
Synthesis, Structure–Activity Relationships, and Parasitological Profiling of Brussonol Derivatives as New Plasmodium falciparum Inhibitors

 ,
,  ,
,  , , and
, , and
Abstract
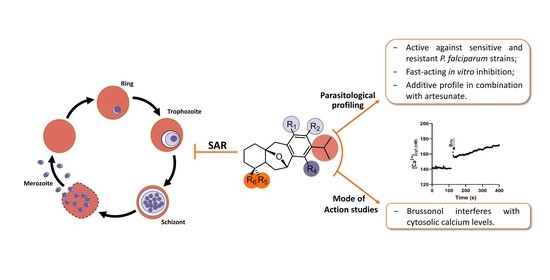
1. Introduction
2. Results
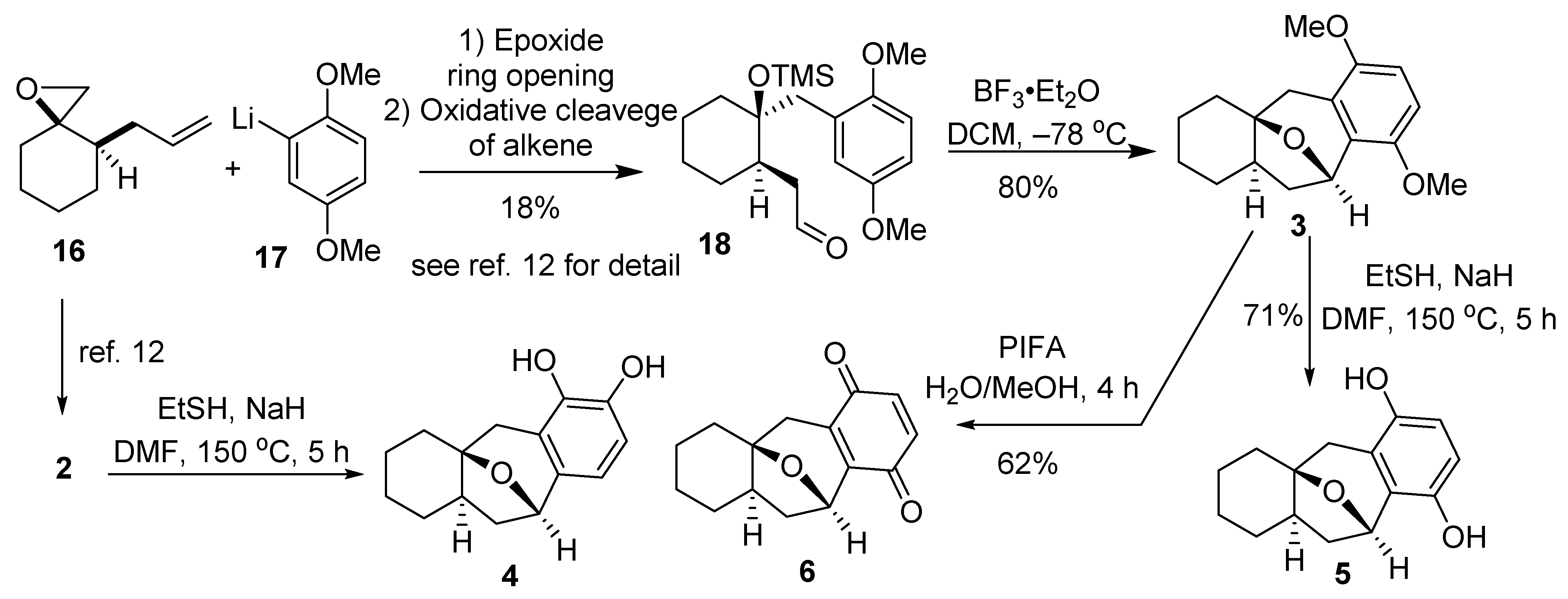
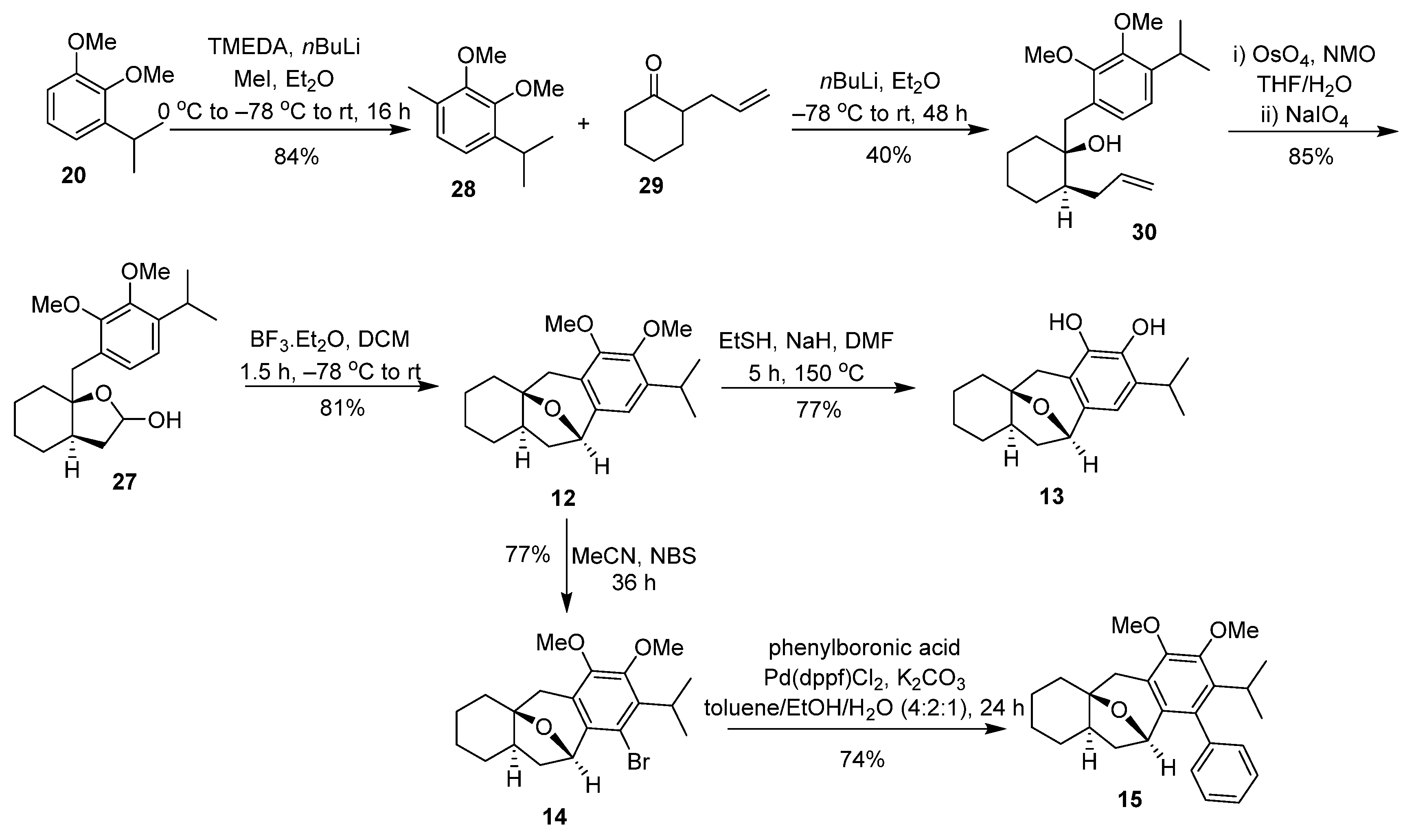
2.1. Synthesis of Brussonol and Derivatives
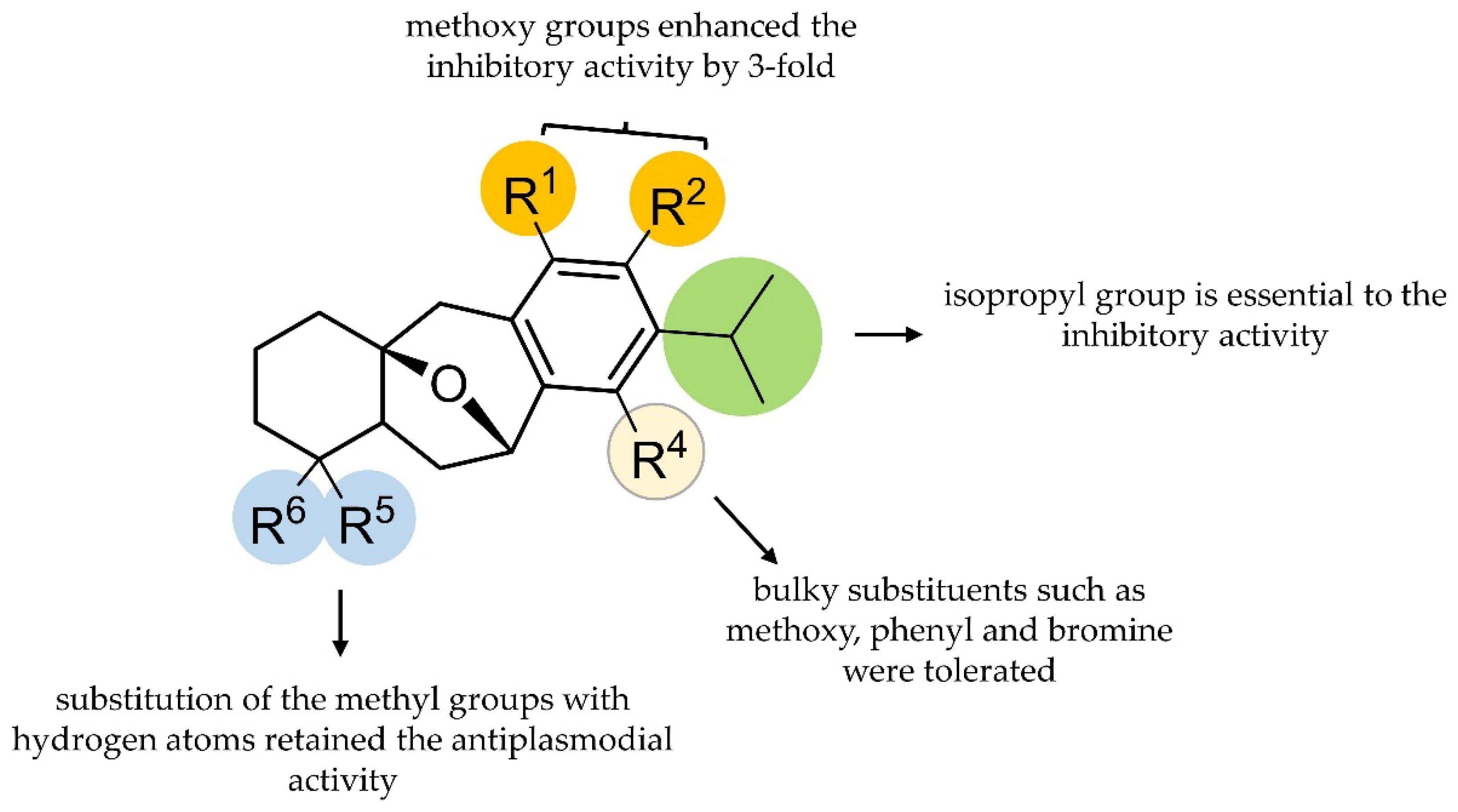
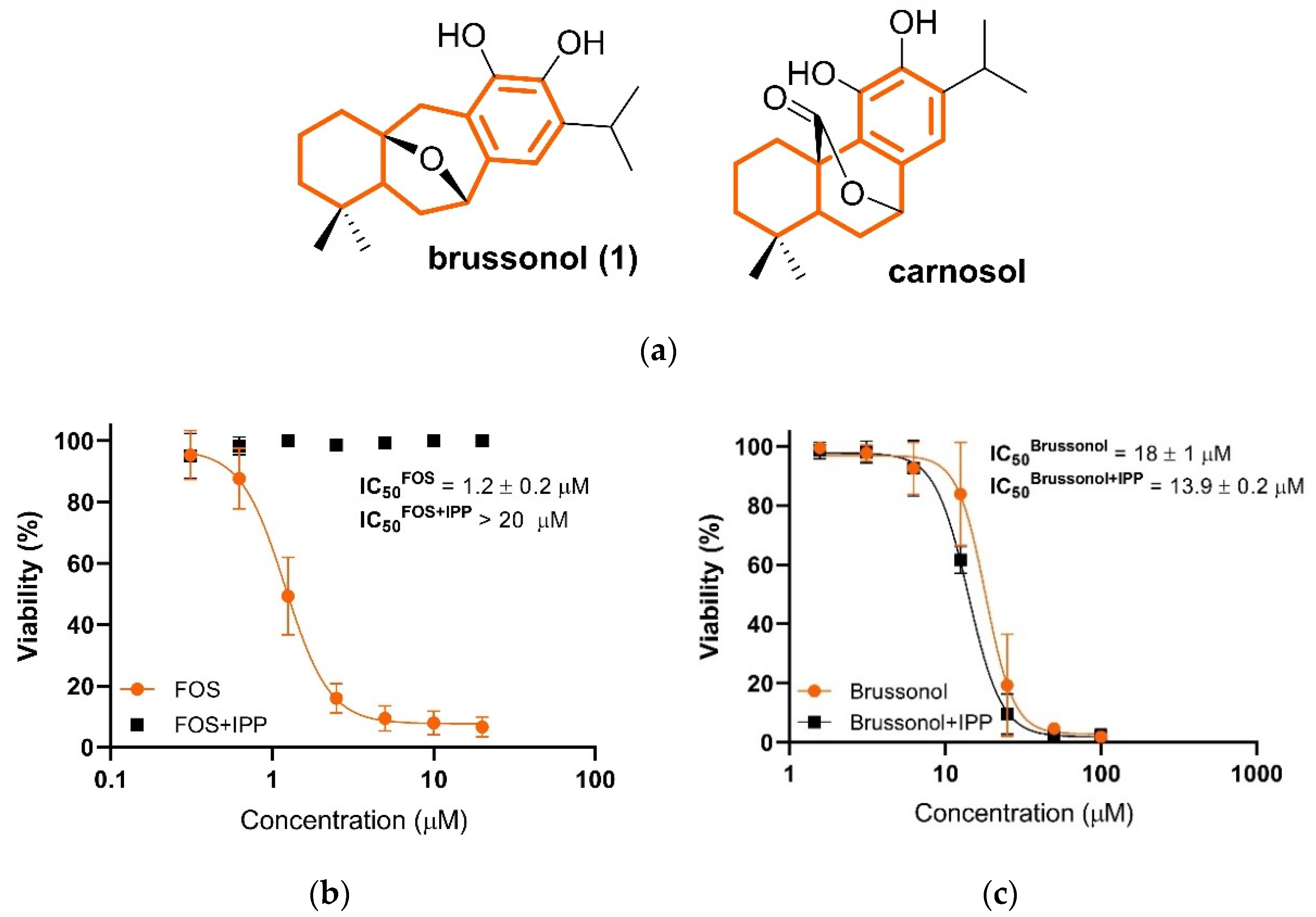
2.2. Brussonol and Derivatives Showed Antiplasmodial Activity and Low Cytotoxic Activity
