
In Silico Design, Synthesis, and Biological Evaluation of Anticancer Arylsulfonamide Endowed with Anti-Telomerase Activity

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Results
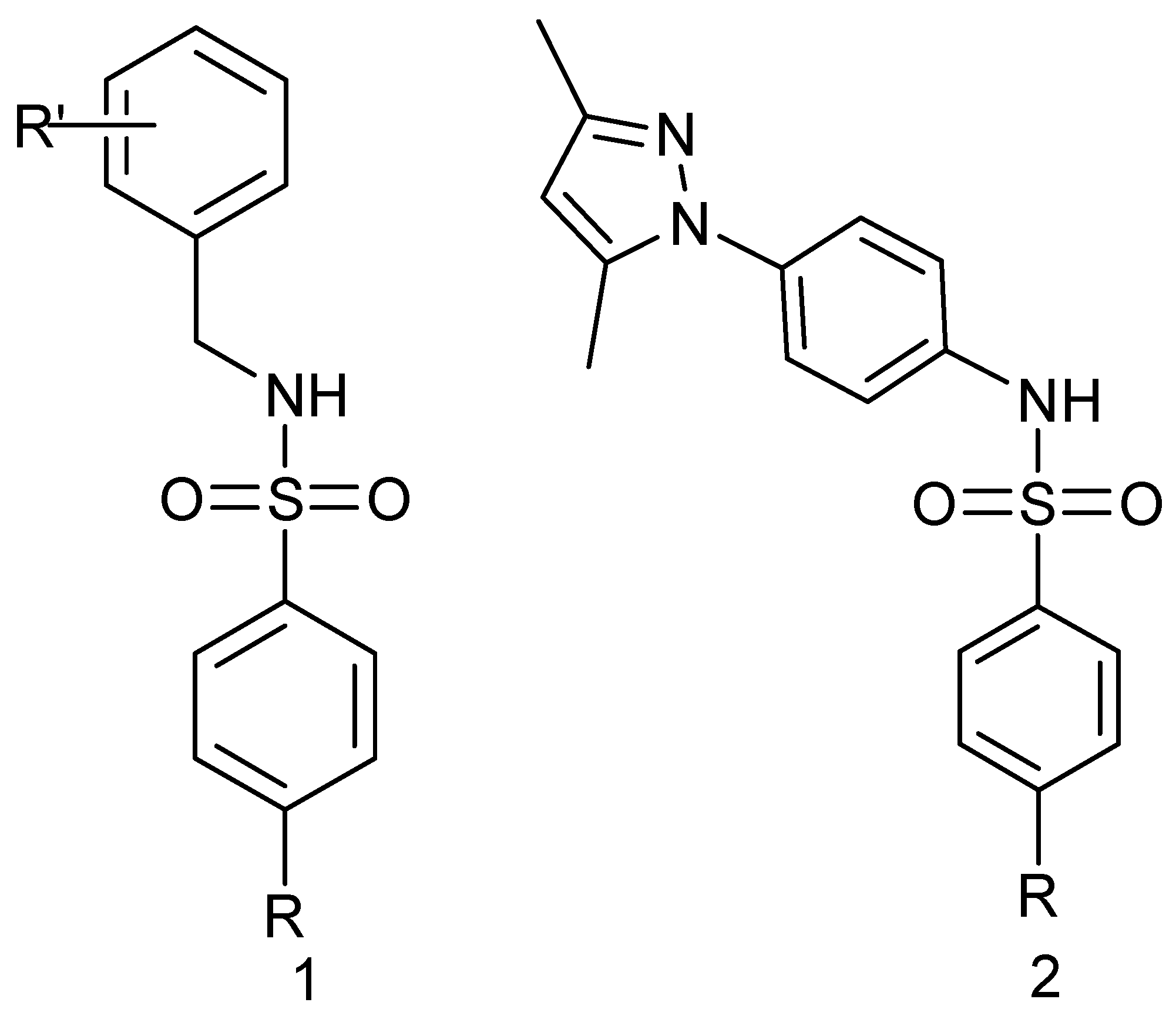
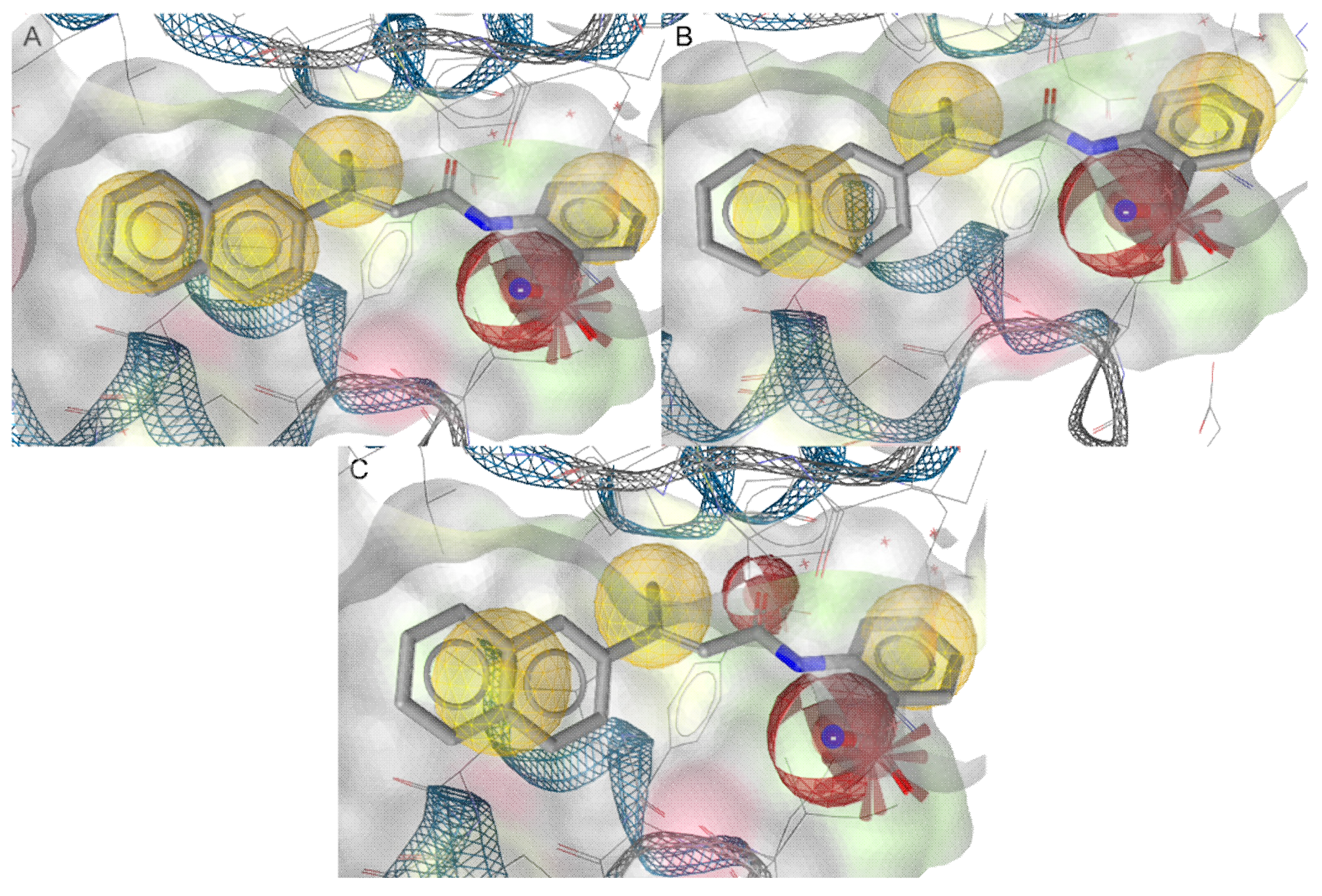
2.1. In Silico Modeling and Virtual Screening
- Four hydrophobic features;
- One H-bond acceptor;
- One anionic feature.
