
Sirtuins and Hypoxia in EMT Control

 ,
,  , ,
, ,  , and
, and
Abstract
:1. EMT
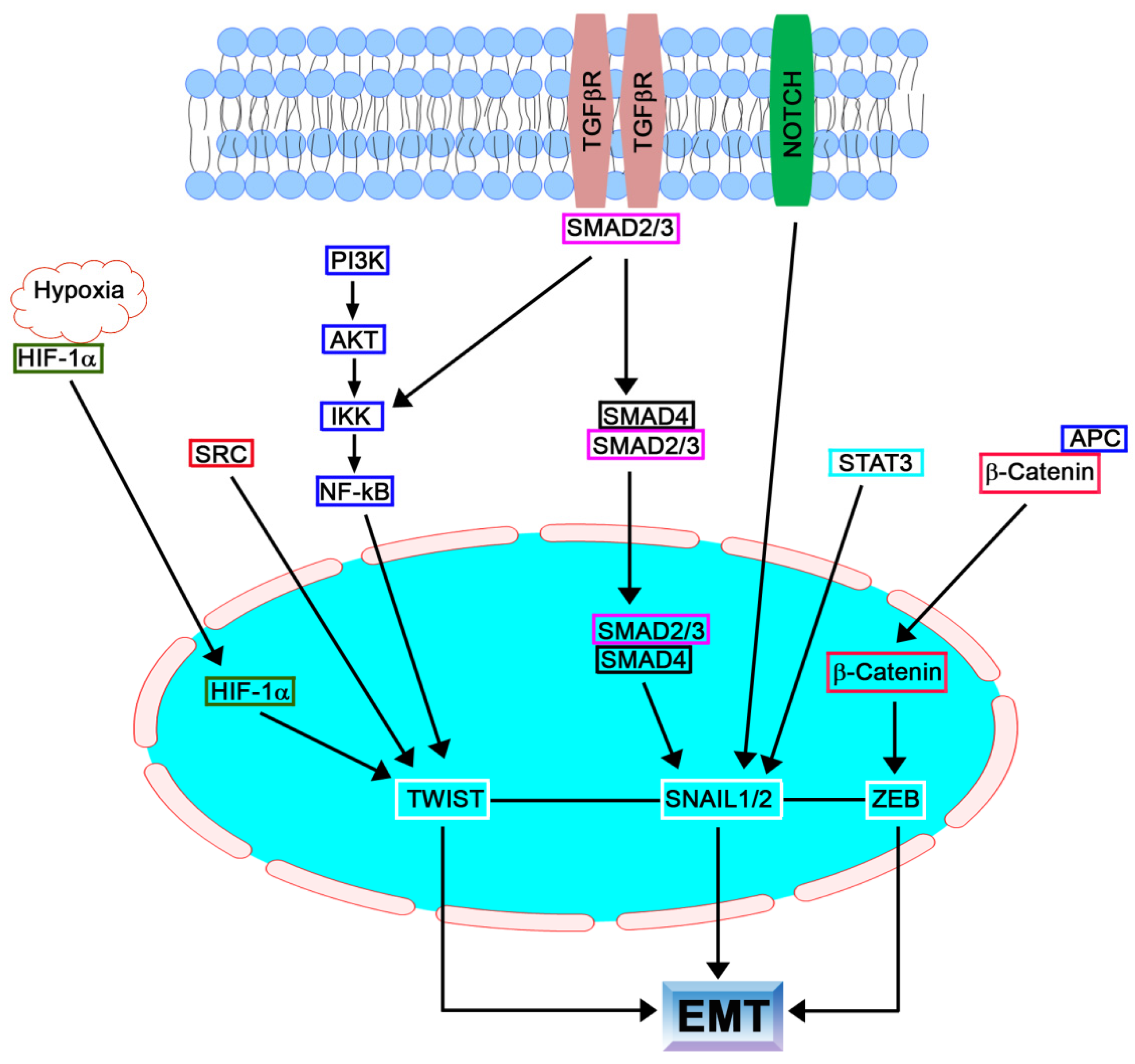
1.1. Introduction
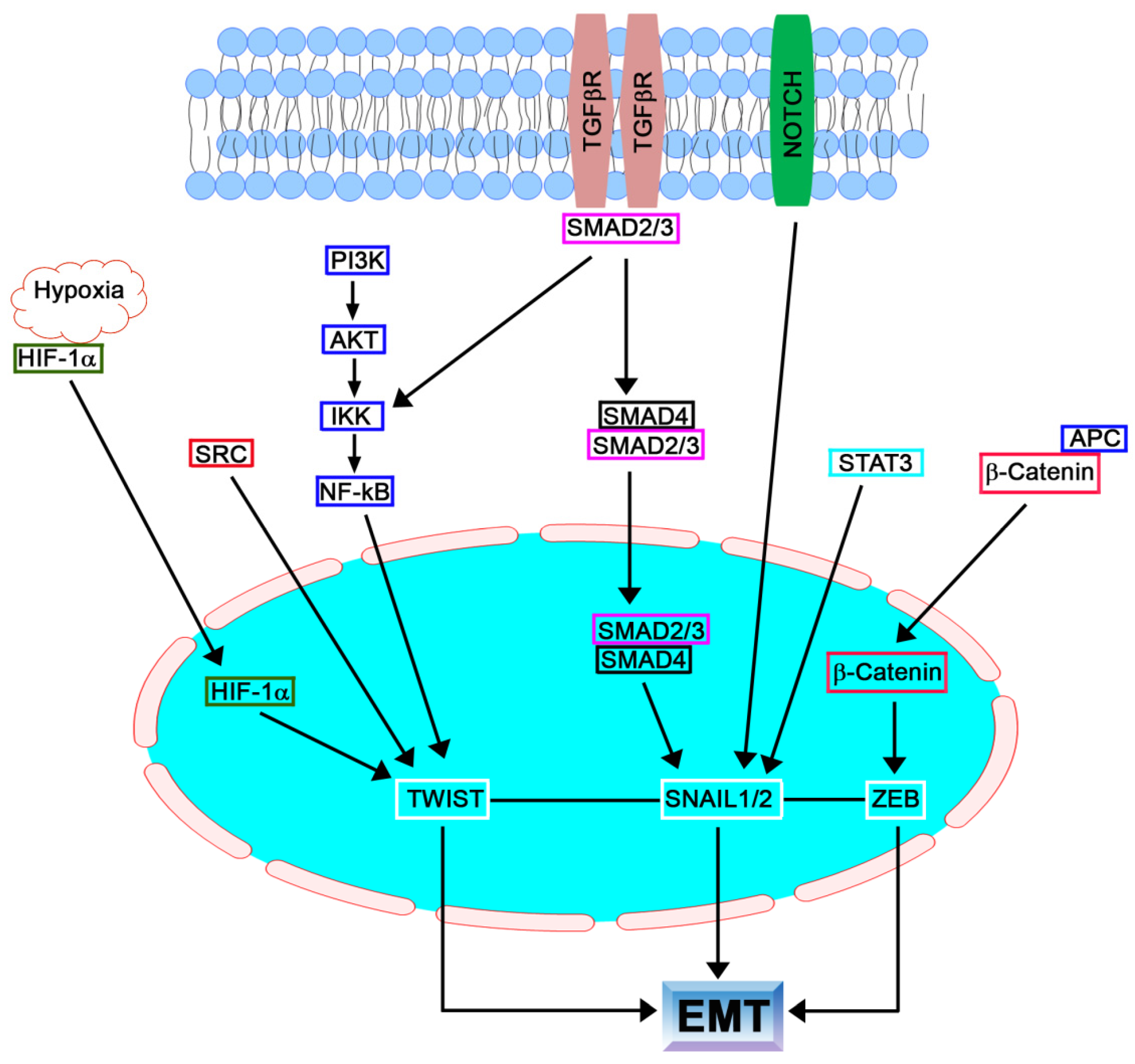
1.2. SNAIL Family
1.3. TGFβ Family
1.4. TWIST Family
1.5. ZEB Family
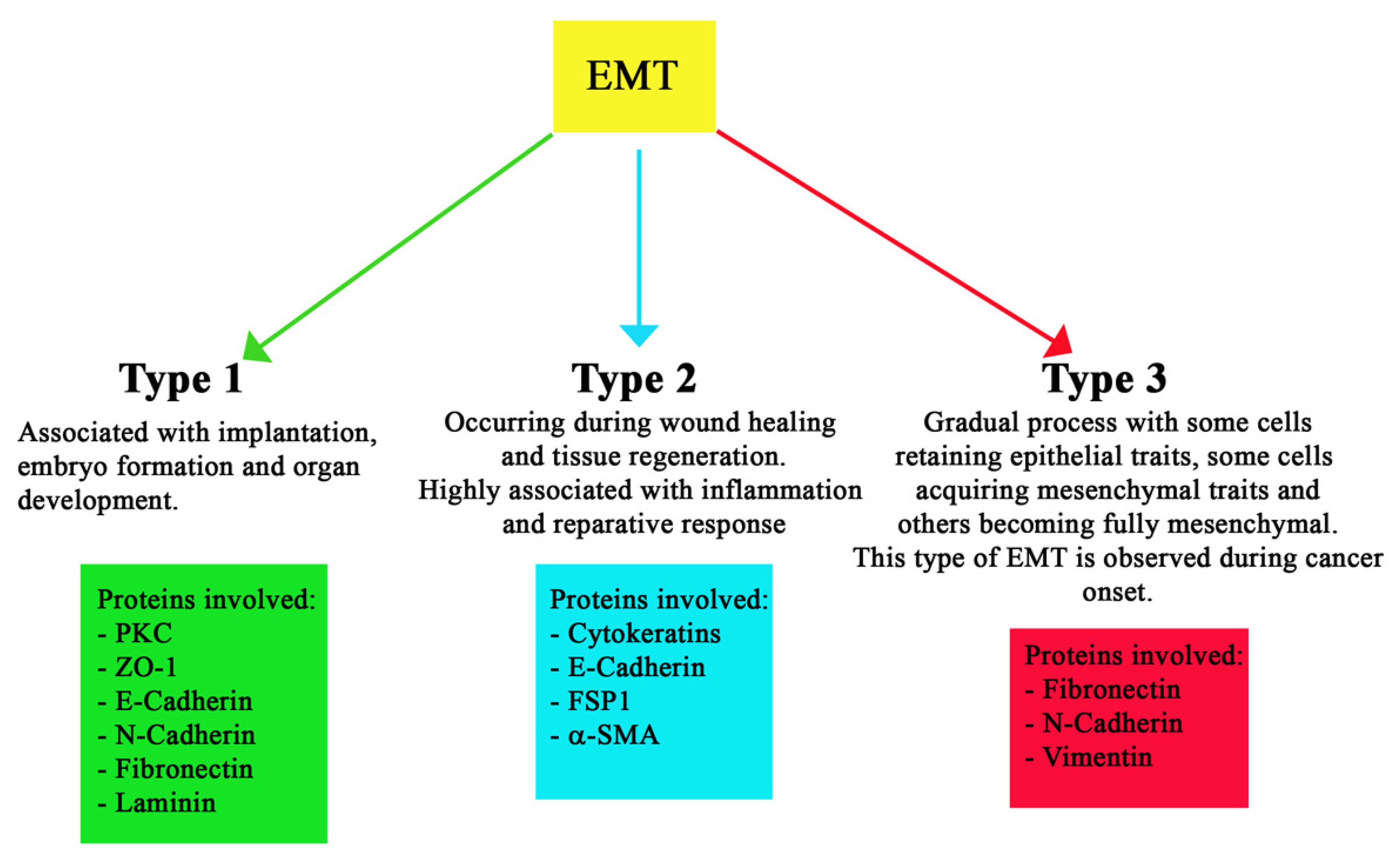
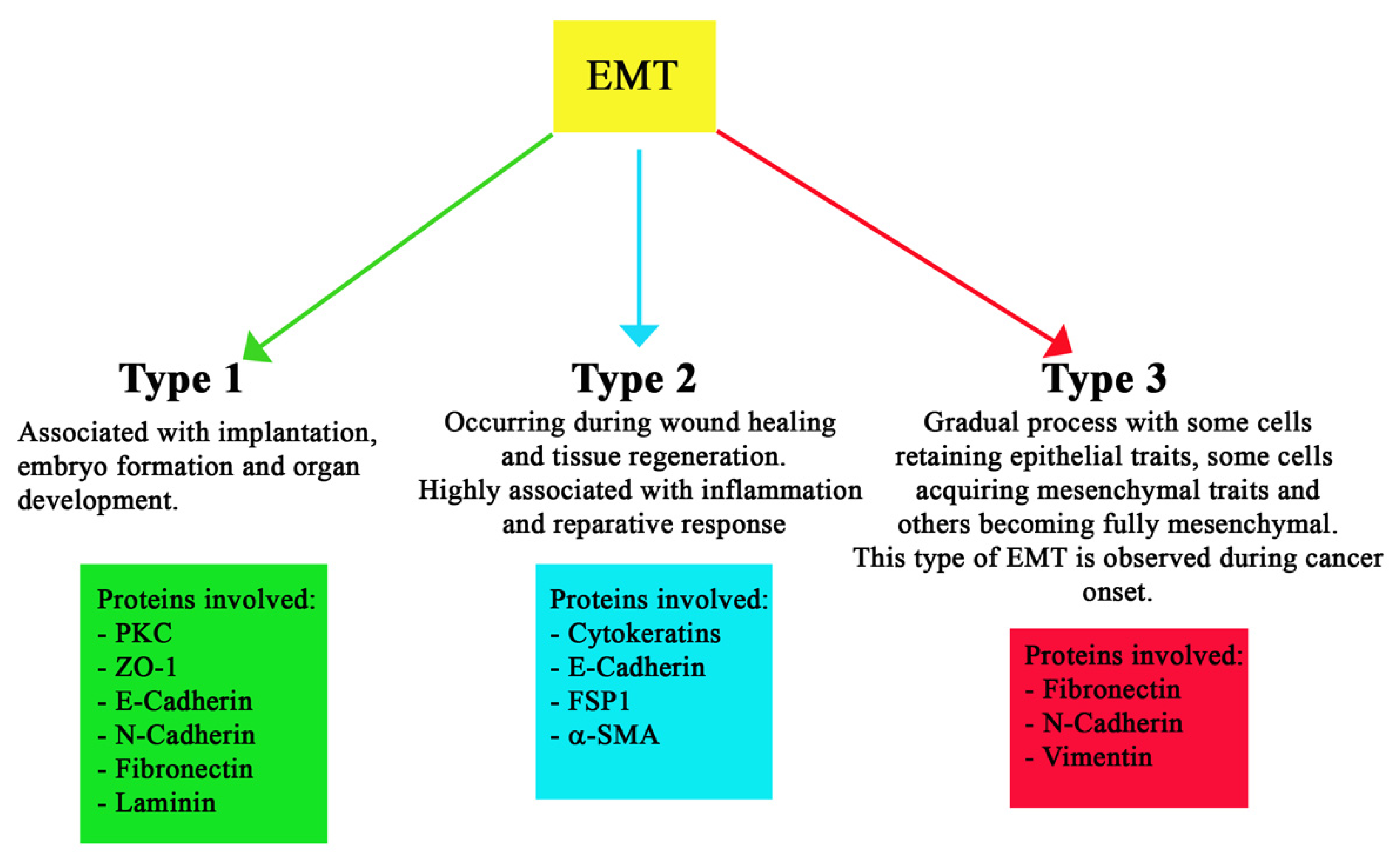
1.6. Different Types of EMT
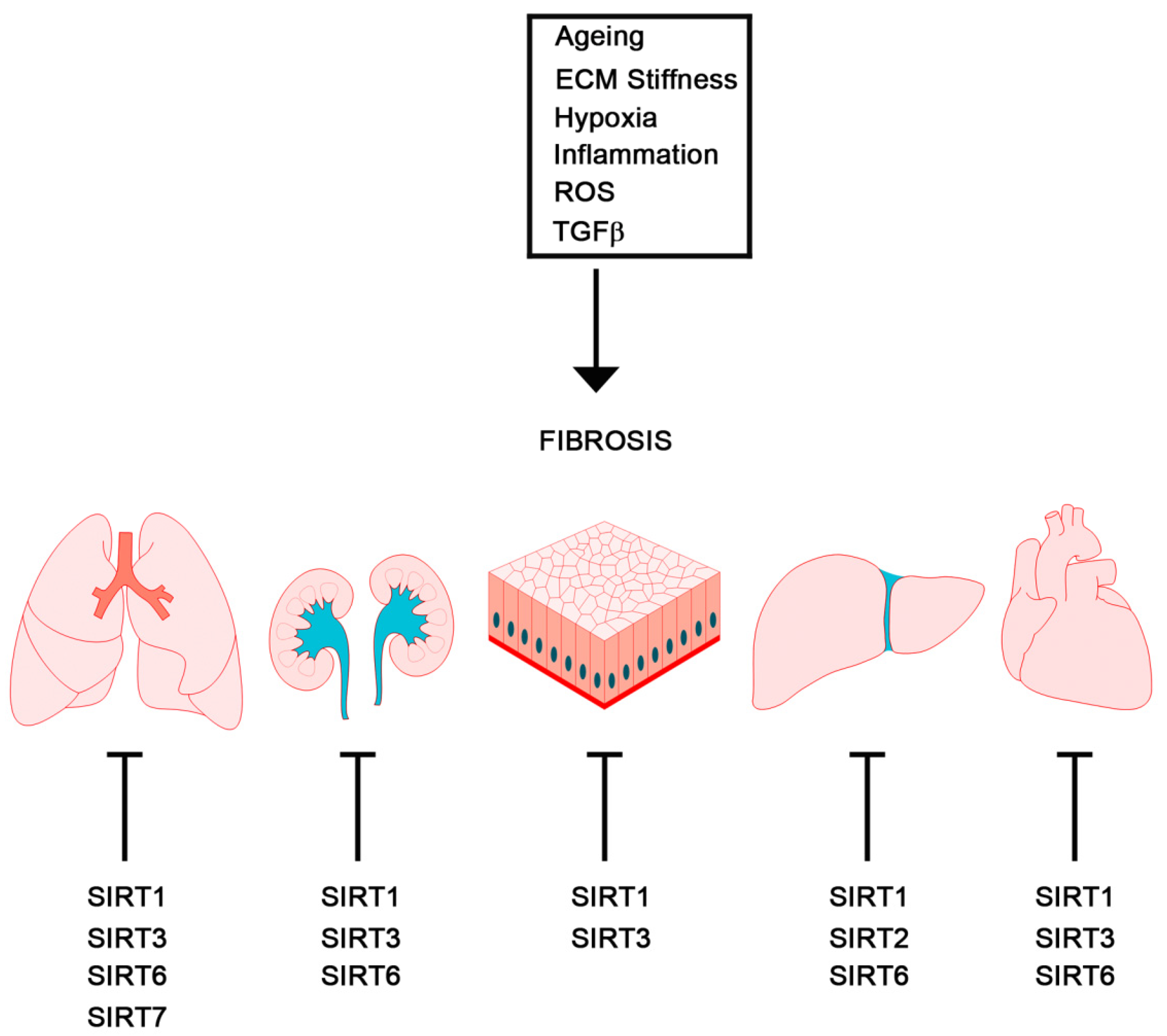
2. EMT in Fibrosis and Cancer
2.1. Fibrosis
2.2. Cancer
2.2.1. EMT Regulators and Cancer Stem Cells
2.2.2. EMT Regulators and Tumor Metastasis
2.2.3. EMT-Associated Transcription Factors and miRNA
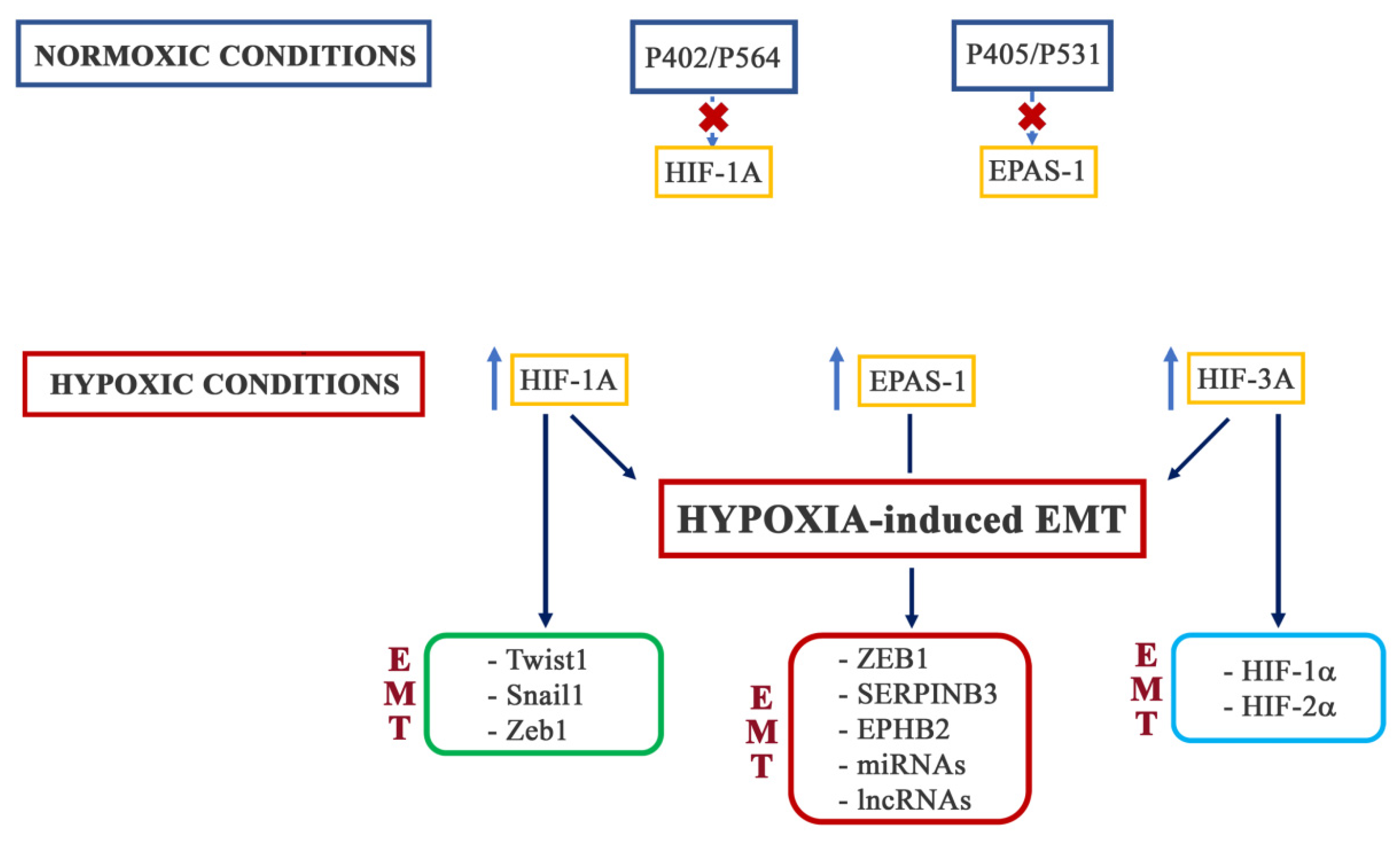
3. Hypoxia-Induced EMT
3.1. HIF-1
3.2. HIF-2
3.3. HIF-3
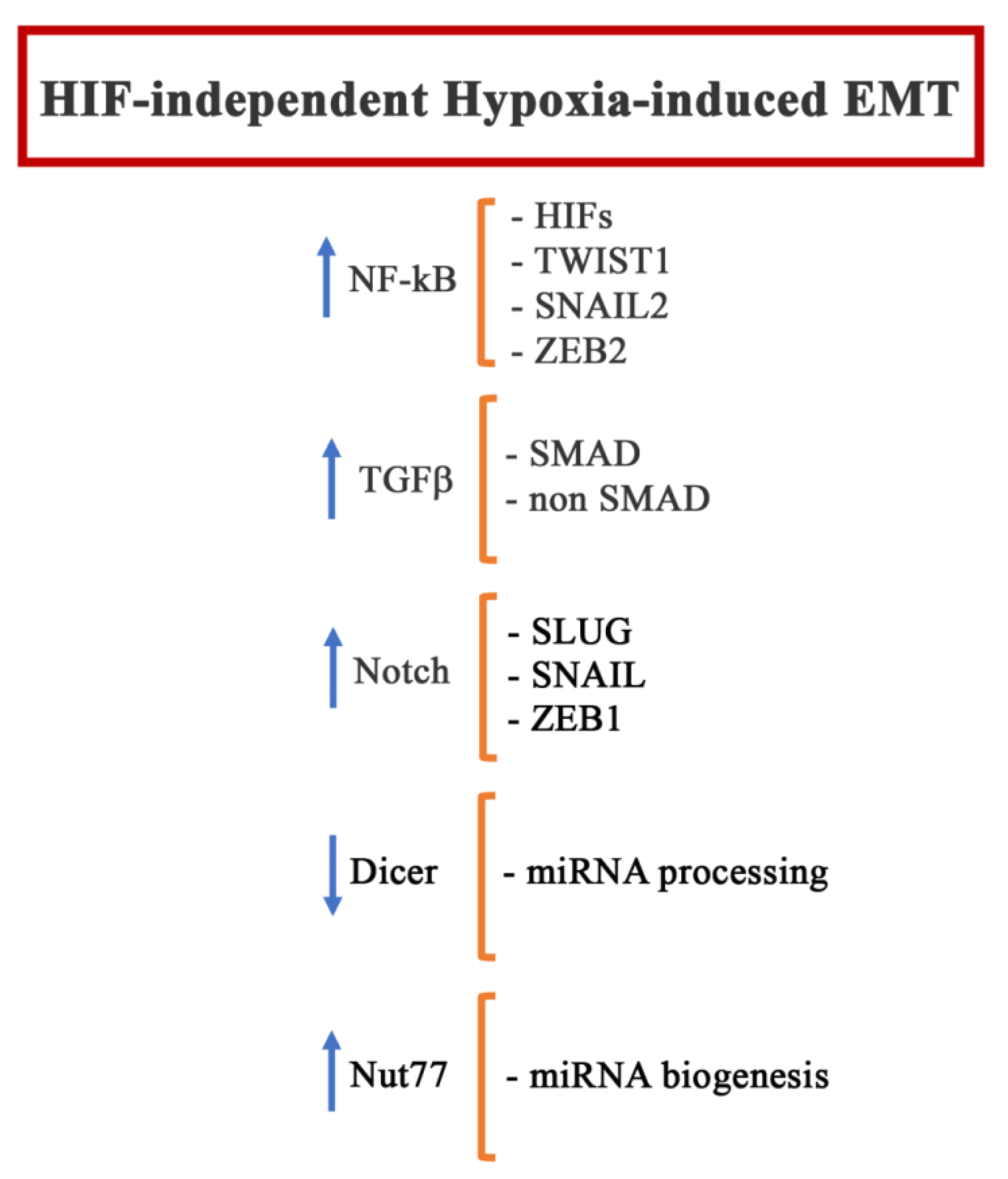
4. HIF-Independent Hypoxia-Induced EMT
4.1. NF-kB
4.2. TGFβ, MAPKs and mTOR
4.3. Notch, AMPK
4.4. Microenvironment
4.5. microRNA
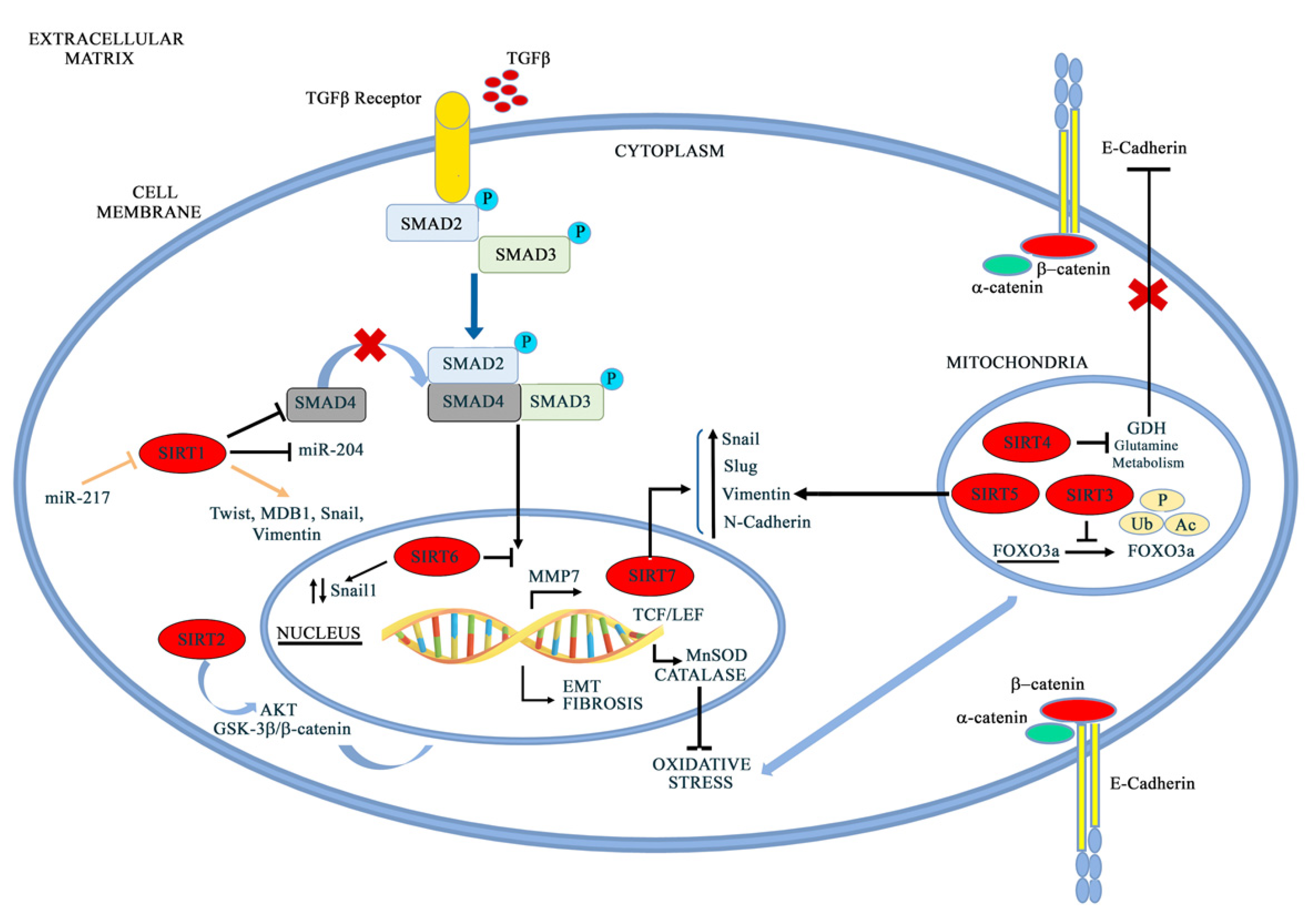
5. Sirtuins in EMT
5.1. SIRT1
5.2. SIRT2
5.3. SIRT3
5.4. SIRT4 and SIRT5
5.5. SIRT6
5.6. SIRT7
6. Sirtuins in Hypoxia
6.1. SIRT1
6.2. SIRT2
6.3. SIRT3
6.4. SIRT6
6.5. SIRT7
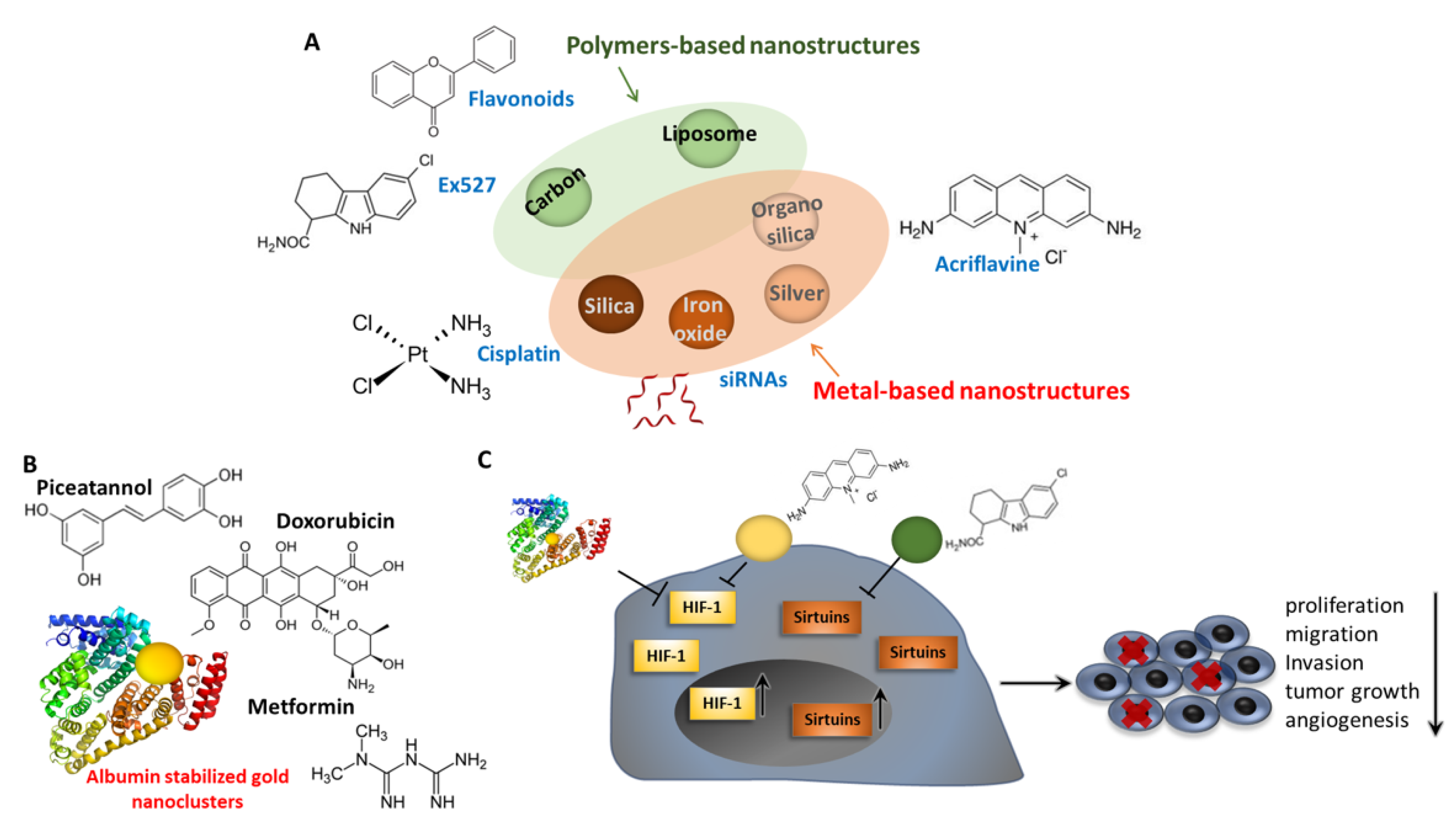
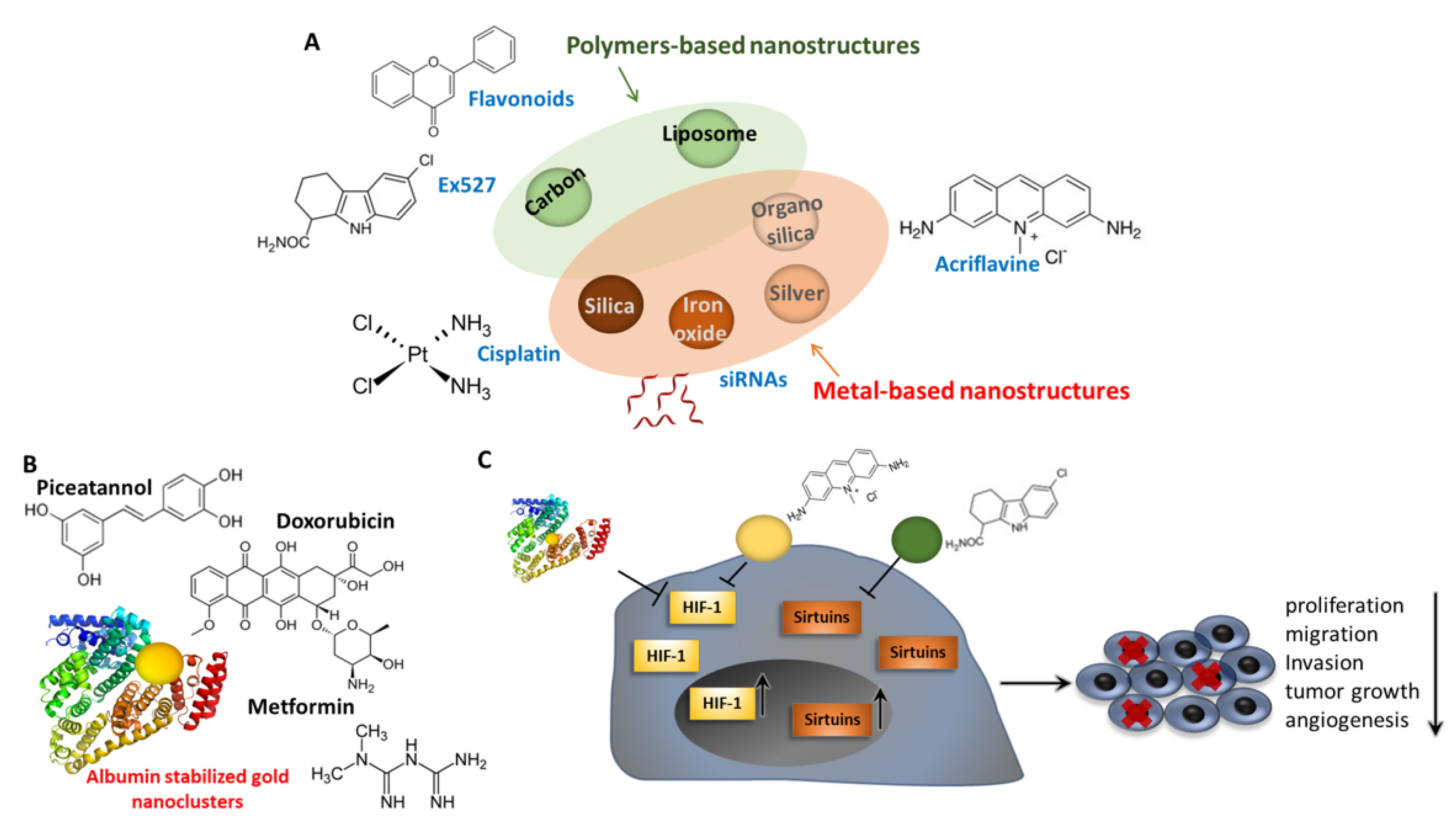
7. Pharmacological Control of EMT through Sirtuins and HIF Modulation: Nanomaterials and Nanomedicine
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thiery, J.P. Epithelial–mesenchymal transitions in development and pathologies. Curr. Opin. Cell Biol. 2003, 15, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Thiery, J.P. Epithelial-mesenchymal transitions: Insights from development. Development 2012, 139, 3471–3486. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. Epithelial Plasticity: A Common Theme in Embryonic and Cancer Cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695.e4. [Google Scholar] [CrossRef]
- Moreno-Bueno, G.; Portillo, F.; Cano, A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene 2008, 27, 6958–6969. [Google Scholar] [CrossRef]
- Ungefroren, H.; Wellner, U.F.; Keck, T.; Lehnert, H.; Marquardt, J.-U. The Small GTPase RAC1B: A Potent Negative Regulator of-and Useful Tool to Study-TGFβ Signaling. Cancers 2020, 12, 3475. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Loh, C.-Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef]
- Georgakopoulos-Soares, I.; Chartoumpekis, D.V.; Kyriazopoulou, V.; Zaravinos, A. EMT Factors and Metabolic Pathways in Cancer. Front. Oncol. 2020, 10, 499. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, B.P. Snail: More than EMT. Cell Adhes. Migr. 2010, 4, 199–203. [Google Scholar] [CrossRef]
- Tang, H.; Massi, D.; Hemmings, B.A.; Mandalà, M.; Hu, Z.; Wicki, A.; Xue, G. AKT-ions with a TWIST between EMT and MET. Oncotarget 2016, 7, 62767–62777. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Sun, Y.; Ma, L. ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015, 14, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Bornes, L.; Belthier, G.; van Rheenen, J. Epithelial-to-Mesenchymal Transition in the Light of Plasticity and Hybrid E/M States. J. Clin. Med. 2021, 10, 2403. [Google Scholar] [CrossRef]
- Brabletz, S.; Schuhwerk, H.; Brabletz, T.; Stemmler, M.P. Dynamic EMT: A multi-tool for tumor progression. EMBO J. 2021, 40, e108647. [Google Scholar] [CrossRef]
- Barrallo-Gimeno, A.; Nieto, M.A. The Snail genes as inducers of cell movement and survival: Implications in development and cancer. Development 2005, 132, 3151–3161. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Lin, T.; Ponn, A.; Hu, X.; Law, B.K.; Lu, J. Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene 2010, 29, 4896–4904. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef]
- Xi, Q.; Wang, Z.; Zaromytidou, A.-I.; Zhang, X.H.F.; Chow-Tsang, L.-F.; Liu, J.X.; Kim, H.; Barlas, A.; Manova-Todorova, K.; Kaartinen, V.; et al. A Poised Chromatin Platform for TGF-β Access to Master Regulators. Cell 2011, 147, 1511–1524. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Massagué, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [PubMed]
- Yuki, R. Aberrant Activation Mechanism of TGF-β Signaling in Epithelial-mesenchymal Transition. Yakugaku Zasshi J. Pharm. Soc. Jpn. 2021, 141, 1229–1234. [Google Scholar] [CrossRef]
- Gal, A.; Sjöblom, T.; Fedorova, L.; Imreh, S.; Beug, H.; Moustakas, A. Sustained TGFβ exposure suppresses Smad and non-Smad signalling in mammary epithelial cells, leading to EMT and inhibition of growth arrest and apoptosis. Oncogene 2008, 27, 1218–1230. [Google Scholar] [CrossRef]
- Geng, X.-Q.; Ma, A.; He, J.-Z.; Wang, L.; Jia, Y.-L.; Shao, G.-Y.; Li, M.; Zhou, H.; Lin, S.-Q.; Ran, J.-H.; et al. Ganoderic acid hinders renal fibrosis via suppressing the TGF-β/Smad and MAPK signaling pathways. Acta Pharmacol. Sin. 2020, 41, 670–677. [Google Scholar] [CrossRef]
- Mercado-Pimentel, M.E.; Runyan, R.B. Multiple Transforming Growth Factor-β Isoforms and Receptors Function during Epithelial-Mesenchymal Cell Transformation in the Embryonic Heart. Cells Tissues Organs 2007, 185, 146–156. [Google Scholar] [CrossRef]
- Nawshad, A.; LaGamba, D.; Hay, E. Transforming growth factor β (TGFβ) signalling in palatal growth, apoptosis and epithelial mesenchymal transformation (EMT). Arch. Oral Biol. 2004, 49, 675–689. [Google Scholar] [CrossRef]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.-C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. [Google Scholar] [CrossRef]
- Zhao, Z.; Rahman, M.A.; Chen, Z.G.; Shin, D.M. Multiple biological functions of Twist1 in various cancers. Oncotarget 2017, 8, 20380–20393. [Google Scholar] [CrossRef]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a Master Regulator of Morphogenesis, Plays an Essential Role in Tumor Metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Tillo, E.; Lazaro, A.; Torrent, R.; Cuatrecasas, M.; Vaquero, E.C.; Castells, A.; Engel, P.; Postigo, A. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 2010, 29, 3490–3500. [Google Scholar] [CrossRef] [PubMed]
- Postigo, A.A.; Depp, J.L.; Taylor, J.J.; Kroll, K.L. Regulation of Smad signaling through a differential recruitment of coactivators and corepressors by ZEB proteins. EMBO J. 2003, 22, 2453–2462. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Scully, K.; Zhu, X.; Cai, L.; Zhang, J.; Prefontaine, G.G.; Krones, A.; Ohgi, K.A.; Zhu, P.; Garcia-Bassets, I.; et al. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature 2007, 446, 882–887. [Google Scholar] [CrossRef]
- Dave, N.; Guaita-Esteruelas, S.; Gutarra, S.; Frias, A.; Beltran, M.; Peiró, S.; de Herreros, A.G. Functional Cooperation between Snail1 and Twist in the Regulation of ZEB1 Expression during Epithelial to Mesenchymal Transition. J. Biol. Chem. 2011, 286, 12024–12032. [Google Scholar] [CrossRef]
- Shirakihara, T.; Saitoh, M.; Miyazono, K. Differential Regulation of Epithelial and Mesenchymal Markers by δEF1 Proteins in Epithelial–Mesenchymal Transition Induced by TGF-β. Mol. Biol. Cell 2007, 18, 3533–3544. [Google Scholar] [CrossRef]
- Long, J.; Zuo, D.; Park, M. Pc2-mediated Sumoylation of Smad-interacting Protein 1 Attenuates Transcriptional Repression of E-cadherin. J. Biol. Chem. 2005, 280, 35477–35489. [Google Scholar] [CrossRef]
- Vićovac, L.; Aplin, J.D. Epithelial-mesenchymal transition during trophoblast differentiation. Cells Tissues Organs 1996, 156, 202–216. [Google Scholar] [CrossRef]
- Stern, C.D. Vertebrate gastrulation. Curr. Opin. Genet. Dev. 1992, 2, 556–561. [Google Scholar] [CrossRef]
- Nakaya, Y.; Sukowati, E.W.; Wu, Y.; Sheng, G. RhoA and microtubule dynamics control cell–basement membrane interaction in EMT during gastrulation. Nat. Cell Biol. 2008, 10, 765–775. [Google Scholar] [CrossRef]
- Flanders, K.C. Smad3 as a mediator of the fibrotic response. Int. J. Exp. Pathol. 2004, 85, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Marconi, G.D.; Fonticoli, L.; Rajan, T.S.; Pierdomenico, S.D.; Trubiani, O.; Pizzicannella, J.; Diomede, F. Epithelial-Mesenchymal Transition (EMT): The Type-2 EMT in Wound Healing, Tissue Regeneration and Organ Fibrosis. Cells 2021, 10, 1587. [Google Scholar] [CrossRef] [PubMed]
- Turley, E.A.; Veiseh, M.; Radisky, D.C.; Bissell, M.J. Mechanisms of Disease: Epithelial–mesenchymal transition—Does cellular plasticity fuel neoplastic progression? Nat. Clin. Pract. Oncol. 2008, 5, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Plieth, D.; Venkov, C.; Xu, C.; Neilson, E.G. The gatekeeper effect of epithelial-mesenchymal transition regulates the frequency of breast cancer metastasis. Cancer Res. 2003, 63, 3386–3394. [Google Scholar]
