
A Review on Parallel Virtual Screening Softwares for High-Performance Computers

 , and
, and
Abstract
1. Introduction
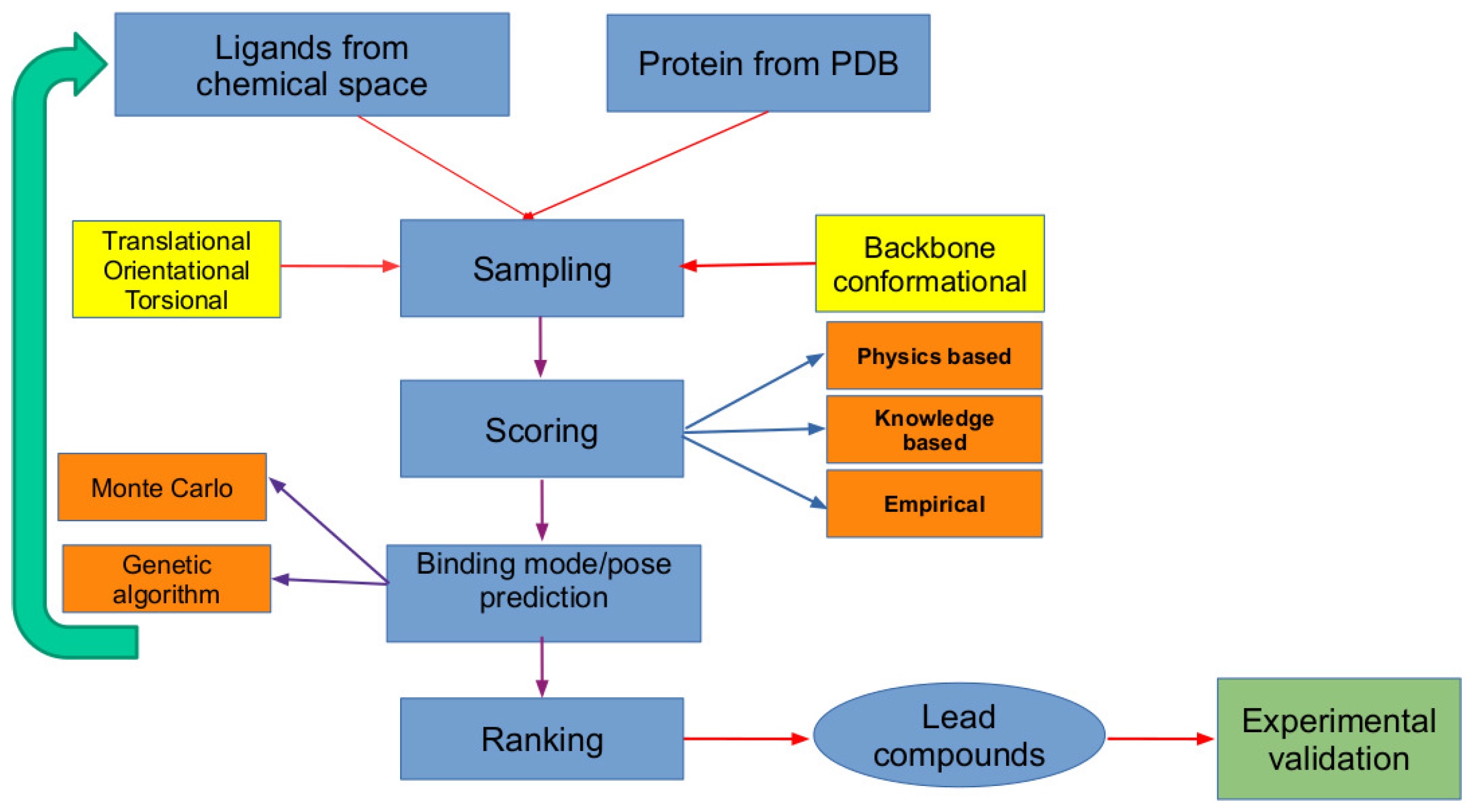
2. The In Silico Virtual Screening Problem
2.1. Scoring Functions
- (i)
- Physics-based (also referred to as force-field based) scoring functions are based on the binding free energies which are the sum of various interactions between protein–ligand subsystems such as van der Waals, electrostatic, hydrogen bonding, solvation energy, and entropic contributions.
- (ii)
- The knowledge-based scoring functions are based on the available protein–ligand complex structural data from which the distributions of different atom–atom pairwise contacts are estimated. The frequency of appearance of different pairwise contacts are used to compute potential mean force which is used for ranking protein–ligand complexes.
- (iii)
- Finally, the empirical scoring functions, as the name implies, are based on empirical fitting of binding affinity data to potential functions whose weights are computed using a reference test system. Modern scoring functions mainly fall into this class, including the machine learning-based approaches built based on the available information on the protein–ligand 3D structures and inhibition/dissociation constants [10].
2.2. Search Algorithms
2.3. Validation of Molecular Docking Approaches
- (i)
- RMSD computed for the predicted binding pose against the crystallographic pose obtained experimentally.
