fully Modified N3′→P5′ Phosphoramidate DNA Dodecamer Duplex. J. Am. Chem. Soc. 1998, 120, 269–283. [Google Scholar] [CrossRef]
- Gryaznov, S.M.; Lloyd, D.H.; Chen, J.; Schultz, R.G.; DeDionsio, L.A.; Ratmeyer, L.; Wilson, W.D. Oligonucleotide N3′→P5′ phosphoramidates. Proc. Natl. Acad. Sci. USA 1995, 92, 5798–5802. [Google Scholar] [CrossRef] [PubMed]
- Sood, A.; Shaw, B.R.; Spielvogel, B.F. Boron-Containing Nucleic Acids. 2′ Synthesis of Oligodeoxynucleoside Boranophosphates. J. Am. Chem. Soc. 1990, 112, 9000–9001. [Google Scholar] [CrossRef]
- Sergueev, D.S.; Shaw, B.R. H-Phosphonate Approach for Solid-Phase Synthesis of Oligodeoxyribonucleoside Boranophosphates and Their Characterization. J. Am. Chem. Soc. 1998, 120, 9417–9427. [Google Scholar] [CrossRef]
- Reynolds, M.A.; Hogrefe, R.I.; Jaeger, J.A.; Schwartz, D.A.; Riley, T.A.; Marvin, W.B.; Daily, W.J.; Vaghefi, M.M.; Beck, T.A.; Knowles, S.K.; et al. Synthesis and thermodynamics of oligonucleotides containing chirally pure RP methylphosphonate linkages. Nucleic Acid Res. 1996, 24, 4584–4591. [Google Scholar] [CrossRef]
- Flür, S.; Micura, R. Chemical synthesis of RNA with site-specific methylphosphonate modifications. Methods 2016, 107, 79–88. [Google Scholar] [CrossRef]
- Clavé, G.; Reverte, M.; Vasseur, J.; Smietana, M. Modified internucleoside linkages for nuclease-resistant oligonucleotides. RSC Chem. Biol. 2021, 2, 94–150. [Google Scholar] [CrossRef]
- Brill, W.K.D.; Caruthers, M.H. Synthesis of nucleoside methylphosphonothioates. Tetrahedron Lett. 1987, 28, 3205–3208. [Google Scholar] [CrossRef]
- Pasmapriya, A.A.; Agrawal, S. Synthesis of Oligodeoxynucleoside methylphosphonothioates. Bioorg. Med. Chem. Lett. 1993, 3, 761–764. [Google Scholar] [CrossRef]
- Wozniak, L.A.; Bukowiecka-Matusiak, M.; Gora, M.; Stec, W.J. One-Pot Synthesis of Dinucleoisde (3′,5′)-Methylphosphonothioates and their Seleno Congeners via the Phosphonotriazolidite Approach. Synlett 2006, 9, 1331–1334. [Google Scholar] [CrossRef]
- Wozniak, L.A.; Gora, M.; Stec, W.J. Chemoselective Activation of Nucleoside 3′-O-Methylphosphonothioates with 1,3,5-Triazinyl Morpholinium Salts. J. Org. Chem. 2007, 72, 8584–8587. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, Y.; Hirose, M.; Hayakawa, M.; Noyori, R. General Synthesis and Binding Affinity of Position-Selective Phosphonodiester- and Phosphotriester-Incorporated Oligodeoxyribonucleotides. J. Org. Chem. 1995, 60, 925–930. [Google Scholar] [CrossRef]
- Meade, B.R.; Gogoi, K.; Hamil, A.S.; Palm-Apergi, C.; Berg, A.; Hagopian, J.C.; Springer, A.D.; Eguchi, A.; Kacsinta, A.D.; Dowdy, C.F.; et al. Efficient delivery of RNAi prodrugs containing reversible charge-neutralizing phosphotriester backbone modifications. Nat. Biotechnol. 2014, 32, 1256–1263. [Google Scholar] [CrossRef]
- Mungall, W.S.; Kaiser, J.K. Carbamate Analogues of Oligonucleotides. J. Org. Chem. 1977, 42, 703–706. [Google Scholar] [CrossRef]