2.3. Brussonol Is a Potent Inhibitor of Resistant P. falciparum Strains
2.4. Brussonol Is a Potent P. knowlesi Inhibitor
2.5. Brussonol Is a Fast-Acting P. falciparum Inhibitor
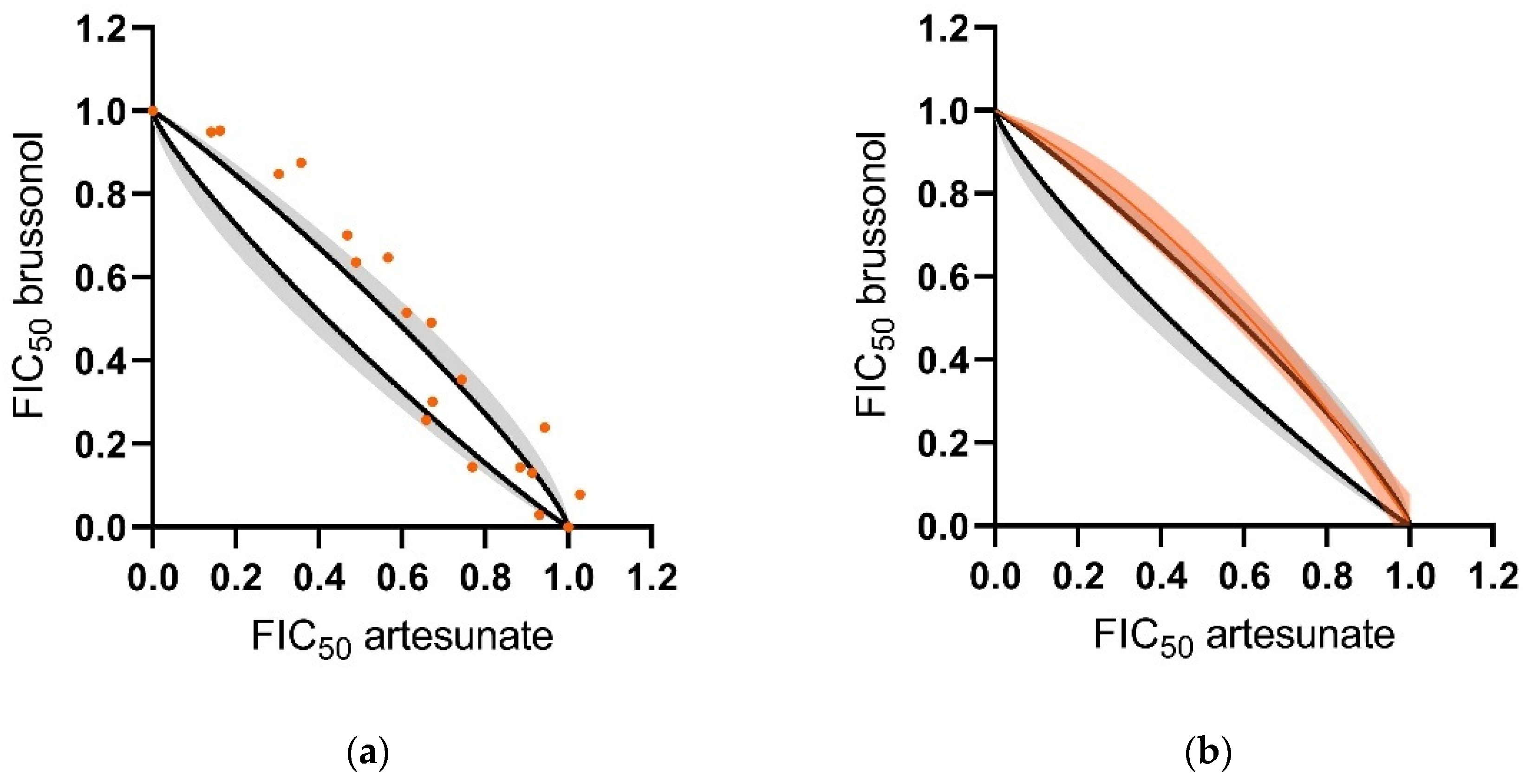
2.6. Brussonol Shows an Additive Combination Profile with Artesunate
2.7. Brussonol Does Not Interfere with Isoprenoid Biosynthesis
2.8. Brussonol Induces [Ca2+]cyt Rise in P. falciparum
3. Discussion
4. Materials and Methods
4.1. Maintenance of In Vitro Culture
4.2. Biological Activity against P. falciparum Blood-Stage Parasites In Vitro
4.3. Cytotoxic Tests Using Immortalized Cells
4.4. Calculation of Selectivity Index (SI)
4.5. In Vitro Evaluation against P. knowlesi
4.6. Resistance Assessment
4.7. Speed-of-Action Assay
4.8. In Vitro Combination with Artesunate
4.9. Chemical Rescue Assay
4.10. [Ca2+]cyt Measurements
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization World Malaria Report 2021; World Health Organization: Geneva, Switzerland, 2021.