- Iwatsuki, M.; Mimori, K.; Yokobori, T.; Ishi, H.; Beppu, T.; Nakamori, S.; Baba, H.; Mori, M. Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer Sci. 2010, 101, 293–299. [Google Scholar] [CrossRef]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef]
- Santoro, A.; Bufo, P.; Russo, G.; Cagiano, S.; Papagerakis, S.; Bucci, P.; Aquino, G.; Longo, F.; Feola, A.; Giordano, A.; et al. Expression and clinical implication of cyclooxygenase-2 and E-cadherin in oral squamous cell carcinomas. Cancer Biol. Ther. 2020, 21, 667–674. [Google Scholar] [CrossRef]
- Pedrosa, A.-R.; Trindade, A.; Carvalho, C.; Graça, J.; Carvalho, S.; Peleteiro, M.C.; Adams, R.H.; Duarte, A. Endothelial Jagged1 promotes solid tumor growth through both pro-angiogenic and angiocrine functions. Oncotarget 2015, 6, 24404–24423. [Google Scholar] [CrossRef]
- Li, M.; Luan, F.; Zhao, Y.; Hao, H.; Zhou, Y.; Han, W.; Fu, X. Epithelial-mesenchymal transition: An emerging target in tissue fibrosis. Exp. Biol. Med. 2016, 241, 1–13. [Google Scholar] [CrossRef]
- Tai, Y.; Woods, E.L.; Dally, J.; Kong, D.; Steadman, R.; Moseley, R.; Midgley, A.C. Myofibroblasts: Function, Formation, and Scope of Moleculare Therapies for Skin Fibrosis. Biomolecules 2021, 11, 1095. [Google Scholar] [CrossRef]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Debnath, P.; Huirem, R.S.; Dutta, P.; Palchaudhuri, S. Epithelial–mesenchymal transition and its transcription factors. Biosci. Rep. 2021, 42, BSR2021117. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Novoa, J.M.; Nieto, M.A. Inflammation and EMT: An alliance towards organ fibrosis and cancer progression. EMBO Mol. Med. 2009, 1, 303–314. [Google Scholar] [CrossRef]
- Farris, A.B.; Colvin, R.B. Renal interstitial fibrosis. Curr. Opin. Nephrol. Hypertens. 2012, 21, 289–300. [Google Scholar] [CrossRef] [PubMed]
- MacKinnon, A.C.; Gibbons, M.A.; Farnworth, S.L.; Leffler, H.; Nilsson, U.J.; Delaine, T.; Simpson, A.J.; Forbes, S.J.; Hirani, N.; Gauldie, J.; et al. Regulation of Transforming Growth Factor-β1–driven Lung Fibrosis by Galectin-3. Am. J. Respir. Crit. Care Med. 2012, 185, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef]
- Gabbiani, G. The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 2003, 200, 500–503. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Ketteler, M.; Border, W.A.; Noble, N.A. Cytokines and L-arginine in renal injury and repair. Am. J. Physiol.-Ren. Physiol. 1994, 267, F197–F207. [Google Scholar] [CrossRef]
- Romero, Y.; Aquino-Gálvez, A. Hypoxia in Cancer and Fibrosis: Part of the Problem and Part of the Solution. Int. J. Mol. Sci. 2021, 22, 8335. [Google Scholar] [CrossRef]
- Sharma, V.; Letson, J.; Furuta, S. Fibrous stroma: Driver and passenger in cancer development. Sci. Signal. 2022, 15, eabg3449. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Bonner, G.; Maeshima, Y.; Colorado, P.; Müller, G.A.; Strutz, F.; Kalluri, R. Renal Fibrosis. Am. J. Pathol. 2001, 159, 1313–1321. [Google Scholar] [CrossRef]
- Zeisberg, M.; Hanai, J.-I.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 counteracts TGF-β1–induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003, 9, 964–968. [Google Scholar] [CrossRef]
- Boutet, A.; De Frutos, C.A.; Maxwell, P.H.; Mayol, M.J.; Romero, J.; Nieto, M.A. Snail activation disrupts tissue homeostasis and induces fibrosis in the adult kidney. EMBO J. 2006, 25, 5603–5613. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef]
- Lim, J.H.; Kim, E.N.; Kim, M.Y.; Chung, S.; Shin, S.J.; Kim, H.W.; Yang, C.W.; Kim, Y.S.; Chang, Y.S.; Park, C.W.; et al. Age-Associated Molecular Changes in the Kidney in Aged Mice. Oxid. Med. Cell. Longev. 2012, 2012, 171383. [Google Scholar] [CrossRef]
- Li, P.; Liu, Y.; Qin, X.; Chen, K.; Wang, R.; Yuan, L.; Chen, X.; Hao, C.; Huang, X. SIRT1 attenuates renal fibrosis by repressing HIF-2α. Cell Death Discov. 2021, 7, 59. [Google Scholar] [CrossRef]
- Zhang, Y.; Wen, P.; Luo, J.; Ding, H.; Cao, H.; He, W.; Zen, K.; Zhou, Y.; Yang, J.; Jiang, L. Sirtuin 3 regulates mitochondrial protein acetylation and metabolism in tubular epithelial cells during renal fibrosis. Cell Death Dis. 2021, 12, 847. [Google Scholar] [CrossRef]
- Yang, X.; Feng, J.; Liang, W.; Zhu, Z.; Chen, Z.; Hu, J.; Yang, D.; Ding, G. Roles of SIRT6 in kidney disease: A novel therapeutic target. Cell. Mol. Life Sci. 2021, 79, 53. [Google Scholar] [CrossRef]
- Chapman, H.A. Epithelial-Mesenchymal Interactions in Pulmonary Fibrosis. Annu. Rev. Physiol. 2011, 73, 413–435. [Google Scholar] [CrossRef] [PubMed]
- Zolak, J.S.; Jagirdar, R.; Surolia, R.; Karki, S.; Oliva, O.; Hock, T.; Guroji, P.; Ding, Q.; Liu, R.-M.; Bolisetty, S.; et al. Pleural Mesothelial Cell Differentiation and Invasion in Fibrogenic Lung Injury. Am. J. Pathol. 2013, 182, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Mo, X.; Cui, W.; Zhang, Z.; Li, D.; Li, L.-C.; Xu, L.; Yao, H.; Gao, J. Nrf2 inhibits epithelial-mesenchymal transition by suppressing snail expression during pulmonary fibrosis. Sci. Rep. 2016, 6, 38646. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, L.; Luo, L.; He, P.; Xiong, A.; Jiang, M.; Liu, Y.; Liu, S.; Ran, Q.; Wu, D.; et al. Characterizing cellular heterogeneity in fibrotic hypersensitivity pneumonitis by single-cell transcriptional analysis. Cell Death Discov. 2022, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Ding, C.; Sang, X.; Peng, M.; Yang, Q.; Ning, Y.; Lv, Q.; Shan, Q.; Hao, M.; Wang, K.; et al. Targeting Sirtuin1 to treat aging-related tissue fibrosis: From prevention to therapy. Pharmacol. Ther. 2021, 229, 107983. [Google Scholar] [CrossRef] [PubMed]
- Akamata, K.; Wei, J.; Bhattacharyya, M.; Cheresh, P.; Bonner, M.Y.; Arbiser, J.L.; Raparia, K.; Gupta, M.P.; Kamp, D.W.; Varga, J. SIRT3 is attenuated in systemic sclerosis skin and lungs, and its pharmacologic activation mitigates organ fibrosis. Oncotarget 2016, 7, 69321–69336. [Google Scholar] [CrossRef]
- Tian, K.; Chen, P.; Liu, Z.; Si, S.; Zhang, Q.; Mou, Y.; Han, L.; Wang, Q.; Zhou, X. Sirtuin 6 inhibits epithelial to mesenchymal transition during idiopathic pulmonary fibrosis via inactivating TGF-β1/Smad3 signaling. Oncotarget 2017, 8, 61011–61024. [Google Scholar] [CrossRef]
- Wyman, A.E.; Noor, Z.; Fishelevich, R.; Lockatell, V.; Shah, N.G.; Todd, N.W.; Atamas, S.P. Sirtuin 7 is decreased in pulmonary fibrosis and regulates the fibrotic phenotype of lung fibroblasts. Am. J. Physiol. Cell. Mol. Physiol. 2017, 312, L945–L958. [Google Scholar] [CrossRef]
- Limana, F.; Zacheo, A.; Mocini, D.; Mangoni, A.; Borsellino, G.; Diamantini, A.; De Mori, R.; Battistini, L.; Vigna, E.; Santini, M.; et al. Identification of Myocardial and Vascular Precursor Cells in Human and Mouse Epicardium. Circ. Res. 2007, 101, 1255–1265. [Google Scholar] [CrossRef]
- Moore, A.W.; McInnes, L.; Kreidberg, J.; Hastie, N.D.; Schedl, A. YAC complementation shows a requirement for Wt1 in the development of epicardium, adrenal gland and throughout nephrogenesis. Development 1999, 126, 1845–1857. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Gupte, M.; Umbarkar, P.; Singh, A.P.; Sui, J.Y.; Force, T.; Lal, H. Entanglement of GSK-3β, β-catenin and TGF-β1 signaling network to regulate myocardial fibrosis. J. Mol. Cell. Cardiol. 2017, 110, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Palomer, X.; Román-Azcona, M.S.; Pizarro-Delgado, J.; Planavila, A.; Villarroya, F.; Valenzuela-Alcaraz, B.; Crispi, F.; Sepúlveda-Martínez, Á.; Miguel-Escalada, I.; Ferrer, J.; et al. SIRT3-mediated inhibition of FOS through histone H3 deacetylation prevents cardiac fibrosis and inflammation. Signal Transduct. Target. Ther. 2020, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Yan, F.; Li, J.; Zhang, C.; Bu, P. SIRT3 attenuates AngII-induced cardiac fibrosis by inhibiting myofibroblasts trans-differentiation via STAT3-NFATc2 pathway. Am. J. Transl. Res. 2017, 9, 3258–3269. [Google Scholar] [PubMed]
