
Discovery of Small Molecules as Membrane-Bound Catechol-O-methyltransferase Inhibitors with Interest in Parkinson’s Disease: Pharmacophore Modeling, Molecular Docking and In Vitro Experimental Validation Studies

 ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Results
2.1. Pharmacophore Modeling
2.2. Database Searching
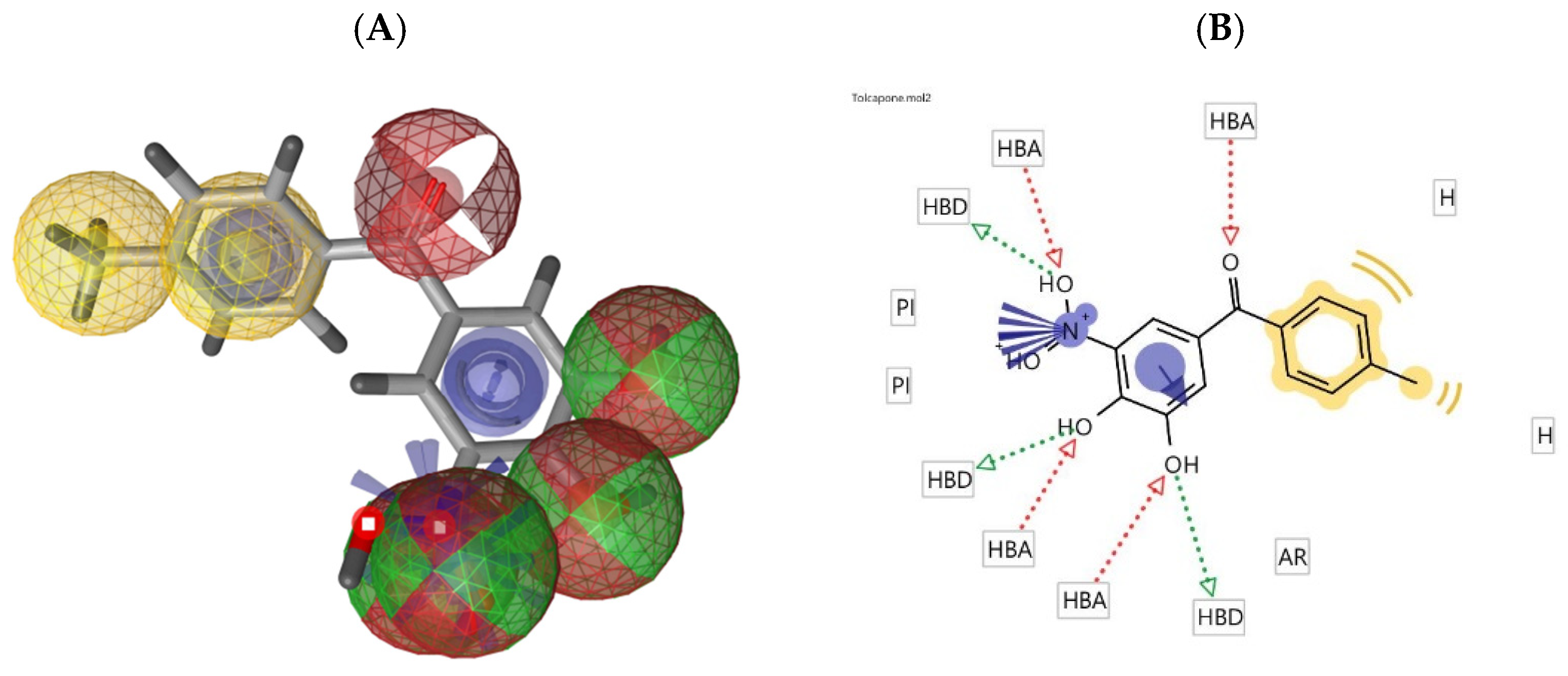
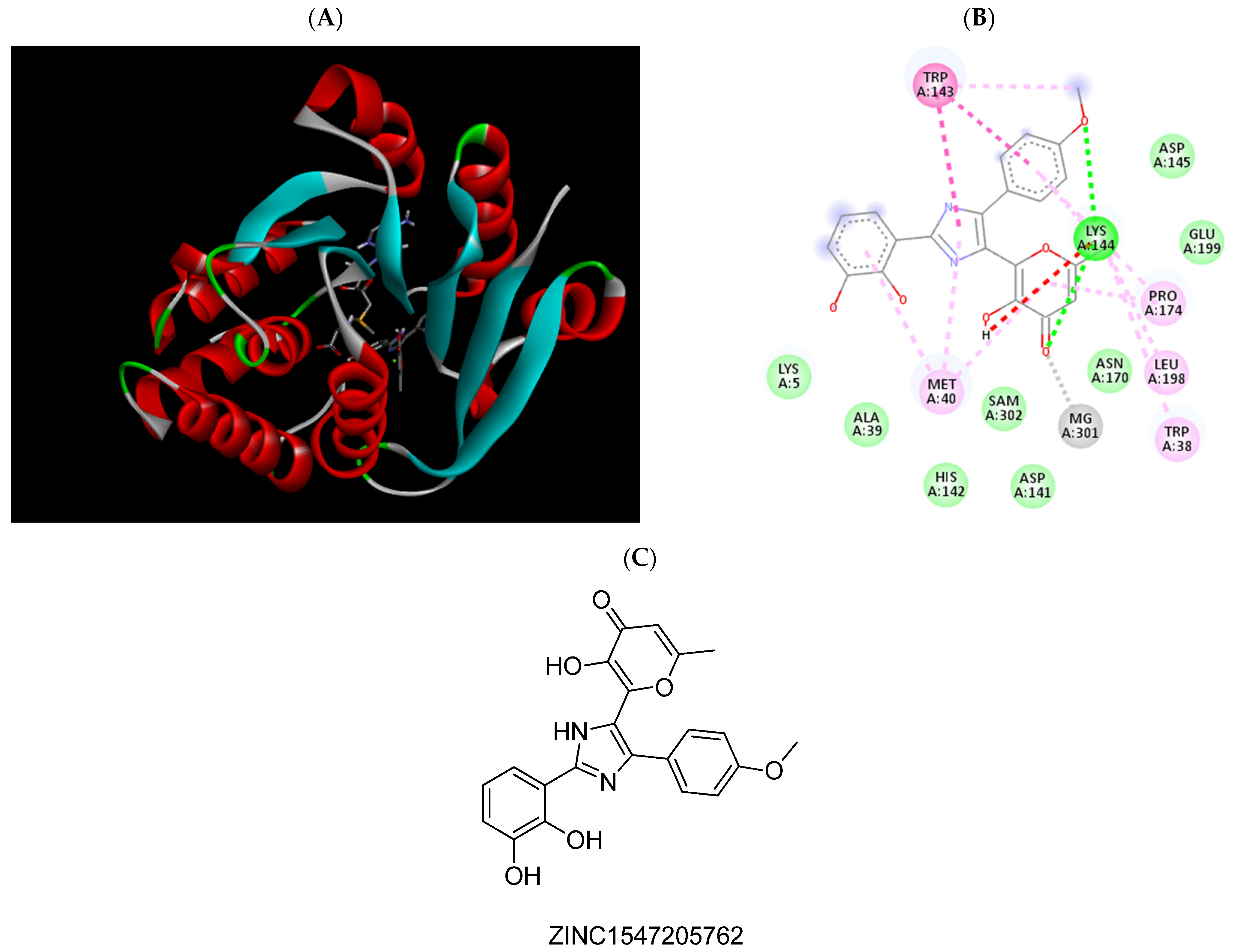
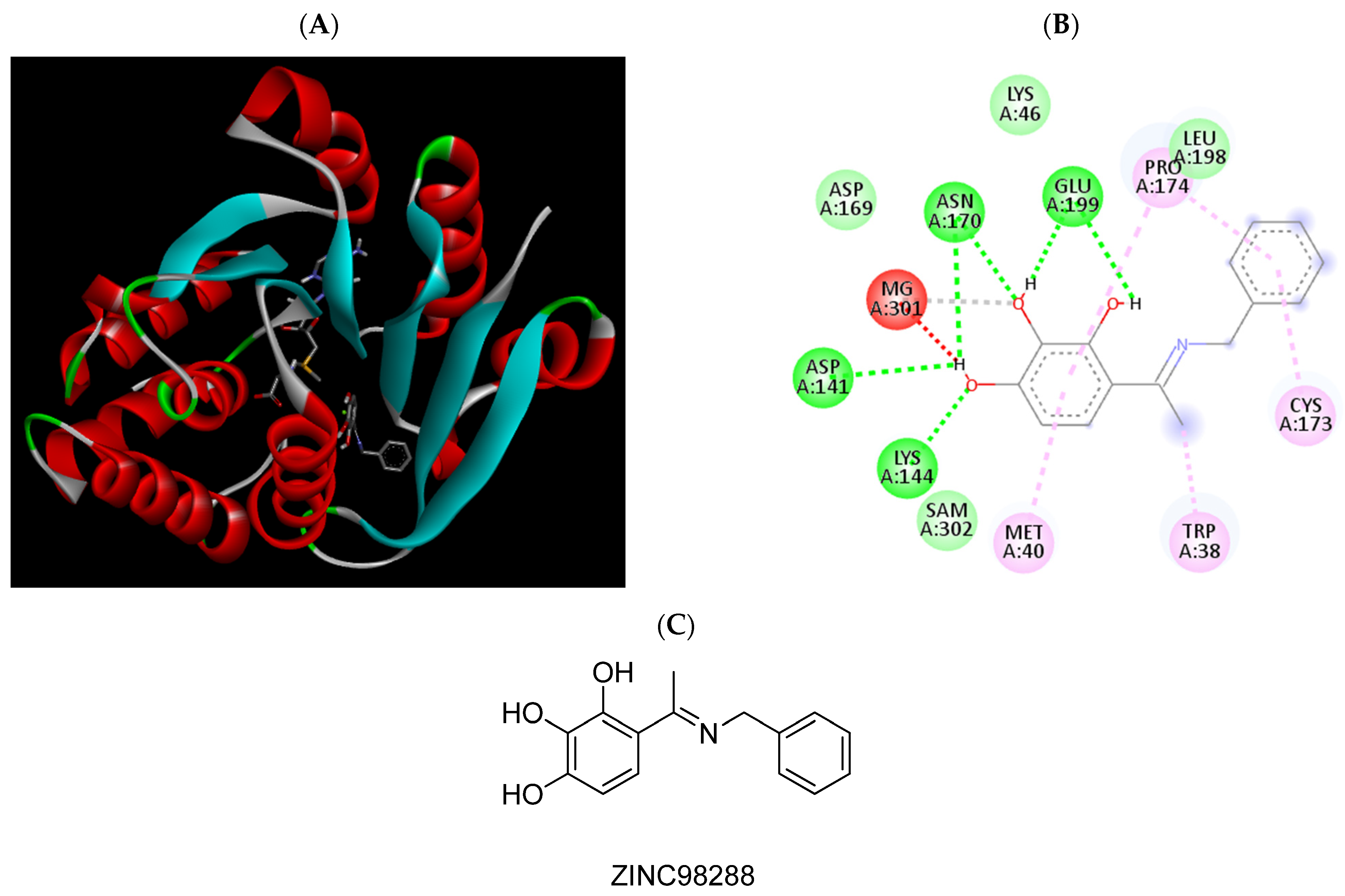
2.3. Molecular Docking Studies
2.4. ADMET Property Prediction
2.5. In-Vitro MBCOMT Assays
2.6. Cytotoxicity Studies
3. Discussion
4. Materials and Methods
4.1. Ligand Selection
4.2. Pharmacophore Generation
4.3. Virtual Screening and ADMET Virtual Filtration
4.4. Molecular Docking
4.5. ADMET Property Analysis
4.6. Materials, Reagents, and Solutions for In-Vitro Studies
4.7. In-Vitro MBCOMT Inhibition Assays
4.7.1. Biosynthesis and Recuperation of MBCOMT
4.7.2. MBCOMT Enzymatic Assay
4.8. Cytotoxicity Studies
4.8.1. Cell Cultures
4.8.2. MTT Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dong, J.; Cui, Y.; Li, S.; Le, W. Current Pharmaceutical Treatments and Alternative Therapies of Parkinson’s Disease. Curr. Neuropharmacol. 2016, 14, 339–355. [Google Scholar] [CrossRef]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef]
- McFarthing, K.; Buff, S.; Rafaloff, G.; Dominey, T.; Wyse, R.K.; Stott, S.R.W. Parkinson’s Disease Drug Therapies in the Clinical Trial Pipeline: 2020. J. Parkinsons Dis. 2020, 10, 757–774. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, J.; Tomchick, R. Enzymatic O-methylation of epinephrine and other catechols. J. Biol. Chem. 1958, 233, 702–705. [Google Scholar] [CrossRef]
- Guldberg, H.C.; Marsden, C.A. Catechol-O-Methyl Transferase: Pharmacological Aspects and Physiological Role. Pharmacol. Rev. 1975, 27, 135–206. [Google Scholar]
- Chen, J.; Lipska, B.K.; Halim, N.; Ma, Q.D.; Matsumoto, M.; Melhem, S.; Kolachana, B.S.; Hyde, T.M.; Herman, M.M.; Apud, J.; et al. Functional analysis of genetic variation in catechol-O-methyltransferase (COMT): Effects on mRNA, protein, and enzyme activity in postmortem human brain. Am. J. Hum. Genet. 2004, 75, 807–821. [Google Scholar] [CrossRef]