- Yeung, S. Malaria-Update on Antimalarial Resistance and Treatment Approaches. Pediatr. Infect. Dis. J. 2018, 37, 367–369. [Google Scholar] [CrossRef]
- Simmons, E.M.; Sarpong, R. Structure, Biosynthetic Relationships and Chemical Synthesis of the Icetexane Diterpenoids. Nat. Prod. Rep. 2009, 26, 1195–1217. [Google Scholar] [CrossRef]
- Fraga, B.M.; Díaz, C.E.; Guadaño, A.; González-Coloma, A. Diterpenes from Salvia Broussonetii Transformed Roots and Their Insecticidal Activity. J. Agric. Food Chem. 2005, 53, 5200–5206. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Yu, Y.J.; Huang, C.G.; Huang, X.Z.; Hu, Q.F.; Yang, G.Y.; bin Wang, H.; Zhang, X.Y.; Li, G.P. Icetexane Diterpenoids from Perovskia Atriplicifolia. Planta Med. 2015, 81, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, N.; Kiuchi, F.; Ito, M.; Honda, G.; Takeda, Y.; Khodzhimatov, O.K.; Ashurmetov, O.A. New Icetexane and 20-Norabietane Diterpenes with Trypanocidal Activity from Dracocephalum Komarovi. J. Nat. Prod. 2003, 66, 128–131. [Google Scholar] [CrossRef] [PubMed]
- El-Lakany, A.M.; Abdel-Kader, M.S.; Sabri, N.N.; Stermitz, F.R. Lanigerol: A New Antimicrobial Icetexane Diterpene from Salvia Lanigera. Planta Med. 1995, 61, 559–560. [Google Scholar] [CrossRef] [PubMed]
- Cezarotto, C.S.; Dorneles, A.; Baldissera, F.G.; da Silva, M.B.; Markoski, M.M.; Júnior, L.C.R.; Peres, A.; Fazolo, T.; Bordignon, S.A.L.; Apel, M.A.; et al. Leishmanicidal and Antichemotactic Activities of Icetexanes from Salvia Uliginosa Benth. Phytomedicine 2019, 58, 152748. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, N.; Kabututu, Z.; Kubata, B.K.; Kiuchi, F.; Ito, M.; Nakajima-Shimada, J.; Aoki, T.; Ohkubo, K.; Fukuzumi, S.; Martin, S.K.; et al. Antichagasic Activity of Komaroviquinone Is Due to Generation of Reactive Oxygen Species Catalyzed by Trypanosoma Cruzi Old Yellow Enzyme. Antimicrob. Agents Chemother. 2005, 49, 5123–5126. [Google Scholar] [CrossRef]
- Tabefam, M.; Farimani, M.M.; Danton, O.; Ramseyer, J.; Kaiser, M.; Ebrahimi, S.N.; Salehi, P.; Batooli, H.; Potterat, O.; Hamburger, M. Antiprotozoal Diterpenes from Perovskia Abrotanoides. Planta Med. 2018, 84, 913–919. [Google Scholar] [CrossRef]
- Esquivel, B.; Bustos-Brito, C.; Sánchez-Castellanos, M.; Nieto-Camacho, A.; Ramírez-Apan, T.; Joseph-Nathan, P.; Quijano, L. Structure, Absolute Configuration, & Antiproliferative Activity of Abietane & Icetexane Diterpenoids from Salvia Ballotiflora. Molecules 2017, 22, 1690. [Google Scholar] [CrossRef]
- Carita, A.; Burtoloso, A.C.B. An Epoxide Ring-Opening Approach for a Short and Stereoselective Synthesis of Icetexane Diterpenoids. Tetrahedron Lett. 2010, 51, 686–688. [Google Scholar] [CrossRef]
- Reddy, K.R.K.K.; Longato, G.B.; de Carvalho, J.E.; Ruiz, A.L.T.G.; Silva, L.F. Populene D Analogs: Design, Concise Synthesis and Antiproliferative Activity. Molecules 2012, 17, 9621–9630. [Google Scholar] [CrossRef] [PubMed]
- Tohma, H.; Morioka, H.; Harayama, Y.; Hashizume, M.; Kita, Y. Novel and Efficient Synthesis of P-Quinones in Water via Oxidative Demethylation of Phenol Ethers Using Hypervalent Iodine(III) Reagents. Tetrahedron Lett. 2001, 42, 6899–6902. [Google Scholar] [CrossRef]
- Ahmad, A.; Burtoloso, A.C.B. Total Synthesis of (±)-Brussonol and (±)-Komaroviquinone via a Regioselective Cross-Electrophile Coupling of Aryl Bromides and Epoxides. Org. Lett. 2019, 21, 6079–6083. [Google Scholar] [CrossRef]
- Maier, M.E.; Bayer, A. A Formal Total Synthesis of Salvadione. Eur. J. Org. Chem. 2006, 17, 4034–4043. [Google Scholar] [CrossRef]
- Padwa, A.; Chughtai, M.J.; Boonsombat, J.; Rashatasakhon, P. A Rh(II)-Catalyzed Cycloaddition Approach toward the Synthesis of Komaroviquinone. Tetrahedron 2008, 64, 4758–4767. [Google Scholar] [CrossRef][Green Version]
- Stevens, R.V.; Bisacchi, G.S. Benzocyclobutenones as Synthons for the Synthesis of C-11 Oxygenated Diterpenoids. Application to the Total Synthesis of (±)-Taxodione. J. Org. Chem. 1982, 47, 2396–2399. [Google Scholar] [CrossRef]