- Three hydrophobic features;
- Two H-bond acceptors;
- One anionic feature.
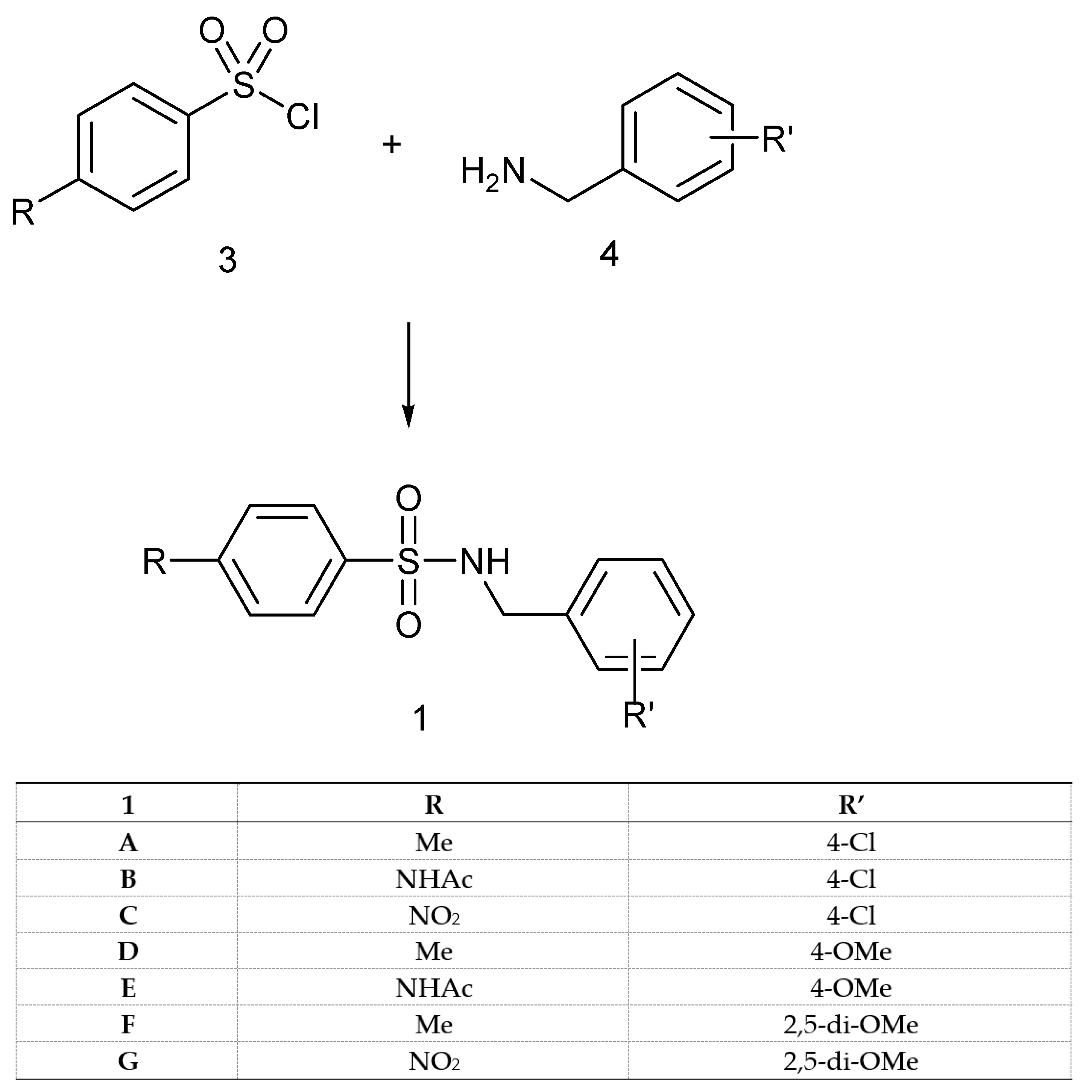
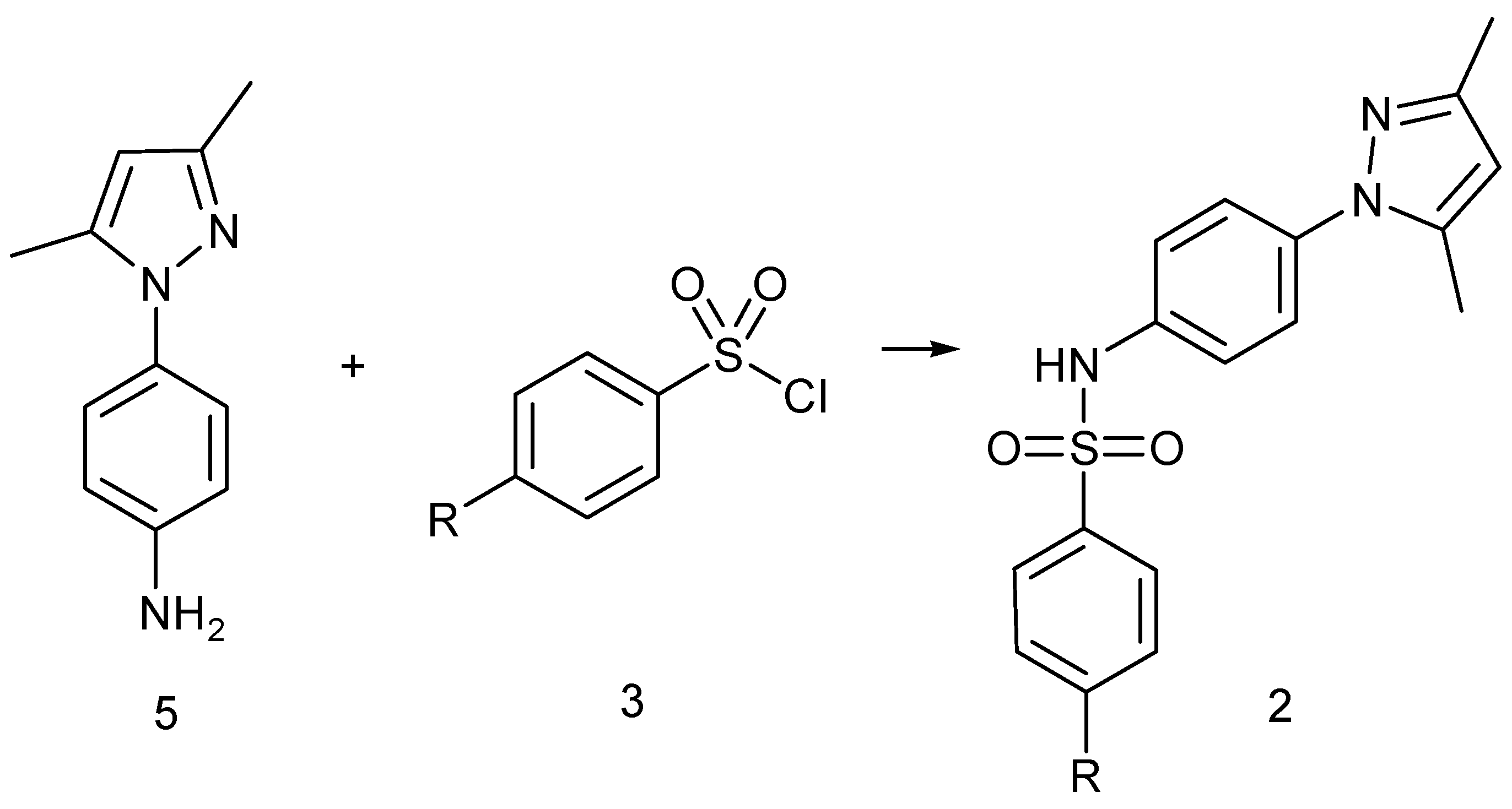
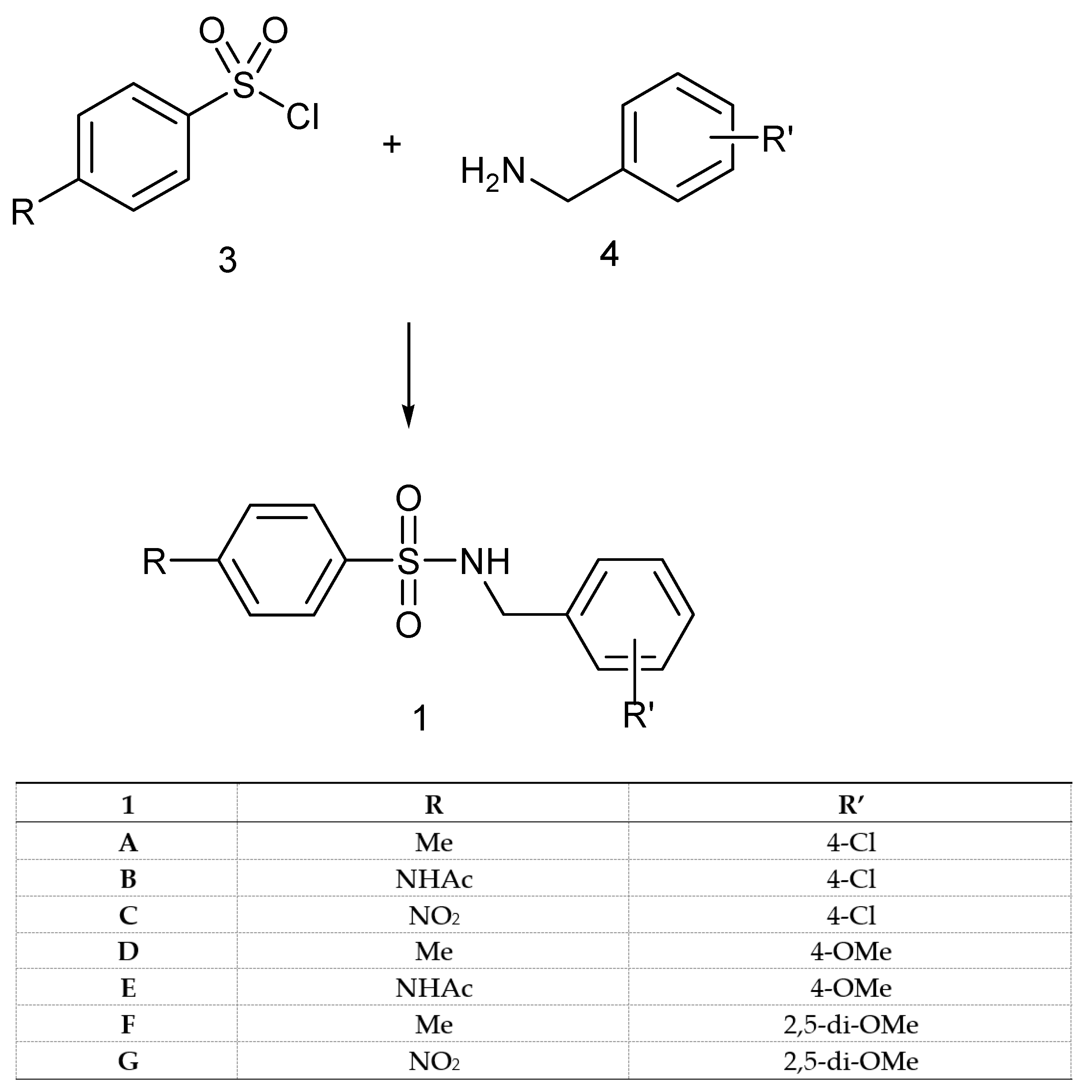
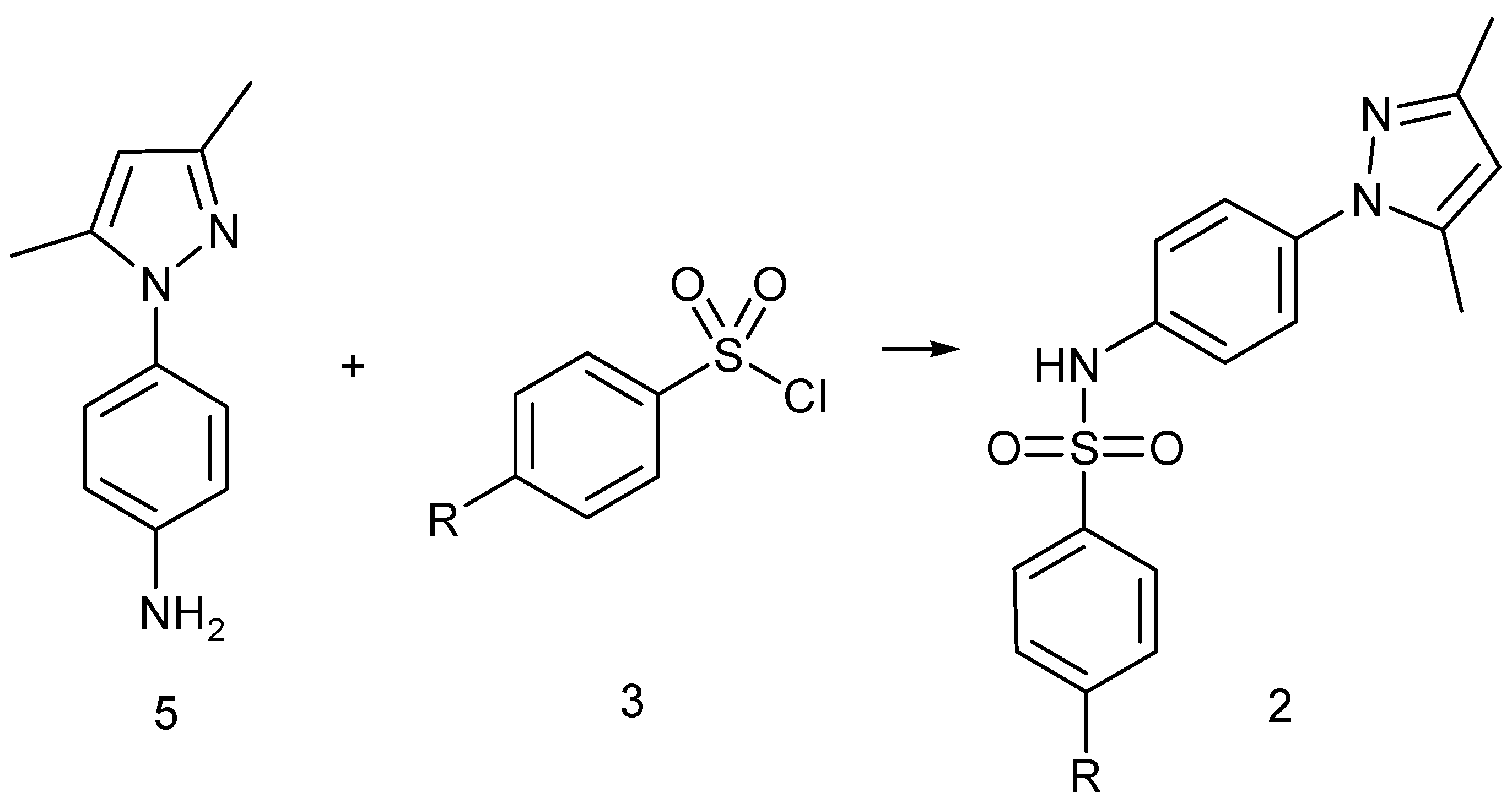
2.2. Synthesis
2.3. Cytotoxic Activity and Selectivity Index
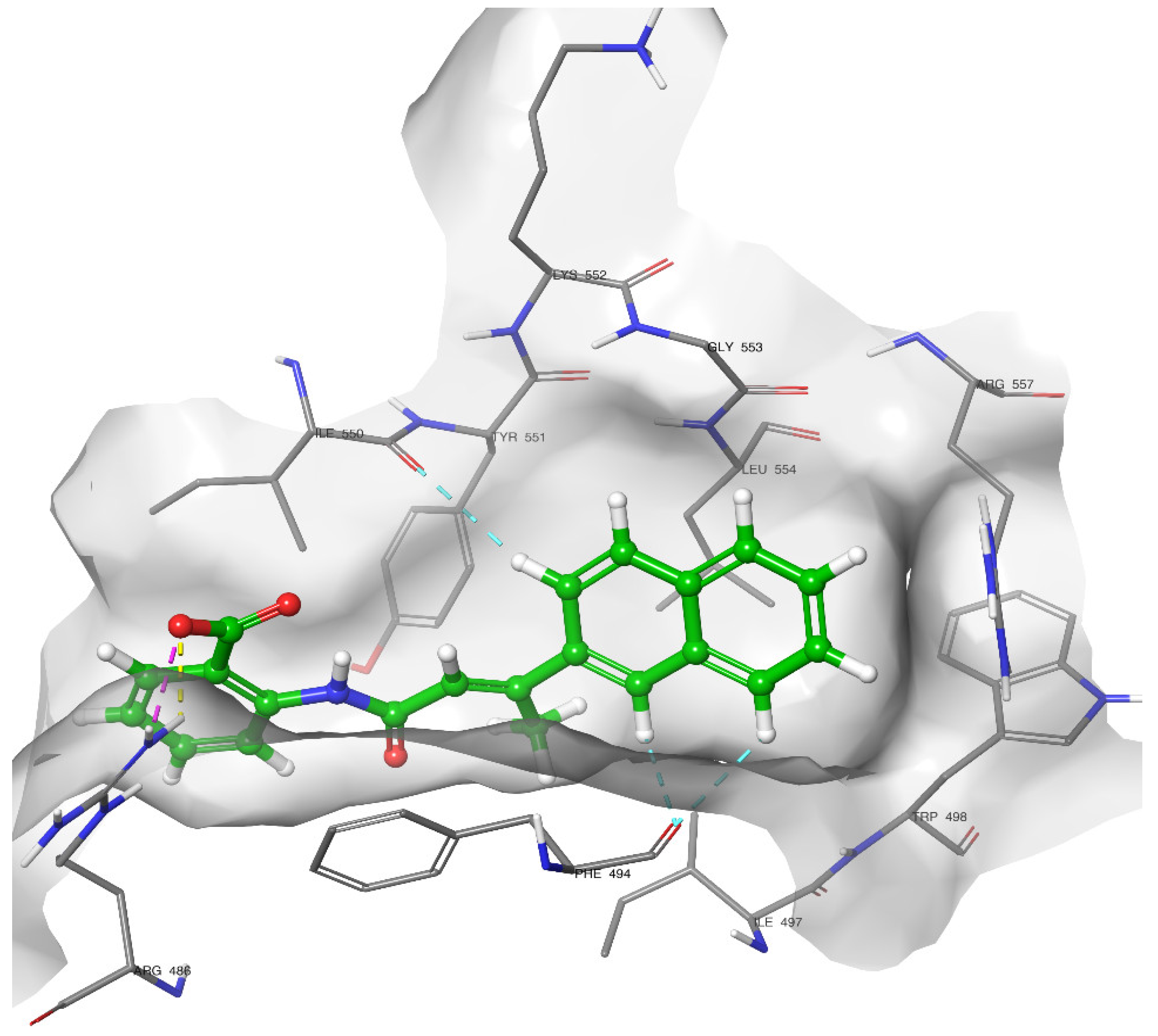