- Pillai, V.B.; Samant, S.; Hund, S.; Gupta, M.; Gupta, M.P. The nuclear sirtuin SIRT6 protects the heart from developing aging-associated myocyte senescence and cardiac hypertrophy. Aging 2021, 13, 12334–12358. [Google Scholar] [CrossRef]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.-P.; Schwabe, R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013, 4, 2823. [Google Scholar] [CrossRef]
- Kaimori, A.; Potter, J.; Kaimori, J.-Y.; Wang, C.; Mezey, E.; Koteish, A. Transforming Growth Factor-β1 Induces an Epithelial-to-Mesenchymal Transition State in Mouse Hepatocytes in Vitro. J. Biol. Chem. 2007, 282, 22089–22101. [Google Scholar] [CrossRef]
- Park, S.; Chung, M.-J.; Son, J.-Y.; Yun, H.H.; Park, J.-M.; Yim, J.-H.; Jung, S.-J.; Lee, S.-H.; Jeong, K.-S. The role of Sirtuin 2 in sustaining functional integrity of the liver. Life Sci. 2021, 285, 119997. [Google Scholar] [CrossRef]
- Zhu, T.; Zhang, L.; Li, C.; Tan, X.; Liu, J.; Li, H.; Fan, Q.; Zhang, Z.; Zhan, M.; Fu, L.; et al. The S100 calcium binding protein A11 promotes liver fibrogenesis by targeting TGF-β signaling. J. Genet. Genom. 2022, 49, 338–349. [Google Scholar] [CrossRef]
- Gazi, H.; Pope, J.E.; Clements, P.; Medsger, T.A.; Martin, R.W.; Merkel, P.A.; Kahaleh, B.; Wollheim, F.A.; Baron, M.; Csuka, M.E.; et al. Outcome measurements in scleroderma: Results from a delphi exercise. J. Rheumatol. 2007, 34, 501–509. [Google Scholar]
- E Postlethwaite, A.; Shigemitsu, H.; Kanangat, S. Cellular origins of fibroblasts: Possible implications for organ fibrosis in systemic sclerosis. Curr. Opin. Rheumatol. 2004, 16, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Nikitorowicz-Buniak, J.; Denton, C.P.; Abraham, D.; Stratton, R. Partially Evoked Epithelial-Mesenchymal Transition (EMT) Is Associated with Increased TGFβ Signaling within Lesional Scleroderma Skin. PLoS ONE 2015, 10, e0134092. [Google Scholar] [CrossRef]
- Yan, C.; Grimm, W.A.; Garner, W.L.; Qin, L.; Travis, T.; Tan, N.; Han, Y.-P. Epithelial to Mesenchymal Transition in Human Skin Wound Healing Is Induced by Tumor Necrosis Factor-α through Bone Morphogenic Protein-2. Am. J. Pathol. 2010, 176, 2247–2258. [Google Scholar] [CrossRef] [PubMed]
- Manetti, M.; Rosa, I.; Fioretto, B.S.; Matucci-Cerinic, M.; Romano, E. Decreased Serum Levels of SIRT1 and SIRT3 Correlate with Severity of Skin and Lung Fibrosis and Peripheral Microvasculopathy in Systemic Sclerosis. J. Clin. Med. 2022, 11, 1362. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Ghosh, A.K.; Chu, H.; Fang, F.; Hinchcliff, M.E.; Wang, J.; Marangoni, R.G.; Varga, J. The Histone Deacetylase Sirtuin 1 Is Reduced in Systemic Sclerosis and Abrogates Fibrotic Responses by Targeting Transforming Growth Factor β Signaling. Arthritis Rheumatol. 2015, 67, 1323–1334. [Google Scholar] [CrossRef]
- Kakarala, M.; Wicha, M.S. Implications of the Cancer Stem-Cell Hypothesis for Breast Cancer Prevention and Therapy. J. Clin. Oncol. 2008, 26, 2813–2820. [Google Scholar] [CrossRef]
- Marotta, L.L.C.; Polyak, K. Cancer stem cells: A model in the making. Curr. Opin. Genet. Dev. 2009, 19, 44–50. [Google Scholar] [CrossRef]
- Cho, M.H.; Park, J.-H.; Choi, H.-J.; Park, M.-K.; Won, H.-Y.; Park, Y.-J.; Lee, C.H.; Oh, S.-H.; Song, Y.-S.; Kim, H.S.; et al. DOT1L cooperates with the c-Myc-p300 complex to epigenetically derepress CDH1 transcription factors in breast cancer progression. Nat. Commun. 2015, 6, 7821. [Google Scholar] [CrossRef]
- Morel, A.-P.; Hinkal, G.W.; Thomas, C.; Fauvet, F.; Courtois-Cox, S.; Wierinckx, A.; Devouassoux-Shisheboran, M.; Treilleux, I.; Tissier, A.; Gras, B.; et al. EMT Inducers Catalyze Malignant Transformation of Mammary Epithelial Cells and Drive Tumorigenesis towards Claudin-Low Tumors in Transgenic Mice. PLoS Genet. 2012, 8, e1002723. [Google Scholar] [CrossRef]
- Ansieau, S.; Bastid, J.; Doreau, A.; Morel, A.-P.; Bouchet, B.P.; Thomas, C.; Fauvet, F.; Puisieux, I.; Doglioni, C.; Piccinin, S.; et al. Induction of EMT by Twist Proteins as a Collateral Effect of Tumor-Promoting Inactivation of Premature Senescence. Cancer Cell 2008, 14, 79–89. [Google Scholar] [CrossRef]
- Ohashi, S.; Natsuizaka, M.; Wong, G.S.; Michaylira, C.Z.; Grugan, K.D.; Stairs, D.B.; Kalabis, J.; Vega, M.E.; Kalman, R.A.; Nakagawa, M.; et al. Epidermal Growth Factor Receptor and Mutant p53 Expand an Esophageal Cellular Subpopulation Capable of Epithelial-to-Mesenchymal Transition through ZEB Transcription Factors. Cancer Res. 2010, 70, 4174–4184. [Google Scholar] [CrossRef] [PubMed]
- DiMeo, T.A.; Anderson, K.; Phadke, P.; Feng, C.; Perou, C.; Naber, S.; Kuperwasser, C. A Novel Lung Metastasis Signature Links Wnt Signaling with Cancer Cell Self-Renewal and Epithelial-Mesenchymal Transition in Basal-like Breast Cancer. Cancer Res. 2009, 69, 5364–5373. [Google Scholar] [CrossRef] [PubMed]
- Tanno, B.; Sesti, F.; Cesi, V.; Bossi, G.; Ferrari-Amorotti, G.; Bussolari, R.; Tirindelli, D.; Calabretta, B.; Raschellà, G. Expression of Slug Is Regulated by c-Myb and Is Required for Invasion and Bone Marrow Homing of Cancer Cells of Different Origin. J. Biol. Chem. 2010, 285, 29434–29445. [Google Scholar] [CrossRef] [PubMed]
- Buyuk, B.; Jin, S.; Ye, K. Epithelial-to-Mesenchymal Transition Signaling Pathways Responsible for Breast Cancer Metastasis. Cell. Mol. Bioeng. 2021, 15, 1–13. [Google Scholar] [CrossRef]
- Satelli, A.; Li, S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell. Mol. Life Sci. 2011, 68, 3033–3046. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Kong, D.; Banerjee, S.; Ahmad, A.; Azmi, A.; Ali, S.; Abbruzzese, J.L.; Gallick, G.E.; Sarkar, F.H. Acquisition of Epithelial-Mesenchymal Transition Phenotype of Gemcitabine-Resistant Pancreatic Cancer Cells Is Linked with Activation of the Notch Signaling Pathway. Cancer Res. 2009, 69, 2400–2407. [Google Scholar] [CrossRef]
- Yang, W.-H.; Lan, H.-Y.; Huang, C.-H.; Tai, S.-K.; Tzeng, C.-H.; Kao, S.-Y.; Wu, K.-J.; Hung, M.-C.; Yang, M.-H. RAC1 activation mediates Twist1-induced cancer cell migration. Nat. Cell Biol. 2012, 14, 366–374. [Google Scholar] [CrossRef]
- Siemens, H.; Jackstadt, R.; Hünten, S.; Kaller, M.; Menssen, A.; Götz, U.; Hermeking, H. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle 2011, 10, 4256–4271. [Google Scholar] [CrossRef]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef]
- Hong, T.; Watanabe, K.; Ta, C.H.; Villarreal-Ponce, A.; Nie, Q.; Dai, X. An Ovol2-Zeb1 Mutual Inhibitory Circuit Governs Bidirectional and Multi-step Transition between Epithelial and Mesenchymal States. PLoS Comput. Biol. 2015, 11, e1004569. [Google Scholar] [CrossRef]
- Semenza, G.L. Heritable disorders of oxygen sensing. Am. J. Med. Genet. Part A 2021, 185, 3334–3339. [Google Scholar] [CrossRef] [PubMed]
- Tirpe, A.A.; Gulei, D.; Ciortea, S.M.; Crivii, C.; Berindan-Neagoe, I. Hypoxia: Overview on Hypoxia-Mediated Mechanisms with a Focus on the Role of HIF Genes. Int. J. Mol. Sci. 2019, 20, 6140. [Google Scholar] [CrossRef] [PubMed]
- Kewley, R.J.; Whitelaw, M.L.; Chapman-Smith, A. The mammalian basic helix–loop–helix/PAS family of transcriptional regulators. Int. J. Biochem. Cell Biol. 2004, 36, 189–204. [Google Scholar] [CrossRef]
- Semenza, G.L.; Agani, F.; Booth, G.; Forsythe, J.; Iyer, N.; Jiang, B.-H.; Leung, S.; Roe, R.; Wiener, C.; Yu, A. Structural and functional analysis of hypoxia-inducible factor 1. Kidney Int. 1997, 51, 553–555. [Google Scholar] [CrossRef]
- Iyer, N.V.; Leung, S.W.; Semenza, G.L. The Human Hypoxia-Inducible Factor 1α Gene:HIF1AStructure and Evolutionary Conservation. Genomics 1998, 52, 159–165. [Google Scholar] [CrossRef] [PubMed]
- van Patot, M.C.T.; Gassmann, M. Hypoxia: Adapting to High Altitude by Mutating EPAS-1, the Gene Encoding HIF-2α. High Alt. Med. Biol. 2011, 12, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.Z.; Moran, S.M.; Hogenesch, J.B.; Wartman, L.; Bradfield, C.A. Molecular Characterization and Chromosomal Locali-zation of a Third Alpha-Class Hypoxia Inducible Factor Subunit, HIF-3alpha. Gene Expr. 1998, 7, 205–213. [Google Scholar]
- Jaakkola, P.; Mole, D.R.; Tian, Y.-M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau Ubiquitylation Complex by O2-Regulated Prolyl Hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Ohh, M.; Park, C.W.; Ivan, M.; Hoffman, M.A.; Kim, T.-Y.; Huang, E.; Pavletich, N.; Chau, V.; Kaelin, W.G. Ubiquitination of hypoxia-inducible factor requires direct binding to the β-domain of the von Hippel–Lindau protein. Nat. Cell Biol. 2000, 2, 423–427. [Google Scholar] [CrossRef]