- (ii)
- Binding free energies/docking energies which are proportional to experimental inhibition/dissociation constants.
2.4. Computational Cost Associated with Virtual Screening

{kind=link}
{kind=link}
{kind=link}
| Chemical Library | NO of Compounds | Features |
|---|---|---|
| Virtual compounds | 10 | Molecular mass ≤ 500 daltons |
| GDB17 [25] | 166 B | 17 heavy atoms of type C, O, N, S and halogens |
| REAL DB (Enamine) [30] | 1.95 B | Synthesizeable compounds M ≤ 500, Slogd ≤ 5, HB ≤ 10, HB ≤ 5 |
| rotatable bond ≤ 10, and TPS ≤ 140 | ||
| ZINC15 [28] | 980 M | Synthesizable, available in ready-to-dock format |
| Pubchem [31] | 90 M | Literature-derived bioactive compounds |
| Chemspider [32] | 63 M | Curated database with chemical structure and physicochemical properties |
| ChEMBL [33] | 2 M | Manually-curated drug-like bioactive molecules |
3. Milestones in Virtual Screening
4. High-Performance Computing
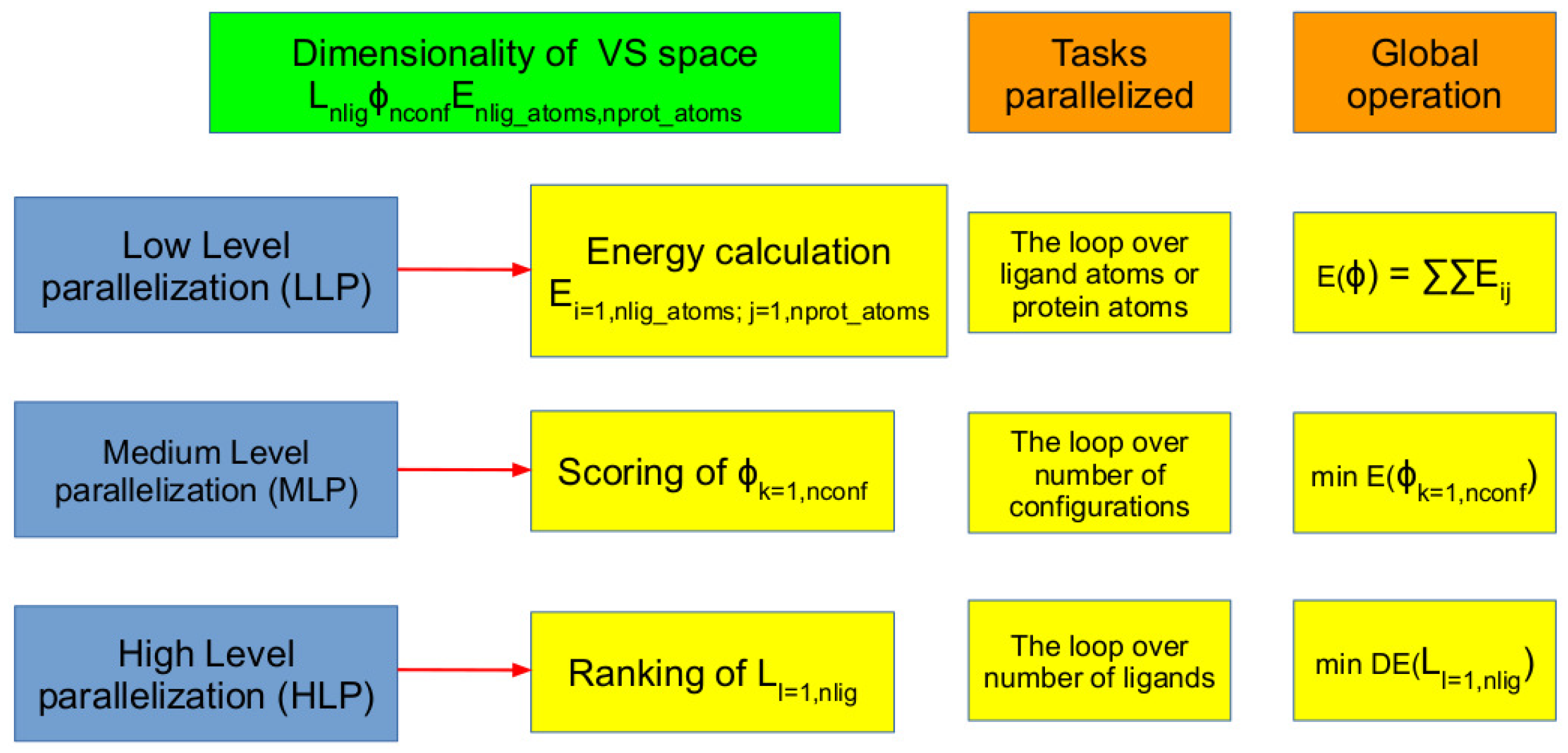
4.1. Parallelization Strategies of Virtual Screening for High-Performance Computers
- (i)
- Sampling over configurational phase space of ligands within the binding site.
