
Target Therapies for Systemic Mastocytosis: An Update

 ,
,
Abstract
:1. Introduction
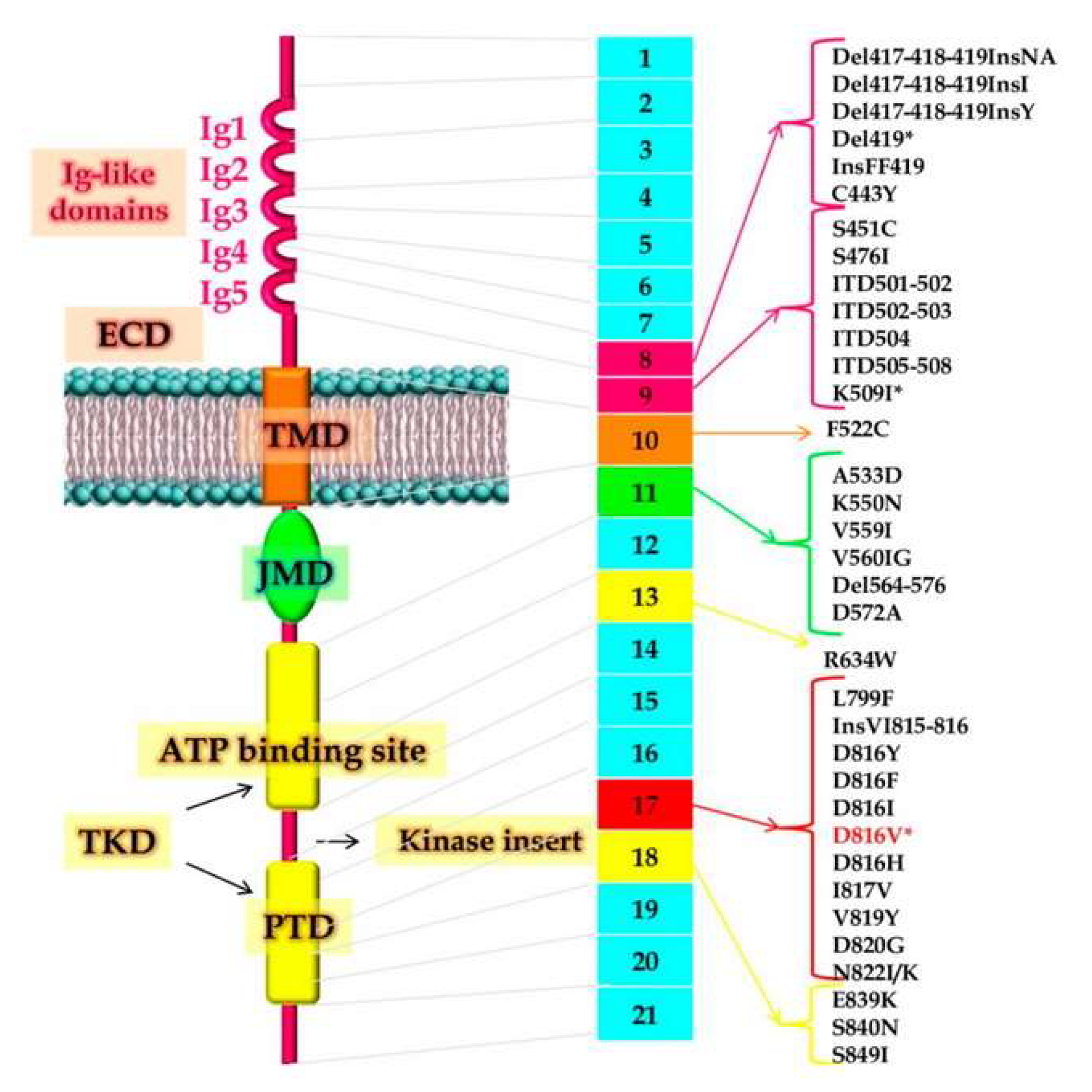
2. Pathogenesis and Molecular Aspects of Systemic Mastocytosis
3. Clinical Presentation and Diagnostic Subtypes
4. Treatment of Systemic Mastocytosis
4.1. Current Treatment Approaches for Mast Cell Mediator-Related Symptoms
4.2. Future Potential Therapeutic Targets for Mast Cell Mediator-Related Symptoms
4.3. Interferon-α (INF-α)
4.4. Chlorodeoxyadenosine (Cladribine or 2-CdA)
4.5. Tyrosine-Kinase Inhibitors
4.5.1. Imatinib Mesylate
4.5.2. Masitinib
4.5.3. Midostaurin
4.5.4. Avapritinib/BLU-285
4.5.5. Other Investigational Tyrosine-Kinase Inhibitors
4.6. The Role of Allogeneic Stem Cell Transplantation in the Age of KIT Inhibitors
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Horny, H.P.; Akin, C.; Metcalfe, D.D.; Escribano, L.; Bennett, J.M.; Valent, P.; Bain, B.J. Mastocytosis. In WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues; Swerdlow, S.H., Ed.; International Agency for Research and Cancer (IARC): Lyon, France, 2017; pp. 62–69. [Google Scholar]
- Pardanani, A. Systemic mastocytosis in adults: 2021 Update on diagnosis, risk stratification and management. Am. J. Hematol. 2021, 96, 508–525. [Google Scholar] [CrossRef] [PubMed]
- Furitsu, T.; Tsujimura, T.; Tono, T.; Ikeda, H.; Kitayama, H.; Koshimizu, U.; Sugahara, H.; Butterfield, J.H.; Ashman, L.K.; Kanayama, Y.; et al. Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of c-kit product. J. Clin. Investig. 1993, 92, 1736–1744. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Montero, A.C.; Jara-Acevedo, M.; Teodosio, C.; Sanchez, M.L.; Nunez, R.; Prados, A.; Aldanondo, I.; Sanchez, L.; Dominguez, M.; Botana, L.M.; et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: A prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood 2006, 108, 2366–2372. [Google Scholar] [CrossRef]
- Galli, S.J.; Borregaard, N.; Wynn, T.A. Phenotypic and functional plasticity of cells of innate immunity: Macrophages, mast cells and neutrophils. Nat. Immunol. 2011, 12, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.J.; Tsai, M. IgE and mast cells in allergic disease. Nat. Med. 2012, 18, 693–704. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Valent, P.; Akin, C. Mast Cells, Mastocytosis, and Related Disorders. N. Engl. J. Med. 2015, 373, 163–172. [Google Scholar] [CrossRef]
- Zhang, B.; Asadi, S.; Weng, Z.; Sismanopoulos, N.; Theoharides, T.C. Stimulated human mast cells secrete mitochondrial components that have autocrine and paracrine inflammatory actions. PLoS ONE 2012, 7, e49767. [Google Scholar] [CrossRef]
- Martelli, M.; Monaldi, C.; De Santis, S.; Bruno, S.; Mancini, M.; Cavo, M.; Soverini, S. Recent Advances in the Molecular Biology of Systemic Mastocytosis: Implications for Diagnosis, Prognosis, and Therapy. Int. J. Mol. Sci. 2020, 21, 3987. [Google Scholar] [CrossRef]