- Corrado, C.; Fontana, S. Hypoxia and HIF Signaling: One Axis with Divergent Effects. Int. J. Mol. Sci. 2020, 21, 5611. [Google Scholar] [CrossRef]
- Tam, S.Y.; Wu, V.W.C.; Law, H.K.W. Hypoxia-Induced Epithelial-Mesenchymal Transition in Cancers: HIF-1α and Beyond. Front. Oncol. 2020, 10, 486. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Ning, X.; Zhang, Y.; Lu, Y.; Nie, Y.; Han, S.; Liu, L.; Du, R.; Xia, L.; He, L.; et al. Hypoxia-inducible factor-1α induces Twist expression in tubular epithelial cells subjected to hypoxia, leading to epithelial-to-mesenchymal transition. Kidney Int. 2009, 75, 1278–1287. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.-J.; Park, S.-A.; Lee, S.-Y.; Cha, Y.N.; Surh, Y.-J. Hypoxia induces epithelial-mesenchymal transition in colorectal cancer cells through ubiquitin-specific protease 47-mediated stabilization of Snail: A potential role of Sox9. Sci. Rep. 2017, 7, 15918. [Google Scholar] [CrossRef]
- Wu, M.-Z.; Tsai, Y.-P.; Yang, M.-H.; Huang, C.-H.; Chang, S.-Y.; Chang, C.-C.; Teng, S.-C.; Wu, K.-J. Interplay between HDAC3 and WDR5 Is Essential for Hypoxia-Induced Epithelial-Mesenchymal Transition. Mol. Cell 2011, 43, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-T.; Wu, K.-J. Epigenetic regulation of epithelial-mesenchymal transition: Focusing on hypoxia and TGF-β signaling. J. Biomed. Sci. 2020, 27, 39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Shi, X.; Peng, Y.; Wu, M.; Zhang, P.; Xie, R.; Wu, Y.; Yan, Q.; Liu, S.; Wang, J. HIF-1α Promotes Epithelial-Mesenchymal Transition and Metastasis through Direct Regulation of ZEB1 in Colorectal Cancer. PLoS ONE 2015, 10, e0129603. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Tanaka, F.; Morita, H.; Hiraki, A.; Hashimoto, S. Hypoxia-induced HIF-1α and ZEB1 are critical for the malignant transformation of ameloblastoma via TGF-β-dependent EMT. Cancer Med. 2019, 8, 7822–7832. [Google Scholar] [CrossRef]
- Nakuluri, K.; Mukhi, D.; Nishad, R.; Saleem, M.A.; Mungamuri, S.K.; Menon, R.K.; Pasupulati, A.K. Hypoxia induces ZEB2 in podocytes: Implications in the pathogenesis of proteinuria. J. Cell. Physiol. 2018, 234, 6503–6518. [Google Scholar] [CrossRef]
- Storci, G.; Sansone, P.; Mari, S.; D’Uva, G.; Tavolari, S.; Guarnieri, T.; Taffurelli, M.; Ceccarelli, C.; Santini, D.; Chieco, P.; et al. TNFalpha up-regulates SLUG via the NF-kappaB/HIF1alpha axis, which imparts breast cancer cells with a stem cell-like phenotype. J. Cell. Physiol. 2010, 225, 682–691. [Google Scholar] [CrossRef]
- Tang, C.; Liu, T.; Wang, K.; Wang, X.; Xu, S.; He, D.; Zeng, J. Transcriptional regulation of FoxM1 by HIF-1α mediates hypoxia-induced EMT in prostate cancer. Oncol. Rep. 2019, 42, 1307–1318. [Google Scholar] [CrossRef]
- Ma, C.; Guo, Y.; Zhang, Y.; Duo, A.; Jia, Y.; Liu, C.; Li, B. PAFAH1B2 is a HIF1a target gene and promotes metastasis in pancreatic cancer. Biochem. Biophys. Res. Commun. 2018, 501, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Colgan, S.P. HIF2 keeps paces in tight hypoxic spaces. Blood 2021, 137, 3323–3324. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cai, S.; Wang, L.; Yang, C.; Zhou, B.; Wang, H. HINT2 downregulation promotes colorectal carcinoma migration and metastasis. Oncotarget 2017, 8, 13521–13531. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, X.; Zhang, Y.; Zhu, D.; Zhang, L.; Li, Y.; Zhu, Y.; Li, D.; Zhou, J. HIF-2α promotes epithelial-mesenchymal transition through regulating Twist2 binding to the promoter of E-cadherin in pancreatic cancer. J. Exp. Clin. Cancer Res. 2016, 35, 26. [Google Scholar] [CrossRef] [PubMed]
- Cannito, S.; Turato, C.; Paternostro, C.; Biasiolo, A.; Colombatto, S.; Cambieri, I.; Quarta, S.; Novo, E.; Morello, E.; Villano, G.; et al. Hypoxia up-regulates SERPINB3 through HIF-2α in human liver cancer cells. Oncotarget 2014, 6, 2206–2221. [Google Scholar] [CrossRef]
- Qiu, W.; Song, S.; Chen, W.; Zhang, J.; Yang, H.; Chen, Y. Hypoxia-Induced EPHB2 Promotes Invasive Potential of Glioblas-toma. Int. J. Clin. Exp. Pathol. 2019, 12, 539–548. [Google Scholar]
- Zhang, K.-D.; Hu, B.; Cen, G.; Yang, Y.-H.; Chen, W.-W.; Guo, Z.-Y.; Wang, X.-F.; Zhao, Q.; Qiu, Z.-J. MiR-301a transcriptionally activated by HIF-2α promotes hypoxia-induced epithelial-mesenchymal transition by targeting TP63 in pancreatic cancer. World J. Gastroenterol. 2020, 26, 2349–2373. [Google Scholar] [CrossRef]
- Yang, F.; Liu, C.; Zhao, G.; Ge, L.; Song, Y.; Chen, Z.; Liu, Z.; Hong, K.; Ma, L. Long non-coding RNA LINC01234 regulates proliferation, migration and invasion via HIF-2α pathways in clear cell renal cell carcinoma cells. PeerJ 2020, 8, e10149. [Google Scholar] [CrossRef]
- Zheng, X.; Zhang, Y.; Liu, Y.; Fang, L.; Li, L.; Sun, J.; Pan, Z.; Xin, W.; Huang, P. HIF-2α activated lncRNA NEAT1 promotes hepatocellular carcinoma cell invasion and metastasis by affecting the epithelial-mesenchymal transition. J. Cell. Biochem. 2018, 119, 3247–3256. [Google Scholar] [CrossRef]
- Kong, X.; Zhao, Y.; Li, X.; Tao, Z.; Hou, M.; Ma, H. Overexpression of HIF-2a-Dependent NEAT1 Promotes the Progression of Non-Small Cell Lung Cancer through miR-1013p/SOX9/Wnt/β-Catenin Signal Pathway. Cell. Physiol. Biochem. 2019, 52, 368–381. [Google Scholar] [CrossRef]
- Hong, C.-F.; Chen, W.-Y.; Wu, C.-W. Upregulation of Wnt signaling under hypoxia promotes lung cancer progression. Oncol. Rep. 2017, 38, 1706–1714. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Kong, Y.; Cao, M.; Zhou, H.; Li, H.; Cui, Y.; Fang, F.; Zhang, W.; Li, J.; Zhu, X.; et al. Decreased expression of acetyl-CoA synthase 2 promotes metastasis and predicts poor prognosis in hepatocellular carcinoma. Cancer Sci. 2017, 108, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.Y.; Lemos, R.; Liu, X.; Powis, G. The Hypoxia-Associated Factor Switches Cells from HIF-1α- to HIF-2α-Dependent Signaling Promoting Stem Cell Characteristics, Aggressive Tumor Growth and Invasion. Cancer Res. 2011, 71, 4015–4027. [Google Scholar] [CrossRef] [PubMed]
- Maynard, M.A.; Qi, H.; Chung, J.; Lee, E.H.L.; Kondo, Y.; Hara, S.; Conaway, R.C.; Conaway, J.; Ohh, M. Multiple Splice Variants of the Human HIF-3α Locus Are Targets of the von Hippel-Lindau E3 Ubiquitin Ligase Complex. J. Biol. Chem. 2003, 278, 11032–11040. [Google Scholar] [CrossRef]
- Makino, Y.; Uenishi, R.; Okamoto, K.; Isoe, T.; Hosono, O.; Tanaka, H.; Kanopka, A.; Poellinger, L.; Haneda, M.; Morimoto, C. Transcriptional Up-regulation of Inhibitory PAS Domain Protein Gene Expression by Hypoxia-inducible Factor 1 (HIF-1). J. Biol. Chem. 2007, 282, 14073–14082. [Google Scholar] [CrossRef]
- Zhang, P.; Lü, L.; Yao, Q.; Li, Y.; Zhou, J.; Liu, Y.; Duan, C. Molecular, functional, and gene expression analysis of zebrafish hypoxia-inducible factor-3α. Am. J. Physiol. Integr. Comp. Physiol. 2012, 303, R1165–R1174. [Google Scholar] [CrossRef]
- Gilmore, T.D. Introduction to NF-κB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef]
- Pires, B.R.B.; Mencalha, A.L.; Ferreira, G.M.; De Souza, W.F.; Morgado-Díaz, J.A.; Maia, A.M.; Corrêa, S.; Abdelhay, E.S.F.W. NF-kappaB Is Involved in the Regulation of EMT Genes in Breast Cancer Cells. PLoS ONE 2017, 12, e0169622. [Google Scholar] [CrossRef]
- Wu, Y.; Deng, J.; Rychahou, P.G.; Qiu, S.; Evers, B.M.; Zhou, B.P. Stabilization of Snail by NF-κB Is Required for Inflammation-Induced Cell Migration and Invasion. Cancer Cell 2009, 15, 416–428. [Google Scholar] [CrossRef]
- Koong, A.C.; Chen, E.Y.; Giaccia, A.J. Hypoxia causes the activation of nuclear factor kappa B through the phosphorylation of I kappa B alpha on tyrosine residues. Cancer Res. 1994, 54, 1425–1430. [Google Scholar] [PubMed]
- Cockman, M.E.; Lippl, K.; Tian, Y.-M.; Pegg, H.B.; Figg, W.D.J.; Abboud, M.I.; Heilig, R.; Fischer, R.; Myllyharju, J.; Schofield, C.J.; et al. Lack of activity of recombinant HIF prolyl hydroxylases (PHDs) on reported non-HIF substrates. eLife 2019, 8, e46490. [Google Scholar] [CrossRef] [PubMed]
- Cockman, M.E.; Lancaster, D.E.; Stolze, I.P.; Hewitson, K.S.; McDonough, M.A.; Coleman, M.L.; Coles, C.H.; Yu, X.; Hay, R.T.; Ley, S.C.; et al. Posttranslational hydroxylation of ankyrin repeats in IκB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH). Proc. Natl. Acad. Sci. USA 2006, 103, 14767–14772. [Google Scholar] [CrossRef]