- Stirchak, E.P.; Summerton, J.E.; Weller, D.D. Uncharged stereoregular nucleic acid analogs: 2. Morpholino nucleoside oligomers with carbamate internucleoside linkages. Nucleic Acid Res. 1989, 17, 6129–6141. [Google Scholar] [CrossRef]
- Seliger, H.; Feger, G. Oligonucleotide Analogues with Dialkyl Silyl Internucleoside Linkages. Nucleosides Nucleotides 1987, 6, 483–484. [Google Scholar] [CrossRef]
- Saha, A.K.; Sardaro, M.; Waychunas, C.; Delecki, D.; Rustny, R.; Cavanaugh, P.; Yawman, A.; Upson, D.A.; Kruse, L.I.; Kutny, R. Diisopropylsilyl-linked oligonucleotide analogs: Solid-phase synthesis and physicochemical properties. J. Org. Chem. 1993, 58, 7827–7831. [Google Scholar] [CrossRef]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-Selective Recognition of DNA by Strand Displacement with a ThymineSubstituted Polyamide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef]
- Uhlmann, E.; Peyman, A.; Breipohl, G.; Will, D.W. PNA: Synthetic Polyamide Nucleic Acids with Unusual Binding Properties. Angew. Chem. Int. Ed. 1998, 37, 2796–2823. [Google Scholar] [CrossRef]
- Das, A.; Pradhan, B. Evolution of Peptide Nucleic Acid with modifications of its backbone and application in Biotechnology. Chem. Biol. Drug Des. 2020, 97, 865–892. [Google Scholar] [CrossRef] [PubMed]
- Lebreton, J.; Mesmaeker, A.; Waldner, A.; Fritsch, V.; Wolf, R.M.; Freie, S.M. Synthesis of Thymidine Dimer Derivatives Containing an Amide Linkage and their Incorporation into Oligodeoxyribonucleotides. Tetrahedron Lett. 1993, 34, 6383–6386. [Google Scholar] [CrossRef]
- Wada, T.; Minamimoto, N.; Inaki, Y.; Inoue, Y. Peptide Ribonucleic Acids (PRNA). 2. A Novel Strategy for Active Control of DNA Recognition through Borate Ester Formation. J. Am. Chem. Soc. 2000, 122, 6900–6910. [Google Scholar] [CrossRef]
- Sato, H.; Hashimoto, Y.; Wada, T.; Inoue, Y. Solid-phase synthesis of peptide ribonucleic acids (PRNA). Tetrahedron 2003, 59, 7871–7878. [Google Scholar] [CrossRef]
- Bege, M.; Bereczki, I.; Molnár, D.J.; Kicsák, M.; Pénzes-Daku, K.; Bereczky, Z.; Ferenc, G.; Kovács, L.; Herczegh, P.; Borbás, A. Synthesis and oligomerization of cysteinyl nucleosides. Org. Biomol. Chem. 2020, 18, 8161–8178. [Google Scholar] [CrossRef]
- Jain, M.L.; Bruice, P.Y.; Szabó, I.E.; Bruice, T.C. Incorporation of Positively Charged Linkages into DNA and RNA Backbones: A Novel Strategy for Antigene and Antisense Agents. Chem. Rev. 2012, 112, 1284–1309. [Google Scholar] [CrossRef]
- Meng, M.; Ducho, C. Oligonucleotide analogues with cationic backbone linkages. Beilstein J. Org. Chem. 2018, 14, 1293–1308. [Google Scholar] [CrossRef]
- Blaskó, A.; Dempcy, R.O.; Minyat, E.E.; Bruice, T.C. Association of Short-Strand DNA Oligomers with Guanidinium-Linked Nucleosides. A Kinetic and Thermodynamic Study. J. Am. Chem. Soc. 1996, 118, 7892–7899. [Google Scholar] [CrossRef]
