
Synthesis, Antiplasmodial, and Antileukemia Activity of Dihydroartemisinin–HDAC Inhibitor Hybrids as Multitarget Drugs

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
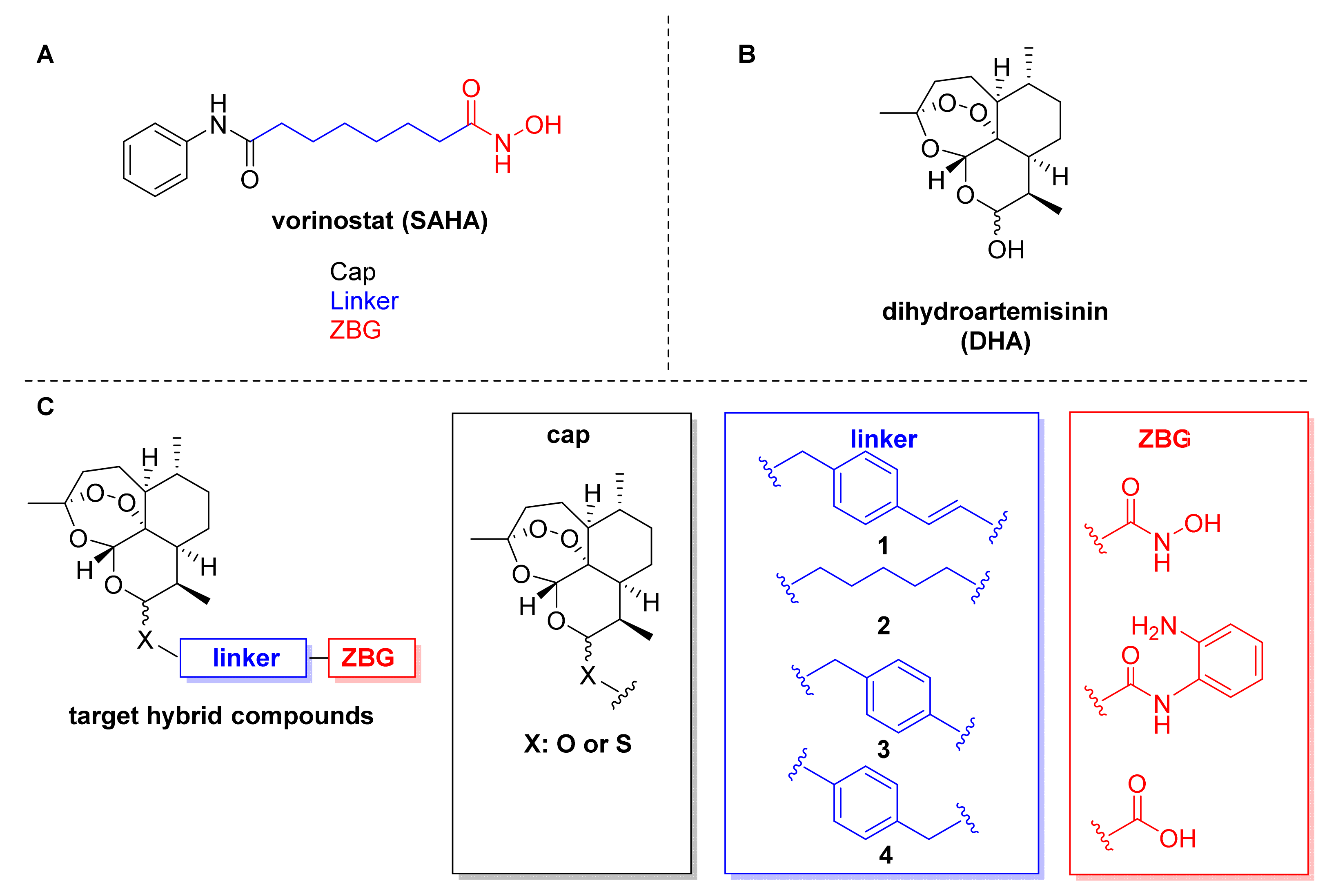
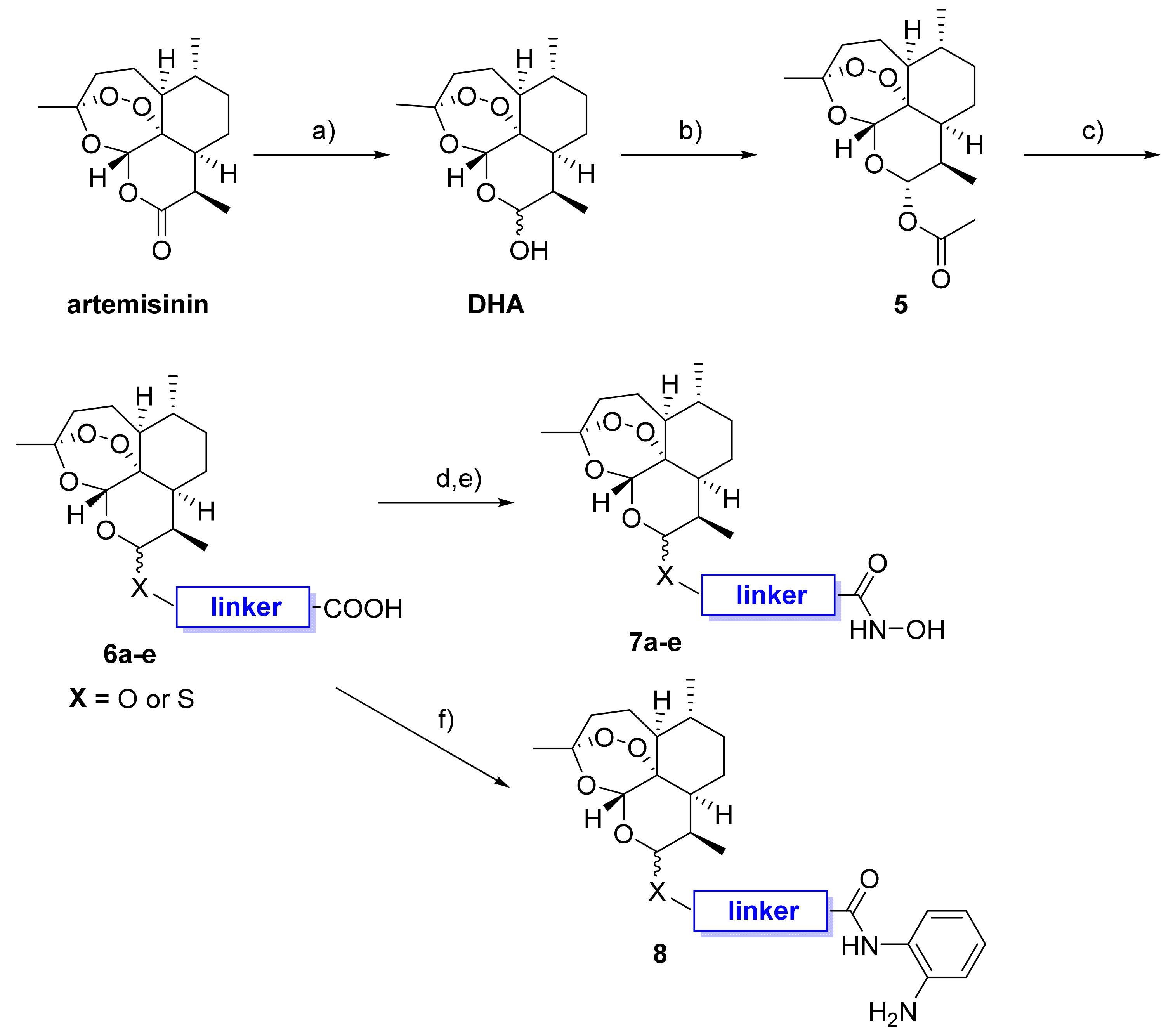
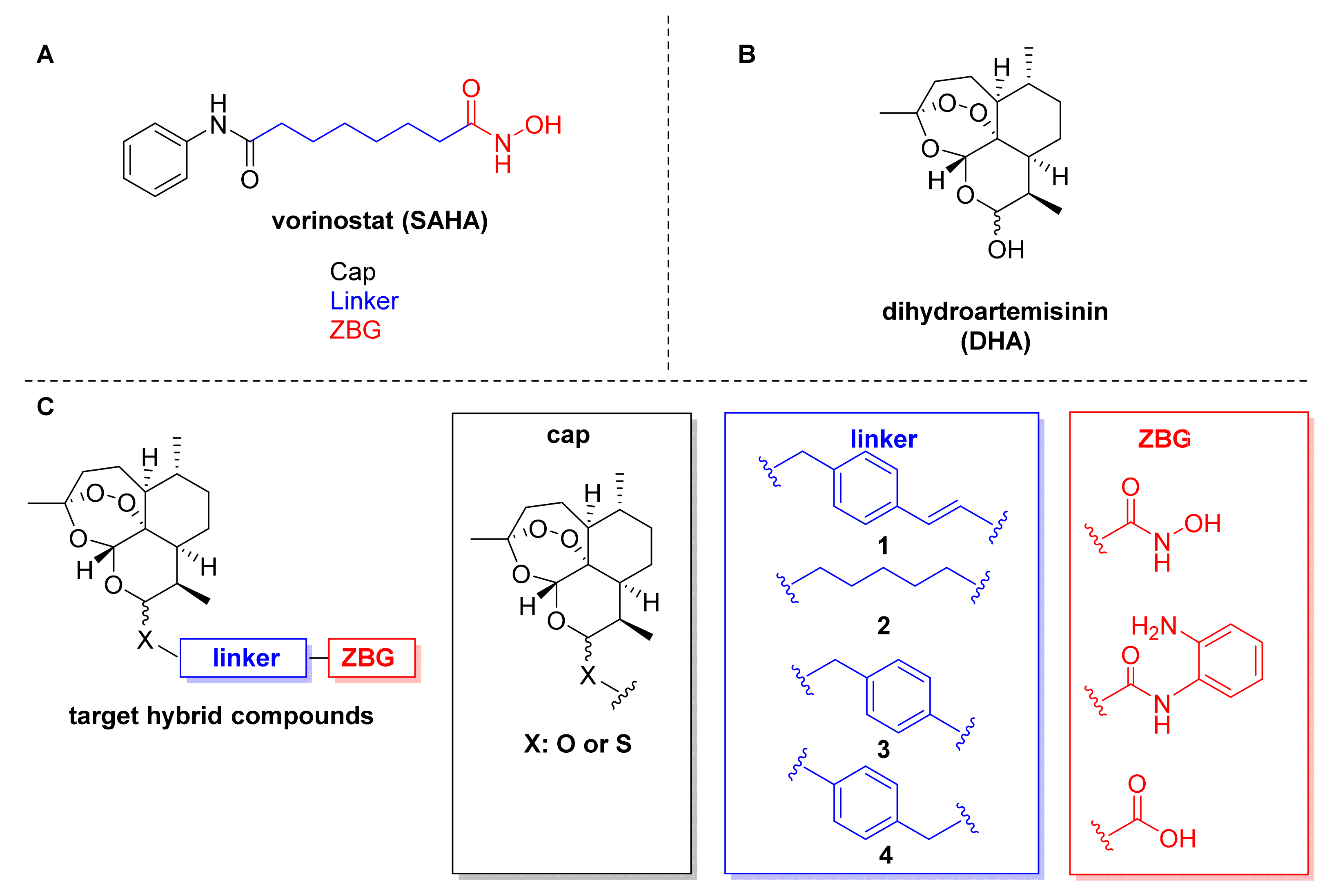
2.1. Design and Synthesis of Dihydroartemisinin–HDACi Hybrids
2.2. HDAC Inhibitory Activities and Selectivity Profiles
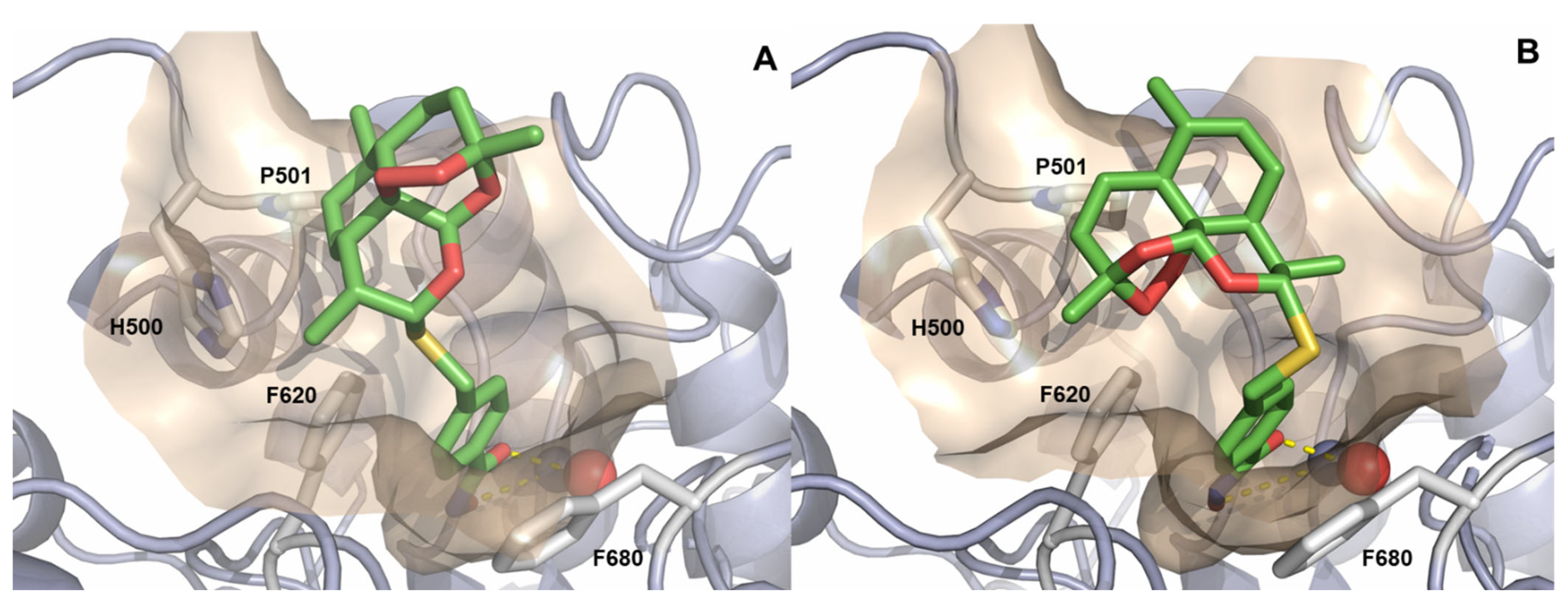
2.3. Docking of (α)-7c and (β)-7c
2.4. Antiplasmodial Properties and Parasite Selectivity
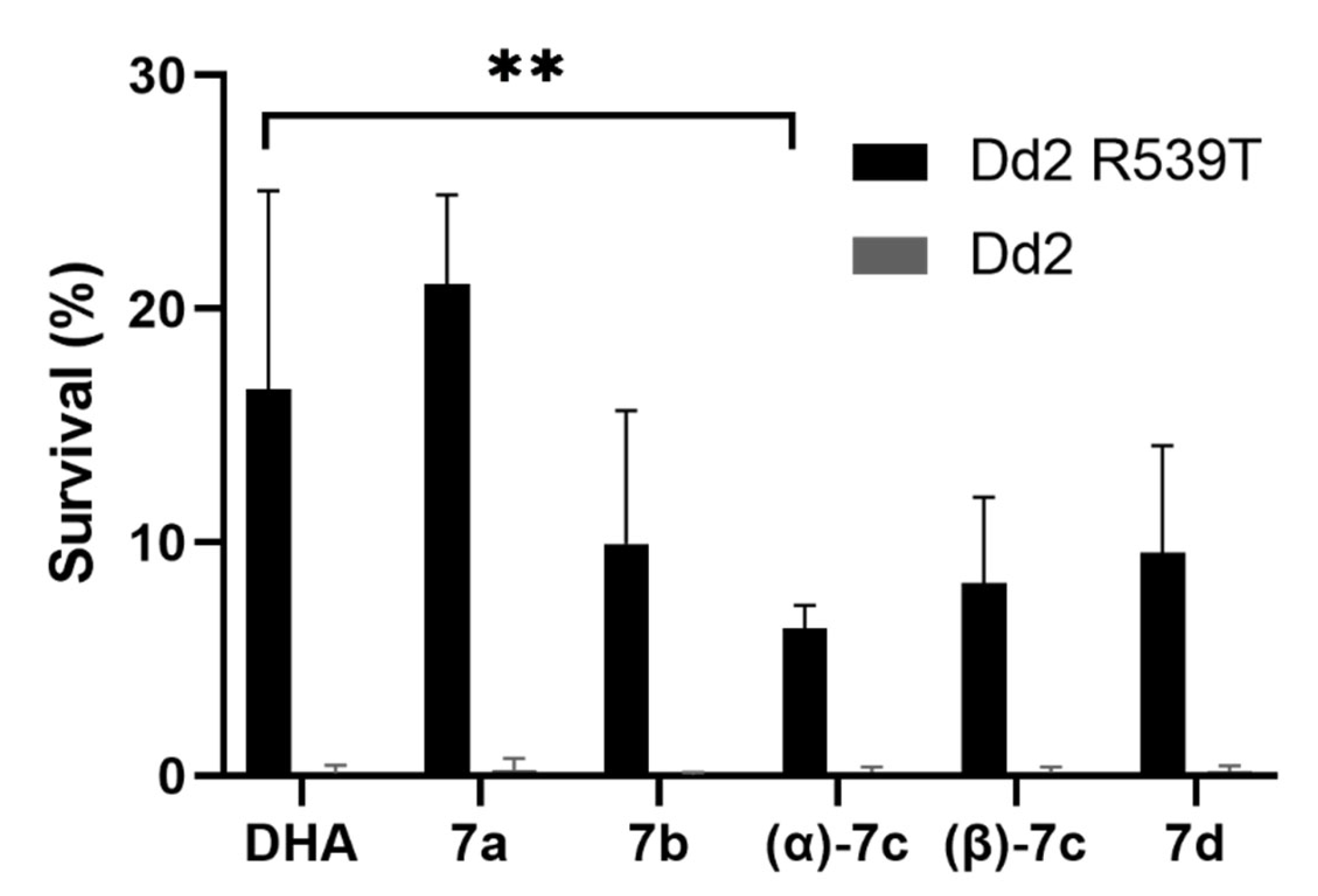
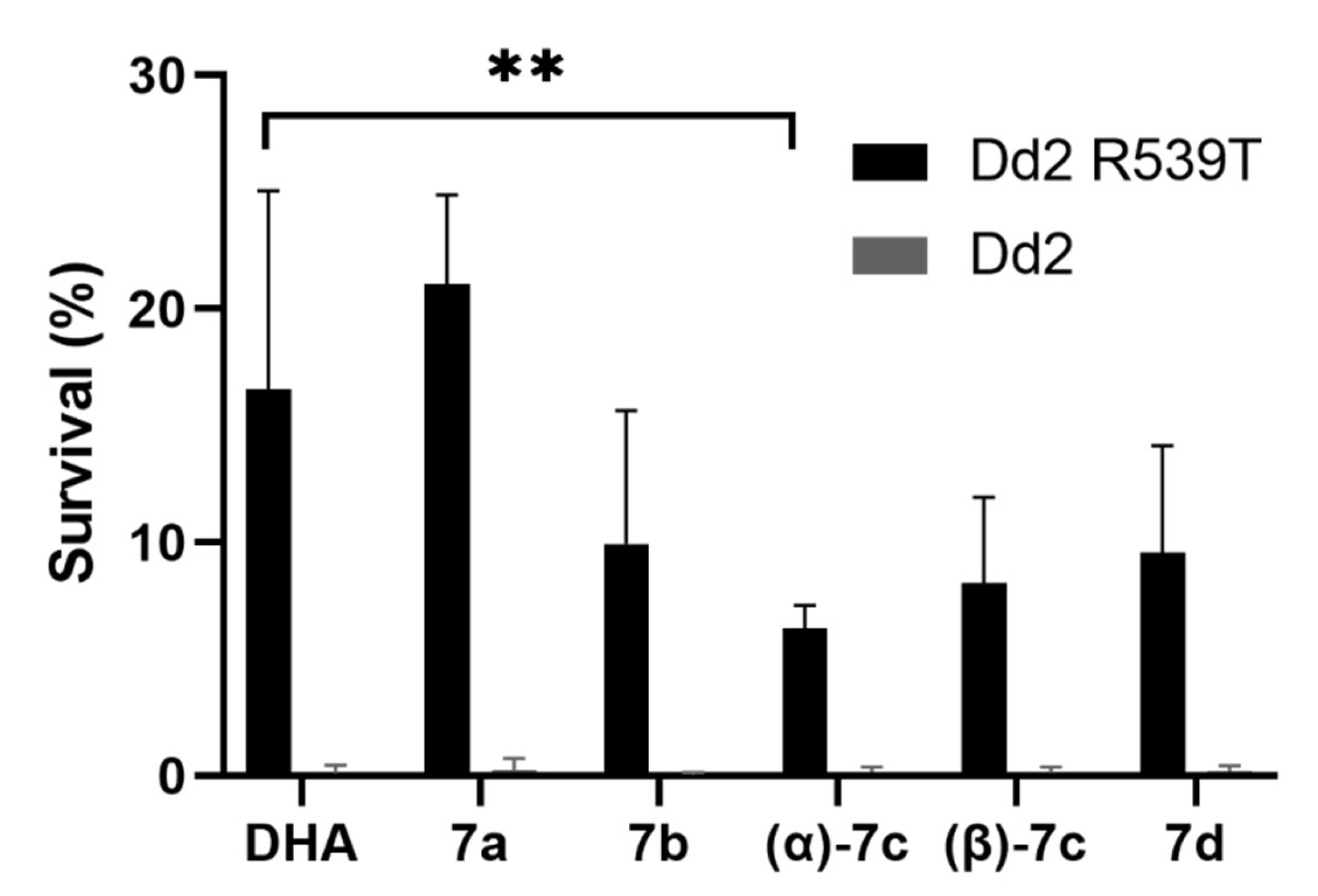
2.5. Ring-Stage Survival Assay
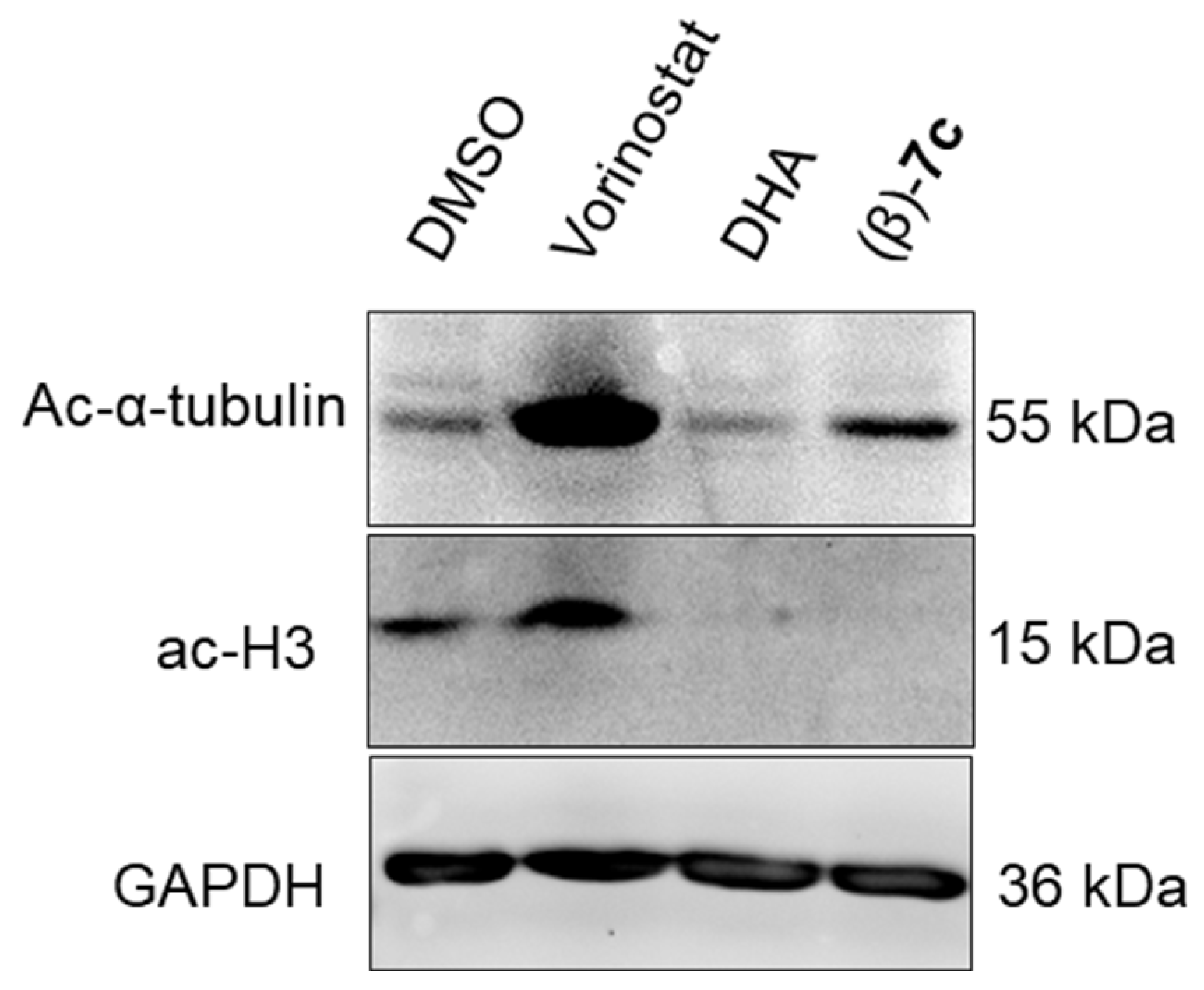
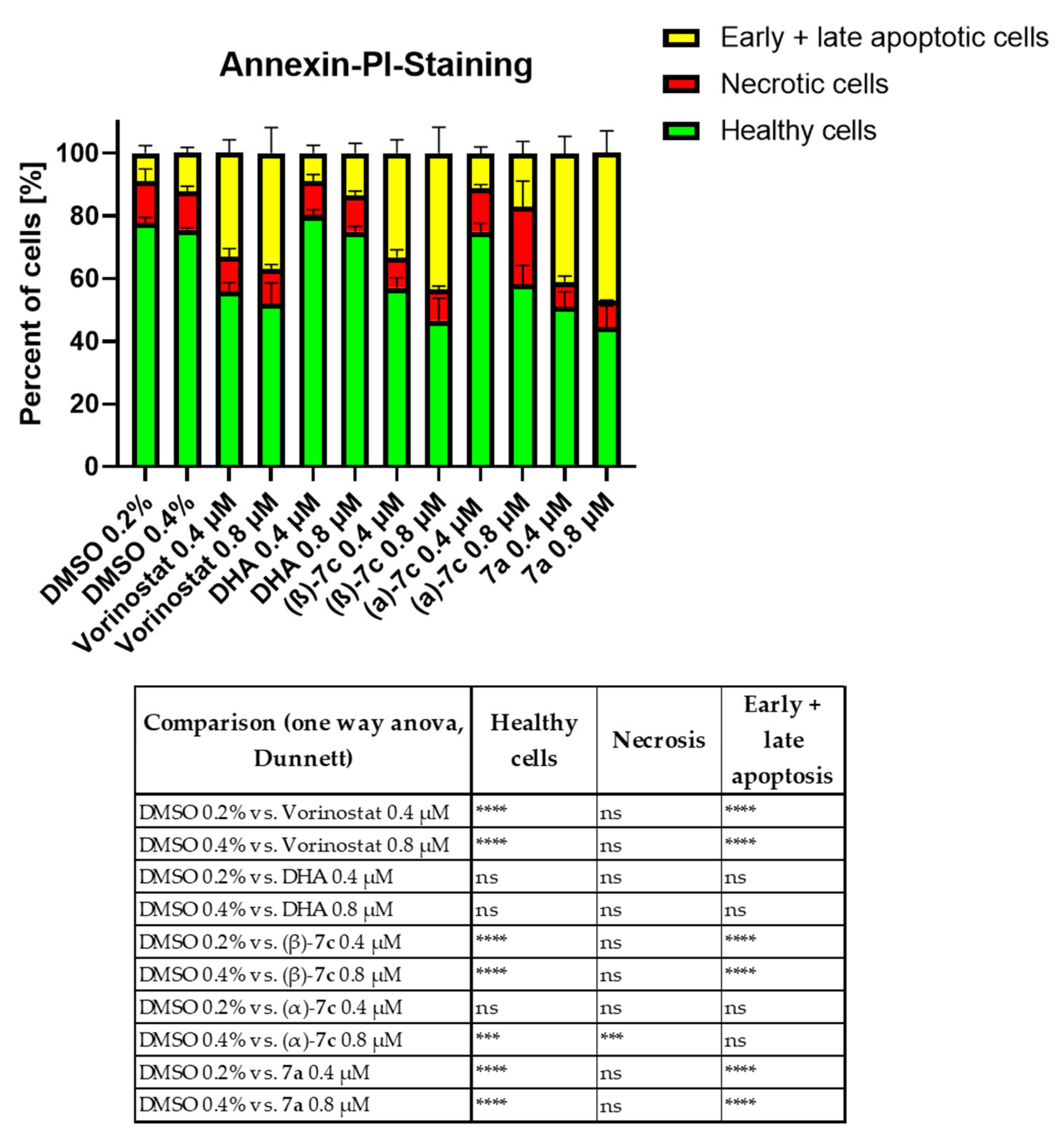
2.6. Antileukemia Properties
3. Materials and Methods
3.1. Chemistry
3.2. General Method for the Synthesis of DHA-Coupled Carboxylic Acids 6a–e
3.2.1. (E)-3-(4-((((3R,5aS,6R,8aS,9R,12R,12aR)-3,6,9-Trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)oxy)methyl)phenyl)acrylic acid (6a)
3.2.2. 6-(((3R,5aS,6R,8aS,9R,12S,12aR)-3,6,9-Trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)thio)hexanoic acid (6b)
3.2.3. 4-((((3R,5aS,6R,8aS,9R,10R,12S,12aR)-3,6,9-Trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)thio)methyl)benzoic acid (6c)
3.2.4. 4-((((3R,5aS,6R,8aS,12R,12aR)-3,6,9-Trimethyldecahydro-12H-3,12-methano[1,2]dioxepino[4,3-i]isochromen-10-yl)oxy)methyl)benzoic acid (6d)
3.3. General Method for the Synthesis of the Hydroxamic Acids 7a–e
3.3.1. (E)-N-Hydroxy-3-(4-((((3R,5aS,6R,8aS,9R,12R,12aR)-3,6,9-trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)oxy)methyl)phenyl)acrylamide (7a)
3.3.2. N-Hydroxy-6-(((3R,5aS,6R,8aS,9R,12S,12aR)-3,6,9-trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)thio)hexanamide (7b)
3.3.3. N-Hydroxy-4-((((3R,5aS,6R,8aS,9R,10R,12S,12aR)-3,6,9-trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)thio)methyl)benzamide ((α)-7c)
3.3.4. N-Hydroxy-4-((((3R,5aS,6R,8aS,9R,10S,12S,12aR)-3,6,9-trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)thio)methyl)benzamide ((β)-7c)
3.3.5. N-Hydroxy-4-((((3R,5aS,6R,8aS,9R,10R,12R,12aR)-3,6,9-trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)oxy)methyl)benzamide (7d)