- Jawhar, M.; Schwaab, J.; Schnittger, S.; Sotlar, K.; Horny, H.P.; Metzgeroth, G.; Müller, N.; Schneider, S.; Naumann, N.; Walz, C.; et al. Molecular profiling of myeloid progenitor cells in multi-mutated advanced systemic mastocytosis identifies KIT D816V as a distinct and late event. Leukemia 2015, 29, 1115–1122. [Google Scholar] [CrossRef]
- Arock, M.; Sotlar, K.; Akin, C.; Broesby-Olsen, S.; Hoermann, G.; Escribano, L.; Kristensen, T.K.; Kluin-Nelemans, H.C.; Hermine, O.; Dubreuil, P.; et al. KIT mutation analysis in mast cell neoplasms: Recommendations of the European Competence Network on Mastocytosis. Leukemia 2015, 29, 1223–1232. [Google Scholar] [CrossRef]
- Jawhar, M.; Schwaab, J.; Schnittger, S.; Meggendorfer, M.; Pfirrmann, M.; Sotlar, K.; Horny, H.-P.; Metzgeroth, G.; Kluger, S.; Naumann, N.; et al. Additional mutations in SRSF2, ASXL1 and/or RUNX1 identify a high-risk group of patients with KIT D816V+ advanced systemic mastocytosis. Leukemia 2016, 30, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Jawhar, M.; Schwaab, J.; Naumann, N.; Horny, H.P.; Sotlar, K.; Haferlach, T.; Metzgeroth, G.; Fabarius, A.; Valent, P.; Hofmann, W.-K.; et al. Response and progression on midostaurin in advanced systemic mastocytosis: KIT D816V and other molecular markers. Blood 2017, 130, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Shah, S.; Mannelli, F.; Elala, Y.C.; Guglielmelli, P.; Lasho, T.L.; Patnaik, M.M.; Gangat, N.; Ketterling, R.P.; Reichard, K.K.; et al. Mayo alliance prognostic system for mastocytosis: Clinical and hybrid clinical-molecular models. Blood Adv. 2018, 2, 2964–2972. [Google Scholar] [CrossRef] [PubMed]
- Jawhar, M.; Schwaab, J.; Alvarez-Twose, I.; Naumann, N.; Lübke, J.; Perkins, C.; Muñoz-González, J.I.; Meggendorfer, M.; Kennedy, V.; Metzgeroth, G.; et al. MARS: Mutation-adjusted risk score for advanced systemic mastocytosis. J. Clin. Oncol. 2019, 37, 2846–2856. [Google Scholar] [CrossRef]
- Sperr, W.R.; Kundi, M.; Alvarez-Twose, I.; van Anrooij, B.; Elberink, J.N.G.O.; Górska, A.; Niedoszytko, M.; Gleixner, K.V.; Hadzijusufovic, E.; Zanotti, R.; et al. International prognostic scoring system for mastocytosis (IPSM): A retrospective cohort study. Lancet Haematol. 2019, 6, e638–e649. [Google Scholar] [CrossRef]
- Martinelli, G.; Mancini, M.; De Benedittis, C.; Rondoni, M.; Papayannidis, C.; Manfrini, M.; Meggendorfer, M.; Calogero, R.; Guadagnuolo, V.; Fontana, M.C.; et al. SETD2 and histone H3 lysine 36 methylation deficiency in advanced systemic mastocytosis. Leukemia 2017, 32, 139–148. [Google Scholar] [CrossRef]
- Lim, K.H.; Tefferi, A.; Lasho, T.L.; Finke, C.; Patnaik, M.; Butterfield, J.H.; McClure, R.F.; Li, C.Y.; Pardanani, A. stemic mastocytosis in 342 consecutive adults: Survival studies and prognostic factors. Blood 2009, 113, 5727–57366. [Google Scholar] [CrossRef]
- Tefferi, A.; Shah, S.; Reichard, K.K.; Hanson, C.A.; Pardanani, A. Smoldering mastocytosis: Survival comparisons with indolent and aggressive mastocytosis. Am. J. Hematol. 2019, 94, E1–E2. [Google Scholar] [CrossRef]