- Schott, B.; Frischknecht, R.; Debska-Vielhaber, G.; John, N.; Behnisch, G.; Düzel, E.; Gundelfinger, E.; Seidenbecher, C. Membrane-Bound Catechol-O-Methyl Transferase in Cortical Neurons and Glial Cells is Intracellularly Oriented. Front. Psychiatry 2010, 1, 142. [Google Scholar] [CrossRef]
- Bonifácio, M.J.; Palma, P.N.; Almeida, L.; Soares-da-Silva, P. Catechol-O-methyltransferase and its inhibitors in Parkinson’s disease. CNS Drug Rev. 2007, 13, 352–379. [Google Scholar] [CrossRef]
- Kiss, L.E.; Soares-da-Silva, P. Medicinal chemistry of catechol O-methyltransferase (COMT) inhibitors and their therapeutic utility. J. Med. Chem. 2014, 57, 8692–8717. [Google Scholar] [CrossRef]
- Najib, J. Entacapone: A catechol-O-methyltransferase inhibitor for the adjunctive treatment of Parkinson’s disease. Clin. Ther. 2001, 23, 802–832. [Google Scholar] [CrossRef]
- Vokurka, P.; Barron, A.; Sumaria, S.; Stockford, L.; Jarman, P.; Bhatia, K.; Farmer, S.; Saifee, T.; Warner, T.; Weil, R.; et al. Opicapone Efficacy and Tolerability in Parkinson’s Disease Patients Reporting Insufficient Benefit/Failure of Entacapone. Mov. Disord. Clin. Pract. 2020, 7, 955–960. [Google Scholar] [CrossRef]
- Vidgren, J. X-ray crystallography of catechol O-methyltransferase: Perspectives for target-based drug development. Adv. Pharmaco. 1998, 42, 328–331. [Google Scholar]
- Vidgren, J.; Svensson, L.A.; Liljas, A. Crystal structure of catechol O-methyltransferase. Nature 1994, 368, 354–358. [Google Scholar] [CrossRef]
- Lautala, P.; Ulmanen, I.; Taskinen, J. Molecular mechanisms controlling the rate and specificity of catechol O-methylation by human soluble catechol O-methyltransferase. Mol. Pharmacol. 2001, 59, 393–402. [Google Scholar] [CrossRef]
- Chen, D.; Wang, C.Y.; Lambert, J.D.; Ai, N.; Welsh, W.J.; Yang, C.S. Inhibition of human liver catechol-O-methyltransferase by tea catechins and their metabolites: Structure-activity relationship and molecular-modeling studies. Biochem. Pharmacol. 2005, 69, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Chen, Z.J.; Jiang, H.D.; Chen, J.Z. Computational studies of the regioselectivities of COMT-catalyzed meta-/para-O methylations of luteolin and quercetin. J. Phys. Chem. B 2014, 118, 470–481. [Google Scholar] [CrossRef]