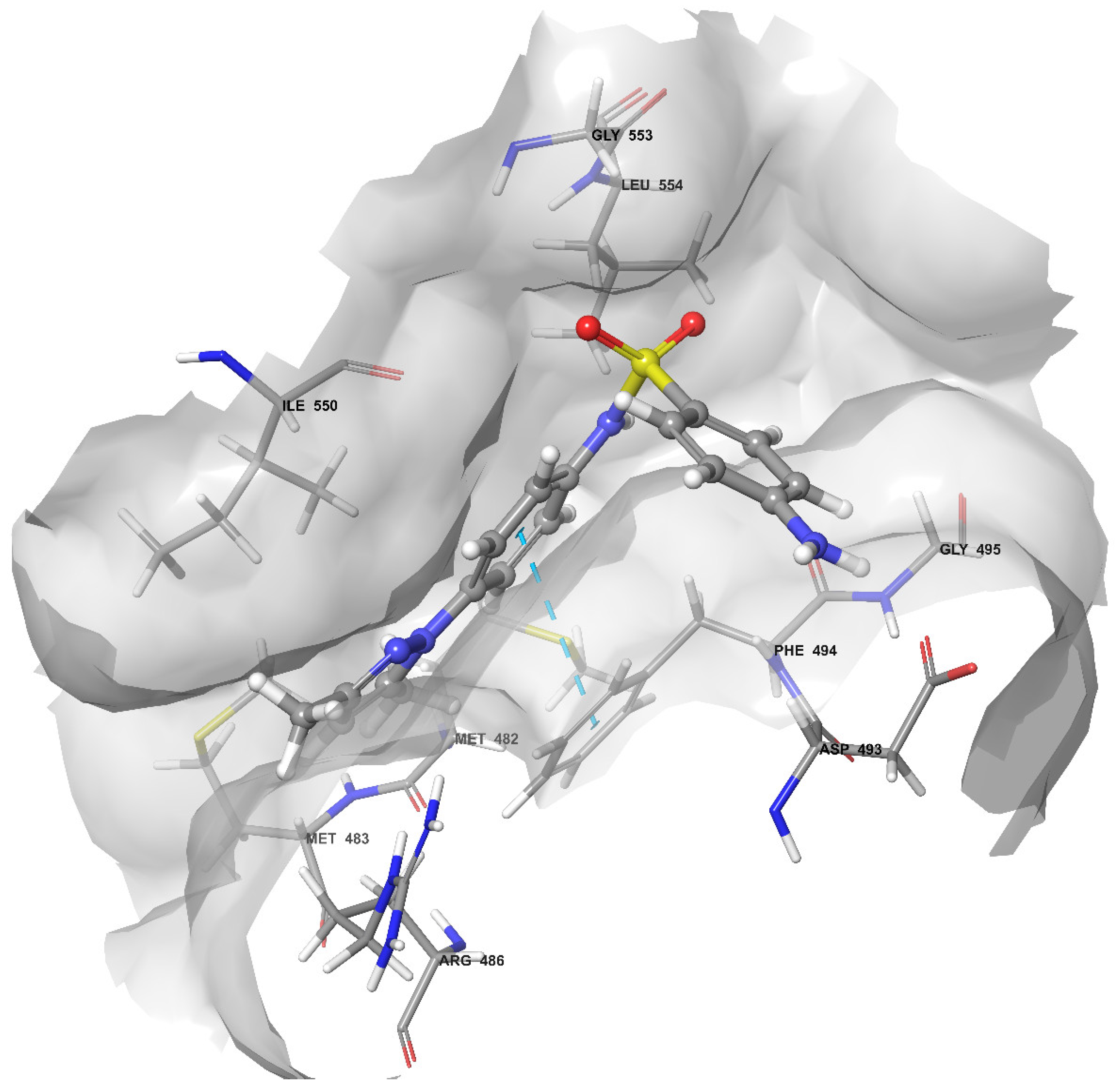
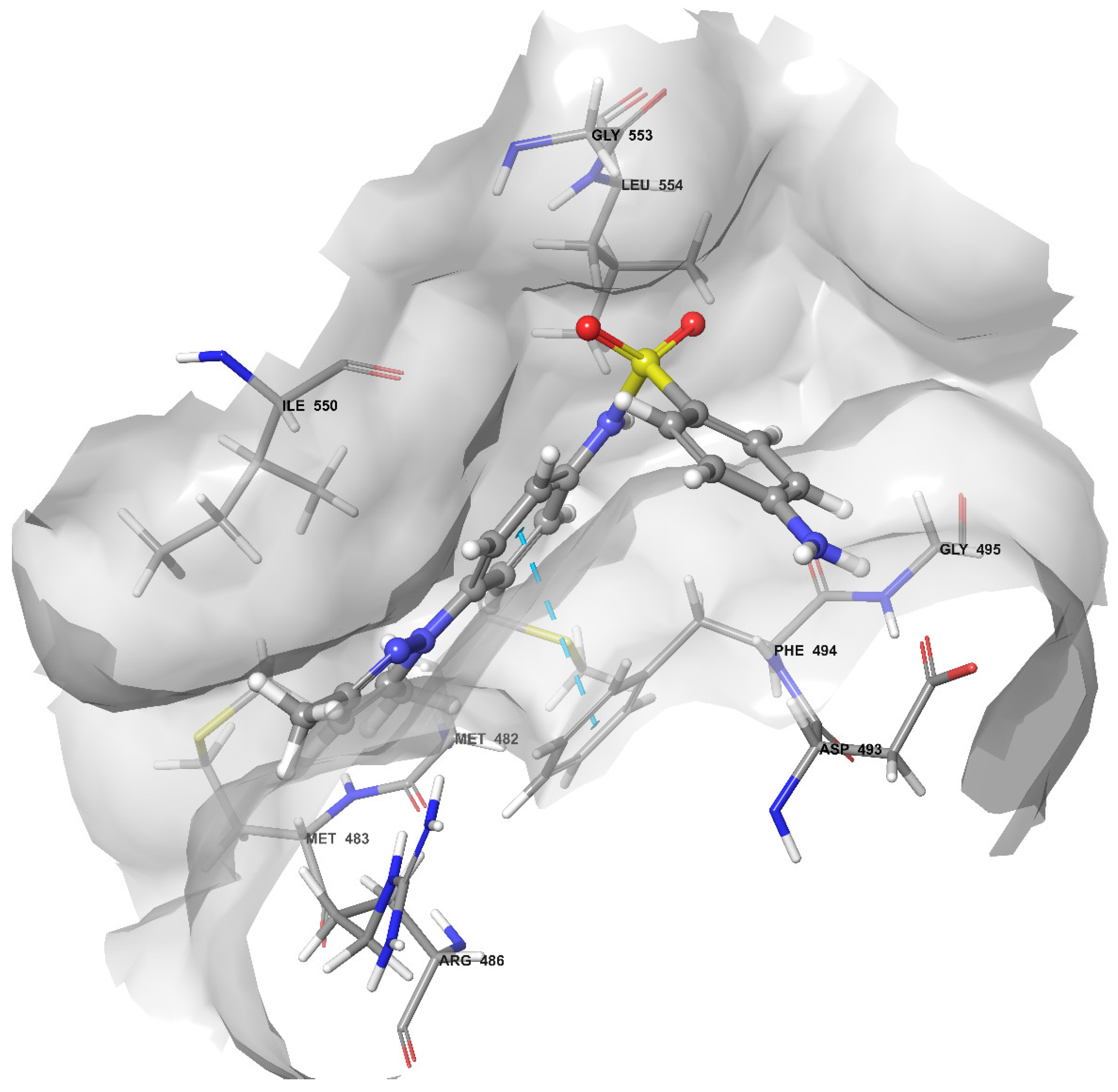
2.4. Docking of Compound 2C
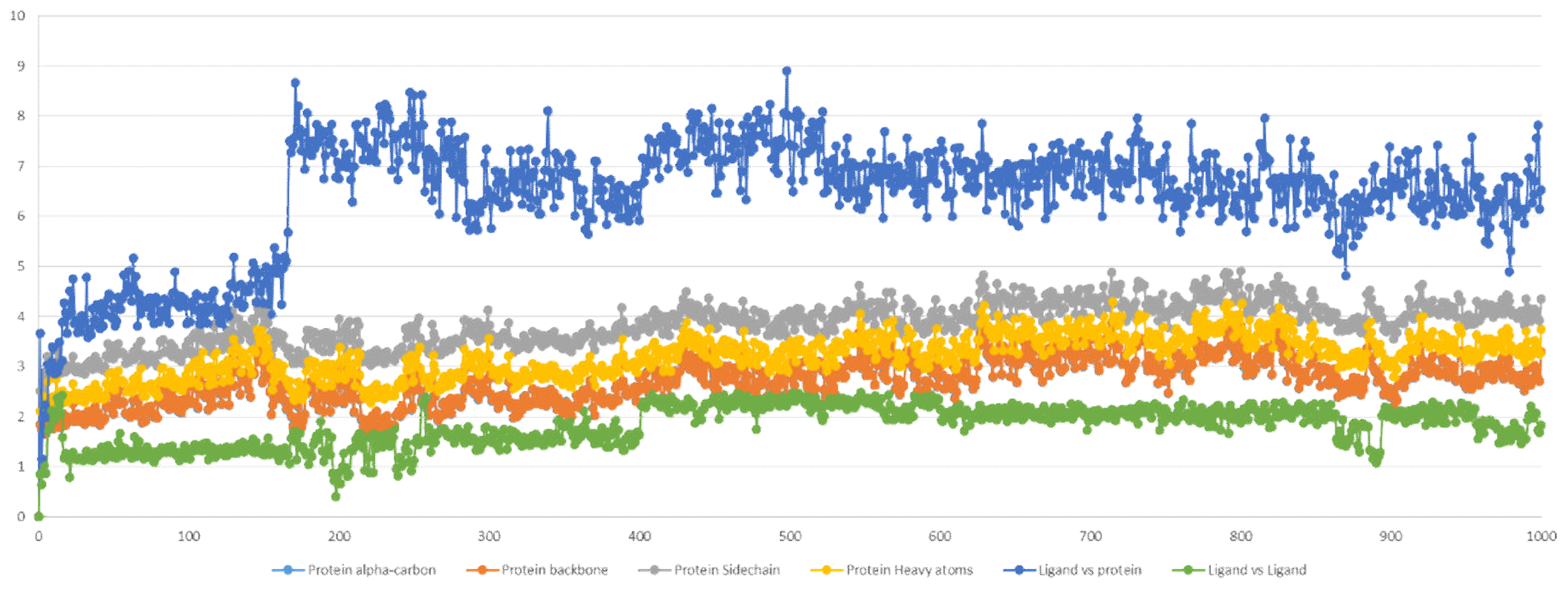
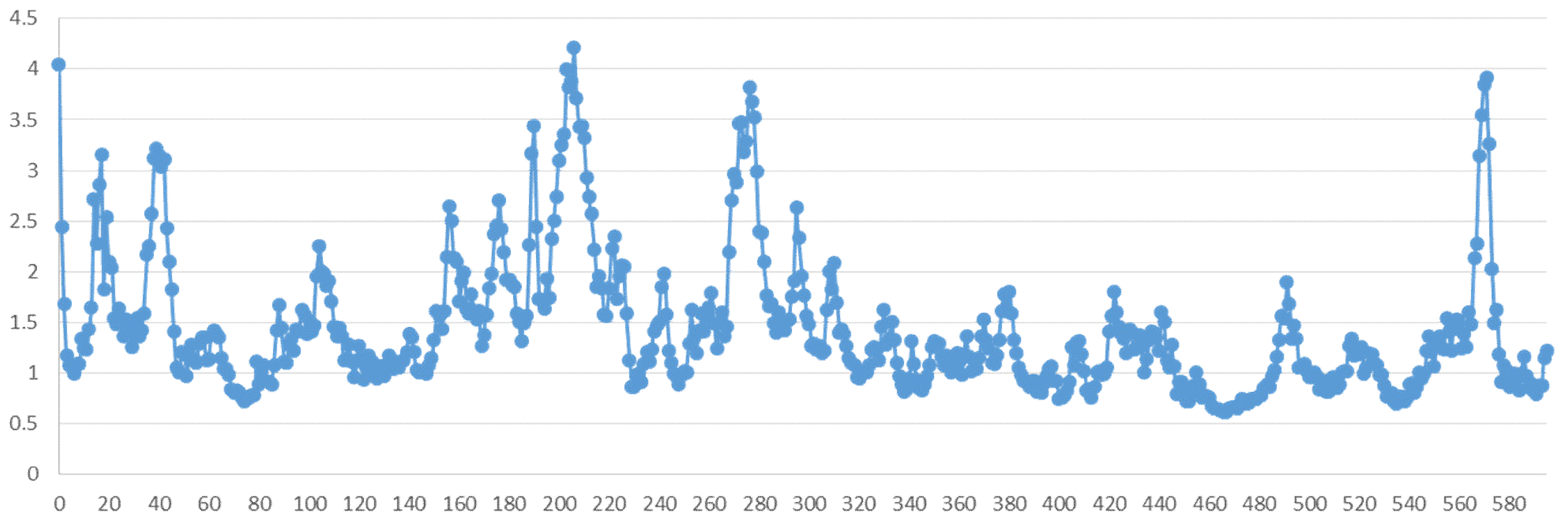
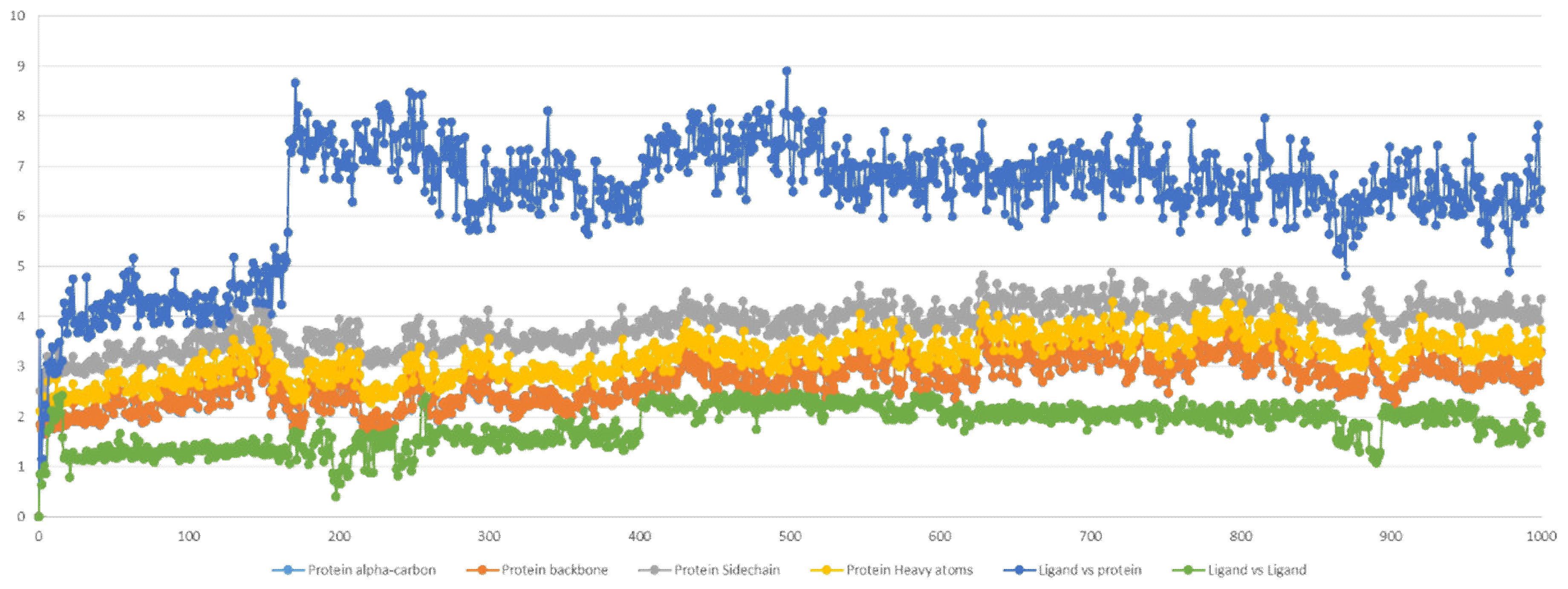
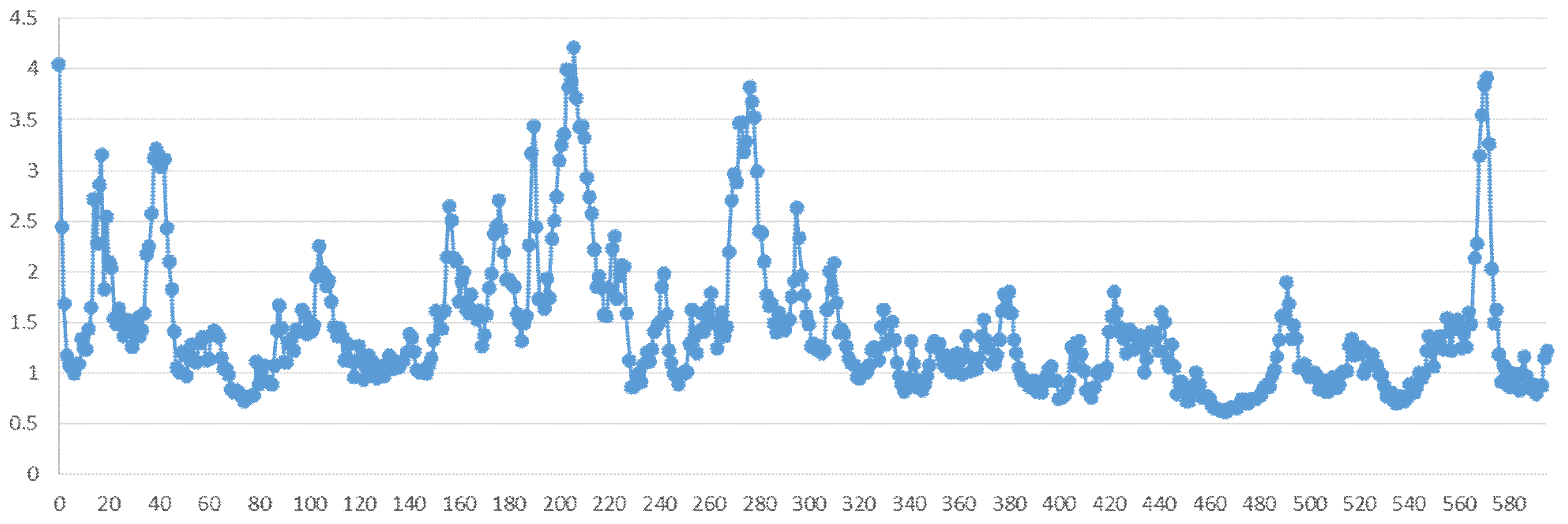
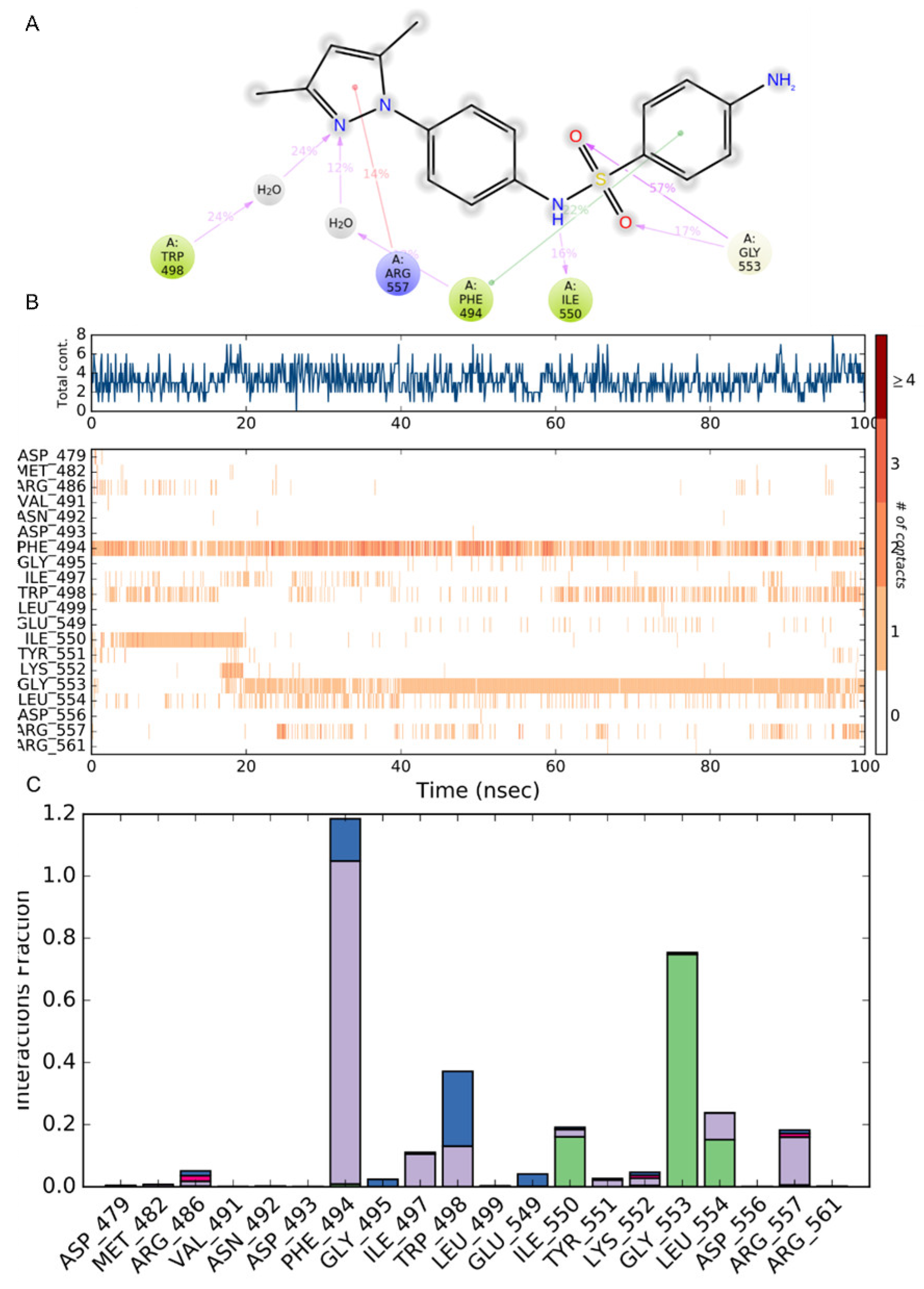