3.3.6. N-Hydroxy-2-(4-(((3R,5aS,6R,8aS,9R,10R,12S,12aR)-3,6,9-trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)thio)phenyl)acetamide ((α)-7e)
3.3.7. N-Hydroxy-2-(4-(((3R,5aS,6R,8aS,9R,10S,12S,12aR)-3,6,9-trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)thio)phenyl)acetamide ((β)-7e)
3.4. (E)-N-(2-Aminophenyl)-3-(4-((((3R,5aS,6R,8aS,9R,12R,12aR)-3,6,9-trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)oxy)methyl)phenyl)acrylamide (8)
3.5. Biological Evaluation
3.5.1. In Vitro Human HDAC1 and 6 Assay
3.5.2. MTT Cell Viability Assay
3.5.3. Cell Culture (Leukemia Cell Lines and Fibroblasts)
3.5.4. CellTiter-Glo Based Cell Viability Assay
3.5.5. Annexin V-PI Staining
3.5.6. Immunoblotting
3.5.7. In Vitro P. falciparum Growth Inhibition Assay
3.5.8. Ring-Stage Survival Assay (0–3 h)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. World Malaria Report 2020—20 Years of Global Progress & Challenges; WHO: Geneva, Switzerland, 2020; pp. 14–15. [Google Scholar]
- Ariey, F.; Witkowski, B.; Amaratunga, C.; Beghain, J.; Langlois, A.-C.; Khim, N.; Kim, S.; Duru, V.; Bouchier, C.; Ma, L.; et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 2014, 505, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Dondorp, A.M.; Nosten, F.; Yi, P.; Das, D.; Phyo, A.P.; Tarning, J.; Lwin, K.M.; Ariey, F.; Hanpithakpong, W.; Lee, S.J.; et al. Artemisinin Resistance in Plasmodium falciparum Malaria. N. Engl. J. Med. 2009, 361, 455–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrows, J.N.; Duparc, S.; Gutteridge, W.E.; van Huijsduijnen, R.H.; Kaszubska, W.; Macintyre, F.; Mazzuri, S.; Möhrle, J.J.; Wells, T.N.C. New developments in anti-malarial target candidate and product profiles. Malar. J. 2017, 16, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, K.T.; Haque, A.; Jones, M.K. HDAC inhibitors in parasitic diseases. Immunol. Cell Biol. 2012, 90, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [Green Version]
- Duffy, M.F.; Selvarajah, S.A.; Josling, G.A.; Petter, M. Epigenetic regulation of the Plasmodium falciparum genome. Brief. Funct. Genom. 2014, 13, 203–216. [Google Scholar] [CrossRef] [Green Version]