- Monteiro, A.F.M.; Viana, J.O.; Nayarisseri, A.; Zondegoumba, E.N.; Mendonça Junior, F.J.B.; Scotti, M.T.; Scotti, L. Computational Studies Applied to Flavonoids against Alzheimer’s and Parkinson’s Diseases. Oxidative Med. Cell. Longev. 2018, 2018, 7912765. [Google Scholar] [CrossRef]
- Palma, P.N.; Bonifácio, M.J.; Loureiro, A.I.; Wright, L.C.; Learmonth, D.A.; Soares-da-Silva, P. Molecular modeling and metabolic studies of the interaction of catechol-O-methyltransferase and a new nitrocatechol inhibitor. Drug Metab. Dispos. 2003, 31, 250–258. [Google Scholar] [CrossRef]
- Palma, P.N.; Rodrigues, M.L.; Archer, M.; Bonifácio, M.J.; Loureiro, A.I.; Learmonth, D.A.; Carrondo, M.A.; Soares-da-Silva, P. Comparative study of ortho- and meta-nitrated inhibitors of catechol-O-methyltransferase: Interactions with the active site and regioselectivity of O-methylation. Mol. Pharmacol. 2006, 70, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Moschovou, K.; Melagraki, G.; Mavromoustakos, T.; Zacharia, L.C.; Afantitis, A. Cheminformatics and virtual screening studies of COMT inhibitors as potential Parkinson’s disease therapeutics. Expert Opin.Drug Discov. 2020, 15, 53–62. [Google Scholar] [CrossRef]
- Lerner, C.; Ruf, A.; Gramlich, V.; Masjost, B.; Zürcher, G.; Jakob-Roetne, R.; Borroni, E.; Diederich, F. X-ray Crystal Structure of a Bisubstrate Inhibitor Bound to the Enzyme Catechol-O-methyltransferase: A Dramatic Effect of Inhibitor Preorganization on Binding Affinity We thank F. Hoffmann-La Roche for generous support of this work. We are grateful to P. Malherbe for the cloning of COMT, P. Caspers for the expression of COMT, A. Cesura for enzyme purification, B. Wipf for fermentation, and H. W. Lahm for sequencing. Angew. Chem. Int. Ed. Engl. 2001, 40, 4040–4042. [Google Scholar]
- Paulini, R.; Lerner, C.; Jakob-Roetne, R.; Zürcher, G.; Borroni, E.; Diederich, F. Bisubstrate inhibitors of the enzyme catechol O-methyltransferase (COMT): Efficient inhibition despite the lack of a nitro group. Chembiochem 2004, 5, 1270–1274. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, Y.M. Comparative Homology Modeling and Ligand Docking Study of Human Catechol-O-Methyltransferase for Antiparkinson Drug Design. Bull. Korean Chem. Soc. 2005, 26, 1695–1700. [Google Scholar] [CrossRef][Green Version]