- (ii)
- Estimating the scoring function for each of the configurations of the chemical compound within the target binding site to identify the most stable binding mode/pose.
- (iii)
- Ranking of compounds with respect to their relative binding potentials.
- (i)
- Low-level parallelization (LLP): Parallelizing the energy calculation.
- (ii)
- Mid-level parallelization (MLP): Parallelizing the conformer evaluations and scoring.
- (iii)
- High-level parallelization (HLP): Parallelizing the ligands evaluations on different computing units.
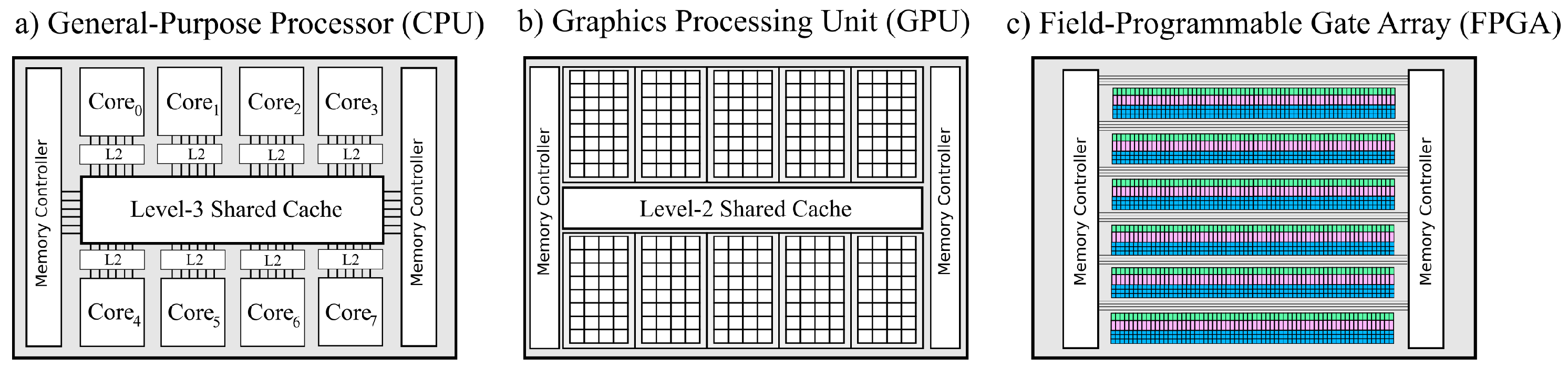
4.2. HPCs and Accelerator Technology as Problem Solvers
- (i)
- In the shared memory architectures, a set of processors use the same memory segment.
- (ii)
- In the case of distributed memory architectures, each computing unit has its own memory.
5. Current Implementation of VS Available for Workstations, Accelerators, and HPCs
5.1. Dock5,6
5.2. DOVIS2.0 VSDocker2.0
5.3. Autodock Vina
5.4. MPAD4
5.5. VinaLC
5.6. VINAMPI
5.7. LiGen Docker-HT
5.8. Geauxdock
5.9. POAP
5.10. GNINA
5.11. AUTODOCK-GPU
5.12. Other VS Tools
6. Emerging Reconfigurable Architectures for Molecular Docking
7. The Advent of Quantum Computing for Molecular Docking
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADMET | Absorption, Distribution, Metabolism, Excretion, Toxicity |
| CUDA | Compute Unified Device Architecture |
| CGRA | Coarse-Grained Reconfigurable Arrays |
| GPU | Graphical processing unit |
| FPGA | Field programmable gate array |
| HPC | High-Performance Computing |
| OS | Operating system |
| PDB | Protein data bank |
| RMSD | Root Mean Square Deviation |
| VS | Virtual screening |
References
- Clark, D.E. What has virtual screening ever done for drug discovery? Expert Opin. Drug Discov. 2008, 3, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.; Grootendorst, P.; Lexchin, J.; Cunningham, C.; Greyson, D. The cost of drug development: A systematic review. Health Policy 2011, 100, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.; Gagnon, J.P. The cost of new drug discovery and development. Discov. Med. 2009, 4, 172–179. [Google Scholar]
- Mullin, R. Drug development costs about $1.7 billion. Chem. Eng. News 2003, 81, 8. [Google Scholar] [CrossRef]
- Rickels, W.; Dovern, J.; Hoffmann, J.; Quaas, M.F.; Schmidt, J.O.; Visbeck, M. Indicators for monitoring sustainable development goals: An application to oceanic development in the European Union. Earths Future 2016, 4, 252–267. [Google Scholar] [CrossRef]
- Petrova, E. Innovation in the pharmaceutical industry: The process of drug discovery and development. In Innovation and Marketing in the Pharmaceutical Industry; Springer: Berlin/Heidelberg, Germany, 2014; pp. 19–81. [Google Scholar]
- Kiriiri, G.K.; Njogu, P.M.; Mwangi, A.N. Exploring different approaches to improve the success of drug discovery and development projects: A review. Future J. Pharm. Sci. 2020, 6, 27. [Google Scholar] [CrossRef]
- Frazier, K. Biopharmaceutical Research & Development: The Process Behind New Medicines; PhRMA: Washington, DC, USA, 2015. [Google Scholar]
- Doman, T.N.; McGovern, S.L.; Witherbee, B.J.; Kasten, T.P.; Kurumbail, R.; Stallings, W.C.; Connolly, D.T.; Shoichet, B.K. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B. J. Med. Chem. 2002, 45, 2213–2221. [Google Scholar] [CrossRef]
- Li, H.; Sze, K.H.; Lu, G.; Ballester, P.J. Machine-learning scoring functions for structure-based drug lead optimization. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2020, 10, e1465. [Google Scholar] [CrossRef]