- Volz, J.C.; Bártfai, R.; Petter, M.; Langer, C.; Josling, G.A.; Tsuboi, T.; Schwach, F.; Baum, J.; Rayner, J.C.; Stunnenberg, H.G.; et al. PfSET10, a Plasmodium falciparum methyltransferase, maintains the active var gene in a poised state during parasite division. Cell Host Microbe 2012, 11, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Coleman, B.I.; Skillman, K.M.; Jiang, R.H.Y.; Childs, L.M.; Altenhofen, L.M.; Ganter, M.; Leung, Y.; Goldowitz, I.; Kafsack, B.F.C.; Marti, M.; et al. A Plasmodium falciparum histone deacetylase regulates antigenic variation and gametocyte conversion. Cell Host Microbe 2014, 16, 177–186. [Google Scholar] [CrossRef] [Green Version]
- Avelar, L.A.A.; Held, J.; Engel, J.A.; Sureechatchaiyan, P.; Hansen, F.K.; Hamacher, A.; Kassack, M.U.; Mordmüller, B.; Andrews, K.T.; Kurz, T. Design and Synthesis of Novel Anti-Plasmodial Histone Deacetylase Inhibitors Containing an Alkoxyamide Connecting Unit. Arch. Pharm. 2017, 350, 1600347. [Google Scholar] [CrossRef]
- Chua, M.J.; Arnold, M.S.J.; Xu, W.; Lancelot, J.; Lamotte, S.; Späth, G.F.; Prina, E.; Pierce, R.J.; Fairlie, D.P.; Skinner-Adams, T.S.; et al. Effect of clinically approved HDAC inhibitors on Plasmodium, Leishmania and Schistosoma parasite growth. Int. J. Parasitol. Drugs Drug Resist. 2017, 7, 42–50. [Google Scholar] [CrossRef] [PubMed]
- De Vreese, R.; De Kock, C.; Smith, P.J.; Chibale, K.; D’Hooghe, M. Exploration of thiaheterocyclic hHDAC6 inhibitors as potential antiplasmodial agents. Future Med. Chem. 2017, 9, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Diedrich, D.; Stenzel, K.; Hesping, E.; Antonova-Koch, Y.; Gebru, T.; Duffy, S.; Fisher, G.; Schöler, A.; Meister, S.; Kurz, T.; et al. One-pot, multi-component synthesis and structure-activity relationships of peptoid-based histone deacetylase (HDAC) inhibitors targeting malaria parasites. Eur. J. Med. Chem. 2018, 158, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Mackwitz, M.K.W.; Hesping, E.; Antonova-Koch, Y.; Diedrich, D.; Woldearegai, T.G.; Skinner-Adams, T.; Clarke, M.; Schöler, A.; Limbach, L.; Kurz, T.; et al. Structure-Activity and Structure-Toxicity Relationships of Peptoid-Based Histone Deacetylase Inhibitors with Dual-Stage Antiplasmodial Activity. ChemMedChem 2019, 14, 912–926. [Google Scholar] [CrossRef]