- Culver, C.; Sundqvist, A.; Mudie, S.; Melvin, A.; Xirodimas, D.; Rocha, S. Mechanism of Hypoxia-Induced NF-κB. Mol. Cell. Biol. 2010, 30, 4901–4921. [Google Scholar] [CrossRef] [PubMed]
- Acloque, H.; Adams, M.S.; Fishwick, K.; Bronner, M.; Nieto, M.A. Epithelial-mesenchymal transitions: The importance of changing cell state in development and disease. J. Clin. Investig. 2009, 119, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Tang, Y.-L.; Liang, X.-H. EMT: A new vision of hypoxia promoting cancer progression. Cancer Biol. Ther. 2011, 11, 714–723. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, F.; ten Dijke, P. Signaling interplay between transforming growth factor-β receptor and PI3K/AKT pathways in cancer. Trends Biochem. Sci. 2013, 38, 612–620. [Google Scholar] [CrossRef]
- Dong, J.; Zhai, B.; Sun, W.; Hu, F.; Cheng, H.; Xu, J. Activation of phosphatidylinositol 3-kinase/AKT/snail signaling pathway contributes to epithelial-mesenchymal transition-induced multi-drug resistance to sorafenib in hepatocellular carcinoma cells. PLoS ONE 2017, 12, e0185088. [Google Scholar] [CrossRef]
- Lau, M.-T.; Leung, P.C. The PI3K/Akt/mTOR signaling pathway mediates insulin-like growth factor 1-induced E-cadherin down-regulation and cell proliferation in ovarian cancer cells. Cancer Lett. 2012, 326, 191–198. [Google Scholar] [CrossRef]
- Dodd, K.M.; Yang, J.; Shen, M.H.; Sampson, J.R.; Tee, A.R. mTORC1 drives HIF-1α and VEGF-A signalling via multiple mechanisms involving 4E-BP1, S6K1 and STAT3. Oncogene 2015, 34, 2239–2250. [Google Scholar] [CrossRef]
- Tam, S.Y.; Wu, V.W.; Law, H.K. JNK Pathway Mediates Low Oxygen Level Induced Epithelial–Mesenchymal Transition and Stemness Maintenance in Colorectal Cancer Cells. Cancers 2020, 12, 224. [Google Scholar] [CrossRef] [PubMed]
- Leong, K.G.; Niessen, K.; Kulic, I.; Raouf, A.; Eaves, C.; Pollet, I.; Karsan, A. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J. Exp. Med. 2007, 204, 2935–2948. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sha, J.; Yang, G.; Huang, X.; Bo, J.; Huang, Y. Activation of Notch pathway is linked with epithelial-mesenchymal transition in prostate cancer cells. Cell Cycle 2017, 16, 999–1007. [Google Scholar] [CrossRef]
- Chen, J.; Imanaka, N.; Griffin, J.D. Hypoxia potentiates Notch signaling in breast cancer leading to decreased E-cadherin expression and increased cell migration and invasion. Br. J. Cancer 2009, 102, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Kemp, B.E. AMPK in Health and Disease. Physiol. Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; Balaji, S.A.; Deshpande, N.; Ranganathan, S.; Pillai, D.M.; Hindupur, S.K.; Rangarajan, A. AMP-activated protein kinase promotes epithelial-mesenchymal transition in cancer cells through Twist1 upregulation. J. Cell Sci. 2018, 131, jcs208314. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, A.C.; Hidalgo, F.; Tonucci, F.M.; Almada, E.; Pariani, A.; LaRocca, M.C.; Favre, C. Metformin and glucose starvation decrease the migratory ability of hepatocellular carcinoma cells: Targeting AMPK activation to control migration. Sci. Rep. 2019, 9, 2815. [Google Scholar] [CrossRef]
- Liu, J.; Song, N.; Huang, Y.; Chen, Y. Irisin inhibits pancreatic cancer cell growth via the AMPK-mTOR pathway. Sci. Rep. 2018, 8, 15247. [Google Scholar] [CrossRef]
- Chou, C.-C.; Lee, K.-H.; Lai, I.-L.; Wang, D.; Mo, X.; Kulp, S.K.; Shapiro, C.L.; Chen, C.-S. AMPK Reverses the Mesenchymal Phenotype of Cancer Cells by Targeting the Akt–MDM2–Foxo3a Signaling Axis. Cancer Res. 2014, 74, 4783–4795. [Google Scholar] [CrossRef]
- Greenburg, G.; Hay, E.D. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J. Cell Biol. 1982, 95, 333–339. [Google Scholar] [CrossRef]
- Hapke, R.Y.; Haake, S.M. Hypoxia-induced epithelial to mesenchymal transition in cancer. Cancer Lett. 2020, 487, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Palucka, A.K.; Coussens, L.M. The basis of oncoimmunology. Cell 2016, 164, 1233–1247. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Muthana, M.; Lewis, C.E. Hypoxia Regulates Macrophage Functions in Inflammation. J. Immunol. 2005, 175, 6257–6263. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A key player in antitumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C569–C579. [Google Scholar] [CrossRef]
- Beucken, T.V.D.; Koch, E.; Chu, K.; Rupaimoole, R.; Prickaerts, P.; Adriaens, M.; Voncken, J.W.; Harris, A.; Buffa, F.; Haider, S.; et al. Hypoxia promotes stem cell phenotypes and poor prognosis through epigenetic regulation of DICER. Nat. Commun. 2014, 5, 5203. [Google Scholar] [CrossRef]
- Shi, Z.; To, S.K.Y.; Zhang, S.; Deng, S.; Artemenko, M.; Zhang, M.; Tang, J.; Zeng, J.-Z.; Wong, A.S. Hypoxia-induced Nur77 activates PI3K/Akt signaling via suppression of Dicer/let-7i-5p to induce epithelial-to-mesenchymal transition. Theranostics 2021, 11, 3376–3391. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, Y.; Dou, C.; Xu, M.; Sun, L.; Wang, L.; Yao, B.; Li, Q.; Yang, W.; Tu, K.; et al. Hypoxia-induced up-regulation of VASP promotes invasiveness and metastasis of hepatocellular carcinoma. Theranostics 2018, 8, 4649–4663. [Google Scholar] [CrossRef]
- Chen, S.-Y.; Teng, S.-C.; Cheng, T.-H.; Wu, K.-J. miR-1236 regulates hypoxia-induced epithelial–mesenchymal transition and cell migration/invasion through repressing SENP1 and HDAC3. Cancer Lett. 2016, 378, 59–67. [Google Scholar] [CrossRef]
- Liu, H.; Chen, C.; Zeng, J.; Zhao, Z.; Hu, Q. MicroRNA-210-3p is transcriptionally upregulated by hypoxia induction and thus promoting EMT and chemoresistance in glioma cells. PLoS ONE 2021, 16, e0253522. [Google Scholar] [CrossRef]
- Kuo, M.-C.; Chang, W.-A.; Wu, L.-Y.; Tsai, Y.-C.; Hsu, Y.-L. Hypoxia-Induced Epithelial-to-Mesenchymal Transition in Proximal Tubular Epithelial Cells through miR-545-3p–TNFSF10. Biomolecules 2021, 11, 1032. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Chen, W.; Chen, X.; Wang, J. Overexpression of STAT4 under hypoxia promotes EMT through miR-200a/STAT4 signal pathway. Life Sci. 2021, 273, 119263. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Liu, X.; Liu, Z.; Zhou, Z.; Wang, Y.; Tu, J.; Li, L.; Bao, H.; Yang, L.; Tu, K. MicroRNA-1296 inhibits metastasis and epithelial-mesenchymal transition of hepatocellular carcinoma by targeting SRPK1-mediated PI3K/AKT pathway. Mol. Cancer 2017, 16, 103. [Google Scholar] [CrossRef] [PubMed]
- Yao, B.; Li, Y.; Wang, L.; Chen, T.; Niu, Y.; Liu, Q.; Liu, Z. MicroRNA-3194-3p inhibits metastasis and epithelial-mesenchymal transition of hepatocellular carcinoma by decreasing Wnt/β-catenin signaling through targeting BCL9. Artif. Cells Nanomed. Biotechnol. 2019, 47, 3885–3895. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Sun, W.; Xia, L.; Zhao, A.; Yu, Y.; Zhao, L.; Wang, H.; Huang, C.; Sun, S. Hypoxia-Induced Down-Regulation of microRNA-34a Promotes EMT by Targeting the Notch Signaling Pathway in Tubular Epithelial Cells. PLoS ONE 2012, 7, e30771. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.A. Phylogenetic Classification of Prokaryotic and Eukaryotic Sir2-like Proteins. Biochem. Biophys. Res. Commun. 2000, 273, 793–798. [Google Scholar] [CrossRef]
- Blander, G.; Guarente, L. The Sir2 Family of Protein Deacetylases. Annu. Rev. Biochem. 2004, 73, 417–435. [Google Scholar] [CrossRef]
- Yamamoto, H.; Schoonjans, K.; Auwerx, J. Sirtuin Functions in Health and Disease. Mol. Endocrinol. 2007, 21, 1745–1755. [Google Scholar] [CrossRef]
- Dali-Youcef, N.; Lagouge, M.; Froelich, S.; Koehl, C.; Schoonjans, K.; Auwerx, J. Sirtuins: The ‘magnificent seven’, function, metabolism and longevity. Ann. Med. 2007, 39, 335–345. [Google Scholar] [CrossRef]
- Michishita, E.; Park, J.Y.; Burneskis, J.M.; Barrett, J.C.; Horikawa, I. Evolutionarily Conserved and Nonconserved Cellular Localizations and Functions of Human SIRT Proteins. Mol. Biol. Cell 2005, 16, 4623–4635. [Google Scholar] [CrossRef]
- Jin, Q.; Yan, T.; Ge, X.; Sun, C.; Shi, X.; Zhai, Q. Cytoplasm-localized SIRT1 enhances apoptosis. J. Cell. Physiol. 2007, 213, 88–97. [Google Scholar] [CrossRef]
- Vaquero, A.; Scher, M.B.; Lee, D.H.; Sutton, A.; Cheng, H.-L.; Alt, F.W.; Serrano, L.; Sternglanz, R.; Reinberg, D. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006, 20, 1256–1261. [Google Scholar] [CrossRef] [PubMed]