2.5. Molecular Dynamics Simulation
2.5.1. Stability Analysis
2.5.2. Residue Mobility and Protein–Ligand Contact Analyses
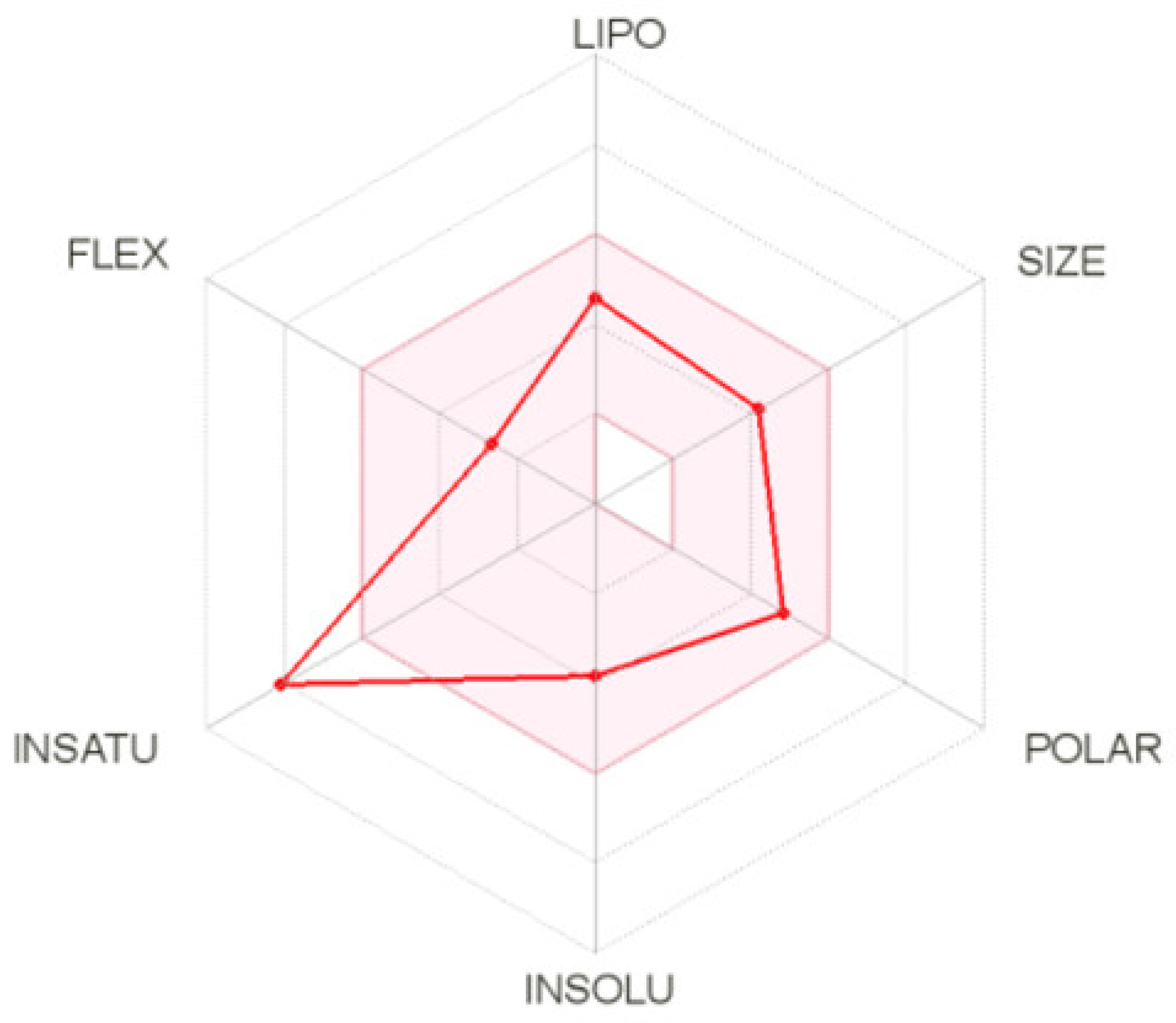
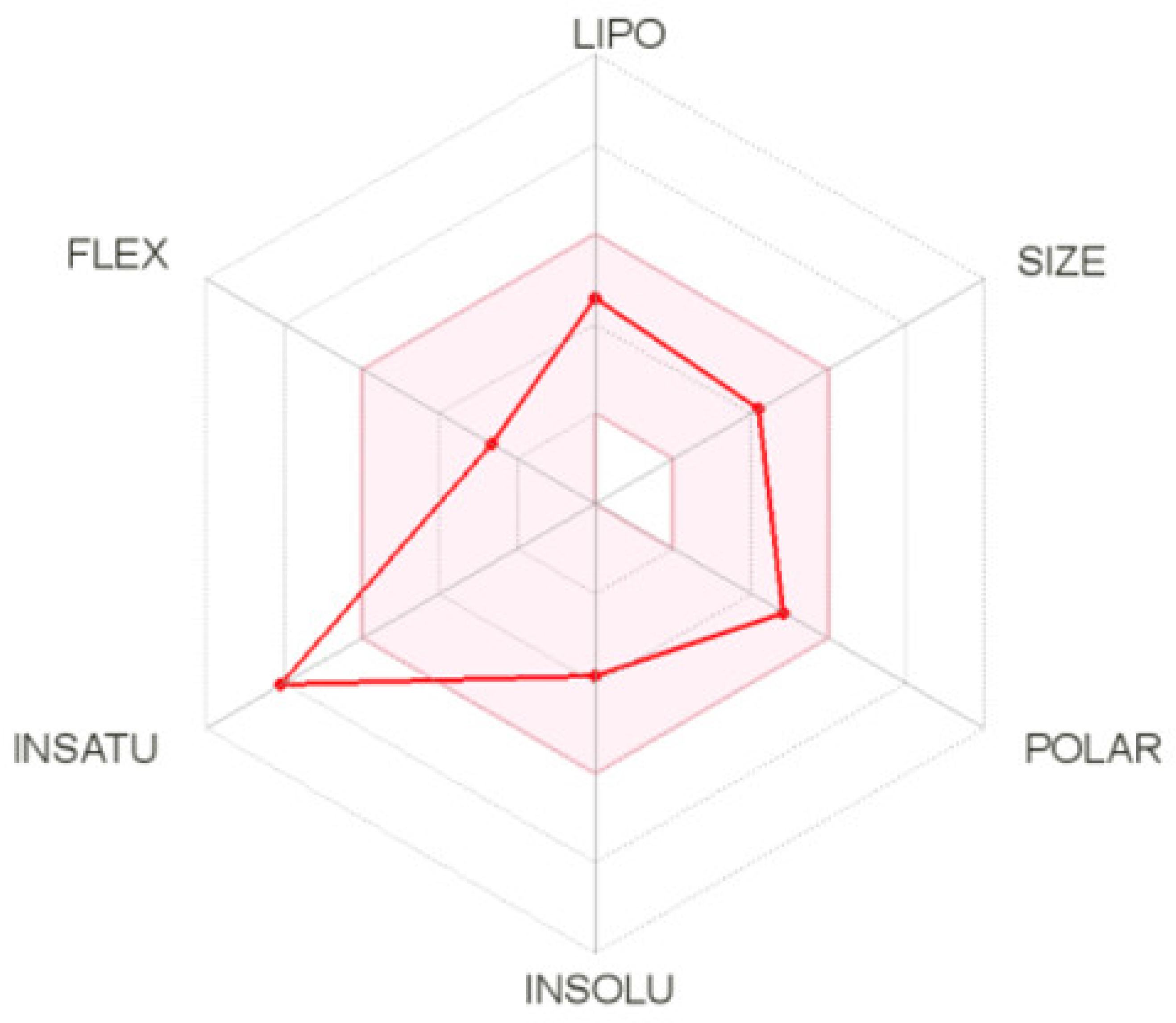
2.5.3. ADME Calculation for Compound 2C
3. Discussion
4. Materials and Methods
4.1. Library and Protein Preparation