- Mackwitz, M.K.W.; Hesping, E.; Eribez, K.; Schöler, A.; Antonova-Koch, Y.; Held, J.; Winzeler, E.A.; Andrews, K.T.; Hansen, F.K. Investigation of the in vitro and in vivo efficacy of peptoid-based HDAC inhibitors with dual-stage antiplasmodial activity. Eur. J. Med. Chem. 2021, 211, 113065. [Google Scholar] [CrossRef]
- Rosini, M. Polypharmacology: The rise of multitarget drugs over combination therapies. Future Med. Chem. 2014, 6, 485–487. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R.; Rankovic, Z. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2019, 62, 420–444. [Google Scholar] [CrossRef]
- Zhang, Z.; Hou, S.; Chen, H.; Ran, T.; Jiang, F.; Bian, Y.; Zhang, D.; Zhi, Y.; Wang, L.; Zhang, L.; et al. Targeting epigenetic reader and eraser: Rational design, synthesis and in vitro evaluation of dimethylisoxazoles derivatives as BRD4/HDAC dual inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 2931–2935. [Google Scholar] [CrossRef]
- Botta, L.; Filippi, S.; Bizzarri, B.M.; Zippilli, C.; Meschini, R.; Pogni, R.; Baratto, M.C.; Villanova, L.; Saladino, R. Synthesis and Evaluation of Artemisinin-Based Hybrid and Dimer Derivatives as Antimelanoma Agents. ACS Omega 2020, 5, 243–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Çapcı, A.; Lorion, M.M.; Wang, H.; Simon, N.; Leidenberger, M.; Silva, M.C.B.; Moreira, D.R.M.; Zhu, Y.; Meng, Y.; Chen, J.Y.; et al. Artemisinin–(Iso)quinoline Hybrids by C−H Activation and Click Chemistry: Combating Multidrug-Resistant Malaria. Angew. Chem. 2019, 131, 13200–13213. [Google Scholar] [CrossRef] [Green Version]
- Ha, V.T.; Kien, V.T.; Le Binh, H.; Tien, V.D.; My, N.T.T.; Nam, N.H.; Baltas, M.; Hahn, H.; Han, B.W.; Thao, D.T.; et al. Design, synthesis and biological evaluation of novel hydroxamic acids bearing artemisinin skeleton. Bioorg. Chem. 2016, 66, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Maolanon, A.R.; Kristensen, H.M.E.; Leman, L.J.; Ghadiri, M.R.; Olsen, C.A. Natural and Synthetic Macrocyclic Inhibitors of the Histone Deacetylase Enzymes. Chembiochem 2017, 18, 5–49. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, J.; Jiang, Q.; Zhang, L.; Song, W. Zinc binding groups for histone deacetylase inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef]
- Gour, R.; Ahmad, F.; Prajapati, S.K.; Giri, S.K.; Karna, S.K.L.; Kartha, K.P.R.; Pokharel, Y.R. Synthesis of novel S-linked dihydroartemisinin derivatives and evaluation of their anticancer activity. Eur. J. Med. Chem. 2019, 178, 552–570. [Google Scholar] [CrossRef]