- Valent, P.; Sotlar, K.; Sperr, W.R.; Escribano, L.; Yavuz, S.; Reiter, A.; George, T.I.; Kluin-Nelemans, H.C.; Hermine, O.; Butterfield, J.H.; et al. Refined diagnostic criteria and classification of mast cell leukemia (MCL) and myelomastocytic leukemia (MML): A consensus proposal. Ann. Oncol. 2014, 25, 1691–1700. [Google Scholar] [CrossRef]
- Jawhar, M.; Schwaab, J.; Meggendorfer, M.; Naumann, N.; Horny, H.P.; Sotlar, K.; Haferlach, T.; Schmitt, K.; Fabarius, A.; Valent, P.; et al. The clinical and molecular diversity of mast cell leukemia with or without associated hematologic neoplasm. Haematologica 2017, 102, 1035–1043. [Google Scholar] [CrossRef]
- Naumann, N.; Jawhar, M.; Schwaab, J.; Kluger, S.; Lübke, J.; Metzgeroth, G.; Popp, H.D.; Khaled, N.; Horny, H.P.; Sotlar, K.; et al. Incidence and prognostic impact of cytogenetic aberrations in patients with systemic mastocytosis. Genes Chromosomes Cancer 2018, 57, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.H.; Pardanani, A.; Butterfield, J.H.; Li, C.; Tefferi, A. Cytoreductive therapy in 108 adults with systemic mastocytosis: Outcome analysis and response prediction during treatment with interferon-alpha, hydroxyurea, imatinib mesylate or 2-chlorodeoxyadenosine. Am. J. Hematol. 2009, 84, 790–794. [Google Scholar] [CrossRef] [PubMed]
- Gotlib, J.; Kluin-Nelemans, H.C.; George, T.I.; Akin, C.; Sotlar, K.; Hermine, O.; Awan, F.T.; Hexner, E.; Mauro, M.J.; Sternberg, D.W.; et al. Efficacy and Safety of Midostaurin in Advanced Systemic Mastocytosis. N. Engl. J. Med. 2016, 374, 2530–2541. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A. How I treat patients with indolent and smoldering mastocytosis (rare conditions but difficult to manage). Blood 2013, 121, 3085–3094. [Google Scholar] [CrossRef]
- Valent, P.; Hartmann, K.; Schwaab, J.; Alvarez-Twose, I.; Brockow, K.; Bonadonna, P.; Hermine, O.; Niedoszytko, M.; Carter, M.; Hoermann, G.; et al. Personalized Management Strategies in Mast Cell Disorders: ECNM-AIM User’s Guide for Daily Clinical Practice. J. Allergy Clin. Immunol. Pract. 2022. Epub ahead of print. [Google Scholar] [CrossRef]
- Broesby-Olsen, S.; Vestergaard, H.; Mortz, C.G.; Jensen, B.; Havelund, T.; Hermann, A.P.; Siebenhaar, F.; Møller, M.B.; Kristensen, T.K.; Bindslev-Jensen, C. Omalizumab prevents anaphylaxis and improves symptoms in systemic mastocytosis: Efficacy and safety observations. Allergy 2018, 73, 230–238. [Google Scholar] [CrossRef]
- Lemal, R.; Fouquet, G.; Terriou, L.; Vaes, M.; Livideanu, C.B.; Frenzel, L.; Barete, S.; Canioni, D.; Lhermitte, L.; Rossignol, J.; et al. Omalizumab therapy for mast cell-mediator symptoms in patients with ISM, CM, MMAS, and MCAS. J. Allergy Clin. Immunol. Pract. 2019, 7, 2387–2395.e3. [Google Scholar] [CrossRef]
- Douglass, J.A.; Carroll, K.; Voskamp, A.; Bourke, P.; Wei, A.; O’Hehir, R.E. Omalizumab is effective in treating systemic mastocytosis in a nonatopic patient. Allergy 2010, 65, 926–927. [Google Scholar] [CrossRef]