- della Ca’, N.; Sassi, G.; Catellani, M. A Direct Palladium-Catalyzed Route to Selectively Substituted Carbazoles through Sequential C-C and C-N Bond Formation: Synthesis of Carbazomycin A. Adv. Synth. Catal. 2008, 350, 2179–2182. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, Y.; Wang, Q. Collective Asymmetric Synthesis of (-)-Antofine, (-)-Cryptopleurine, (-)-Tylophorine, and (-)-Tylocrebrine with Tert- Butanesulfinamide as a Chiral Auxiliary. J. Org. Chem. 2014, 79, 3348–3357. [Google Scholar] [CrossRef]
- Martinez-Solorio, D.; Jennings, M.P. Convergent Formal Syntheses of (±)-Brussonol and (±)-Abrotanone via an Intramolecular Marson-Type Cyclization. Org. Lett. 2009, 11, 189–192. [Google Scholar] [CrossRef]
- Katsuno, K.; Burrows, J.N.; Duncan, K.; van Huijsduijnen, R.H.; Kaneko, T.; Kita, K.; Mowbray, C.E.; Schmatz, D.; Warner, P.; Slingsby, B.T. Hit and Lead Criteria in Drug Discovery for Infectious Diseases of the Developing World. Nat. Rev. Drug Discov. 2015, 14, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Chugh, M.; Scheurer, C.; Sax, S.; Bilsland, E.; van Schalkwyk, D.A.; Wicht, K.J.; Hofmann, N.; Sharma, A.; Bashyam, S.; Singh, S.; et al. Identification and Deconvolution of Cross-Resistance Signals from Antimalarial Compounds Using Multidrug-Resistant Plasmodium Falciparum Strains. Antimicrob. Agents Chemother. 2015, 59, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Yusof, R.; Lau, Y.L.; Mahmud, R.; Fong, M.Y.; Jelip, J.; Ngian, H.U.; Mustakim, S.; Mat Hussin, H.; Marzuki, N.; Mohd Ali, M. High Proportion of Knowlesi Malaria in Recent Malaria Cases in Malaysia. Malar. J. 2014, 13, 163. [Google Scholar] [CrossRef] [PubMed]
- Co, E.M.A.; Dennull, R.A.; Reinbold, D.D.; Waters, N.C.; Johnson, J.D. Assessment of Malaria in Vitro Drug Combination Screening and Mixed-Strain Infections Using the Malaria Sybr Green I-Based Fluorescence Assay. Antimicrob. Agents Chemother. 2009, 53, 2557–2563. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Grabovsky, Y.; Tallarida, R.J. Isobolographic Analysis for Combinations of a Full and Partial Agonist: Curved Isoboles. J. Pharmacol. Exp. Ther. 2004, 310, 981–986. [Google Scholar] [CrossRef]
- Macías-Alonso, M.; Andrés, L.S.; Córdova-Guerrero, I.; Estolano-Cobián, A.; Díaz-Rubio, L.; Marrero, J.G. Inhibition of Squalene Synthase of Rat Liver by Abietane Diterpenes Derivatives. Nat. Prod. Res. 2019, 35, 2972–2976. [Google Scholar] [CrossRef]
- Cerqueira, N.M.F.S.A.; Oliveira, E.F.; Gesto, D.S.; Santos-Martins, D.; Moreira, C.; Moorthy, H.N.; Ramos, M.J.; Fernandes, P.A. Cholesterol Biosynthesis: A Mechanistic Overview. Biochemistry 2016, 55, 5483–5506. [Google Scholar] [CrossRef]
- Guggisberg, A.M.; Amthor, R.E.; Odom, A.R. Isoprenoid Biosynthesis in Plasmodium Falciparum. Eukaryot. Cell 2014, 13, 1348–1359. [Google Scholar] [CrossRef]
- Edwards, R.L.; Brothers, R.C.; Wang, X.; Maron, M.I.; Ziniel, P.D.; Tsang, P.S.; Kraft, T.E.; Hruz, P.W.; Williamson, K.C.; Dowd, C.S.; et al. MEPicides: Potent Antimalarial Prodrugs Targeting Isoprenoid Biosynthesis. Sci. Rep. 2017, 7, 8400. [Google Scholar] [CrossRef]
- Yeh, E.; DeRisi, J.L. Chemical Rescue of Malaria Parasites Lacking an Apicoplast Defines Organelle Function in Blood-Stage Plasmodium Falciparum. PLoS Biol. 2011, 9, e1001138. [Google Scholar] [CrossRef]
- Bowman, J.D.; Merino, E.F.; Brooks, C.F.; Striepen, B.; Carlier, P.R.; Cassera, M.B. Antiapicoplast and Gametocytocidal Screening to Identify the Mechanisms of Action of Compounds within the Malaria Box. Antimicrob. Agents Chemother. 2014, 58, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Herrera, Z.; Ebert, D.; Baska, K.; Cho, S.H.; DeRisi, J.L.; Yeh, E. A Chemical Rescue Screen Identifies a Plasmodium Falciparum Apicoplast Inhibitor Targeting MEP Isoprenoid Precursor Biosynthesis. Antimicrob. Agents Chemother. 2015, 59, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Gisselberg, J.E.; Herrera, Z.; Orchard, L.M.; Llinás, M.; Yeh, E. Specific Inhibition of the Bifunctional Farnesyl/Geranylgeranyl Diphosphate Synthase in Malaria Parasites via a New Small-Molecule Binding Site. Cell Chem. Biol. 2018, 25, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Backman, T.W.H.; Cao, Y.; Girke, T. ChemMine Tools: An Online Service for Analyzing and Clustering Small Molecules. Nucleic Acids Res. 2011, 39, W486–W491. [Google Scholar] [CrossRef]