- Ontoria, J.M.; Paonessa, G.; Ponzi, S.; Ferrigno, F.; Nizi, E.; Biancofiore, I.; Malancona, S.; Graziani, R.; Roberts, D.; Willis, P.; et al. Discovery of a Selective Series of Inhibitors of Plasmodium falciparum HDACs. ACS Med. Chem. Lett. 2016, 7, 454–459. [Google Scholar] [CrossRef]
- Hai, Y.; Christianson, D.W. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol. 2016, 12, 741–747. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Status Report on Artemesinin and ACT Resistance; WHO: Geneva, Switzerland, 2015. [Google Scholar]
- Ashley, E.A.; Dhorda, M.; Fairhurst, R.M.; Amaratunga, C.; Lim, P.; Suon, S.; Sreng, S.; Anderson, J.M.; Mao, S.; Sam, B.; et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2014, 371, 411–423. [Google Scholar] [CrossRef] [Green Version]
- Suresh, N.; Haldar, K. Mechanisms of artemisinin resistance in Plasmodium falciparum malaria. Curr. Opin. Pharmacol. 2018, 42, 46–54. [Google Scholar] [CrossRef]
- Lam, N.S.; Long, X.; Wong, J.W.; Griffin, R.C.; Doery, J.C.G. Artemisinin and its derivatives: A potential treatment for leukemia. Anticancer Drugs 2019, 30, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Presser, A.; Feichtinger, A.; Buzzi, S. A simplified and scalable synthesis of artesunate. Mon. Chem. 2017, 148, 63–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdeljac, N.; Bussmann, K.; Schöler, A.; Hansen, F.K.; Gilmour, R. Fluorinated Analogues of the Histone Deacetylase Inhibitor Vorinostat (Zolinza): Validation of a Chiral Hybrid Bioisostere, BITE. ACS Med. Chem. Lett. 2019, 10, 1336–1340. [Google Scholar] [CrossRef] [PubMed]
- Selg, C.; Schöler, A.; Schliehe-Diecks, J.; Hanl, M.; Sinatra, L.; Borkhardt, A.; Sárosi, M.B.; Bhatia, S.; Hey-Hawkins, E.; Hansen, F.K. Borinostats: Solid-phase synthesis of carborane-capped histone deacetylase inhibitors with a tailor-made selectivity profile. Chem. Sci. 2021, 12, 11873–11881. [Google Scholar] [CrossRef] [PubMed]
- Straimer, J.; Gnädig, N.F.; Witkowski, B.; Amaratunga, C.; Duru, V.; Ramadani, A.P.; Dacheux, M.; Khim, N.; Zhang, L.; Lam, S.; et al. Drug resistance. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science 2015, 347, 428–431. [Google Scholar] [CrossRef] [Green Version]