- Arya, D.P.; Bruice, T.C. Triple-helix formation of DNA oligomers with methylthiourealinked nucleosides (DNmt): A kinetic and thermodynamic analysis. Proc. Natl. Acad. Sci. USA 1999, 96, 4384–4389. [Google Scholar] [CrossRef]
- Schmidtgall, B.; Kuepper, A.; Meng, M.; Grossmann, T.N.; Ducho, C. Oligonucleotides with Cationic Backbone and Their Hybridization with DNA: Interplay of Base Pairing and Electrostatic Attraction. Chem. Eur. J. 2017, 24, 1544–1553. [Google Scholar] [CrossRef] [PubMed]
- Schmidtgall, B.; Spork, A.P.; Wachowius, F.; Höbartner, C.; Ducho, C. Synthesis and properties of DNA oligonucleotides with a zwitterionic backbone structure. Chem. Commun. 2014, 50, 13742–13745. [Google Scholar] [CrossRef] [PubMed]
- Pattanayak, S.; Khatra, H.; Saha, S.; Sinha, S. A cationic morpholino antisense oligomer conjugate: Synthesis, cellular uptake and inhibition of Gli1 in the hedgehog signalling pathway. RSC Adv. 2014, 4, 1951–1954. [Google Scholar] [CrossRef]
- Warren, T.K.; Shurtleff, A.C.; Bavari, S. Advanced morpholino oligomers: A novel approach to antiviral therapy. Antivir. Res. 2012, 94, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Meng, M.; Zhang, J.; Liu, A.; Reuschel, S.; Sazani, P.; Wong, M. Quantitative determination of AVI-7100 (Radavirsen), a phosphorodiamidate morpholino oligomer (PMOplus®), in human plasma using LC–MS/MS. Bioanalysis 2017, 9, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Debreczeni, N.; Bege, M.; Herczeg, M.; Bereczki, I.; Batta, G.; Herczegh, P.; Borbás, A. Tightly linked morpholino-nucleoside chimeras: New, compact cationic oligonucleotide analogues. Org. Biomol. Chem. 2021, 19, 8711–8721. [Google Scholar] [CrossRef]
- Ni, S.; Zhou, Z.; Pan, Y.; Yu, Y.; Li, F.; Liu, J.; Wang, L.; Wu, X.; Li, D.; Wan, Y.; et al. Recent Progress in Aptamer Discoveries and Modifications for Therapeutic Applications. ACS Appl. Mater. Interfaces 2021, 13, 9500–9519. [Google Scholar] [CrossRef]
- WHO. International Nonproprietary Names (INN) for Biological and Biotechnological Substances (A Review); WHO Document Production Services, WHO/EMP/RHT/TSN/2016.1; WHO: Geneva, Switzerland, 2016. [Google Scholar]
- Munro, M.; Yadavalli, T.; Fonteh, C.; Arfeen, S.; Lobo-Chan, A. Cytomegalovirus Retinitis in HIV and Non-HIV Individuals. Microorganisms 2020, 8, 55. [Google Scholar] [CrossRef]
- Paulus, C.; Nevels, M. The Human Cytomegalovirus Major Immediate-Early Proteins as Antagonists of Intrinsic and Innate Antiviral Host Responses. Viruses 2009, 1, 760–779. [Google Scholar] [CrossRef]
- Azad, R.F.; Driver, V.B.; Tanaka, K.; Crooke, R.M.; Anderson, K.P. Antiviral Activity of a Phosphorothioate Oligonucleotide Complementary to RNA of the Human Cytomegalovirus Major Immediate-Early Region. Antimicrob. Agents Chemother. 1993, 37, 1945–1954. [Google Scholar] [CrossRef]
- Anderson, K.P.; Fox, M.C.; Driver, V.B.; Martin, M.J.; Azad, R.F. Inhibition of Human Cytomegalovirus Immediate-Early Gene Expression by an Antisense Oligonucleotide Complementary to Immediate-Early RNA. Antimicrob. Agents Chemother. 1996, 40, 2004–2011. [Google Scholar] [CrossRef] [PubMed]