- DesJarlais, R.L.; Sheridan, R.P.; Seibel, G.L.; Dixon, J.S.; Kuntz, I.D.; Venkataraghavan, R. Using shape complementarity as an initial screen in designing ligands for a receptor binding site of known three-dimensional structure. J. Med. Chem. 1988, 31, 722–729. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Cosconati, S.; Forli, S.; Perryman, A.L.; Harris, R.; Goodsell, D.S.; Olson, A.J. Virtual screening with AutoDock: Theory and practice. Expert Opin. Drug Discov. 2010, 5, 597–607. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Jaghoori, M.M.; Bleijlevens, B.; Olabarriaga, S.D. 1001 Ways to run AutoDock Vina for virtual screening. J. Comput.-Aided Mol. Des. 2016, 30, 237–249. [Google Scholar] [CrossRef]
- McNutt, A.T.; Francoeur, P.; Aggarwal, R.; Masuda, T.; Meli, R.; Ragoza, M.; Sunseri, J.; Koes, D.R. GNINA 1.0: Molecular docking with deep learning. J. Cheminform. 2021, 13, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Poongavanam, V.; Namasivayam, V.; Vanangamudi, M.; Al Shamaileh, H.; Veedu, R.N.; Kihlberg, J.; Murugan, N.A. Integrative approaches in HIV-1 non-nucleoside reverse transcriptase inhibitor design. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1328. [Google Scholar] [CrossRef]
- Dittrich, J.; Schmidt, D.; Pfleger, C.; Gohlke, H. Converging a knowledge-based scoring function: DrugScore2018. J. Chem. Inf. Model. 2018, 59, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Beccari, A.R.; Cavazzoni, C.; Beato, C.; Costantino, G. LiGen: A High Performance Workflow for Chemistry Driven De Novo Design; ACS Publications: Washington, DC, USA, 2013. [Google Scholar]
- Nayeem, A.; Vila, J.; Scheraga, H.A. A comparative study of the simulated-annealing and Monte Carlo-with-minimization approaches to the minimum-energy structures of polypeptides: [Met]-enkephalin. J. Comput. Chem. 1991, 12, 594–605. [Google Scholar] [CrossRef]
- Kennedy, J.; Eberhart, R. Particle swarm optimization. In Proceedings of the ICNN’95-International Conference on Neural Networks, Perth, Australia, 27 November–1 December 1995; Volume 4, pp. 1942–1948. [Google Scholar]
- Bursulaya, B.D.; Totrov, M.; Abagyan, R.; Brooks, C.L. Comparative study of several algorithms for flexible ligand docking. J. Comput.-Aided Mol. Des. 2003, 17, 755–763. [Google Scholar] [CrossRef]
- Vieira, T.F.; Sousa, S.F. Comparing AutoDock and Vina in ligand/decoy discrimination for virtual screening. Appl. Sci. 2019, 9, 4538. [Google Scholar] [CrossRef]
- Walters, W.P. Virtual chemical libraries: Miniperspective. J. Med. Chem. 2018, 62, 1116–1124. [Google Scholar] [CrossRef]
- Graff, D.E.; Shakhnovich, E.I.; Coley, C.W. Accelerating high-throughput virtual screening through molecular pool-based active learning. Chem. Sci. 2021, 12, 7866–7881. [Google Scholar] [CrossRef] [PubMed]
- Ruddigkeit, L.; Van Deursen, R.; Blum, L.C.; Reymond, J.L. Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17. J. Chem. Inf. Model. 2012, 52, 2864–2875. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Lyu, J.; Wang, S.; Balius, T.E.; Singh, I.; Levit, A.; Moroz, Y.S.; O’Meara, M.J.; Che, T.; Algaa, E.; Tolmachova, K.; et al. Ultra-large library docking for discovering new chemotypes. Nature 2019, 566, 224–229. [Google Scholar] [CrossRef]
- Shivanyuk, A.; Ryabukhin, S.; Tolmachev, A.; Bogolyubsky, A.; Mykytenko, D.; Chupryna, A.; Heilman, W.; Kostyuk, A. Enamine real database: Making chemical diversity real. Chem. Today 2007, 25, 58–59. [Google Scholar]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Williams, A.J.; Tkachenko, V.; Golotvin, S.; Kidd, R.; McCann, G. ChemSpider-building a foundation for the semantic web by hosting a crowd sourced databasing platform for chemistry. J. Cheminform. 2010, 2, O16. [Google Scholar] [CrossRef]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef]
- Martin, Y.C. Accomplishments and challenges in integrating software for computer-aided ligand design in drug discovery. Perspect. Drug Discov. Des. 1995, 3, 139–150. [Google Scholar] [CrossRef]
- Martin, Y.C. 3D database searching in drug design. J. Med. Chem. 1992, 35, 2145–2154. [Google Scholar] [CrossRef]