- Carter, M.C.; Robyn, J.A.; Bressler, P.B.; Walker, J.C.; Shapiro, G.G.; Metcalfe, D.D. Omalizumab for the treatment of unprovoked anaphylaxis in patients with systemic mastocytosis. J. Allergy Clin. Immunol. 2007, 119, 1550–1551. [Google Scholar] [CrossRef]
- Kumar, C.; Zito, P.M. Omalizumab; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Jendoubi, F.; Gaudenzio, N.; Gallini, A.; Negretto, M.; Paul, C.; Bulai Livideanu, C. Omalizumab in the treatment of adult patients with mastocytosis: A systematic review. Clin. Exp. Allergy 2020, 50, 654–661. [Google Scholar] [CrossRef]
- Distler, M.; Maul, J.T.; Steiner, U.C.; Jandus, P.; Kolios, A.G.A.; Murer, C.; Graf, N.; Seebach, J.D.; Pichler, W.J.; Navarini, A.A.; et al. Efficacy of omalizumab in mastocytosis: Allusive indication obtained from a prospective, double-blind, multicenter study (XOLMA study). Dermatology 2020, 236, 529–539. [Google Scholar] [CrossRef]
- Caughey, G.H. Mast cell proteases as pharmacological targets. Eur. J. Pharmacol. 2016, 778, 44–55. [Google Scholar] [CrossRef]
- Atiakshin, D.; Buchwalow, I.; Tiemann, M. Mast cell chymase: Morphofunctional characteristics. Histochem. Cell Biol. 2019, 152, 253–269. [Google Scholar] [CrossRef]
- Kosanovic, D.; Luitel, H.; Dahal, B.K.; Cornitescu, T.; Janssen, W.; Danser, A.H.; Garrelds, I.M.; De Mey, J.G.; Fazzi, G.; Schiffers, P.; et al. Chymase: A multifunctional player in pulmonary hypertension associated with lung fibrosis. Eur. Respir. J. 2015, 46, 1084–1094. [Google Scholar] [CrossRef]
- Takai, S.; Jin, D. Chymase as a Possible Therapeutic Target for Amelioration of Non-Alcoholic Steatohepatitis. Int. J. Mol. Sci. 2020, 21, 7543. [Google Scholar] [CrossRef]
- Atiakshin, D.; Buchwalow, I.; Samoilova, V.; Tiemann, M. Tryptase as a polyfunctional component of mast cells. Histochem. Cell Biol. 2018, 149, 461–477. [Google Scholar] [CrossRef]
- Yu, M.; Tsai, M.; Tam, S.Y.; Jones, C.; Zehnder, J.; Galli, S.J. Mast cells can promote the development of multiple features of chronic asthma in mice. J. Clin. Investig. 2006, 116, 1633–1641. [Google Scholar] [CrossRef]
- Ammendola, M.; Sacco, R.; Sammarco, G.; Piardi, T.; Zuccala, V.; Patruno, R.; Zullo, A.; Zizzo, N.; Nardo, B.; Marech, I.; et al. Mast cells positive to tryptase, endothelial cells positive to protease-activated receptor-2, and microvascular density correlate among themselves in hepatocellular carcinoma patients who have undergone surgery. OncoTargets Ther. 2016, 9, 4465–4471. [Google Scholar] [CrossRef]
- Ammendola, M.; Sacco, R.; Zuccala, V.; Luposella, M.; Patruno, R.; Gadaleta, P.; Zizzo, N.; Gadaleta, C.D.; De Sarro, G.; Sammarco, G.; et al. Mast cells density positive to tryptase correlate with microvascular density in both primary gastric cancer tissue and loco-regional lymph node metastases from patients that have undergone radical surgery. Int. J. Mol. Sci. 2016, 17, 1905. [Google Scholar] [CrossRef]
- Maun, H.R.; Jackman, J.K.; Choy, D.F.; Loyet, K.M.; Staton, T.L.; Jia, G.; Dressen, A.; Hackney, J.A.; Bremer, M.; Walters, B.T.; et al. An Allosteric Anti-tryptase Antibody for the Treatment of Mast Cell-Mediated Severe Asthma. Cell 2019, 179, 417–431.e19. [Google Scholar] [CrossRef]