- Ellermann, M.; Jakob-Roetne, R.; Lerner, C.; Borroni, E.; Schlatter, D.; Roth, D.; Ehler, A.; Rudolph, M.G.; Diederich, F. Molecular recognition at the active site of catechol-o-methyltransferase: Energetically favorable replacement of a water molecule imported by a bisubstrate inhibitor. Angew. Chem. Int. Ed. Engl. 2009, 48, 9092–9096. [Google Scholar] [CrossRef] [PubMed]
- Ellermann, M.; Paulini, R.; Jakob-Roetne, R.; Lerner, C.; Borroni, E.; Roth, D.; Ehler, A.; Schweizer, W.B.; Schlatter, D.; Rudolph, M.G.; et al. Molecular recognition at the active site of catechol-O-methyltransferase (COMT): Adenine replacements in bisubstrate inhibitors. Chemistry 2011, 17, 6369–6381. [Google Scholar] [CrossRef] [PubMed]
- Deb, S.; Phukan, B.C.; Mazumder, M.K.; Dutta, A.; Paul, R.; Bhattacharya, P.; Sandhir, R.; Borah, A. Garcinol, a multifaceted sword for the treatment of Parkinson’s disease. Neurochem. Int. 2019, 128, 50–57. [Google Scholar] [CrossRef]
- Jatana, N.; Sharma, A.; Latha, N. Pharmacophore modeling and virtual screening studies to design potential COMT inhibitors as new leads. J. Mol. Graph. Model. 2013, 39, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Czarnota, S.; Johannissen, L.O.; Baxter, N.J.; Rummel, F.; Wilson, A.L.; Cliff, M.J.; Levy, C.W.; Scrutton, N.S.; Waltho, J.P.; Hay, S. Equatorial Active Site Compaction and Electrostatic Reorganization in Catechol-O-methyltransferase. ACS Catal. 2019, 9, 4394–4401. [Google Scholar] [CrossRef]
- Cruz-Vicente, P.; Passarinha, L.A.; Silvestre, S.; Gallardo, E. Recent Developments in New Therapeutic Agents against Alzheimer and Parkinson Diseases: In-Silico Approaches. Molecules 2021, 26, 2193. [Google Scholar] [CrossRef]
- Ma, Z.; Liu, H.; Wu, B. Structure-based drug design of catechol-O-methyltransferase inhibitors for CNS disorders. Br. J. Clin. Pharmacol. 2014, 77, 410–420. [Google Scholar] [CrossRef]
- Wang, X.; Song, K.; Li, L.; Chen, L. Structure-Based Drug Design Strategies and Challenges. Curr. Top. Med.Chem. 2018, 18, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Pedro, A.Q.; Correia, F.F.; Santos, F.M.; Espírito-Santo, G.; Gonçalves, A.M.; Bonifácio, M.J.; Queiroz, J.A.; Passarinha, L.A. Biosynthesis and purification of histidine-tagged human soluble catechol-O-methyltransferase. J. Chem. Technol. Biotechnol. 2016, 91, 3035–3044. [Google Scholar] [CrossRef]