- Glaser, J.; Vermaas, J.V.; Rogers, D.M.; Larkin, J.; LeGrand, S.; Boehm, S.; Baker, M.B.; Scheinberg, A.; Tillack, A.F.; Thavappiragasam, M.; et al. High-throughput virtual laboratory for drug discovery using massive datasets. Int. J. High Perform. Comput. Appl. 2021, 35, 452–468. [Google Scholar] [CrossRef]
- Gorgulla, C.; Boeszoermenyi, A.; Wang, Z.F.; Fischer, P.D.; Coote, P.W.; Das, K.M.P.; Malets, Y.S.; Radchenko, D.S.; Moroz, Y.S.; Scott, D.A.; et al. An open-source drug discovery platform enables ultra-large virtual screens. Nature 2020, 580, 663–668. [Google Scholar] [CrossRef] [PubMed]
- LeGrand, S.; Scheinberg, A.; Tillack, A.F.; Thavappiragasam, M.; Vermaas, J.V.; Agarwal, R.; Larkin, J.; Poole, D.; Santos-Martins, D.; Solis-Vasquez, L.; et al. GPU-accelerated drug discovery with docking on the summit supercomputer: Porting, optimization, and application to COVID-19 research. In Proceedings of the 11th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, Virtual, 21–24 September 2020; pp. 1–10. [Google Scholar]
- OpenEye Scientific, GigaDocking™—Structure Based Virtual Screening of over 1 Billion Molecules Webinar. Available online: https://www.eyesopen.com/ (accessed on 1 August 2020).
- Acharya, A.; Agarwal, R.; Baker, M.B.; Baudry, J.; Bhowmik, D.; Boehm, S.; Byler, K.G.; Chen, S.; Coates, L.; Cooper, C.J.; et al. Supercomputer-based ensemble docking drug discovery pipeline with application to COVID-19. J. Chem. Inf. Model. 2020, 60, 5832–5852. [Google Scholar] [CrossRef] [PubMed]
- Gadioli, D.; Vitali, E.; Ficarelli, F.; Latini, C.; Manelfi, C.; Talarico, C.; Silvano, C.; Cavazzoni, C.; Palermo, G.; Beccari, A.R. EXSCALATE: An extreme-scale in-silico virtual screening platform to evaluate 1 trillion compounds in 60 h on 81 PFLOPS supercomputers. arXiv 2021, arXiv:2110.11644. [Google Scholar]
- Dong, D.; Xu, Z.; Zhong, W.; Peng, S. Parallelization of molecular docking: A review. Curr. Top. Med. Chem. 2018, 18, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Perez-Sanchez, H.; Wenzel, W. Optimization methods for virtual screening on novel computational architectures. Curr. Comput.-Aided Drug Des. 2011, 7, 44–52. [Google Scholar] [CrossRef]
- Stone, J.E.; Phillips, J.C.; Freddolino, P.L.; Hardy, D.J.; Trabuco, L.G.; Schulten, K. Accelerating molecular modeling applications with graphics processors. J. Comput. Chem. 2007, 28, 2618–2640. [Google Scholar] [CrossRef]
- Harvey, M.; De Fabritiis, G. An implementation of the smooth particle mesh Ewald method on GPU hardware. J. Chem. Theory Comput. 2009, 5, 2371–2377. [Google Scholar] [CrossRef]
- Guerrero, G.D.; Pérez-Sánchez, H.; Wenzel, W.; Cecilia, J.M.; García, J.M. Effective parallelization of non-bonded interactions kernel for virtual screening on gpus. In Proceedings of the 5th International Conference on Practical Applications of Computational Biology & Bioinformatics (PACBB 2011), Salamanca, Spain, 6–8 April 2011; Springer: Berlin/Heidelberg, Germany, 2011; pp. 63–69. [Google Scholar]
- Moustakas, D.T.; Lang, P.T.; Pegg, S.; Pettersen, E.; Kuntz, I.D.; Brooijmans, N.; Rizzo, R.C. Development and validation of a modular, extensible docking program: DOCK 5. J. Comput.-Aided Mol. Des. 2006, 20, 601–619. [Google Scholar] [CrossRef]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Kuntz, I.D.; Zou, X. Pairwise GB/SA scoring function for structure-based drug design. J. Phys. Chem. B 2004, 108, 5453–5462. [Google Scholar] [CrossRef]
- Hawkins, G.D.; Cramer, C.J.; Truhlar, D.G. Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J. Phys. Chem. 1996, 100, 19824–19839. [Google Scholar] [CrossRef]
- Grant, J.A.; Pickup, B.T.; Nicholls, A. A smooth permittivity function for Poisson–Boltzmann solvation methods. J. Comput. Chem. 2001, 22, 608–640. [Google Scholar] [CrossRef]