- di Marino, D.; D’Annessa, I.; Coletta, A.; Via, A.; Tramontano, A. Characterization of the Differences in the Cyclopiazonic Acid Binding Mode to Mammalian and P. Falciparum Ca2+ Pumps: A Computational Study. Proteins Struct. Funct. Bioinform. 2015, 83, 564–574. [Google Scholar] [CrossRef]
- Arnou, B.; Montigny, C.; Morth, J.P.; Nissen, P.; Jaxel, C.; Møller, J.V.; le Maire, M. The Plasmodium Falciparum Ca2+-ATPase PfATP6: Insensitive to Artemisinin, but a Potential Drug Target. Proc. Biochem. Soc. Trans. 2011, 39, 823–831. [Google Scholar] [CrossRef]
- Pandey, K.; Ferreira, P.E.; Ishikawa, T.; Nagai, T.; Kaneko, O.; Yahata, K. Ca2+ Monitoring in Plasmodium Falciparum Using the Yellow Cameleon-Nano Biosensor. Sci. Rep. 2016, 6, 23454. [Google Scholar] [CrossRef]
- Borges-Pereira, L.; Thomas, S.J.; dos Anjos e Silva, A.L.; Bartlett, P.J.; Thomas, A.P.; Garcia, C.R.S. The Genetic Ca2+ Sensor GCaMP3 Reveals Multiple Ca2+ Stores Differentially Coupled to Ca2+ Entry in the Human Malaria Parasite Plasmodium Falciparum. J. Biol. Chem. 2020, 295, 14998–15012. [Google Scholar] [CrossRef]
- Forte, B.; Ottilie, S.; Plater, A.; Campo, B.; Dechering, K.J.; Gamo, F.J.; Goldberg, D.E.; Istvan, E.S.; Lee, M.; Lukens, A.K.; et al. Prioritization of Molecular Targets for Antimalarial Drug Discovery. ACS Infect. Dis. 2021, 7, 2764–2776. [Google Scholar] [CrossRef]
- Ding, X.C.; Ubben, D.; Wells, T.N. A Framework for Assessing the Risk of Resistance for Anti-Malarials in Development. Malar. J. 2012, 11, 292. [Google Scholar] [CrossRef]
- Burrows, J.N.; Leroy, D.; Lotharius, J.; Waterson, D. Challenges in Antimalarial Drug Discovery. Future Med. Chem. 2011, 3, 1401–1412. [Google Scholar] [CrossRef] [PubMed]
- le Manach, C.; Paquet, T.; Gonzàlez Cabrera, D.; Younis, Y.; Taylor, D.; Wiesner, L.; Lawrence, N.; Schwager, S.; Waterson, D.; Witty, M.J.; et al. Medicinal Chemistry Optimization of Antiplasmodial Imidazopyridazine Hits from High Throughput Screening of a SoftFocus Kinase Library: Part 2. J. Med. Chem. 2014, 57, 8839–8848. [Google Scholar] [CrossRef] [PubMed]
- Foucquier, J.; Guedj, M. Analysis of Drug Combinations: Current Methodological Landscape. Pharmacol. Res. Perspect. 2015, 3, e00149. [Google Scholar] [CrossRef] [PubMed]
- van der Pluijm, R.W.; Amaratunga, C.; Dhorda, M.; Dondorp, A.M. Triple Artemisinin-Based Combination Therapies for Malaria—A New Paradigm? Trends Parasitol. 2021, 37, 15–24. [Google Scholar] [CrossRef]
- Aguiar, A.C.C.; Panciera, M.; Simao Dos Santos, E.F.; Singh, M.K.; Garcia, M.L.; de Souza, G.E.; Nakabashi, M.; Costa, J.L.; Garcia, C.R.S.; Oliva, G.; et al. Discovery of Marinoquinolines as Potent and Fast-Acting Plasmodium Falciparum Inhibitors with in Vivo Activity. J. Med. Chem. 2018, 61, 5547–5568. [Google Scholar] [CrossRef] [PubMed]
- de Souza, J.O.; Almeida, S.M.; Souza, G.E.; Zanini, C.L.; da Silva, E.M.; Calit, J.; Bargieri, D.Y.; Amporndanai, K.; Antonyuk, S.; Hasnain, S.S.; et al. Parasitological Profiling Shows 4(1H)-Quinolone Derivatives as New Lead Candidates for Malaria. Eur. J. Med. Chem. Rep. 2021, 3, 100012. [Google Scholar] [CrossRef]
- Parra, L.L.L.; Bertonha, A.F.; Severo, I.R.M.; Aguiar, A.C.C.; de Souza, G.E.; Oliva, G.; Guido, R.V.C.; Grazzia, N.; Costa, T.R.; Miguel, D.C.; et al. Isolation, Derivative Synthesis, and Structure-Activity Relationships of Antiparasitic Bromopyrrole Alkaloids from the Marine Sponge Tedania Brasiliensis. J. Nat. Prod. 2018, 81, 188–202. [Google Scholar] [CrossRef]
- de Souza, G.E.; Bueno, R.V.; de Souza, J.O.; Zanini, C.L.; Cruz, F.C.; Oliva, G.; Guido, R.V.C.; Aguiar, A.C.C. Antiplasmodial Profile of Selected Compounds from Malaria Box: In Vitro Evaluation, Speed of Action and Drug Combination Studies. Malar. J. 2019, 18, 447. [Google Scholar] [CrossRef]