- Scher, M.B.; Vaquero, A.; Reinberg, D. SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 2007, 21, 920–928. [Google Scholar] [CrossRef] [PubMed]
- van de Ven, R.A.; Santos, D.; Haigis, M.C. Mitochondrial Sirtuins and Molecular Mechanisms of Aging. Trends Mol. Med. 2017, 23, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Vachharajani, V.T.; Liu, T.; Wang, X.; Hoth, J.J.; Yoza, B.K.; McCall, C.E. Sirtuins Link Inflammation and Metabolism. J. Immunol. Res. 2016, 2016, 8167273. [Google Scholar] [CrossRef]
- Jęśko, H.; Wencel, P.; Strosznajder, R.; Strosznajder, J. Sirtuins and Their Roles in Brain Aging and Neurodegenerative Disorders. Neurochem. Res. 2016, 42, 876–890. [Google Scholar] [CrossRef]
- Tang, B.L. Sirtuins as modifiers of Parkinson’s disease pathology. J. Neurosci. Res. 2017, 95, 930–942. [Google Scholar] [CrossRef]
- Winnik, S.; Auwerx, J.; Sinclair, D.; Matter, C.M. Protective effects of sirtuins in cardiovascular diseases: From bench to bedside. Eur. Heart J. 2015, 36, 3404–3412. [Google Scholar] [CrossRef]
- Zhou, S.; Tang, X.; Chen, H.-Z. Sirtuins and Insulin Resistance. Front. Endocrinol. 2018, 9, 748. [Google Scholar] [CrossRef]
- Zhu, S.; Dong, Z.; Ke, X.; Hou, J.; Zhao, E.; Zhang, K.; Wang, F.; Yang, L.; Xiang, Z.; Cui, H. The roles of sirtuins family in cell metabolism during tumor development. Semin. Cancer Biol. 2018, 57, 59–71. [Google Scholar] [CrossRef]
- D’Onofrio, N.; Servillo, L.; Balestrieri, M.L. SIRT1 and SIRT6 Signaling Pathways in Cardiovascular Disease Protection. Antioxid. Redox Signal. 2018, 28, 711–732. [Google Scholar] [CrossRef]
- Yang, H.; Bi, Y.; Xue, L.; Wang, J.; Lu, Y.; Zhang, Z.; Chen, X.; Chu, Y.; Yang, R.; Wang, R.; et al. Multifaceted Modulation of SIRT1 in Cancer and Inflammation. Crit. Rev. Oncog. 2015, 20, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Cantor, J.R.; Sabatini, D.M. Cancer Cell Metabolism: One Hallmark, Many Faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef] [PubMed]
- Simic, P.; Williams, E.O.; Bell, E.L.; Gong, J.J.; Bonkowski, M.; Guarente, L. SIRT1 Suppresses the Epithelial-to-Mesenchymal Transition in Cancer Metastasis and Organ Fibrosis. Cell Rep. 2013, 3, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Byles, V.; Zhu, L.; Lovaas, J.D.; Chmilewski, L.K.; Wang, J.; Faller, D.V.; Dai, Y. SIRT1 induces EMT by cooperating with EMT transcription factors and enhances prostate cancer cell migration and metastasis. Oncogene 2012, 31, 4619–4629. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Zhu, S.; Wang, B.; Li, X.; Liu, Y.; Qin, Q.; Gong, Q.; Niu, Y.; Xiang, C.; Chen, J.; et al. Chronic pancreatitis and pancreatic cancer demonstrate active epithelial–mesenchymal transition profile, regulated by miR-217-SIRT1 pathway. Cancer Lett. 2014, 355, 184–191. [Google Scholar] [CrossRef]
- Xu, J.; Zhu, W.; Xu, W.; Yao, W.; Zhang, B.; Xu, Y.; Ji, S.; Liu, C.; Long, J.; Ni, Q.; et al. Up-Regulation of MBD1 Promotes Pancreatic Cancer Cell Epithelial-Mesenchymal Transition and Invasion by Epigenetic down-Regulation of E-Cadherin. Curr. Mol. Med. 2013, 13, 387–400. [Google Scholar]
- Hao, C.; Zhu, P.-X.; Yang, X.; Han, Z.-P.; Jiang, J.-H.; Zong, C.; Zhang, X.-G.; Liu, W.-T.; Zhao, Q.-D.; Fan, T.-T.; et al. Overexpression of SIRT1 promotes metastasis through epithelial-mesenchymal transition in hepatocellular carcinoma. BMC Cancer 2014, 14, 978. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, X.; Chen, P. MiR-204 down regulates SIRT1 and reverts SIRT1-induced epithelial-mesenchymal transition, anoikis resistance and invasion in gastric cancer cells. BMC Cancer 2013, 13, 290. [Google Scholar] [CrossRef]
- Sun, T.; Jiao, L.; Wang, Y.; Yu, Y.; Ming, L. SIRT1 induces epithelial-mesenchymal transition by promoting autophagic degradation of E-cadherin in melanoma cells. Cell Death Dis. 2018, 9, 136. [Google Scholar] [CrossRef]
- Shi, L.; Tang, X.; Qian, M.; Liu, Z.; Meng, F.; Fu, L.; Wang, Z.; Zhu, W.-G.; Huang, J.-D.; Zhou, Z.; et al. A SIRT1-centered circuitry regulates breast cancer stemness and metastasis. Oncogene 2018, 37, 6299–6315. [Google Scholar] [CrossRef]
- Chen, J.; Chan, A.W.; To, K.-F.; Chen, W.; Zhang, Z.; Ren, J.; Song, C.; Cheung, Y.-S.; Lai, P.B.; Cheng, S.-H.; et al. SIRT2 overexpression in hepatocellular carcinoma mediates epithelial to mesenchymal transition by protein kinase B/glycogen synthase kinase-3β/β-catenin signaling. Hepatology 2013, 57, 2287–2298. [Google Scholar] [CrossRef] [PubMed]
- Cheon, M.G.; Kim, W.; Choi, M.; Kim, J.-E. AK-1, a specific SIRT2 inhibitor, induces cell cycle arrest by downregulating Snail in HCT116 human colon carcinoma cells. Cancer Lett. 2015, 356, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Quan, Y.; Xia, W. SIRT3 inhibits prostate cancer metastasis through regulation of FOXO3A by suppressing Wnt/β-catenin pathway. Exp. Cell Res. 2018, 364, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, P.; Karthikeyan, A.; Lu, J.; Ling, E.-A.; Dheen, S. Sirtuin 3 regulates Foxo3a-mediated antioxidant pathway in microglia. Neuroscience 2015, 311, 398–414. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef]
- Dey, P.; Ström, A.; Gustafsson, J.Å. Estrogen receptor β upregulates FOXO3a and causes induction of apoptosis through PUMA in prostate cancer. Oncogene 2014, 33, 4213–4225. [Google Scholar] [CrossRef]
- Kypta, R.; Waxman, J. Wnt/β-catenin signalling in prostate cancer. Nat. Rev. Urol. 2012, 9, 418–428. [Google Scholar] [CrossRef]
- Dong, X.-C.; Jing, L.-M.; Wang, W.-X.; Gao, Y.-X. Down-regulation of SIRT3 promotes ovarian carcinoma metastasis. Biochem. Biophys. Res. Commun. 2016, 475, 245–250. [Google Scholar] [CrossRef]
- He, P.; Li, Z.; Yue, Z.; Gao, H.; Feng, G.; Wang, P.; Huang, Y.; Luo, W.; Hong, H.; Liang, L.; et al. SIRT3 prevents angiotensin II-induced renal tubular epithelial-mesenchymal transition by ameliorating oxidative stress and mitochondrial dysfunction. Mol. Cell. Endocrinol. 2018, 460, 1–13. [Google Scholar] [CrossRef]
- Carvajal, G.; Rodriguez-Vita, J.; Rodrigues-Díez, R.; Sanchez-Lopez, E.; Rupérez, M.; Cartier, C.; Esteban, V.; Ortiz, A.; Egido, J.; Mezzano, S.A.; et al. Angiotensin II activates the Smad pathway during epithelial mesenchymal transdifferentiation. Kidney Int. 2008, 74, 585–595. [Google Scholar] [CrossRef]
- Burns, W.; Thomas, M. Angiotensin II and Its Role in Tubular Epithelial to Mesenchymal Transition Associated with Chronic Kidney Disease. Cells Tissues Organs 2011, 193, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Djamali, A.; Reese, S.; Yracheta, J.; Oberley, T.; Hullett, D.; Becker, B. Epithelial-to-Mesenchymal Transition and Oxidative Stress in Chronic Allograft Nephropathy. Am. J. Transplant. 2005, 5, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Rhyu, D.Y.; Yang, Y.; Ha, H.; Lee, G.T.; Song, J.S.; Uh, S.-T.; Lee, H.B. Role of Reactive Oxygen Species in TGF-β1-Induced Mitogen-Activated Protein Kinase Activation and Epithelial-Mesenchymal Transition in Renal Tubular Epithelial Cells. J. Am. Soc. Nephrol. 2005, 16, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Jia, Z.; Guo, X.; Yang, T. Aldosterone induces epithelial-mesenchymal transition via ROS of mitochondrial origin. Am. J. Physiol. Physiol. 2007, 293, F723–F731. [Google Scholar] [CrossRef]
- Guo, D.; Song, X.; Guo, T.; Gu, S.; Chang, X.; Su, T.; Yang, X.; Liang, B.; Huang, D. Vimentin acetylation is involved in SIRT5-mediated hepatocellular carcinoma migration. Am. J. Cancer Res. 2018, 8, 2453–2466. [Google Scholar]
- Ye, H.; Duan, M. Downregulation of FOXO6 in breast cancer promotes epithelial–mesenchymal transition and facilitates migration and proliferation of cancer cells. Cancer Manag. Res. 2018, 10, 5145–5156. [Google Scholar] [CrossRef]
- Min, L.; Ji, Y.; Bakiri, L.; Qiu, Z.; Cen, J.; Chen, X.; Chen, L.; Scheuch, H.; Zheng, H.; Qin, L.; et al. Liver cancer initiation is controlled by AP-1 through SIRT6-dependent inhibition of survivin. Nat. Cell Biol. 2012, 14, 1203–1211. [Google Scholar] [CrossRef]
- Mostoslavsky, R.; Chua, K.F.; Lombard, D.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Mostoslavsky, G.; Franco, S.; Murphy, M.M.; et al. Genomic Instability and Aging-like Phenotype in the Absence of Mammalian SIRT6. Cell 2006, 124, 315–329. [Google Scholar] [CrossRef]