- Mukherjee, S.; Balius, T.E.; Rizzo, R.C. Docking validation resources: Protein family and ligand flexibility experiments. J. Chem. Inf. Model. 2010, 50, 1986–2000. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhou, Q.; Li, B.; Wang, Y.; Luan, Z.; Qian, D.; Li, H. GPU acceleration of Dock6’s Amber scoring computation. In Advances in Computational Biology; Springer: Berlin/Heidelberg, Germany, 2010; pp. 497–511. [Google Scholar]
- Jiang, X.; Kumar, K.; Hu, X.; Wallqvist, A.; Reifman, J. DOVIS 2.0: An efficient and easy to use parallel virtual screening tool based on AutoDock 4.0. Chem. Cent. J. 2008, 2, 18. [Google Scholar] [CrossRef]
- Prakhov, N.D.; Chernorudskiy, A.L.; Gainullin, M.R. VSDocker: A tool for parallel high-throughput virtual screening using AutoDock on Windows-based computer clusters. Bioinformatics 2010, 26, 1374–1375. [Google Scholar] [CrossRef]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef]
- Norgan, A.P.; Coffman, P.K.; Kocher, J.P.A.; Katzmann, D.J.; Sosa, C.P. Multilevel parallelization of AutoDock 4.2. J. Cheminform. 2011, 3, 1–9. [Google Scholar] [CrossRef]
- Zhang, X.; Wong, S.E.; Lightstone, F.C. Message passing interface and multithreading hybrid for parallel molecular docking of large databases on petascale high performance computing machines. J. Comput. Chem. 2013, 34, 915–927. [Google Scholar] [CrossRef]
- Ellingson, S.R.; Smith, J.C.; Baudry, J. VinaMPI: Facilitating multiple receptor high-throughput virtual docking on high-performance computers. J. Comput. Chem. 2013, 34, 2212–2221. [Google Scholar] [CrossRef] [PubMed]
- Beato, C.; Beccari, A.R.; Cavazzoni, C.; Lorenzi, S.; Costantino, G. Use of experimental design to optimize docking performance: The case of ligendock, the docking module of ligen, a new de novo design program. J. Chem. Inf. Model. 2013, 53, 1503–1517. [Google Scholar] [CrossRef] [PubMed]
- Gadioli, D.; Palermo, G.; Cherubin, S.; Vitali, E.; Agosta, G.; Manelfi, C.; Beccari, A.R.; Cavazzoni, C.; Sanna, N.; Silvano, C. Tunable approximations to control time-to-solution in an HPC molecular docking Mini-App. J. Supercomput. 2021, 77, 841–869. [Google Scholar] [CrossRef]
- Vitali, E.; Gadioli, D.; Palermo, G.; Beccari, A.; Silvano, C. Accelerating a geometric approach to molecular docking with OpenACC. In Proceedings of the 6th International Workshop on Parallelism in Bioinformatics, Barcelona, Spain, 23 September 2018; pp. 45–51. [Google Scholar]
- Markidis, S.; Gadioli, D.; Vitali, E.; Palermo, G. Understanding the I/O Impact on the Performance of High-Throughput Molecular Docking. In Proceedings of the IEEE/ACM Sixth International Parallel Data Systems Workshop (PDSW), St. Louis, MO, USA, 15 November 2021. [Google Scholar]
- Fang, Y.; Ding, Y.; Feinstein, W.P.; Koppelman, D.M.; Moreno, J.; Jarrell, M.; Ramanujam, J.; Brylinski, M. GeauxDock: Accelerating structure-based virtual screening with heterogeneous computing. PLoS ONE 2016, 11, e0158898. [Google Scholar] [CrossRef]
- Samdani, A.; Vetrivel, U. POAP: A GNU parallel based multithreaded pipeline of open babel and AutoDock suite for boosted high throughput virtual screening. Comput. Biol. Chem. 2018, 74, 39–48. [Google Scholar] [CrossRef]
- Santos-Martins, D.; Solis-Vasquez, L.; Tillack, A.F.; Sanner, M.F.; Koch, A.; Forli, S. Accelerating AutoDock4 with GPUs and gradient-based local search. J. Chem. Theory Comput. 2021, 17, 1060–1073. [Google Scholar] [CrossRef] [PubMed]
- Hartshorn, M.J.; Verdonk, M.L.; Chessari, G.; Brewerton, S.C.; Mooij, W.T.; Mortenson, P.N.; Murray, C.W. Diverse, high-quality test set for the validation of protein-ligand docking performance. J. Med. Chem. 2007, 50, 726–741. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Han, L.; Liu, Z.; Wang, R. Comparative assessment of scoring functions on an updated benchmark: 2. Evaluation methods and general results. J. Chem. Inf. Model. 2014, 54, 1717–1736. [Google Scholar] [CrossRef]