- Yang, T.; Ottilie, S.; Istvan, E.S.; Godinez-Macias, K.P.; Lukens, A.K.; Baragaña, B.; Campo, B.; Walpole, C.; Niles, J.C.; Chibale, K.; et al. MalDA, Accelerating Malaria Drug Discovery. Trends Parasitol. 2021, 37, 493–507. [Google Scholar] [CrossRef]
- de Oliveira, L.S.; Alborghetti, M.R.; Carneiro, R.G.; Bastos, I.M.D.; Amino, R.; Grellier, P.; Charneau, S. Calcium in the Backstage of Malaria Parasite Biology. Front. Cell. Infect. Microbiol. 2021, 11, 708834. [Google Scholar] [CrossRef]
- Moore, C.M.; Wang, J.; Lin, Q.; Ferreira, P.; Avery, M.A.; Elokely, K.; Staines, H.M.; Krishna, S. Selective Inhibition of Plasmodium Falciparum ATPase 6 by Artemisinins and Identification of New Classes of Inhibitors after Expression in Yeast. Antimicrob. Agents Chemother. 2022, 66, e0207921. [Google Scholar] [CrossRef] [PubMed]
- Eckstein-Ludwig, U.; Webb, R.J.; van Goethem, I.D.A.; East, J.M.; Lee, A.G.; Kimura, M.; O’Neill, P.M.; Bray, P.G.; Ward, S.A.; Krishna, S. Artemisinins Target the SERCA of Plasmodium Falciparum. Nature 2003, 424, 957–961. [Google Scholar] [CrossRef] [PubMed]
- Cardi, D.; Pozza, A.; Arnou, B.; Marchal, E.; Clausen, J.D.; Andersen, J.P.; Krishna, S.; Møller, J.V.; le Maire, M.; Jaxel, C. Purified E255L Mutant SERCA1a and Purified PfATP6 Are Sensitive to SERCA-Type Inhibitors but Insensitive to Artemisinins. J. Biol. Chem. 2010, 285, 26406–26416. [Google Scholar] [CrossRef]
- Gazarini, M.L.; Sigolo, C.A.; Markus, R.P.; Thomas, A.P.; Garcia, C.R. Antimalarial Drugs Disrupt Ion Homeostasis in Malarial Parasites. Mem. Inst. Oswaldo Cruz 2007, 102, 329–334. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Moraes Barros, R.R.; Gibson, T.J.; Kite, W.A.; Sá, J.M.; Wellems, T.E. Comparison of Two Methods for Transformation of Plasmodium Knowlesi: Direct Schizont Electroporation and Spontaneous Plasmid Uptake from Plasmid-Loaded Red Blood Cells. Mol. Biochem. Parasitol. 2017, 218, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Trager, W.; Jensen, J.B. Human Malaria Parasites in Continuous Culture. Science 1976, 193, 673–675. [Google Scholar] [CrossRef]
- Zeeman, A.-M.; Voorberg-van der Wel, A.; Kocken, C.H.M. Ex Vivo Culture of Plasmodium Vivax and Plasmodium Cynomolgi and In Vitro Culture of Plasmodium Knowlesi Blood Stages. Methods Mol. Biol. 2013, 923, 35–49. [Google Scholar] [CrossRef]
- Lambros, C.; Vanderberg, J.P. Synchronization of Plasmodium Falciparum Erythrocytic Stages in Culture. J. Parasitol. 1979, 65, 418–420. [Google Scholar] [CrossRef]
- Vossen, M.G.; Pferschy, S.; Chiba, P.; Noedl, H. The SYBR Green I Malaria Drug Sensitivity Assay: Performance in Low Parasitemia Samples. Am. J. Trop. Med. Hyg. 2010, 82, 398–401. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Moraes Barros, R.R.; Thawnashom, K.; Gibson, T.J.; Armistead, J.S.; Caleon, R.L.; Kaneko, M.; Kite, W.A.; Mershon, J.P.; Brockhurst, J.K.; Engels, T.; et al. Activity of Plasmodium Vivax Promoter Elements in Plasmodium Knowlesi, and a Centromere-Containing Plasmid That Expresses NanoLuc throughout the Parasite Life Cycle. Malar. J. 2021, 20, 247. [Google Scholar] [CrossRef] [PubMed]
- ter Terkuile, F.; White, N.J.; Holloway, P.; Pasvol, G.; Krishna, S. Plasmodium falciparum: In vitro studies of the pharmacodynamic properties of drugs used for the treatment of severe malaria. Exp. Parasitol. 1993, 76, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Fivelman, Q.L.; Adagu, I.S.; Warhurst, D.C. Modified Fixed-Ratio Isobologram Method for Studying in Vitro Interactions between Atovaquone and Proguanil or Dihydroartemisinin against Drug-Resistant Strains of Plasmodium Falciparum. Antimicrob. Agents Chemother. 2004, 48, 4097–4102. [Google Scholar] [CrossRef]
- Williams, D.B.G.; Lawton, M. Drying of organic solvents: Quantitative evaluation of the efficiency of several desiccants. J. Org. Chem. 2010, 75, 8351–8354. [Google Scholar] [CrossRef]