- Noedl, H.; Bronnert, J.; Yingyuen, K.; Attlmayr, B.; Kollaritsch, H.; Fukuda, M. Simple histidine-rich protein 2 double-site sandwich enzyme-linked immunosorbent assay for use in malaria drug sensitivity testing. Antimicrob. Agents Chemother. 2005, 49, 3575–3577. [Google Scholar] [CrossRef] [Green Version]
- De Carvalho, L.P.; Sandri, T.L.; de Melo, E.J.T.; Fendel, R.; Kremsner, P.G.; Mordmüller, B.; Held, J. Ivermectin Impairs the Development of Sexual and Asexual Stages of Plasmodium falciparum In Vitro. Antimicrob. Agents Chemother. 2019, 63, e00085-19. [Google Scholar] [CrossRef] [Green Version]
- Ritz, C.; Baty, F.; Streibig, J.C.; Gerhard, D. Dose-Response Analysis Using R. PLoS ONE 2015, 10, e0146021. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. A Language and Environment for Statistical Computing; Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Witkowski, B.; Amaratunga, C.; Khim, N.; Sreng, S.; Chim, P.; Kim, S.; Lim, P.; Mao, S.; Sopha, C.; Sam, B.; et al. Novel phenotypic assays for the detection of artemisinin-resistant Plasmodium falciparum malaria in Cambodia: In-vitro and ex-vivo drug-response studies. Lancet Infect. Dis. 2013, 13, 1043–1049. [Google Scholar] [CrossRef] [Green Version]
- Witkowski, B.; Menard, D.; Amaratunga, C.; Fairhurst, R.M. Ring-Stage Survival Assays (RSA) to Evaluate the In-Vitro and Ex-Vivo Susceptibility of Plasmodium falciparum to Artemisinins; Procedure RSAv1; Institut Pasteur du Cambodge—National Institutes of Health: Phnom Penh, Cambodia, 2015; Available online: https://www.wwarn.org/sites/default/files/INV10-Standard-Operating-Procedure-Ring-Stage-Survival-Assays.pdf (accessed on 16 February 2022).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Compound | X | Linker | hHDAC1 IC50 [µM] b | hHDAC6 IC50 [µM] b | SI1/6 |
| 6a | O | 1 | >10 c | >10 c | / |
| 7a | O | 1 | 0.546 ± 0.0003 | 0.187 ± 0.004 | 3 |
| 7b | S | 2 | 0.430 ± 0.046 | 0.045 ± 0.005 | 10 |
| (α)-7c | S | 3 | 2.00 ± 0.200 | 0.036 ± 0.008 | 56 |
| (β)-7c | S | 3 | 2.79 ± 0.14 | 0.041 ± 0.009 | 68 |
| 7d | O | 3 | 2.49 ± 0.240 | 0.014 ± 0.002 | 178 |
| (α)-7e | S | 4 | >10 c | 1.101 ± 0.075 | >9 |
| (β)-7e | S | 4 | >10 c | 1.002 ± 0.130 | >10 |