- Geng, C.; Zhang, C.; Zhang, J.; Gao, P.; He, M.; Li, Y. Overexpression of Sirt6 is a novel biomarker of malignant human colon carcinoma. J. Cell. Biochem. 2018, 119, 3957–3967. [Google Scholar] [CrossRef]
- Li, Z.; Huang, J.; Shen, S.; Ding, Z.; Luo, Q.; Chen, Z.; Lu, S. SIRT6 drives epithelial-to-mesenchymal transition and metastasis in non-small cell lung cancer via snail-dependent transrepression of KLF4. J. Exp. Clin. Cancer Res. 2018, 37, 323. [Google Scholar] [CrossRef]
- Han, L.L.; Jia, L.; Wu, F.; Huang, C. Sirtuin6 (SIRT6) Promotes the EMT of Hepatocellular Carcinoma by Stimulating Autophagic Degradation of E-Cadherin. Mol. Cancer Res. 2019, 17, 2267–2280. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Zhang, S.; Wu, S.; Zhu, Q.; Li, W. MiR-770 promotes oral squamous cell carcinoma migration and invasion by regulating the Sirt7/Smad4 pathway. IUBMB Life 2021, 73, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhu, D.; Qin, S. SIRT7 suppresses the epithelial-to-mesenchymal transition in oral squamous cell carcinoma metastasis by promoting SMAD4 deacetylation. J. Exp. Clin. Cancer Res. 2018, 37, 148. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ye, X.; Chen, R.; Gao, Q.; Zhao, D.; Ling, C.; Qian, Y.; Xu, C.; Tao, M.; Xie, Y. Sirtuin 7 promotes non-small cell lung cancer progression by facilitating G1/S phase and epithelial-mesenchymal transition and activating AKT and ERK1/2 signaling. Oncol. Rep. 2020, 44, 959–972. [Google Scholar] [CrossRef]
- Deng, Z.; Wang, X.; Long, X.; Liu, W.; Xiang, C.; Bao, F.; Wang, D. Sirtuin 7 promotes colorectal carcinoma proliferation and invasion through the inhibition of E-cadherin. Exp. Ther. Med. 2018, 15, 2333–2342. [Google Scholar] [CrossRef]
- Monteiro-Reis, S.; Lameirinhas, A.; Miranda-Gonçalves, V.; Felizardo, D.; Dias, P.C.; Oliveira, J.; Graça, I.; Gonçalves, C.S.; Costa, B.M.; Henrique, R.; et al. Sirtuins’ Deregulation in Bladder Cancer: SIRT7 Is Implicated in Tumor Progression through Epithelial to Mesenchymal Transition Promotion. Cancers 2020, 12, 1066. [Google Scholar] [CrossRef]
- Malik, S.; Villanova, L.; Tanaka, S.; Aonuma, M.; Roy, N.; Berber, E.; Pollack, J.R.; Michishita-Kioi, E.; Chua, K.F. SIRT7 inactivation reverses metastatic phenotypes in epithelial and mesenchymal tumors. Sci. Rep. 2015, 5, 9841. [Google Scholar] [CrossRef]
- Solomon, J.P.; Hansel, D.E. The Emerging Molecular Landscape of Urothelial Carcinoma. Surg. Pathol. Clin. 2016, 9, 391–404. [Google Scholar] [CrossRef]
- Barber, M.F.M.; Michishita-Kioi, E.; Xi, Y.; Tasselli, L.; Kioi, M.; Moqtaderi, Z.; Tennen, R.I.; Paredes, S.; Young, N.L.; Chen, K.; et al. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 2012, 487, 114–118. [Google Scholar] [CrossRef]
- Lim, J.-H.; Lee, Y.-M.; Chun, Y.-S.; Chen, J.; Kim, J.-E.; Park, J.-W. Sirtuin 1 Modulates Cellular Responses to Hypoxia by Deacetylating Hypoxia-Inducible Factor 1α. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef]
- Dioum, E.M.; Chen, R.; Alexander, M.S.; Zhang, Q.; Hogg, R.T.; Gerard, R.D.; Garcia, J.A. Regulation of Hypoxia-Inducible Factor 2α Signaling by the Stress-Responsive Deacetylase Sirtuin 1. Science 2009, 324, 1289–1293. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Dioum, E.M.; Hogg, R.T.; Gerard, R.D.; Garcia, J.A. Hypoxia Increases Sirtuin 1 Expression in a Hypoxia-inducible Factor-dependent Manner. J. Biol. Chem. 2011, 286, 13869–13878. [Google Scholar] [CrossRef] [PubMed]
- Aventaggiato, M.; Vernucci, E.; Barreca, F.; Russo, M.A.; Tafani, M. Sirtuins’ control of autophagy and mitophagy in cancer. Pharmacol. Ther. 2020, 221, 107748. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Li, H.; Chen, J.; Iwasaki, Y.; Kubota, T.; Matsuoka, M.; Shen, A.; Chen, Q.; Xu, Y. PIASy mediates hypoxia-induced SIRT1 transcriptional repression and epithelial-to-mesenchymal transition in ovarian cancer cells. J. Cell Sci. 2013, 126, 3939–3947. [Google Scholar] [CrossRef] [PubMed]
- Branco-Price, C.; Zhang, N.; Schnelle, M.; Evans, C.; Katschinski, D.M.; Liao, D.; Ellies, L.; Johnson, R.S. Endothelial Cell HIF-1α and HIF-2α Differentially Regulate Metastatic Success. Cancer Cell 2012, 21, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Sahlgren, C.; Gustafsson, M.V.; Jin, S.; Poellinger, L.; Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. USA 2008, 105, 6392–6397. [Google Scholar] [CrossRef]
- Firestein, R.; Blander, G.; Michan, S.; Oberdoerffer, P.; Ogino, S.; Campbell, J.; Bhimavarapu, A.; Luikenhuis, S.; de Cabo, R.; Fuchs, C.; et al. The SIRT1 Deacetylase Suppresses Intestinal Tumorigenesis and Colon Cancer Growth. PLoS ONE 2008, 3, e2020. [Google Scholar] [CrossRef]
- Kaitsuka, T.; Matsushita, M.; Matsushita, N. SIRT2 inhibition activates hypoxia-inducible factor 1α signaling and mediates neuronal survival. Biochem. Biophys. Res. Commun. 2020, 529, 957–962. [Google Scholar] [CrossRef]
- Geng, H.; Liu, Q.; Xue, C.; David, L.L.; Beer, T.M.; Thomas, G.V.; Dai, M.-S.; Qian, D.Z. HIF1α Protein Stability Is Increased by Acetylation at Lysine 709. J. Biol. Chem. 2012, 287, 35496–35505. [Google Scholar] [CrossRef]
- Semenza, G.L. Hydroxylation of HIF-1: Oxygen Sensing at the Molecular Level. Physiology 2004, 19, 176–182. [Google Scholar] [CrossRef]
- Hiratsuka, M.; Inoue, T.; Toda, T.; Kimura, N.; Shirayoshi, Y.; Kamitani, H.; Watanabe, T.; Ohama, E.; Tahimic, C.G.; Kurimasa, A.; et al. Proteomics-based identification of differentially expressed genes in human gliomas: Down-regulation of SIRT2 gene. Biochem. Biophys. Res. Commun. 2003, 309, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.-S.; Park, J.-H.; Heo, J.-Y.; Jing, K.; Han, J.; Min, K.-N.; Kim, C.; Koh, G.Y.; Lim, K.; Kang, G.-Y.; et al. SIRT2 regulates tumour hypoxia response by promoting HIF-1α hydroxylation. Oncogene 2014, 34, 1354–1362. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Emerling, B.M.; Ricoult, S.J.H.; Guarente, L.P. SirT3 suppresses hypoxia inducible factor 1α and tumor growth by inhibiting mitochondrial ROS production. Oncogene 2011, 30, 2986–2996. [Google Scholar] [CrossRef] [PubMed]
- Finley, L.W.; Carracedo, A.; Lee, J.; Souza, A.; Egia, A.; Zhang, J.; Teruya-Feldstein, J.; Moreira, P.; Cardoso, S.M.; Clish, C.; et al. SIRT3 Opposes Reprogramming of Cancer Cell Metabolism through HIF1α Destabilization. Cancer Cell 2011, 19, 416–428. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008, 15, 678–685. [Google Scholar] [CrossRef]
- Zhong, L.; Durso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.; Nir, T.; et al. The Histone Deacetylase Sirt6 Regulates Glucose Homeostasis via Hif1α. Cell 2010, 140, 280–293. [Google Scholar] [CrossRef]
- Carmeliet, P.; Dor, Y.; Herbert, J.-M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.; et al. Role of HIF-1α in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998, 394, 485–490. [Google Scholar] [CrossRef]
- Arany, Z.; Huang, L.E.; Eckner, R.; Bhattacharya, S.; Jiang, C.; Goldberg, M.A.; Bunn, H.F.; Livingston, D.M. An essential role for p300/CBP in the cellular response to hypoxia. Proc. Natl. Acad. Sci. USA 1996, 93, 12969–12973. [Google Scholar] [CrossRef]
- Kallio, P.J.; Okamoto, K.; O’Brien, S.; Carrero, P.; Makino, Y.; Tanaka, H.; Poellinger, L. Signal transduction in hypoxic cells: Inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. EMBO J. 1998, 17, 6573–6586. [Google Scholar] [CrossRef]
- Sebastian, C.; Zwaans, B.M.; Silberman, D.M.; Gymrek, M.; Goren, A.; Zhong, L.; Ram, O.; Truelove, J.; Guimaraes, A.R.; Toiber, D.; et al. The Histone Deacetylase SIRT6 Is a Tumor Suppressor that Controls Cancer Metabolism. Cell 2012, 151, 1185–1199. [Google Scholar] [CrossRef]
- Yang, Z.; Yu, W.; Huang, R.; Ye, M.; Min, Z. SIRT6/HIF-1α axis promotes papillary thyroid cancer progression by inducing epithelial–mesenchymal transition. Cancer Cell Int. 2019, 19, 17. [Google Scholar] [CrossRef] [PubMed]
- Hubbi, M.E.; Hu, H.; Kshitiz; Gilkes, D.M.; Semenza, G.L. Sirtuin-7 Inhibits the Activity of Hypoxia-inducible Factors. J. Biol. Chem. 2013, 288, 20768–20775. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Witzigmann, D.; Thomson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; van der Meel, R. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021, 16, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gao, D.-Y.; Huang, L. In vivo delivery of miRNAs for cancer therapy: Challenges and strategies. Adv. Drug Deliv. Rev. 2015, 81, 128–141. [Google Scholar] [CrossRef]