- Geary, R.S.; Henry, S.P.; Grillone, L.R. Fomivirsen Clinical Pharmacology and Potential Drug Interactions. Clin. Pharmacokinet. 2002, 41, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Vitravene Study Group. A randomized controlled clinical trial of intravitreous fomivirsen for treatment of newly diagnosed peripheral cytomegalovirus retinitis in patients with AIDS. Am. J. Ophtalmol. 2002, 133, 467–474. [Google Scholar]
- Perry, C.M.; Balfour, J.A.B. Fomivirsen. Drugs 1999, 57, 375–380. [Google Scholar] [CrossRef]
- Stein, C.A.; Castanotto, D. FDA-Approved Oligonucleotide Therapies in 2017. Mol. Ther. 2017, 25, 1069–1075. [Google Scholar] [CrossRef]
- Pugazhendhi, A.; Hubbell, M.; Jairam, P.; Ambati, B. Neovascular Macular Degeneration: A Review of Etiology, Risk Factors, and Recent Advances in Research and Therapy. Int. J. Mol. Sci. 2021, 22, 1170. [Google Scholar] [CrossRef]
- Kourlas, H.; Schiller, D.S. Pegaptanib Sodium for the Treatment of Neovascular Age-Related Macular Degeneration: A Review. Clin. Ther. 2006, 28, 36–44. [Google Scholar] [CrossRef]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as Therapeutics. Nat. Rev. Drug. Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef]
- Ito, M.K. ISIS 301012 Gene Therapy for Hypercholesterolemia: Sense, Antisense, or Nonsense? Ann. Pharmacother. 2007, 41, 1669–1678. [Google Scholar] [CrossRef]
- Geary, R.S.; Baker, B.F.; Crooke, S.T. Clinical and Preclinical Pharmacokinetics and Pharmacodynamics of Mipomersen (Kynamro®): A Second-Generation Antisense Oligonucleotide Inhibitor of Apolipoprotein B. Clin Pharm. 2015, 54, 133–146. [Google Scholar] [CrossRef]
- Raal, F.J.; Santos, R.D.; Blom, D.J.; Marais, A.D.; Charng, M.; Cromwell, W.C.; Lachmann, R.H.; Gaudet, D.; Tan, J.L.; Chasan-Taber, S.; et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: A randomised, double-blind, placebo-controlled trial. Lancet 2010, 375, 998–1006. [Google Scholar] [CrossRef]
- Ricotta, D.N.; Frishman, W. Mipomersen: A Safe and Effective Antisense Therapy Adjunct to Statins in Patients with Hypercholesterolemia. Cardiol. Rev. 2012, 20, 90–95. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency: Refusal of the Marketing Authorisation for Kynamro (Mipomersen) EMA/792736/2012, 13 December 2012. Available online: https://www.ema.europa.eu/en/documents/smop-initial/questions-answers-refusal-marketing-authorisation-kynamro-mipomersen_en.pdf (accessed on 19 July 2022).
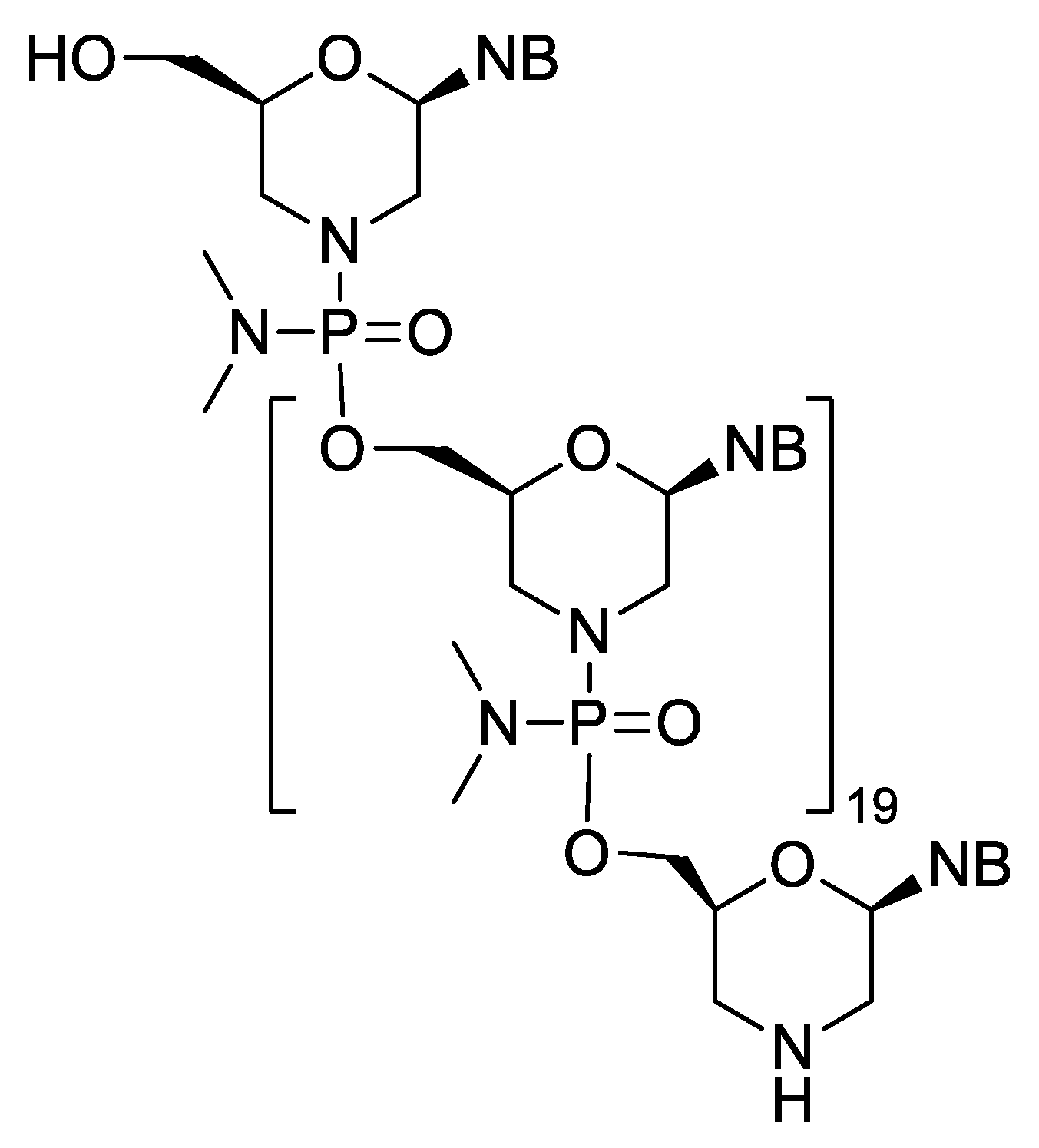
- Kinali, M.; Arechavala-Gomeza, V.; Feng, L.; Cirak, S.; Hunt, D.; Adkin, C.; Guglieri, M.; Ashton, E.; Abbs, S.; Nihoyannopoulos, P.; et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: A single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neruol. 2009, 8, 918–928. [Google Scholar] [CrossRef]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acid Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef]