| 8 | O | 1 | 41% @3.33 µM d | >10 c | / |
| DHA | - | - | >10 c | >10 c | / |
| Vorinostat | - | - | 0.107 ± 0.013 | 0.032 ± 0.008 | 3 |
| Compound | X | Linker | Pf3D7 IC50 [nM] b | PfDd2 IC50 [nM] b | PfDd2 R539T IC50 [nM] b | A2780 IC50 [µM] c | SIA2780/Pf 3D7 | SIA2780/Pf Dd2 |
|---|---|---|---|---|---|---|---|---|
| 6a | O | 1 | 5.9 ± 0.70 | 5.8 ± 2.8 | n.d. | 3.38 ± 0.21 | 573 | 583 |
| 7a | O | 1 | 2.6 ± 1.4 | 1.7 ± 0.92 | 1.9 ± 1.4 | 1.95 ± 0.52 | 750 | 1147 |
| 7b | S | 2 | 5.2 ± 0.4 | 6.6 ± 2.5 | 3.0 ± 1.2 | 1.52 ± 0.05 | 292 | 230 |
| (α)-7c | S | 3 | 3.3 ± 1.4 | 3.4 ± 0.12 | 3.3 ± 2.2 | 1.10 ± 0.25 | 333 | 324 |
| (β)-7c | S | 3 | 3.6 ± 1.1 | 3.2 ± 0.02 | 2.6 ± 2.0 | 0.88 ± 0.13 | 244 | 275 |
| 7d | O | 3 | 2.8 ± 1.3 | 2.0 ± 0.7 | 1.2 ± 0.8 | 1.31 ± 0.32 | 468 | 655 |
| (α)-7e | S | 4 | 3.1 ± 0.7 | 1.8 ± 0.8 | n.d. | 1.83 ± 0.10 | 590 | 1017 |
| (β)-7e | S | 4 | 2.6 ± 1.3 | 1.6 ± 0.8 | n.d. | 2.09 ± 0.42 | 804 | 1306 |
| 8 | O | 1 | 2.5 ± 1.4 | 2.5 ± 0.14 | n.d. | 1.47 ± 0.98 | 588 | 588 |
| DHA | - | - | 3.0 ± 2.7 | 1.8 ± 0.02 | n.d. | 0.97 ± 0.07 | 323 | 539 |
| Vorinostat | - | - | 241.8 ± 33.4 | 424.9 ± 1.3 | n.d. | 1.09 ± 0.41 | 5 | 3 |
| Compound | X | Linker | K562 b IC50 [µM] | HL60 c IC50 [µM] | NALM6 d IC50 [µM] | HPBALL e IC50 [µM] | MOLM13 c IC50 [µM] |
|---|---|---|---|---|---|---|---|
| 6a | O | 1 | 4.01 ± 0.60 | 2.53 ± 0.56 | 1.31 ± 0.11 | 23.86 ± 4.69 | 0.68 ± 0.09 |
| 7a | O | 1 | 0.31 ± 0.06 | 1.33 ± 0.14 | 0.15 ± 0.01 | 1.36 ± 0.03 | 0.22 ± 0.04 |
| 7b | S | 2 | 0.91 ± 0.32 | 2.07 ± 0.03 | 0.26 ± 0.01 | 2.41 ± 0.38 | 0.58 ± 0.19 |
| (α)-7c | S | 3 | 0.62 ± 0.06 | 1.02 ± 0.27 | 0.28 ± 0.05 | 6.72 ± 0.44 | 0.33 ± 0.11 |
| (β)-7c | S | 3 | 0.41 ± 0.11 | 2.34 ± 0.46 | 0.25 ± 0.01 | 12.33 ± 0.57 | 0.38 ± 0.07 |
| 7d | O | 3 | 1.00 ± 0.36 | 2.41 ± 0.45 | 0.36 ± 0.05 | 6.81 ± 0.54 | 0.39 ± 0.05 |
| (α)-7e | S | 4 | 0.77 ± 0.24 | 1.62 ± 0.12 | 0.44 ± 0.01 | 3.34 ± 0.37 | 0.45 ± 0.18 |
| (β)-7e | S | 4 | 1.71 ± 0.33 | 2,76 ± 0.62 | 0.77 ± 0.08 | 20.57 ± 6.13 | 0.64 ± 0.17 |
| 8 | O | 1 | 0.69 ± 0.11 | 1.64 ± 0.17 | 0.34 ± 0.03 | 1.69 ± 0.19 | 0.49 ± 0.08 |
| DHA | - | - | 2.40 ± 0.66 | 3.57 ± 1.04 | 2.50 ± 0.16 | 9.93 ± 1.51 | 0.69 ± 0.17 |
| Vorinostat | - | - | 0.28 ± 0.15 | 0.22 ± 0.05 | 0.63 ± 0.06 | 0.59 ± 0.05 | 0.20 ± 0.07 |