- Lyne, P.D. Structure-based virtual screening: An overview. Drug Discov. Today 2002, 7, 1047–1055. [Google Scholar] [CrossRef]
- Marze, N.A.; Roy Burman, S.S.; Sheffler, W.; Gray, J.J. Efficient flexible backbone protein–protein docking for challenging targets. Bioinformatics 2018, 34, 3461–3469. [Google Scholar] [CrossRef]
- Jain, A.N. Surflex: Fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.; Blackburn, E.; Sheng, Y.; Harding, S.; Hsin, K.Y.; Kan, D.; Shave, S.; Walkinshaw, M. Ligand discovery and virtual screening using the program LIDAEUS. Br. J. Pharmacol. 2008, 153, S55–S67. [Google Scholar] [CrossRef]
- Capuccini, M.; Ahmed, L.; Schaal, W.; Laure, E.; Spjuth, O. Large-scale virtual screening on public cloud resources with Apache Spark. J. Cheminform. 2017, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Vetter, J.S.; DeBenedictis, E.P.; Conte, T.M. Architectures for the post-Moore era. IEEE Micro 2017, 37, 6–8. [Google Scholar] [CrossRef]
- Podobas, A.; Sano, K.; Matsuoka, S. A survey on coarse-grained reconfigurable architectures from a performance perspective. IEEE Access 2020, 8, 146719–146743. [Google Scholar] [CrossRef]
- Nane, R.; Sima, V.M.; Pilato, C.; Choi, J.; Fort, B.; Canis, A.; Chen, Y.T.; Hsiao, H.; Brown, S.; Ferrandi, F.; et al. A survey and evaluation of FPGA high-level synthesis tools. IEEE Trans. Comput.-Aided Des. Integr. Circuits Syst. 2015, 35, 1591–1604. [Google Scholar] [CrossRef]
- Czajkowski, T.S.; Aydonat, U.; Denisenko, D.; Freeman, J.; Kinsner, M.; Neto, D.; Wong, J.; Yiannacouras, P.; Singh, D.P. From OpenCL to high-performance hardware on FPGAs. In Proceedings of the 22nd international conference on field programmable logic and applications (FPL), Oslo, Norway, 29–31 August 2012; pp. 531–534. [Google Scholar]
- Podobas, A. Accelerating parallel computations with openmp-driven system-on-chip generation for fpgas. In Proceedings of the IEEE 8th International Symposium on Embedded Multicore/Manycore SoCs, Aizu-Wakamatsu, Japan, 23–25 September 2014; pp. 149–156. [Google Scholar]
- Pechan, I.; Fehér, B.; Bérces, A. FPGA-based acceleration of the AutoDock molecular docking software. In Proceedings of the 6th Conference on Ph.D. Research in Microelectronics & Electronics, Berlin, Germany, 18–21 July 2010; pp. 1–4. [Google Scholar]
- Solis-Vasquez, L.; Koch, A. A case study in using opencl on fpgas: Creating an open-source accelerator of the autodock molecular docking software. In Proceedings of the FSP Workshop 2018; Fifth International Workshop on FPGAs for Software Programmers, Dublin, Ireland, 31 August 2018; pp. 1–10. [Google Scholar]
- Solis Vasquez, L. Accelerating Molecular Docking by Parallelized Heterogeneous Computing—A Case Study of Performance, Quality of Results, and Energy-Efficiency Using CPUs, GPUs, and FPGAs. Ph.D. Thesis, Technische Universität, Darmstadt, Germany, 2019. [Google Scholar] [CrossRef]
- Majumder, T.; Pande, P.P.; Kalyanaraman, A. Hardware accelerators in computational biology: Application, potential, and challenges. IEEE Des. Test 2013, 31, 8–18. [Google Scholar] [CrossRef]
- Pechan, I.; Fehér, B. Hardware Accelerated Molecular Docking: A Survey. Bioinformatics, Horacio Pérez-Sánchez, IntechOpen. 2012, Volume 133. Available online: https://www.intechopen.com/chapters/41236 (accessed on 1 August 2020). [CrossRef]
- Castelvecchi, D. Quantum computers ready to leap out of the lab in 2017. Nat. News 2017, 541, 9. [Google Scholar] [CrossRef]
- Banchi, L.; Fingerhuth, M.; Babej, T.; Ing, C.; Arrazola, J.M. Molecular docking with Gaussian boson sampling. Sci. Adv. 2020, 6, eaax1950. [Google Scholar] [CrossRef]
- Mato, K.; Mengoni, R.; Ottaviani, D.; Palermo, G. Quantum Molecular Unfolding. arXiv 2021, arXiv:2107.13607. [Google Scholar]



| NO | Year | Target | No of Compounds | Docking Tool |
|---|---|---|---|---|