- Vieira-Coelho, M.A.; Soares-da-Silva, P. Effects of tolcapone upon soluble and membrane-bound brain and liver catechol-O-methyltransferase. Brain Res. 1999, 821, 69–78. [Google Scholar] [CrossRef]
- Hitge, R.; Smit, S.; Petzer, A.; Petzer, J.P. Evaluation of nitrocatechol chalcone and pyrazoline derivatives as inhibitors of catechol-O-methyltransferase and monoamine oxidase. Bioorg. Med. Chem. Lett. 2020, 30, 127188. [Google Scholar] [CrossRef]
- Matias, M.; Campos, G.; Silvestre, S.; Falcão, A.; Alves, G. Early preclinical evaluation of dihydropyrimidin(thi)ones as potential anticonvulsant drug candidates. Eur. J. Pharm. Sci. 2017, 102, 264–274. [Google Scholar] [CrossRef]
- Haasio, K. Toxicology and safety of COMT inhibitors. Int. Rev. Neurobiol. 2010, 95, 163–189. [Google Scholar]
- Kiss, L.E.; Ferreira, H.S.; Torrão, L.; Bonifácio, M.J.; Palma, P.N.; Soares-da-Silva, P.; Learmonth, D.A. Discovery of a long-acting, peripherally selective inhibitor of catechol-O-methyltransferase. J. Med. Chem. 2010, 53, 3396–3411. [Google Scholar] [CrossRef]
- Antonini, A.; Abbruzzese, G.; Barone, P.; Bonuccelli, U.; Lopiano, L.; Onofrj, M.; Zappia, M.; Quattrone, A. COMT inhibition with tolcapone in the treatment algorithm of patients with Parkinson’s disease (PD): Relevance for motor and non-motor features. Neuropsychiatr. Dis. Treat. 2008, 4, 1–9. [Google Scholar] [CrossRef]
- Borgulya, J.; Bruderer, H.; Bernauer, K.; Zürcher, G.; Prada, M.D. Catechol-O-methyltransferase-inhibiting pyrocatechol derivatives: Synthesis and structure-activity studies. Helv. Chim. Acta 1989, 72, 952–968. [Google Scholar] [CrossRef]
- Backstrom, R.; Honkanen, E.; Pippuri, A.; Kairisalo, P.; Pystynen, J.; Heinola, K.; Nissinen, E.; Linden, I.B.; Mannisto, P.T. Synthesis of some novel potent and selective catechol O-methyltransferase inhibitors. J. Med. Chem. 1989, 32, 841–846. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Modeling 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Chen, Z.; Li, H.L.; Zhang, Q.J.; Bao, X.G.; Yu, K.Q.; Luo, X.M.; Zhu, W.L.; Jiang, H.L. Pharmacophore-based virtual screening versus docking-based virtual screening: A benchmark comparison against eight targets. Acta Pharmacol. Sin. 2009, 30, 1694–1708. [Google Scholar] [CrossRef]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Modeling 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv. Drug Deliv. Rev. 2016, 101, 34–41. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Pedro, A.Q.; Oppolzer, D.J.; Bonifácio, M.J.; Maia, C.J.; Queiroz, J.A.; Passarinha, L.A. Evaluation of MutS and Mut+Pichia pastoris Strains for Membrane-Bound Catechol-O-Methyltransferase Biosynthesis. Appl. Biochem. Biotechnol. 2015, 175, 3840–3855. [Google Scholar] [CrossRef]
- Pedro, A.Q.; Rf, S.; Oppolzer, D.J.; Santos, F.M.; La, R.; Gonçalves, A.M.; Bonifácio, M.J.; Queiroz, J.A.; Gallardo, E.; Passarinha, L.A. An Improved HPLC Method for Quantification of Metanephrine with Coulometric Detection. J. Chromatogr. Sep. Tech. 2014, 5, 17–24. [Google Scholar]
- Canário, C.; Matias, M.; Brito, V.; Santos, A.O.; Falcão, A.; Silvestre, S.; Alves, G. New Estrone Oxime Derivatives: Synthesis, Cytotoxic Evaluation and Docking Studies. Molecules 2021, 26, 2687. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, A.; Carcaillon, L.; Kab, S.; Moisan, F. Epidemiology of Parkinson’s disease. Rev. Neurol. 2016, 172, 14–26. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
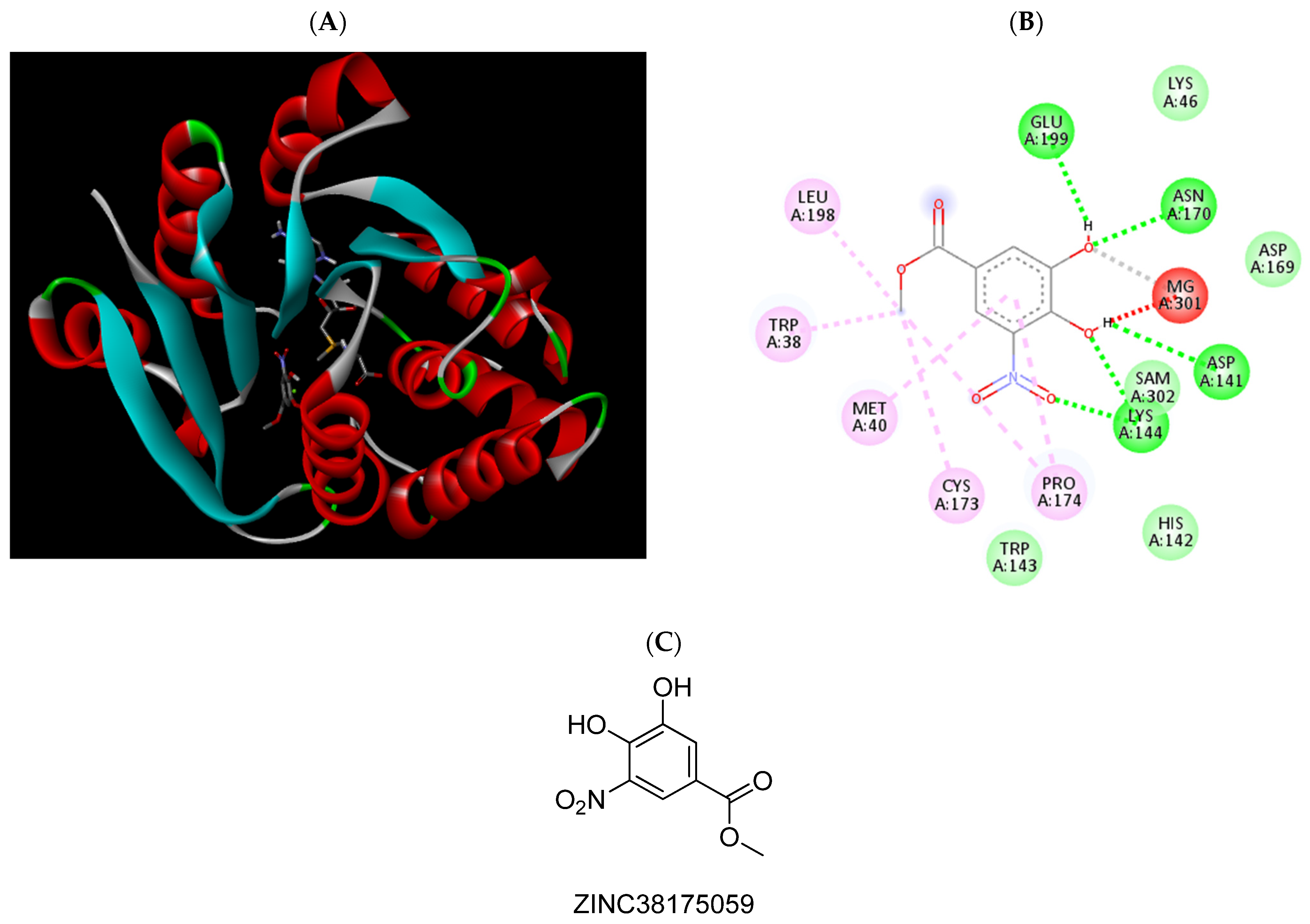{kind=link}
{kind=link}
{kind=link}
{kind=link}
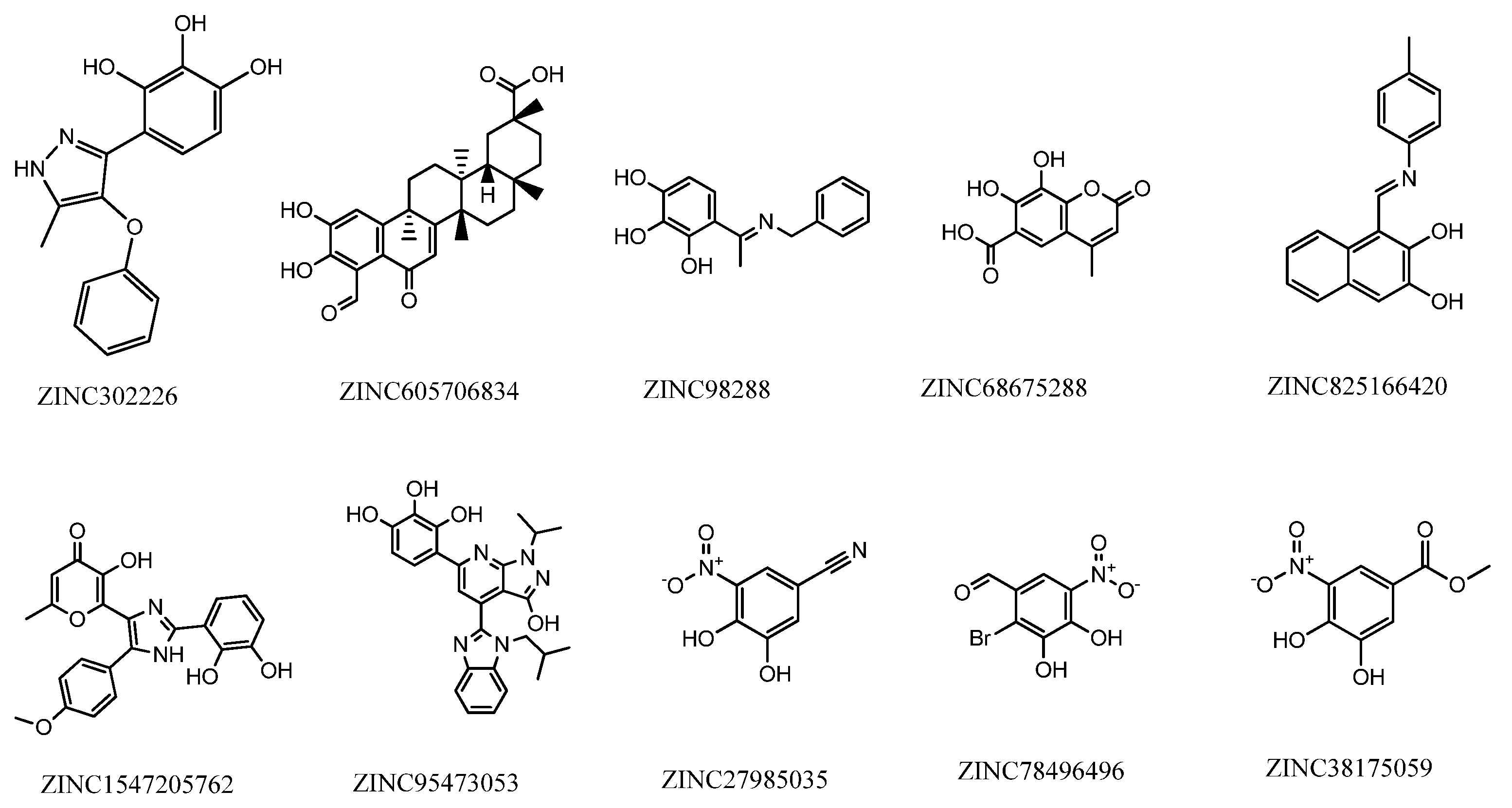
| Compound | Binding Energy | Main Interactions |
|---|---|---|
| ZINC825166420 | −7.48 | Met40, Pro174, Glu199 |
| ZINC1547205762 | −7.39 | Trp38, Met40, Pro174, Mg2+ |
| ZINC98288 | −6.98 | Trp38, Met40, Lys144, Pro174, Glu199 |
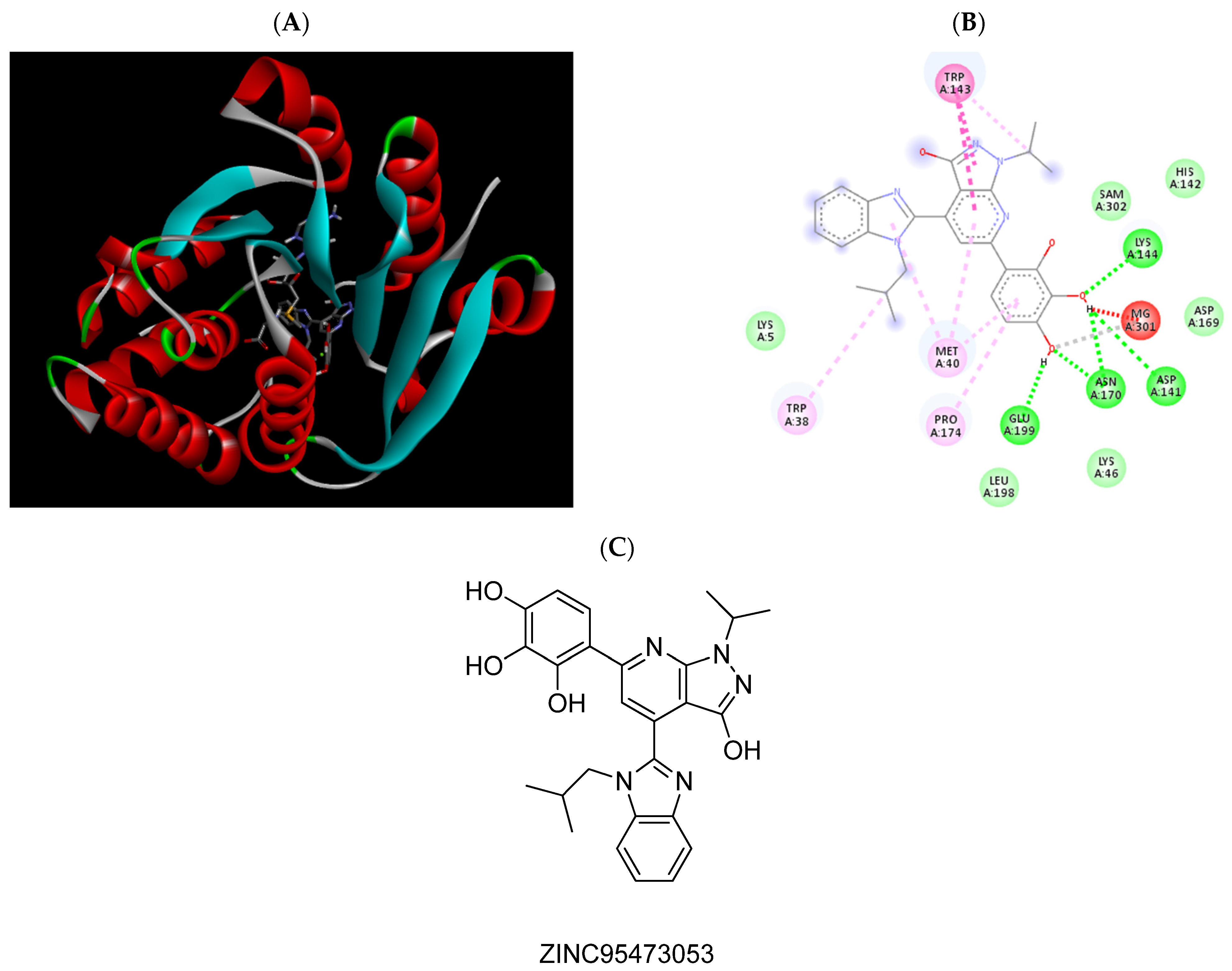
| ZINC95473053 | −6.79 | Trp38, Met40, Asp141, Trp143, Lys144, Asn170, Pro174, Glu199, Mg2+ |
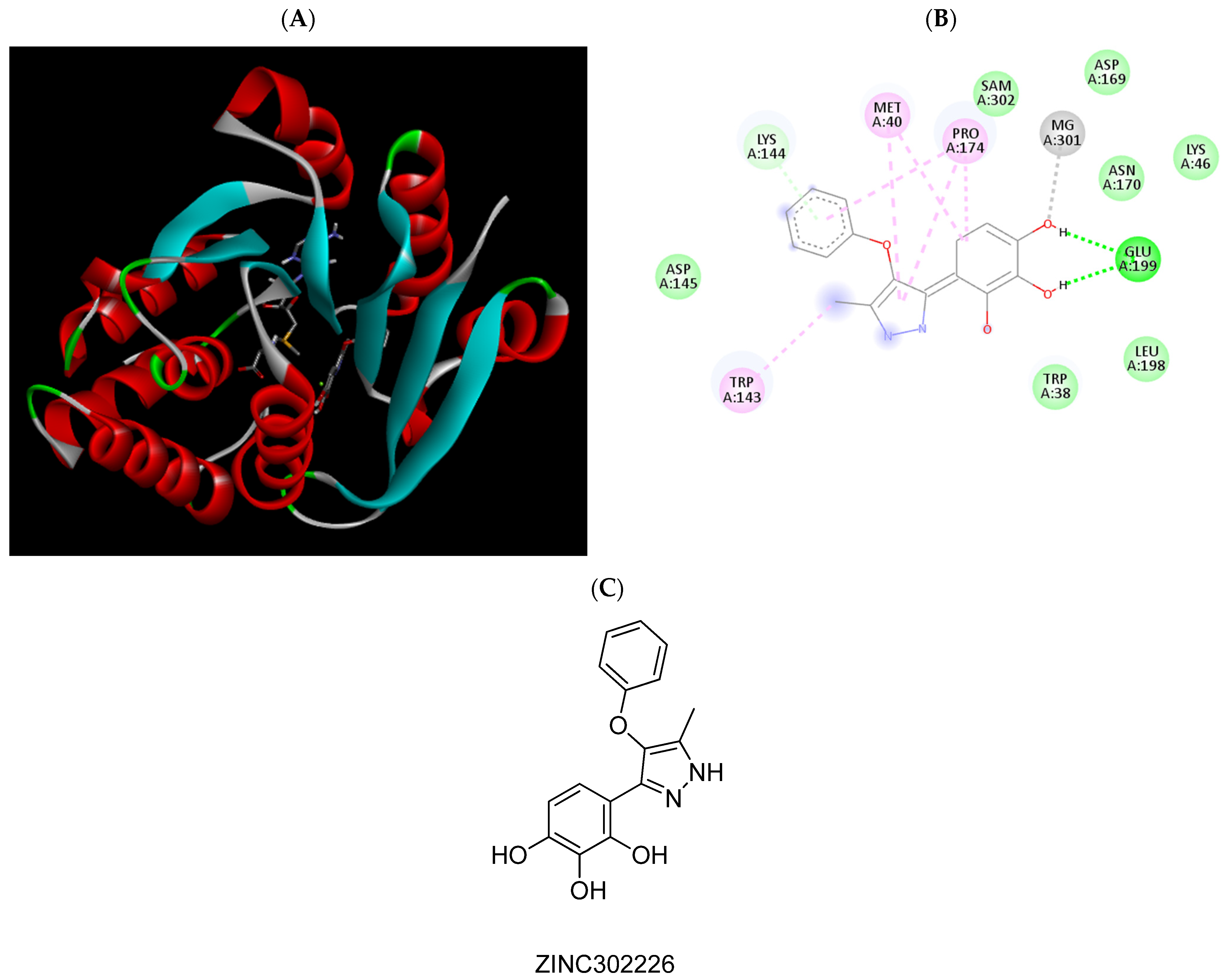
| ZINC302226 | −6.63 | Met40, Pro174, Glu199, Mg2+ |
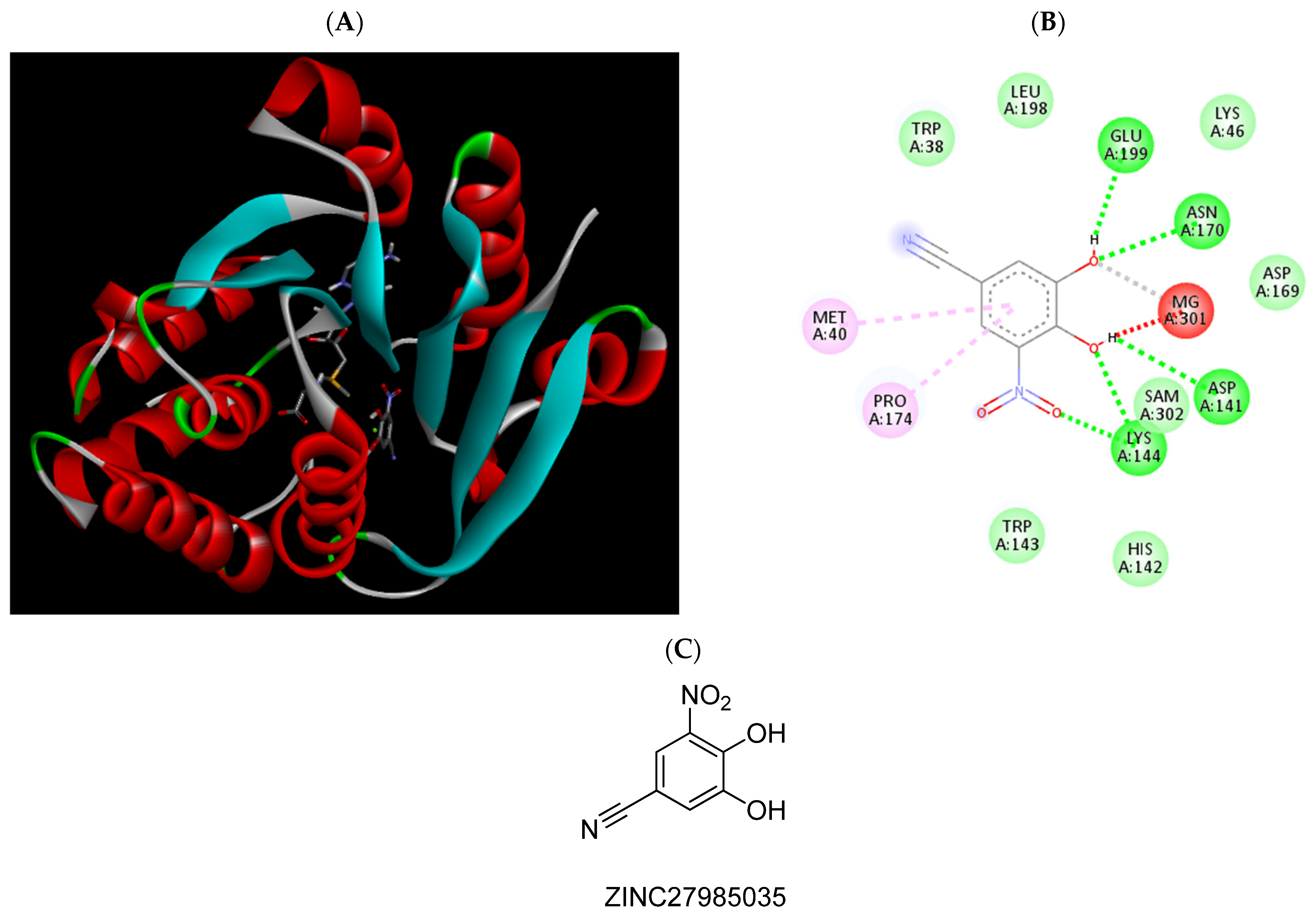
| ZINC27985035 | −6.26 | Met40, Asp141, Lys144, Asn170, Pro174, |
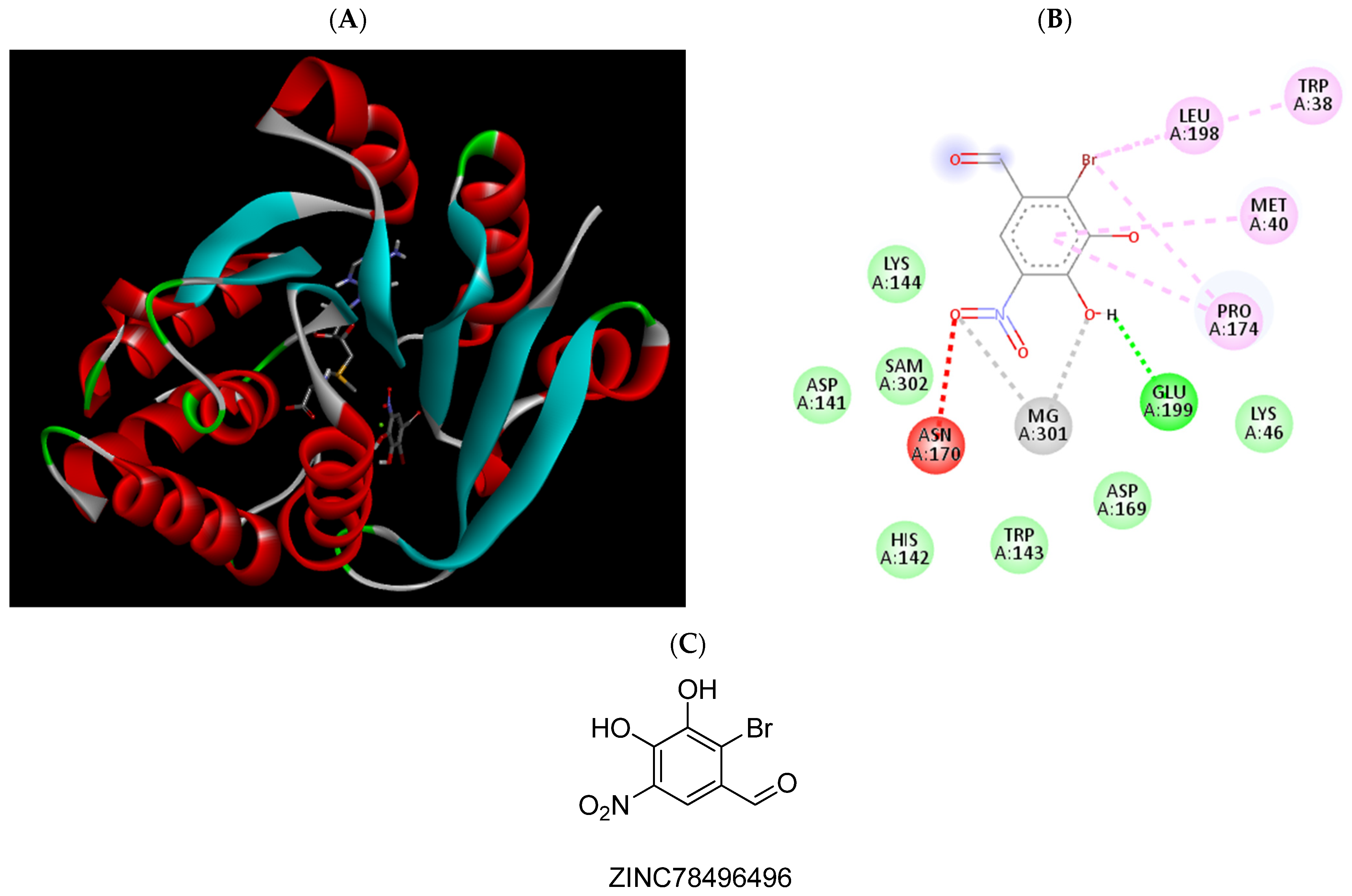
| ZINC78496496 | −6.12 | Trp38, Met40, Pro174, Leu198, Glu199, Mg2+ |