- Haider, N.; Fatima, S.; Taha, M.; Rizwanullah, M.; Firdous, J.; Ahmad, R.; Mazhar, F.; Khan, M.A. Nanomedicines in Diagnosis and Treatment of Cancer: An Update. Curr. Pharm. Des. 2020, 26, 1216–1231. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.Z.; Siddiqui, F.A. Nanomedicine and drug delivery: A mini review. Int. Nano Lett. 2014, 4, 94. [Google Scholar] [CrossRef]
- Cordani, M.; Strippoli, R.; Somoza, Á. Nanomaterials as Inhibitors of Epithelial Mesenchymal Transition in Cancer Treatment. Cancers 2019, 12, 25. [Google Scholar] [CrossRef]
- Gang, W.; JunJie, W.; HongMing, T.; HuaFu, Z.; Jing, W. Role of SIRT1-mediated mitochondrial and Akt pathways in glioblastoma cell death induced by Cotinus coggygria flavonoid nanoliposomes. Int. J. Nanomed. 2015, 10, 5005–5023. [Google Scholar] [CrossRef]
- Wang, G.; Wang, J.J.; Wang, Y.Z.; Feng, S.; Jing, G.; Fu, X.L. Myricetin nanoliposomes induced SIRT3-mediated glycolytic metabolism leading to glioblastoma cell death. Artif. Cells Nanomed. Biotechnol. 2018, 46, S180–S191. [Google Scholar] [CrossRef]
- Wu, X.; He, X.; Wang, K.; Xie, C.; Zhou, B.; Qing, Z. Ultrasmall near-infrared gold nanoclusters for tumor fluorescence imaging in vivo. Nanoscale 2010, 2, 2244–2249. [Google Scholar] [CrossRef]
- Choi, K.Y.; Liu, G.; Lee, S.; Chen, X. Theranostic nanoplatforms for simultaneous cancer imaging and therapy: Current approaches and future perspectives. Nanoscale 2012, 4, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Latorre, A.; Latorre, A.; Castellanos, M.; Lafuente-Gómez, N.; Diaz, C.R.; Crespo-Barreda, A.; Lecea, M.; Cordani, M.; Martín-Duque, P.; Somoza, Á. Albumin-based nanostructures for uveal melanoma treatment. Nanomed. Nanotechnol. Biol. Med. 2021, 35, 102391. [Google Scholar] [CrossRef] [PubMed]
- Gradishar, W.J. Albumin-bound paclitaxel: A next-generation taxane. Expert Opin. Pharmacother. 2006, 7, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Zhang, S.; Guo, X.; Li, Y.; Ren, J.; Zhou, H.; Du, B.; Zhou, J. A traceable nanoplatform for enhanced chemo-photodynamic therapy by reducing oxygen consumption. Nanomed. Nanotechnol. Biol. Med. 2019, 20, 101978. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.; Wang, X.; Nie, S.; Chen, Z.; Shin, D.M. Therapeutic Nanoparticles for Drug Delivery in Cancer. Clin. Cancer Res. 2008, 14, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Somoza, Á. Targeting autophagy using metallic nanoparticles: A promising strategy for cancer treatment. Cell. Mol. Life Sci. 2019, 76, 1215–1242. [Google Scholar] [CrossRef]
- Yu, M.; Guo, G.; Huang, L.; Deng, L.; Chang, C.-S.; Achyut, B.R.; Canning, M.; Xu, N.; Arbab, A.S.; Bollag, R.J.; et al. CD73 on cancer-associated fibroblasts enhanced by the A2B-mediated feedforward circuit enforces an immune checkpoint. Nat. Commun. 2020, 11, 515. [Google Scholar] [CrossRef]
- Hajizadeh, F.; Ardebili, S.M.; Moornani, M.B.; Masjedi, A.; Atyabi, F.; Kiani, M.; Namdar, A.; Karpisheh, V.; Izadi, S.; Baradaran, B.; et al. Silencing of HIF-1α/CD73 axis by siRNA-loaded TAT-chitosan-spion nanoparticles robustly blocks cancer cell progression. Eur. J. Pharmacol. 2020, 882, 173235. [Google Scholar] [CrossRef]
- Karpisheh, V.; Afjadi, J.F.; Afjadi, M.N.; Haeri, M.S.; Sough, T.S.A.; Asl, S.H.; Edalati, M.; Atyabi, F.; Masjedi, A.; Hajizadeh, F.; et al. Inhibition of HIF-1α/EP4 axis by hyaluronate-trimethyl chitosan-SPION nanoparticles markedly suppresses the growth and development of cancer cells. Int. J. Biol. Macromol. 2021, 167, 1006–1019. [Google Scholar] [CrossRef]
- Zhang, X.; He, C.; Liu, X.; Chen, Y.; Zhao, P.; Chen, C.; Yan, R.; Li, M.; Fan, T.; Altine, B.; et al. One-pot synthesis of a microporous organosilica-coated cisplatin nanoplatform for HIF-1-targeted combination cancer therapy. Theranostics 2020, 10, 2918–2929. [Google Scholar] [CrossRef]
- Duan, W.-X.; He, M.-D.; Mao, L.; Qian, F.-H.; Li, Y.-M.; Pi, H.-F.; Liu, C.; Chen, C.; Lu, Y.-H.; Cao, Z.-W.; et al. NiO nanoparticles induce apoptosis through repressing SIRT1 in human bronchial epithelial cells. Toxicol. Appl. Pharmacol. 2015, 286, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Nyga, A.; Hart, A.; Tetley, T.D. Importance of the HIF pathway in cobalt nanoparticle-induced cytotoxicity and inflammation in human macrophages. Nanotoxicology 2015, 9, 905–917. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, L.; Zhao, W.; Yu, N.; Cheng, M.; Su, M.; Hu, J.; Wu, X.; Du, H.; Wang, M. The Role of Apoptosis Pathway in the Cytotoxicity Induced by Fresh and Aged Zinc Oxide Nanoparticles. Nanoscale Res. Lett. 2021, 16, 129. [Google Scholar] [CrossRef] [PubMed]
- Piret, J.-P.; Jacques, D.; Audinot, J.-N.; Mejia, J.; Boilan, E.; Noël, F.; Fransolet, M.; Demazy, C.; Lucas, S.; Saout, C.; et al. Copper(ii) oxide nanoparticles penetrate into HepG2 cells, exert cytotoxicity via oxidative stress and induce pro-inflammatory response. Nanoscale 2012, 4, 7168–7184. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.-K.; Gurunathan, S.; Kang, M.-H.; Han, J.W.; Das, J.; Choi, Y.-J.; Kwon, D.-N.; Cho, S.-G.; Park, C.; Seo, H.G.; et al. Hypoxia-mediated autophagic flux inhibits silver nanoparticle-triggered apoptosis in human lung cancer cells. Sci. Rep. 2016, 6, 21688. [Google Scholar] [CrossRef]
- Sironval, V.; Palmai-Pallag, M.; Vanbever, R.; Huaux, F.; Mejia, J.; Lucas, S.; Lison, D.; Brule, S.V.D. HIF-1α is a key mediator of the lung inflammatory potential of lithium-ion battery particles. Part. Fibre Toxicol. 2019, 16, 35. [Google Scholar] [CrossRef]
- Inkielewicz-Stepniak, I.; Niska, K.; Pyszka, K.; Tukaj, C.; Woźniak, M.; Radomski, M. Titanium dioxide nanoparticles enhance production of superoxide anion and alter the antioxidant system in human osteoblast cells. Int. J. Nanomed. 2015, 10, 1095–1107. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sirtuins | EMT/Hypoxia Related Targets | Enzymatic Activity | Effects | References |
|---|---|---|---|---|
| SIRT1 | HIF-1α | Deacetylation on Lys674 | p300-HIF-1α Transcriptional Activity inhibition HIF-1α decreased activity Epithelial-like phenotype maintenance | [240,241,242,243] |
| HIF-2α | Deacetylation | HIF-2α Increased Activity Metastastic process inhibition | [244,245] | |
| Notch | Notch decreased activity | [246,247] | ||
| β-catenin | Deacetylation | Wnt-dependent EMT inhibition | [246,247] | |
| SIRT2 | HIF-1α | Deacetylation on Lys709 | HIF-1α hydroxylation and degradation increase Transcriptional Activity inhibition Decreased Stability | [248,249,250,251] |
| NF-kB | Deacetylation | NF-kB related genes suppression | [252] | |
| SIRT3 | MnSOD/IDH2 | Deacetylation | HIF-1α indirect inactivation/hydroxylation/proteasomal degradation Cancer growth, angiogenesis, and metastasis inhibition | [253,254,255] |
| SIRT6 | HIF-1α | acH3K9 Deacetylation (Chromatin Regulation) | p300 recruitment inhibition HIF-1α transcriptional activity inhibition (glucose uptake and glycolysis decrease/oxidative phosphorylation increase) HIF-1α stability increase in PTC with cancer progression | [256,257,258,259,260,261] |
| SIRT7 | HIF-1α HIF-2α | Deacetylation-Independent Activity | Reduced stability Transcriptional Activity inhibition | [262] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aventaggiato, M.; Barreca, F.; Sansone, L.; Pellegrini, L.; Russo, M.A.; Cordani, M.; Tafani, M. Sirtuins and Hypoxia in EMT Control. Pharmaceuticals 2022, 15, 737. https://doi.org/10.3390/ph15060737
Aventaggiato M, Barreca F, Sansone L, Pellegrini L, Russo MA, Cordani M, Tafani M. Sirtuins and Hypoxia in EMT Control. Pharmaceuticals. 2022; 15(6):737. https://doi.org/10.3390/ph15060737
Chicago/Turabian StyleAventaggiato, Michele, Federica Barreca, Luigi Sansone, Laura Pellegrini, Matteo A. Russo, Marco Cordani, and Marco Tafani. 2022. "Sirtuins and Hypoxia in EMT Control" Pharmaceuticals 15, no. 6: 737. https://doi.org/10.3390/ph15060737
APA StyleAventaggiato, M., Barreca, F., Sansone, L., Pellegrini, L., Russo, M. A., Cordani, M., & Tafani, M. (2022). Sirtuins and Hypoxia in EMT Control. Pharmaceuticals, 15(6), 737. https://doi.org/10.3390/ph15060737