- Rymut, S.M.; Sukumaran, S.; Sperinde, G.; Bremer, M.; Galanter, J.; Yoshida, K.; Smith, J.; Banerjee, P.; Sverkos, V.; Cai, F.; et al. Dose-dependent inactivation of airway tryptase with a novel dissociating anti-tryptase antibody (MTPS9579A) in healthy participants: A randomized trial. Clin. Transl. Sci. 2022, 15, 451–463. [Google Scholar] [CrossRef]
- Alvarado, D.; Maurer, M.; Gedrich, R.; Seibel, S.B.; Murphy, M.B.; Crew, L.; Goldstein, J.; Crocker, A.; Vitale, L.A.; Morani, P.A.; et al. Anti-KIT monoclonal antibody CDX-0159 induces profound and durable mast cell suppression in a healthy volunteer study. Allergy 2022. Epub ahead of print. [Google Scholar] [CrossRef]
- Casassus, P.; Caillat-Vigneron, N.; Martin, A.; Simon, J.; Gallais, V.; Beaudry, P.; Eclache, V.; Laroche, L.; Lortholary, P.; Raphaël, M.; et al. Treatment of adult systemic mastocytosis with interferon-α: Results of a multicentre phase II trial on 20 patients. Br. J. Haematol. 2002, 119, 1090–1097. [Google Scholar] [CrossRef]
- Simon, J.; Lortholary, O.; Caillat-Vigneron, N.; Raphaël, M.; Martin, A.; Brière, J.; Barète, S.; Hermine, O.; Casassus, P. Interest of interferon alpha in systemic mastocytosis. The French experience and review of the literature. Pathol. Biol. 2004, 52, 294–299. [Google Scholar] [CrossRef]
- Pardanani, A.; Hoffbrand, A.V.; Butterfield, J.H.; Tefferi, A. Treatment of systemic mast cell disease with 2-chlorodeoxyadenosine. Leuk. Res. 2004, 28, 127–131. [Google Scholar] [CrossRef]
- Barete, S.; Lortholary, O.; Damaj, G.; Hirsch, I.; Chandesris, M.O.; Elie, C.; Hamidou, M.; Durieu, I.; Suarez, F.; Grosbois, B.; et al. Long-term efficacy and safety of cladribine (2-CdA) in adult patients with mastocytosis. Blood 2015, 126, 1009–1016. [Google Scholar] [CrossRef]
- Flynn, J.P.; Gerriets, V. Imatinib; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Ma, Y.; Zeng, S.; Metcalfe, D.D.; Akin, C.; Dimitrijevic, S.; Butterfield, J.H.; McMahon, G.; Longley, B.J. The c-KIT mutation causing human mastocytosis is resistant to STI571 and other KIT kinase inhibitors; kinases with enzymatic site mutations show different inhibitor sensitivity profiles than wild-type kinases and those with regulatory-type mutations. Blood 2002, 99, 1741–1744. [Google Scholar] [CrossRef]
- Frost, M.J.; Ferrao, P.T.; Hughes, T.P.; Ashman, L.K. Juxtamembrane mutant V560GKit is more sensitive to Imatinib (STI571) compared with wild-type c-Kit whereas the kinase domain mutant D816VKit is resistant. Mol. Cancer Ther. 2002, 1, 1115–1124. [Google Scholar]
- Droogendijk, H.J.; Kluin-Nelemans, H.J.; van Doormaal, J.J.; Oranje, A.P.; van de Loosdrecht, A.A.; van Daele, P.L. Imatinib mesylate in the treatment of systemic mastocytosis: A phase II trial. Cancer 2006, 107, 345–351. [Google Scholar] [CrossRef]