- Jiao, Z.W.; Tu, Y.Q.; Zhang, Q.; Liu, W.X.; Zhang, S.Y.; Wang, S.H.; Zhang, F.M.; Jiang, S. Tandem C-H oxidation/cyclization/rearrangement and its application to asymmetric syntheses of (-)-brussonol and (-)-przewalskine E. Nat. Commun. 2015, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Majetich, G.; Zhang, Y.; Tian, X.; Britton, J.E.; Li, Y.; Phillips, R. Synthesis of (±)- and (+)-perovskone. Tetrahedron 2011, 67, 10129–10146. [Google Scholar] [CrossRef]
- Fernandes, R.A.; Mulay, S.V.; Chavan, V.P. A concise total synthesis of arizonins B1 and C1. Tetrahedron Asymmetry 2013, 24, 1548–1555. [Google Scholar] [CrossRef]
- Huy, N.T.; Uyen, D.T.; Maeda, A.; Trang, D.T.; Oida, T.; Harada, S.; Kamei, K. Simple colorimetric inhibition assay of heme crystallization for high-throughput screening of antimalarial compounds. Antimicrob. Agents Chemother. 2007, 51, 350–353. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
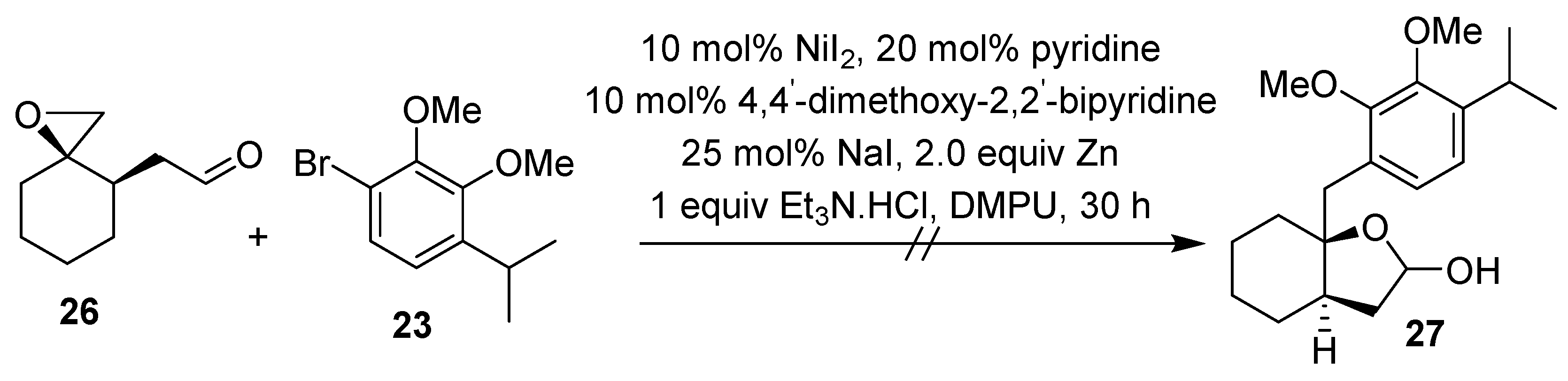{kind=link}
{kind=link}
 | |||||||||
| Code | R1 | R2 | R3 | R4 | R5 | R6 | IC50Pf (µM) | IC50HepG2 (µM) | SI 1 |
| 1 | OH | OH | iPr | H | Me | Me | 16 ± 2 | 67 ± 4 | 4 |
| 2 | OMe | OMe | H | H | H | H | >10 | ND 2 | ND 2 |
| 3 | OMe | H | H | OMe | H | H | >10 | ND 2 | ND 2 |
| 4 | OH | OH | H | H | H | H | >10 | ND 2 | ND 2 |
| 5 | OH | H | H | OH | H | H | >10 | ND 2 | ND 2 |
| 6 |  | >10 | ND 2 | ND 2 | |||||
| 7 | OMe | OMe | iPr | H | Me | Me | 5.2 ± 0.4 | >192 | >37 |
| 8 | OMe | OMe | iPr | OMe | Me | Me | 5 ± 2 | 170 ± 38 | 34 |
| 9 | OMe | OMe | H | H | Me | Me | >10 | ND 2 | ND 2 |
| 10 | OMe | OMe | iPr | Br | Me | Me | 5.6 ± 0.1 | >400 | >70 |
| 11 | OMe | OMe | iPr | Ph | Me | Me | 5.8 ± 0.2 | >12 | >2 |
| 12 | OMe | OMe | iPr | H | H | H | 5.6 ± 0.8 | >400 | >62 |
| 13 | OH | OH | iPr | H | H | H | >10 | ND 2 | ND 2 |
| 14 | OMe | OMe | iPr | Br | H | H | 5.65 ± 0.05 | >25 | >4 |
| 15 | OMe | OMe | iPr | Ph | H | H | 5.89 ± 0.02 | >50 | >8 |
| Pf Strain | IC50brussonol (µM) | RI | IC50PYR (µM) | RI | IC50ATV (µM) | RI | IC50ART (µM) | RI | IC50MMV848 (µM) | RI |
|---|---|---|---|---|---|---|---|---|---|---|
| 3D7 | 13 ± 2 | 1 | 0.06 ± 0.01 | 1 | 0.0007 ± 0.0003 | 1 | 0.008 ± 0.004 | 1 | 0.13 ± 0.02 | 1 |
| K1 | 6.6 ± 0.1 | 0.5 | >10 | >166 | NT | NT | 0.007 ± 0.003 | 1 | NT | NT |
| Dd2 | 7 ± 2 | 0.5 | >10 | >166 | NT | NT | 0.011 ± 0.008 | 1 | NT | NT |
| TM90C6B | 7 ± 2 | 0.5 | NT | NT | 3 ± 1 | 4285 | 0.006 ± 0.002 | 1 | NT | NT |
| 3D7r_MMV848 | 12 ± 3 | 1 | NT | NT | NT | NT | 0.013 ± 0.008 | 2 | 2.4 ± 0.4 | 18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbosa, C.S.; Ahmad, A.; Maluf, S.E.C.; Moura, I.M.R.; Souza, G.E.; Guerra, G.A.H.; Barros, R.R.M.; Gazarini, M.L.; Aguiar, A.C.C.; Burtoloso, A.C.B.; et al. Synthesis, Structure–Activity Relationships, and Parasitological Profiling of Brussonol Derivatives as New Plasmodium falciparum Inhibitors. Pharmaceuticals 2022, 15, 814. https://doi.org/10.3390/ph15070814
Barbosa CS, Ahmad A, Maluf SEC, Moura IMR, Souza GE, Guerra GAH, Barros RRM, Gazarini ML, Aguiar ACC, Burtoloso ACB, et al. Synthesis, Structure–Activity Relationships, and Parasitological Profiling of Brussonol Derivatives as New Plasmodium falciparum Inhibitors. Pharmaceuticals. 2022; 15(7):814. https://doi.org/10.3390/ph15070814
Chicago/Turabian StyleBarbosa, Camila S., Anees Ahmad, Sarah El Chamy Maluf, Igor M. R. Moura, Guilherme E. Souza, Giovanna A. H. Guerra, Roberto R. Moraes Barros, Marcos L. Gazarini, Anna C. C. Aguiar, Antonio C. B. Burtoloso, and et al. 2022. "Synthesis, Structure–Activity Relationships, and Parasitological Profiling of Brussonol Derivatives as New Plasmodium falciparum Inhibitors" Pharmaceuticals 15, no. 7: 814. https://doi.org/10.3390/ph15070814
APA StyleBarbosa, C. S., Ahmad, A., Maluf, S. E. C., Moura, I. M. R., Souza, G. E., Guerra, G. A. H., Barros, R. R. M., Gazarini, M. L., Aguiar, A. C. C., Burtoloso, A. C. B., & Guido, R. V. C. (2022). Synthesis, Structure–Activity Relationships, and Parasitological Profiling of Brussonol Derivatives as New Plasmodium falciparum Inhibitors. Pharmaceuticals, 15(7), 814. https://doi.org/10.3390/ph15070814