- Lim, K.R.Q.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Dev. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef]
- Dowling, J.J. Eteplirsen therapy for Duchenne muscular dystrophy: Skipping to the front of the line. Nat. Rev. Neurol. 2016, 12, 675–676. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 2017, 27, 1–3. [Google Scholar] [CrossRef]
- European Medicines Agency: Refusal of the Marketing Authorisation for Exondys (Eteplirsen), EMA/621972/2018, 21 September 2018. Available online: https://www.ema.europa.eu/en/documents/smop-initial/questions-answers-refusal-marketing-authorisation-exondys-eteplirsen-outcome-re-examination_en.pdf (accessed on 19 July 2022).
- Kilanowska, A.; Studzinska, S. In vivo and in vitro studies of antisense oligonucleotides—A review. RSC Adv. 2020, 10, 34501–34516. [Google Scholar] [CrossRef]
- Richardson, P.; Aggarwal, S.; Topaloglu, O.; Villa, K.F.; Corbacioglu, S. Systematic review of defibrotide studies in the treatment of veno-occlusive disease/sinusoidal obstruction syndrome (VOD/SOS). Bone Marrow Transplant. 2019, 54, 1951–1962. [Google Scholar] [CrossRef]
- Pescador, R.; Capuzzi, L.; Mantovani, M.; Fulgenzi, A.; Ferrero, M.E. Defibrotide: Properties and clinical use of an old/new drug. Vasc. Pharmacol. 2013, 59, 1–10. [Google Scholar] [CrossRef]
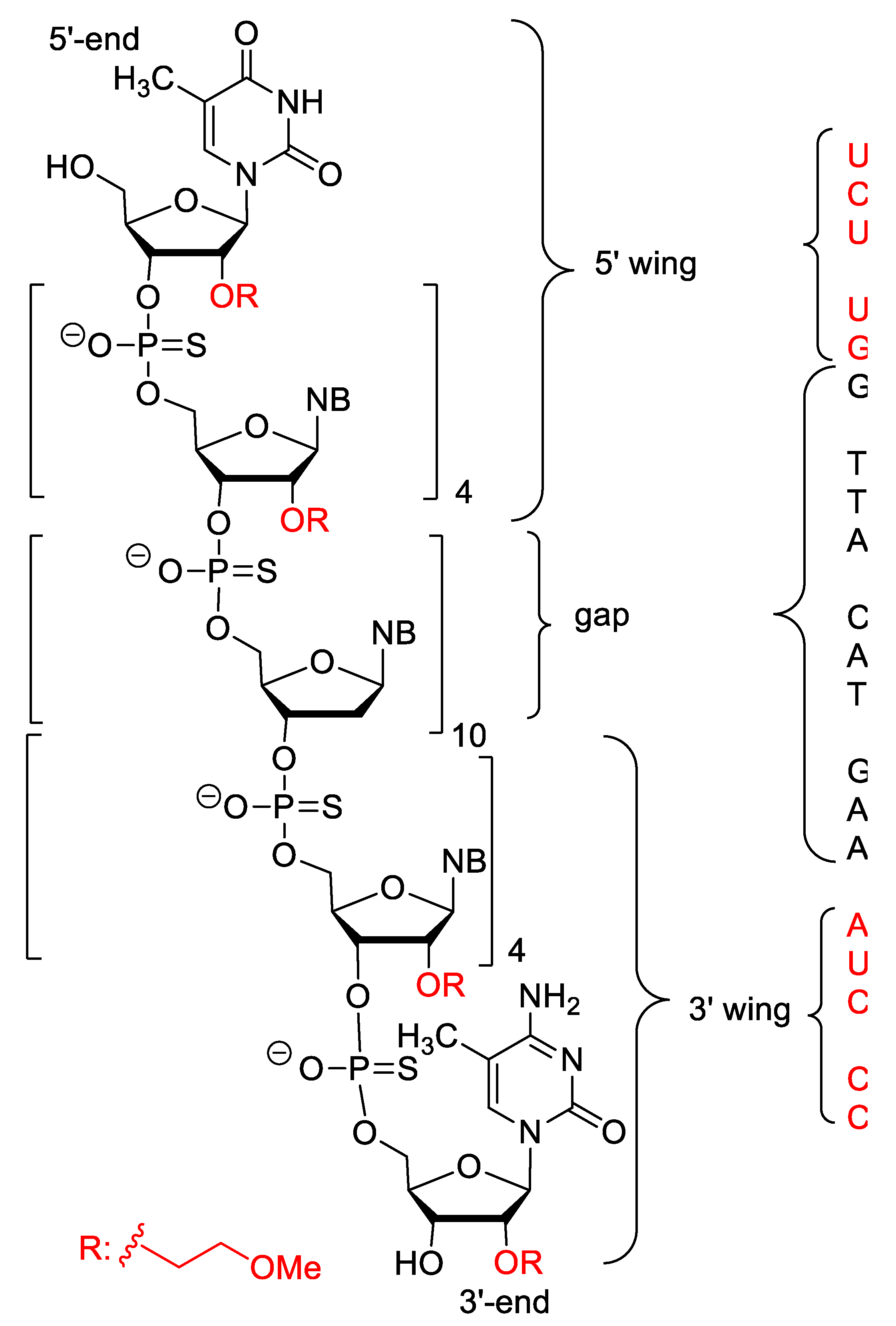
- Neil, E.E.; Bisaccia, E.K. Nusinersen: A Novel Antisense Oligonucleotide for the Treatment of Spinal Muscular Atrophy. J. Pediatr. Pharmacol. Ther. 2019, 24, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Chiriboga, C.A.; Swoboda, K.J.; Darras, B.T.; Iannaccone, S.T.; Montes, J.; De Vivo, D.C.; Norris, D.A.; Bennett, C.F.; Bishop, K.M. Results from a phase 1 study of nusinersen (ISIS-SMNRx) in children with spinal muscular atrophy. Neurology 2016, 86, 890–897. [Google Scholar] [CrossRef]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- Duffy, K.; Arangundy-Franklin, S.; Holliger, P. Modified nucleic acids: Replication, evolution, and next-generation therapeutics. BMC Biol. 2020, 18, 112. [Google Scholar] [CrossRef]
- European Medicines Agency: EPAR Summary for the Public, EMA/736370/2017. Available online: https://www.ema.europa.eu/en/documents/overview/spinraza-epar-summary-public_en.pdf (accessed on 19 July 2022).
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2018, 387, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Albrechtsen, S.S.; Born, A.P.; Boesen, M.S. Nusinersen treatment of spinal muscular atrophy-a systematic review. Dan. J. Med. 2020, 67, A02200100. [Google Scholar]
- European Medicines Agency: Assesment Report Onpattro, EMA/554262/2018, 26 July 2018. Available online: https://www.ema.europa.eu/en/documents/assessment-report/onpattro-epar-public-assessment-report_.pdf (accessed on 19 July 2022).