- Pagano, L.; Valentini, C.G.; Caira, M.; Rondoni, M.; Van Lint, M.T.; Candoni, A.; Allione, B.; Cattaneo, C.; Marbello, L.; Caramatti, C.; et al. Advanced mast cell disease: An Italian Hematological Multicenter experience. Int. J. Hematol. 2009, 88, 483–488. [Google Scholar] [CrossRef]
- Vega-Ruiz, A.; Cortes, J.E.; Sever, M.; Manshouri, T.; Quintás-Cardama, A.; Luthra, R.; Kantarjian, H.M.; Verstovsek, S. Phase II study of imatinib mesylate as therapy for patients with systemic mastocytosis. Leuk. Res. 2009, 33, 1481–1484. [Google Scholar] [CrossRef]
- Pardanani, A.; Elliott, M.; Reeder, T.; Li, C.Y.; Baxter, E.J.; Cross, N.C.; Tefferi, A. Imatinib for systemic mast-cell disease. Lancet 2003, 362, 535–537. [Google Scholar] [CrossRef]
- Alvarez-Twose, I.; Matito, A.; Morgado, J.M.; Sánchez-Muñoz, L.; Jara-Acevedo, M.; García-Montero, A.; Mayado, A.; Caldas, C.; Teodósio, C.; Muñoz-González, J.I.; et al. Imatinib in systemic mastocytosis: A phase IV clinical trial in patients lacking exon 17 KIT mutations and review of the literature. Oncotarget 2016, 8, 68950–68963. [Google Scholar] [CrossRef]
- Dubreuil, P.; Letard, S.; Ciufolini, M.; Gros, L.; Humbert, M.; Castéran, N.; Borge, L.; Hajem, B.; Lermet, A.; Sippl, W.; et al. Masitinib (AB1010), a Potent and Selective Tyrosine Kinase Inhibitor Targeting KIT. PLoS ONE 2009, 4, e7258. [Google Scholar] [CrossRef]
- Paul, C.; Sans, B.; Suarez, F.; Casassus, P.; Barete, S.; Lanternier, F.; Grandpeix-Guyodo, C.; Dubreuil, P.; Palmérini, F.; Mansfield, C.D.; et al. Masitinib for the treatment of systemic and cutaneous mastocytosis with handicap: A phase 2a study. Am. J. Hematol. 2010, 85, 921–925. [Google Scholar] [CrossRef]
- Lortholary, O.; Chandesris, M.O.; Livideanu, C.B.; Paul, C.; Guillet, G.; Jassem, E.; Niedoszytko, M.; Barete, S.; Verstovsek, S.; Grattan, C.; et al. Masitinib for treatment of severely symptomatic indolent systemic mastocytosis: A randomised, placebo-controlled, phase 3 study. Lancet 2017, 389, 612–620. [Google Scholar] [CrossRef]
- Tzogani, K.; Yu, Y.; Meulendijks, D.; Herberts, C.; Hennik, P.; Verheijen, R.; Wangen, T.; Dahlseng Håkonsen, G.; Kaasboll, T.; Dalhus, M.; et al. European Medicines Agency review of midostaurin (Rydapt) for the treatment of adult patients with acute myeloid leukaemia and systemic mastocytosis. ESMO Open 2019, 4, e000606. [Google Scholar] [CrossRef]
- Fabrro, D.; Ruetz, S.; Bodis, S.; Pruschy, M.; Csermak, K.; Man, A.; Campochiaro, P.; Wood, J.; O’Reilly, T.; Meyer, T. PKC412–a protein kinase inhibitor with a broad therapeutic potential. Anticancer Drug Des. 2000, 15, 17–28. [Google Scholar]
- DeAngelo, D.J.; George, T.I.; Linder, A.; Langford, C.; Perkins, C.; Ma, J.; Westervelt, P.; Merker, J.D.; Berube, C.; Coutre, S.; et al. Efficacy and safety of midostaurin in patients with advanced systemic mastocytosis: 10-year median follow-up of a phase II trial. Leukemia 2018, 32, 470–478. [Google Scholar] [CrossRef]
- Chandesris, M.-O.; Damaj, G.; Canioni, D.; Brouzes, C.; Lhermitte, L.; Hanssens, K.; Frenzel, L.; Cherquaoui, Z.; Durieu, I.; Durupt, S.; et al. Midostaurin in Advanced Systemic Mastocytosis. N. Engl. J. Med. 2016, 374, 2605–2606. [Google Scholar] [CrossRef]