| ZINC38175059 | −6.04 | Trp38, Met40, Asp141, Lys144, Asn170, Pro174, Leu198, Glu199 |
| ZINC605706834 | −6.24 | Trp38, Trp143, Pro174 |
| ZINC68675288 | −5.90 | Met40, Asn170, Pro174, Glu199 |
| Compound | Intestinal Absorption (%) | PgP Substrate | PgP I/II Inhibitor | BBB Permeability (logBB) | CNS Permeability (log PS) | CYP2D6 Substrate | CYP3A4 Substrate | CYP1A2 Inhibitor | CYP2C19 Inhibitor | CYP2C9 Inhibitor | CYP2D6 Inhibitor | AMES Toxicity | LD50 (mol/kg) | LOAEL | Hepatoxicity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ZINC825166420 | 91.048 | Yes | No/No | −0.013 | −1.625 | No | Yes | Yes | Yes | Yes | No | Yes | 2.563 | 2.278 | Yes |
| ZINC1547205762 | 84.913 | Yes | No/No | −1.601 | −3.777 | No | No | Yes | Yes | Yes | Yes | No | 2.346 | 2.323 | No |
| ZINC98288 | 88.908 | Yes | No/No | −0.722 | −2.205 | No | No | Yes | Yes | No | No | No | 2.017 | 2.085 | No |
| ZINC95473053 | 97.282 | Yes | Yes/Yes | −1.467 | −3.022 | No | Yes | Yes | Yes | Yes | Yes | Yes | 2.444 | 2.511 | No |
| ZINC302226 | 69.066 | Yes | No/No | −1.669 | −2.427 | No | No | Yes | No | Yes | No | No | 2.67 | 3.036 | No |
| ZINC27985035 | 76.88 | Yes | No/No | −0.356 | −2.587 | No | No | No | No | No | No | Yes | 2.044 | 2.407 | No |
| ZINC78496496 | 77.717 | Yes | No/No | −0.603 | −2.601 | No | No | No | Yes | No | No | Yes | 2.154 | 2.329 | No |
| ZINC38175059 | 71.419 | Yes | No/No | −0.569 | −2.78 | No | No | No | No | No | No | Yes | 1.804 | 2.158 | No |
| ZINC605706834 | 63.604 | Yes | No/No | −0.813 | −2.724 | No | No | No | No | No | No | No | 2.655 | 2.538 | No |
| ZINC68675288 | 53.332 | Yes | No/No | −1.278 | −4.074 | No | No | No | No | No | No | No | 2.474 | 2.537 | No |
| Sample | Compound | IC50 (nM) | Reference |
|---|---|---|---|
| Recombinant SCOMT | 3,5-DNC | 13.26 (10.7 to 16.44) | [32] |
| Entacapone | 4.224 (2.949 to 6.050) | ||
| Brain MBCOMT | Tolcapone | 2 (1 to 2) | [33] |
| Liver MBCOMT | Tolcapone | 123 (52 to 292) | |
| Liver SCOMT | Pyrazoline derivate | 48 | [34] |
| Entacapone | 230 | ||
| Recombinant MBCOMT | ZINC302226 | 1083 (879.9 to 1333) | This work |
| ZINC98288 | 943.8 (816 to 1092) | ||
| ZINC825166420 | 1538 (1254 to 1888) | ||