- Hoy, S.M. Patisiran, First Global Approval. Drugs 2018, 78, 1625–1631. [Google Scholar] [CrossRef]
- Zhang, X.; Goel, V.; Robbie, G.J. Pharmacokinetics of Patisiran, the First Approved RNA Interference Therapy in Patients with Hereditary Transthyretin-Mediated Amyloidosis. J. Clin. Pharmacol. 2020, 60, 573–585. [Google Scholar] [CrossRef]
- Urits, I.; Swanson, D.; Swett, M.C.; Patel, A.; Berardino, K.; Amgalan, A.; Berger, A.A.; Kassem, H.; Kaye, A.D.; Viswanath, O. A Review of Patisiran (ONPATTRO®) for the Treatment of Polyneuropathy in People with Hereditary Transthyretin Amyloidosis. Neurol. Ther. 2020, 9, 301–315. [Google Scholar] [CrossRef]
- Kristen, A.V.; Ajroud-Driss, S.; Conceicao, I.; Gorevic, P.; Kyriakides, T.; Obici, L. Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegener. Dis. Manag. 2019, 9, 5–23. [Google Scholar] [CrossRef]
- European Medicines Agency: Assesment Report Tegsedi, EMA/411876/2018, 31 May 2018. Available online: https://www.ema.europa.eu/en/documents/assessment-report/tegsedi-epar-public-assessment-report_en.pdf (accessed on 19 July 2022).
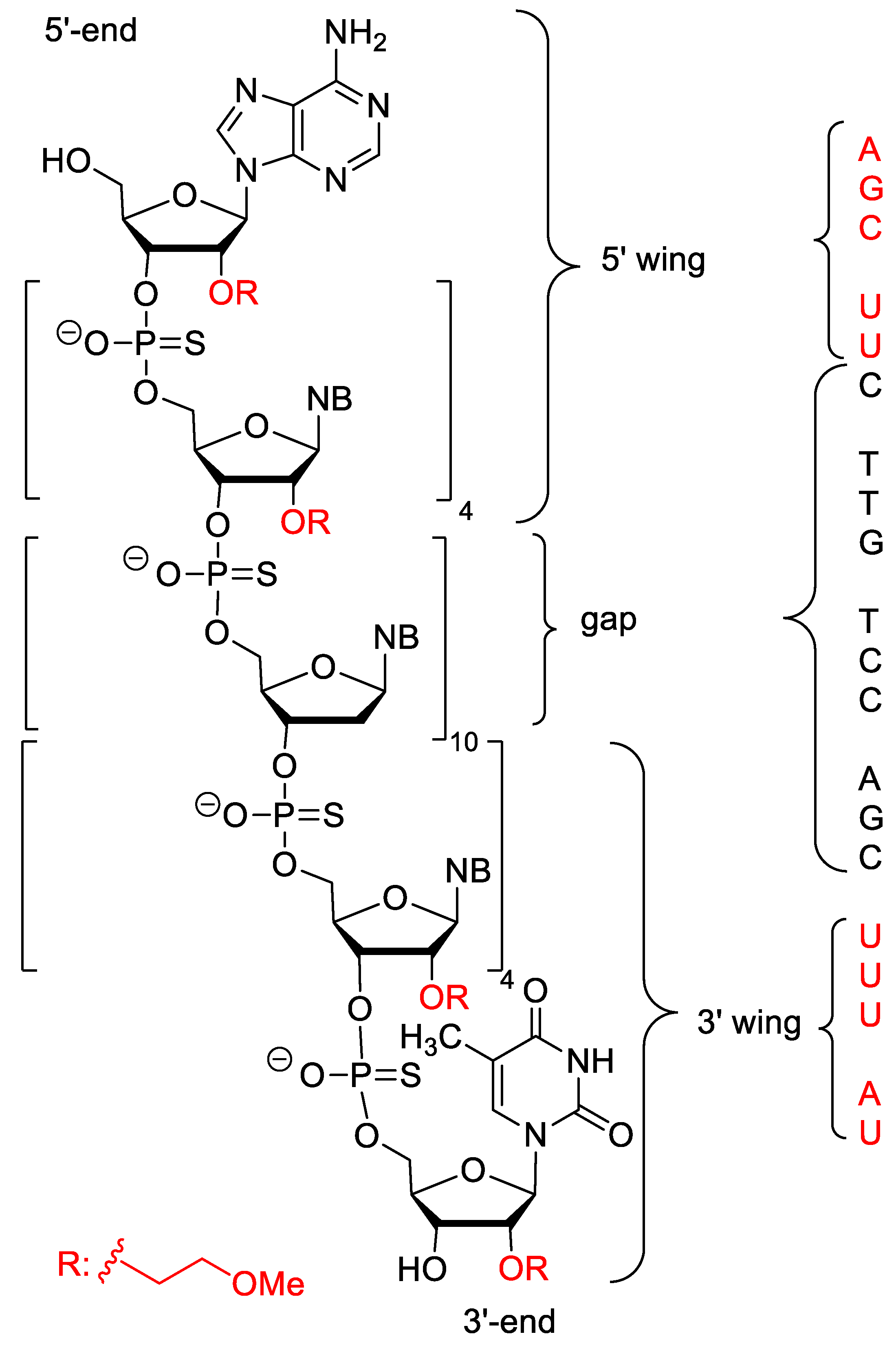
- Mathew, V.; Wang, A.K. Inotersen: New promise for the treatment of hereditary transthyretin amyloidosis. Drug Des. Dev. Ther. 2019, 13, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.Z.; Collins, J.W.; Hall, S.; Ackermann, E.J.; Geary, R.S.; Monia, B.P.; Henry, S.P.; Wang, Y. Population Pharmacokinetic–Pharmacodynamic Modeling of Inotersen, an Antisense Oligonucleotide for Treatment of Patients with Hereditary Transthyretin Amyloidosis. Nucleic Acid Ther. 2020, 30, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Dasgupta, N.R.; Monia, B.P. Inotersen (transthyretin-specific antisense oligonucleotide) for treatment of transthyretin amyloidosis. Neurodegener. Dis. Manag. 2019, 9, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hu, C.; El Achkar, C.M.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Soucy, K.A.; et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N. Engl. J. Med. 2019, 381, 1644–1652. [Google Scholar] [CrossRef]
- Jensen, T.L.; Gotzsche, C.R.; Woldbye, D.P.D. Current and Future Prospects Gene Therapy for Rare Genetic Diseases Affecting the Brain and Spinal Cord. Front. Mol. Neurosci. 2021, 14, 695937. [Google Scholar] [CrossRef]
- Khorkova, O.; Hsiao, J.; Wahlestedt, C. Nucleic Acid-Based Therapeutics in Orphan Neurological Disorders: Recent Developments. Front. Mol. Biosci. 2021, 8, 643681. [Google Scholar] [CrossRef]
- Graham, M.J.; Lee, R.G.; Bell, T.A.; Fu, W.; Mullick, A.E.; Alexander, V.J.; Singleton, W.; Viney, N.; Geary, R.; Su, J.; et al. Antisense Oligonucleotide Inhibition of Apolipoprotein C-III Reduces Plasma Triglycerides in Rodents, Nonhuman Primates and Humans. Circ. Res. 2013, 112, 1479–1490. [Google Scholar] [CrossRef]