- Reiter, A.; Kluin-Nelemans, H.C.; George, T.; Akin, C.; DeAngelo, D.J.; Hermine, O.; Awan, F.; Hexner, E.; Mauro, M.; Schwaab, J.; et al. Pooled Survival Analysis Of Midostaurin Clinical Study Data (D2201 + A2213) In Patients With Advanced Systemic Mastocytosis (Advsm) Compared With Historical Controls. Haematologica 2017, 102, 321–323. [Google Scholar]
- Lübke, J.; Schwaab, J.; Naumann, N.; Horny, H.P.; Weiß, C.; Metzgeroth, G.; Kreil, S.; Cross, N.C.P.; Sotlar, K.; Fabarius, A.; et al. Superior Efficacy of Midostaurin Over Cladribine in Advanced Systemic Mastocytosis: A Registry-Based Analysis. J. Clin. Oncol. 2022, 40, 1783–1794. [Google Scholar] [CrossRef] [PubMed]
- Van Anrooij, B.; Oude Elberink, J.N.G.; Span, L.F.R.; de Monchy, J.G.R.; Rosati, S.; Mulder, A.B.; Kluin-Nelemans, J.C. Midostaurin in patients with indolent systemic mastocytosis: An open-label phase 2 trial. J. Allergy Clin. Immunol. 2018, 142, 1006–1008.e7. [Google Scholar] [CrossRef] [PubMed]
- Farrukh, F.; Gangat, N.; Shah, M.V.; Litzow, M.R.; Elliott, M.A.; Begna, K.; Hook, C.C.; Tefferi, A.; Pardanani, A. Midostaurin therapy for indolent and smoldering systemic mastocytosis: Retrospective review of Mayo Clinic experience. Am. J. Hematol. 2022, 97, E138–E140. [Google Scholar] [CrossRef]
- Evans, E.K.; Gardino, A.K.; Kim, J.L.; Hodous, B.L.; Shutes, A.; Davis, A.; Zhu, X.J.; Schmidt-Kittler, O.; Wilson, D.; Wilson, K.; et al. A precision therapy against cancers driven by KIT/PDGFRA mutations. Sci. Transl. Med. 2017, 9, eaao1690. [Google Scholar] [CrossRef]
- DeAngelo, D.J.; Radia, D.H.; George, T.I.; Robinson, W.A.; Quiery, A.T.; Drummond, M.W.; Bose, P.; Hexner, E.O.; Winton, E.F.; Horny, H.P.; et al. Safety and efficacy of avapritinib in advanced systemic mastocytosis: The phase 1 EXPLORER trial. Nat. Med. 2021, 27, 2183–2191. [Google Scholar] [CrossRef]
- Gotlib, J.; Reiter, A.; Radia, D.H.; Deininger, M.W.; George, T.I.; Panse, J.; Vannucchi, A.M.; Platzbecker, U.; Alvarez-Twose, I.; Mital, A.; et al. Efficacy and safety of avapritinib in advanced systemic mastocytosis: Interim analysis of the phase 2 PATHFINDER trial. Nat. Med. 2021, 27, 2192–2199. [Google Scholar] [CrossRef]
- Akin, C.; Elberink, H.O.; Gotlib, J.; Sabato, V.; Hartmann, K.; Broesby-Olsen, S.; Castells, M.; Tashi, T.; Heaney, M.L.; George, T.I.; et al. Pioneer Part 2: A Randomized, Double-Blind, Placebo-Controlled, Phase 2 Study to Evaluate Safety and Efficacy of Avapritinib in Indolent Systemic Mastocytosis. Blood 2020, 136, 41–42. [Google Scholar] [CrossRef]
- Dhillon, S. Ripretinib: First Approval. Drugs 2020, 80, 1133–1138. [Google Scholar] [CrossRef]
- Dave, N.; Devlin, M.; Rodstrom, J.; Yu, B.; Foley, M.; He, K.; Rassmussen, S.; Boral, A.; Dong Si, T. Safety and pharmacokinetics of BLU-263, a next-generation KIT inhibitor, in healthy volunteers. AACR 2021, 81 (Suppl. 13). [Google Scholar] [CrossRef]