| 1 | 2019 | enzyme AmpC | 99 M | Dock3.7 |
| 2 | 2019 | D4 dopamine receptor | 138 M | Dock3.7 |
| 3 | 2019 | Purine Nucleoside Phosphorylase | 1.43 B | Orion |
| 4 | 2019 | Heat Shock Protein 90 | 1.43 B | Orion |
| 5 | 2020 | KEAP1 | 1.4 B | Quickvina2 |
| 6 | 2021 | Mpro | 1.37 B | Autodock-GPU |
| 7 | 2021 | 12 SARS-CoV-2 Proteins | 71.6 B | LiGen |
| Software | Parallelization Segment | Programming Language | Scoring Function | Minimization | Multi Thread | Multi Node | GPU |
|---|---|---|---|---|---|---|---|
| Dock 5,6 | Conformational search | C++, C, Fortran77, MPI | Physics-based and hybrid | Monte Carlo | Yes | Yes | No |
| DOVIS2.0 | Ligand screening | C++, Perl, Python | Physics-based | Monte Carlo&GA | Yes | Yes | No |
| Autodock Vina | Conformational search | C++, OpenMP | Hybrid | Monte Carlo | Yes | No | No |
| VSDocker | Ligand screening | C++, Perl, Python | Physics-based | Monte Carlo&GA | Yes | Yes | No |
| MPAD4 | Ligand screening, Conformational search | C++, MPI, OpenMP | Physics-based | Lamarckian GA | Yes | Yes | No |
| VinaLC | Ligand screening, Conformational search | C++, MPI, OpenMP | Hybrid | Monte Carlo | Yes | Yes | No |
| VinaMPI | Ligand screening, Conformational search | C++, MPI, OpenMP | Hybrid | Monte Carlo | Yes | Yes | No |
| Ligen Docker-HT | Ligand screening, Conformational search | C++, MPI, CUDA | Empirical | Deterministic | Yes | Yes | Yes |
| GeauxDock | Ligand screening, Conformational search | C++, OpenMP, CUDA | Physics- and knowledge-based | Monte Carlo | Yes | Yes | Yes |
| POAP | Ligand screening | bash | Same as parent docking software | Same as parent Docking software | Yes | Yes | No |
| GNINA | Conformational search | C++ | Empirical and CNN ML | Monte Carlo | Yes | Yes | Yes |
| Autodock-GPU | Ligand screening | C++ and OpenCL | Physics-based | MC/ LGA | Yes | No | Yes |
| No | Year | Parallel VS | Source |
|---|---|---|---|
| 1 | 2006 | Dock5&6 | http://dock.docking.org/ (1 August 2021) |
| 2 | 2008 | DOVIS2.0 | http://www.bioanalysis.org/downloads/DOVIS-2.0.1-installer.tar.gz (15 December 2021) |
| 3 | 2009 | Autodock Vina | http://vina.scripps.edu/ (1 August 2021) |
| 4 | 2010 | VSDocker | http://www.bio.nnov.ru/projects/vsdocker2/ (15 December 2021) |
| 5 | 2011 | MPAD4 | http//autodock.scripps.edu/downloads/multilevel-parallel-autodock4.2 (15 August 2021) |
| 6 | 2013 | vinaMPI | https://github.com/mokarrom/mpi-vina (1 June 2021) |
| 7 | 2013 | vinaLC | https://github.com/XiaohuaZhangLLNL/VinaLC (10 June 2021) |
| 8 | 2016 | GeauxDock | http://www.brylinski.org/geauxdock (20 June 2021) |
| 9 | 2018 | POAP | https://github.com/inpacdb/POAP (21 June 2021) |
| 10 | 2021 | Autodock-GPU | https://github.com/ccsb-scripps/AutoDock-GPU (10 September 2021) |
| 11 | 2021 | GNINA | https://github.com/gnina/gnina (1 November 2021) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murugan, N.A.; Podobas, A.; Gadioli, D.; Vitali, E.; Palermo, G.; Markidis, S. A Review on Parallel Virtual Screening Softwares for High-Performance Computers. Pharmaceuticals 2022, 15, 63. https://doi.org/10.3390/ph15010063
Murugan NA, Podobas A, Gadioli D, Vitali E, Palermo G, Markidis S. A Review on Parallel Virtual Screening Softwares for High-Performance Computers. Pharmaceuticals. 2022; 15(1):63. https://doi.org/10.3390/ph15010063
Chicago/Turabian StyleMurugan, Natarajan Arul, Artur Podobas, Davide Gadioli, Emanuele Vitali, Gianluca Palermo, and Stefano Markidis. 2022. "A Review on Parallel Virtual Screening Softwares for High-Performance Computers" Pharmaceuticals 15, no. 1: 63. https://doi.org/10.3390/ph15010063
APA StyleMurugan, N. A., Podobas, A., Gadioli, D., Vitali, E., Palermo, G., & Markidis, S. (2022). A Review on Parallel Virtual Screening Softwares for High-Performance Computers. Pharmaceuticals, 15(1), 63. https://doi.org/10.3390/ph15010063