- Esan, O.; Wierzbicki, A.S. Volanesorsen in the Treatment of Familial Chylomicronemia Syndrome or Hypertriglyceridaemia: Design, Development and Place in Therapy. Drug Des. Devel. Ther. 2020, 14, 2623–2636. [Google Scholar] [CrossRef]
- Guoni-Berthold, I.; Alexander, V.J.; Yang, Q.; Hurh, E.; Steinhagen-Thiessen, E.; Moriarty, P.M.; Hughes, S.G.; Gaudet, G.; Hegele, R.A.; O’Dea, L.; et al. Efficacy and safety of volanesorsen in patients with multifactorial chylomicronaemia (COMPASS): A multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2021, 9, 264–275. [Google Scholar] [CrossRef]
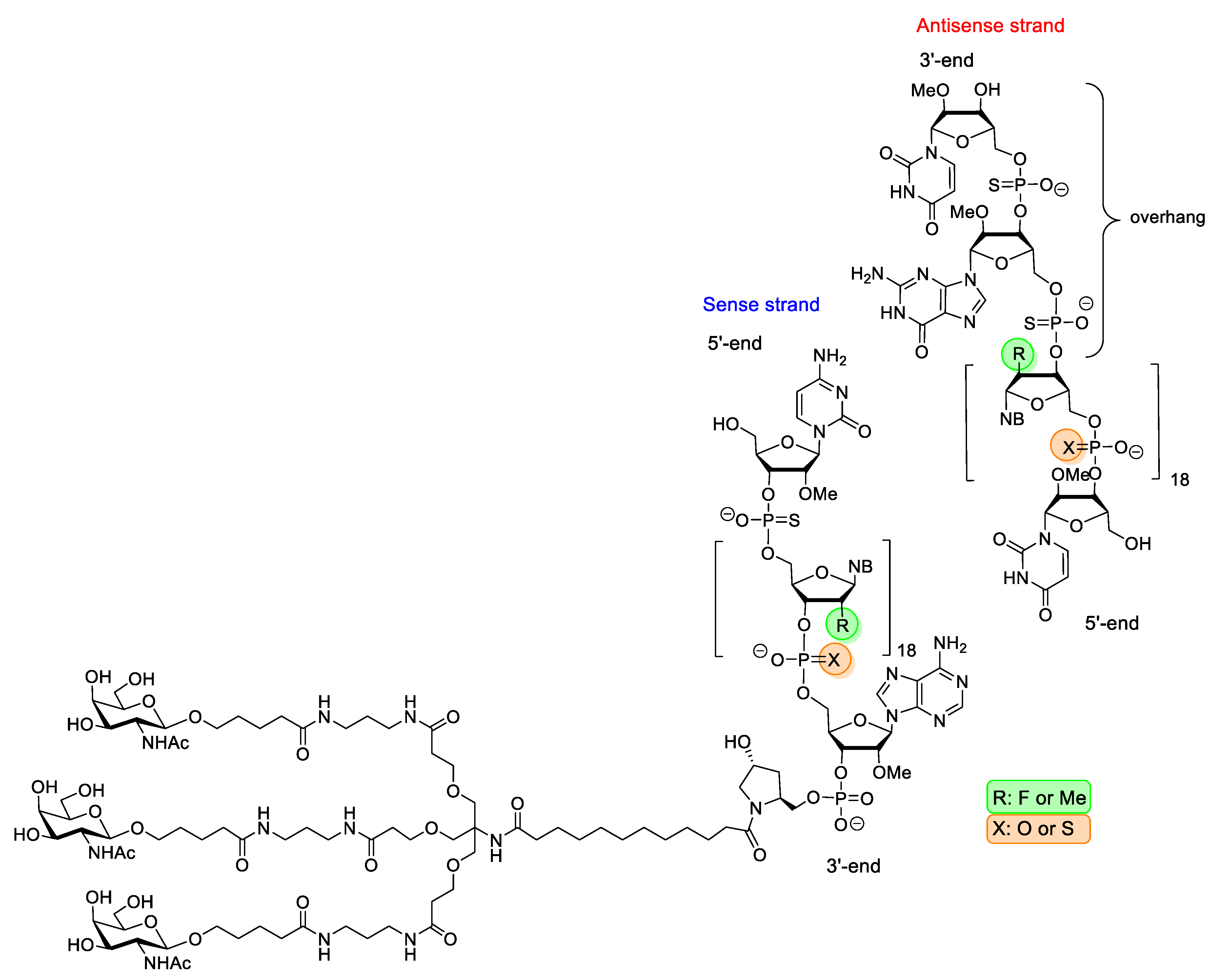
- European Medicines Agency: Assesment Report Givlaari, EMA/CHMP/70703/2020, 30 January 2020. Available online: https://www.ema.europa.eu/en/documents/assessment-report/givlaari-epar-public-assessment-report_en.pdf (accessed on 19 July 2022).
- Scott, L.J. Givosiran: First Approval. Drugs 2020, 80, 335–339. [Google Scholar] [CrossRef]
- Syed, Y.Y. Givosiran: A Review in Acute Hepatic Phorphyria. Drugs 2021, 81, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Dzierlaga, K.; Yokota, T. Optimization of antisense-mediated exon skipping for Duchenne muscular dystrophy. Gene Ther. 2020, 27, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y. Golodirsen: First Approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Corey, D.R. The 10th Oligonucleotide Therapy Approved: Golodirsen for Duchenne Muscular Dystrophy. Nucleic Acid Ther. 2020, 30, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.E.; Schnell, F.J.; Akana, C.; El-Husayni, S.H.; Desjardins, C.A.; Morgan, J.; Charleston, J.S.; Sardone, V.; Domingos, J.; Dickson, G.; et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology 2020, 94, e2270–e2282. [Google Scholar] [CrossRef]
- Dhillon, S. Viltolarsen: First Approval. Drugs 2020, 80, 1027–1031. [Google Scholar] [CrossRef]
- Clemens, P.R.; Rao, V.K.; Connolly, A.M.; Harper, A.D.; Mah, J.K.; Smith, E.C.; McDonald, C.M.; Zaidman, C.M.; Morgenroth, L.P.; Osaki, H.; et al. Safety, Tolerability, and Efficacy of Viltolarsen in Boys with Duchenne Muscular Dystrophy Amenable to Exon 53 Skipping A Phase 2 Randomized Clinical Trial. JAMA Neurol. 2020, 77, 982–991. [Google Scholar] [CrossRef]
- European Medicines Agency: Assesment Report Leqvio, EMA/696912/2020, 15 October 2020. Available online: https://www.ema.europa.eu/en/documents/assessment-report/leqvio-epar-public-assessment-report_en.pdf (accessed on 19 July 2022).
- Kulkarni, J.A.; Witzigmann, D.; Thomson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; van der Meel, R. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021, 16, 630–643. [Google Scholar] [CrossRef]
- Lamb, Y.N. Inclisiran: First Approval. Drugs 2021, 81, 389–395. [Google Scholar] [CrossRef]
- European Medicines Agency: Assesment Report Oxlumo, EMA/568312/2020, 15 October 2020. Available online: https://www.ema.europa.eu/en/documents/assessment-report/oxlumo-epar-public-assessment-report_en.pdf (accessed on 19 July 2022).