- Guarnieri, A.; Chicarelli, M.; Cable, L.; Bouhana, K.; Sullivan, F.; Ball, H.; Sachs, J.; Winski, S.; Robinson, J. Preclinical Data with KIT D816V Inhibitor Bezuclastinib (CGT9486) Demonstrates High Selectivity and Minimal Brain Penetrance. Blood 2021, 138 (Suppl. S1), 4595. [Google Scholar] [CrossRef]
- Ustun, C.; Reiter, A.; Scott, B.L.; Nakamura, R.; Damaj, G.; Kreil, S.; Shanley, R.; Hogan, W.J.; Perales, M.A.; Shore, T.; et al. Hematopoietic stem-cell transplantation for advanced systemic mastocytosis. J. Clin. Oncol. 2014, 32, 3264–3274. [Google Scholar] [CrossRef] [PubMed]
- Ustun, C.; Gotlib, J.; Popat, U.; Artz, A.; Litzow, M.; Reiter, A.; Nakamura, R.; Kluin-Nelemans, H.C.; Verstovsek, S.; Gajewski, J.; et al. Consensus Opinion on Allogeneic Hematopoietic Cell Transplantation in Advanced Systemic Mastocytosis. Biol. Blood Marrow Transplant. 2016, 22, 1348–1356. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Drugs | Type of Study | Patients | Outcomes |
|---|---|---|---|
| Imatinib [52] | Phase II | 12 ISM/SSM + 2 ASM | CR 7%, major response 36% |
| Imatinib [54] | Phase II | 11 ISM + 4 ASM + 5 SM-AHN | CR 5% |
| Masitinib [58] | Phase II | 25 cutaneous mastocytosis or SM with disabilities associated with mediator-related symptoms | ORR 56% |
| Masitinib [59] | Phase III, placebo controlled | 135 ISM/cutaneous mastocytosis | ORR 18.7% |
| Midostaurin [62] | Phase II | 3 ASM + 17 SM-AHN + 6 MCL | ORR 69%, median OS 40 months |
| Midostaurin [24] | Phase II | 16 ASM + 57 SM-AHN + 16 MCL | ORR 60%, median OS 28.7 months |
| Midostaurin [66] | Phase II | 20 ISM patients with severe mediator-related symptoms | 35% and 38% reduction in severity of symptoms, at 12 and 24 months, respectively |
| Avapritinib [70] | Phase II | 9 ASM + 43 SM-AHN + 10 MCL | ORR 75% in 32 response-evaluable patients (CR 19%) |
| Avapritinib [71] | Phase II, randomized, double-blind, placebo-controlled | 204 ISM | Reduction in total symptoms score at 16 weeks 30% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sciumè, M.; De Magistris, C.; Galli, N.; Ferretti, E.; Milesi, G.; De Roberto, P.; Fabris, S.; Grifoni, F.I. Target Therapies for Systemic Mastocytosis: An Update. Pharmaceuticals 2022, 15, 738. https://doi.org/10.3390/ph15060738
Sciumè M, De Magistris C, Galli N, Ferretti E, Milesi G, De Roberto P, Fabris S, Grifoni FI. Target Therapies for Systemic Mastocytosis: An Update. Pharmaceuticals. 2022; 15(6):738. https://doi.org/10.3390/ph15060738
Chicago/Turabian StyleSciumè, Mariarita, Claudio De Magistris, Nicole Galli, Eleonora Ferretti, Giulia Milesi, Pasquale De Roberto, Sonia Fabris, and Federica Irene Grifoni. 2022. "Target Therapies for Systemic Mastocytosis: An Update" Pharmaceuticals 15, no. 6: 738. https://doi.org/10.3390/ph15060738
APA StyleSciumè, M., De Magistris, C., Galli, N., Ferretti, E., Milesi, G., De Roberto, P., Fabris, S., & Grifoni, F. I. (2022). Target Therapies for Systemic Mastocytosis: An Update. Pharmaceuticals, 15(6), 738. https://doi.org/10.3390/ph15060738