| ZINC27985035 | 17.6 (13.53 to 22.96) | ||
| ZINC78496496 | 470 (401.4 to 550.4) |
| Compound | N27 | NHDF |
|---|---|---|
| ZINC302226 | 52.50 | >100 |
| ZINC98288 | 69.78 | >100 |
| ZINC825166420 | 16.98 | 12.14 |
| ZINC27985035 | 61.26 | 40.31 |
| ZINC78496496 | >100 | 92.90 |
| 5-FU | 4.28 | 5.16 |
| Compound | Concentrations for Cytotoxicity Assays (µM) | Concentrations for Bioactivity Assays (µM) |
|---|---|---|
| ZINC302226 | 0.1 to 100 | 0.125 to 10 |
| ZINC98288 | 0.0315 to 10 | |
| ZINC 825166420 | 0.125 to 20 | |
| ZINC27985035 | 0.0078 to 0.5 | |
| ZINC78496496 | 0.125 to 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruz-Vicente, P.; Gonçalves, A.M.; Ferreira, O.; Queiroz, J.A.; Silvestre, S.; Passarinha, L.A.; Gallardo, E. Discovery of Small Molecules as Membrane-Bound Catechol-O-methyltransferase Inhibitors with Interest in Parkinson’s Disease: Pharmacophore Modeling, Molecular Docking and In Vitro Experimental Validation Studies. Pharmaceuticals 2022, 15, 51. https://doi.org/10.3390/ph15010051
Cruz-Vicente P, Gonçalves AM, Ferreira O, Queiroz JA, Silvestre S, Passarinha LA, Gallardo E. Discovery of Small Molecules as Membrane-Bound Catechol-O-methyltransferase Inhibitors with Interest in Parkinson’s Disease: Pharmacophore Modeling, Molecular Docking and In Vitro Experimental Validation Studies. Pharmaceuticals. 2022; 15(1):51. https://doi.org/10.3390/ph15010051
Chicago/Turabian StyleCruz-Vicente, Pedro, Ana M. Gonçalves, Octávio Ferreira, João A. Queiroz, Samuel Silvestre, Luís A. Passarinha, and Eugenia Gallardo. 2022. "Discovery of Small Molecules as Membrane-Bound Catechol-O-methyltransferase Inhibitors with Interest in Parkinson’s Disease: Pharmacophore Modeling, Molecular Docking and In Vitro Experimental Validation Studies" Pharmaceuticals 15, no. 1: 51. https://doi.org/10.3390/ph15010051
APA StyleCruz-Vicente, P., Gonçalves, A. M., Ferreira, O., Queiroz, J. A., Silvestre, S., Passarinha, L. A., & Gallardo, E. (2022). Discovery of Small Molecules as Membrane-Bound Catechol-O-methyltransferase Inhibitors with Interest in Parkinson’s Disease: Pharmacophore Modeling, Molecular Docking and In Vitro Experimental Validation Studies. Pharmaceuticals, 15(1), 51. https://doi.org/10.3390/ph15010051