- Scott, L.J.; Keam, S.J. Lumasiran: First Approval. Drugs 2021, 81, 277–282. [Google Scholar] [CrossRef]
- Shirley, M. Casimersen: First Approval. Drugs 2021, 81, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.U.; Parida, S.; Lingaraju, M.C.; Kesavan, M.; Kumar, D.; Singh, R.K. Drug repurposing approach to fight COVID-19. Pharmacol. Rep. 2020, 72, 1479–1508. [Google Scholar] [CrossRef] [PubMed]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of COVID-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Bernal, A.J.; da Silva, M.M.G.; Musungaie, D.B.; Kovalchuk, E.; Gonzalez, A.; Reyes, V.D.; Martín-Quirós, A.; Caraco, Y.; Williams-Diaz, A.; Brown, M.L.; et al. Molnupiravir for Oral Treatment of COVID-19 in Nonhospitalized Patients. N. Engl. J. Med. 2022, 386, 509–520. [Google Scholar] [CrossRef]
- Yuki, K.; Fujiogi, M.; Koutsogiannaki, S. COVID-19 pathophysiology: A review. Clin. Immunol. 2020, 215, 108427–108434. [Google Scholar] [CrossRef]
- Nance, K.D.; Meier, J.L. Modifications in an Emergency: The Role of N1-Methylpseudouridine in COVID-19 Vaccines. ACS Cent. Sci. 2021, 7, 748–756. [Google Scholar] [CrossRef]
- Khehra, N.; Padda, I.; Jaferi, U.; Atwal, H.; Narain, S.; Parmar, M.S. Tozinameran (BNT162b2) Vaccine: The Journey from Preclinical Research to Clinical Trials and Authorization. AAPS Pharm. Sci. Tech. 2021, 22, 172–181. [Google Scholar] [CrossRef]
- Lamb, Y.N. BNT162b2 mRNA COVID-19 Vaccine: First Approval. Drugs 2021, 81, 495–501. [Google Scholar] [CrossRef]
- Chaudhary, N.; Weissman, D.; Whitehead, K.A. mRNA vaccines for infectious diseases: Principles, delivery and clinical translation. Nat. Rev. Drug Discov. 2021, 20, 817–838. [Google Scholar] [CrossRef]
- Kon, E.; Elia, U.; Peer, D. Principles for designing an optimal mRNA lipid nanoparticle vaccine. Curr. Opin. Biotechnol. 2021, 73, 329–336. [Google Scholar] [CrossRef]
- Doudna, J.A. The promise and challenge of therapeutic genome editing. Nature 2020, 578, 229–236. [Google Scholar] [CrossRef] [PubMed]
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| INN Name | Approval | Chemical Structure | Mechanism of Action | Disease |
|---|---|---|---|---|
| Fomivirsen (Vitravene) | FDA 1998 EMA 1999 | PS | antisense | cytomegalovirus retinitis |
| Pegaptanib (Macugen) | FDA 2004 | 2′-O-Me, 2′-F, PEG-conjugate, 3′-inverted nucleotide | aptamer | age related macula degeneration |
| Mipomersen (Kynamro) | FDA 2013 | PS, 2′-O-MOE gapmer, 5-Me-C | antisense | familial hypercholesterolaemia |
| Eteplirsen (Exondys 51) | FDA 2016 | PMO | antisense, splicing modulation | Duchenne muscular dystrophy |
| Defibrotide (Defitelio) | FDA 2016 | mixture of ds and ss ODNs with 50 bp average length | aptamer, complex | Sinusoidal obstruction syndrome |
| Nusinersen (Spinraza) | FDA 2016 EMA 2017 | 2′-O-MOE, PS, 5-Me-C | antisense, splicing modulation | Spinal muscular atrophy |
| Patisiran (Onpattro) | FDA 2018 EMA 2018 | 2′-OMe | RNA interference | Hereditary transthyretin mediated amyloidosis |
| Inotersen (Tegsedi) | FDA 2018 EMA 2018 | PS, 2′-O-MOE, 5-Me-C | antisense | Hereditary transthyretin mediated amyloidosis |
| Milasen | FDA 2017 | 2′-O-MOE | antisense, splicing modulation | Batten’s disease (patient-costumized) |
| Volanesorsen (Waylivra) | EMA 2019 | PS, 2′-O-MOE, 5-Me-C | antisense | Familial chylomicronemia |
| Givosiran (Givlaari) | FDA 2019 EMA 2020 | PS, 2′-F, 2′-OMe, GalNAc-conjugate | RNA interference | Acute hepatic porphyria |
| Golodirsen (Vyondys 53) | FDA 2019 | PMO | antisense, splicing modulation | Duchenne muscular dystrophy |
| Viltolarsen (Viltepso) | FDA 2020 | PMO | antisense, splicing modulation | Duchenne muscular dystrophy |
| Casimersen (Amondys 45) | FDA 2021 | PMO | antisense, splicing modulation | Duchenne muscular dystrophy |
| Inclisiran (Leqvio) | EMA 2020 | PS, 2′-F, 2′-OMe, GalNAc-conjugate | RNA interference | primary hypercholesterolaemia |
| Lumasiran (Oxlumo) | FDA 2020 EMA 2020 | PS, 2′-F, 2′-OMe, GalNAc-conjugate | RNA interference | primary hyperoxaluria |
| Tozinameran (Comirnaty) | FDA 2020 EMA 2020 | m1ψ, 2′-OMe, 5′-cap | mRNA vaccine | COVID-19 |
| Elasomeran (Soikevax) | FDA 2020 EMA 2020 | m1ψ, 5′-cap | mRNA vaccine | COVID-19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bege, M.; Borbás, A. The Medicinal Chemistry of Artificial Nucleic Acids and Therapeutic Oligonucleotides. Pharmaceuticals 2022, 15, 909. https://doi.org/10.3390/ph15080909
Bege M, Borbás A. The Medicinal Chemistry of Artificial Nucleic Acids and Therapeutic Oligonucleotides. Pharmaceuticals. 2022; 15(8):909. https://doi.org/10.3390/ph15080909
Chicago/Turabian StyleBege, Miklós, and Anikó Borbás. 2022. "The Medicinal Chemistry of Artificial Nucleic Acids and Therapeutic Oligonucleotides" Pharmaceuticals 15, no. 8: 909. https://doi.org/10.3390/ph15080909
APA StyleBege, M., & Borbás, A. (2022). The Medicinal Chemistry of Artificial Nucleic Acids and Therapeutic Oligonucleotides. Pharmaceuticals, 15(8), 909. https://doi.org/10.3390/ph15080909