| Compound | Healthy Fibroblast 1 | Healthy Fibroblast 2 |
|---|---|---|
| IC50 [µM] | IC50 [µM] | |
| 7a | 13.10 ± 0.10 | 6.34 ± 0.79 |
| (α)-7c | 12.69 ± 0.12 | 7.41 ± 1.40 |
| (β)-7c | 14.67 ± 3.42 | 17.75 ± 6.04 |
| DHA | >25 | >25 |
| Vorinostat | 4.55 ± 0.80 | 3.23 ± 0.66 |
| Method A | Method B | Method C | Method D | Method E | |||||
|---|---|---|---|---|---|---|---|---|---|
| tR [min] | A [%] | tR [min] | A [%] | tR [min] | A [%] | tR [min] | A [%] | tR [min] | A [%] |
| 0 | 5 | 0 | 5 | 0 | 5 | 0 | 5 | 0 | 5 |
| 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| 15 | 60 | 20 | 95 | 12 | 95 | 17 | 50 | 17 | 50 |
| 35 | 60 | 25 | 95 | 22 | 95 | 37 | 50 | 45 | 50 |
| 37 | 5 | 26 | 5 | 23 | 5 | 39 | 5 | 47 | 5 |
| 42 | 5 | 31 | 5 | 28 | 5 | 44 | 5 | 52 | 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
von Bredow, L.; Schäfer, T.M.; Hogenkamp, J.; Tretbar, M.; Stopper, D.; Kraft, F.B.; Schliehe-Diecks, J.; Schöler, A.; Borkhardt, A.; Bhatia, S.; et al. Synthesis, Antiplasmodial, and Antileukemia Activity of Dihydroartemisinin–HDAC Inhibitor Hybrids as Multitarget Drugs. Pharmaceuticals 2022, 15, 333. https://doi.org/10.3390/ph15030333
von Bredow L, Schäfer TM, Hogenkamp J, Tretbar M, Stopper D, Kraft FB, Schliehe-Diecks J, Schöler A, Borkhardt A, Bhatia S, et al. Synthesis, Antiplasmodial, and Antileukemia Activity of Dihydroartemisinin–HDAC Inhibitor Hybrids as Multitarget Drugs. Pharmaceuticals. 2022; 15(3):333. https://doi.org/10.3390/ph15030333
Chicago/Turabian Stylevon Bredow, Lukas, Thomas Martin Schäfer, Julian Hogenkamp, Maik Tretbar, Daniel Stopper, Fabian B. Kraft, Julian Schliehe-Diecks, Andrea Schöler, Arndt Borkhardt, Sanil Bhatia, and et al. 2022. "Synthesis, Antiplasmodial, and Antileukemia Activity of Dihydroartemisinin–HDAC Inhibitor Hybrids as Multitarget Drugs" Pharmaceuticals 15, no. 3: 333. https://doi.org/10.3390/ph15030333
APA Stylevon Bredow, L., Schäfer, T. M., Hogenkamp, J., Tretbar, M., Stopper, D., Kraft, F. B., Schliehe-Diecks, J., Schöler, A., Borkhardt, A., Bhatia, S., Held, J., & Hansen, F. K. (2022). Synthesis, Antiplasmodial, and Antileukemia Activity of Dihydroartemisinin–HDAC Inhibitor Hybrids as Multitarget Drugs. Pharmaceuticals, 15(3), 333. https://doi.org/10.3390/ph15030333