4.2. Docking Studies
4.3. MYSHAPE Approach
4.4. MM-GBSA Free Energy Calculations
4.5. Molecular Dynamic Simulations
4.6. ADME Prediction
4.7. Chemistry
4.7.1. General Methods for the Preparation of N-(R’-Benzyl)-4-R-benzenesulfonamides of Type 1
N-(4-Chlorobenzyl)-4-methylbenzenesulfonamide (1A)
N-(4-Chlorobenzyl)-4-acetylaminobenzenesulfonamide (1B)
N-(4-Chlorobenzyl)-4-nitrobenzenesulfonamide (1C)
N-(4-Methoxybenzyl)-4-methylbenzenesulfonamide (1D)
N-(4-Methoxybenzyl)-4-acetylaminobenzenesulfonamide (1E)
N-(2,5-Dimethoxybenzyl)-4-methylbenzenesulfonamide (1F)
N-(2,5-Dimethoxybenzyl)-4-nitrobenzenesulfonamide (1G)
4.7.2. Preparation of 1-(4-Aminophenyl)-3,5-dimethylpyrazole
4.7.3. General Method for the Preparation of N-[4-(3,5-Dimethyl-1H-pyrazol-1-yl)phenyl]-4-R-benzenesulfonamide of Type 2
N-[4-(3,5-Dimethylpyrazol-1-yl)phenyl]-4-methylbenzenesulfonamide (2A)
N-[4-(3,5-Dimethylpyrazol-1-yl)phenyl]-4-nitrobenzenesulfonamide (2B)
Preparation of N-[4-(3,5-Dimethylpyrazol-1-yl)phenyl]-4-aminobenzenesulfonamide (2C)
4.8. Cell Proliferation Assay
Cell Culturing and MTT Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cong, Y.-S.; Wright, W.E.; Shay, J.W. Human Telomerase and Its Regulation. Microbiol. Mol. Biol. Rev. 2002, 66, 407–425. [Google Scholar] [CrossRef] [Green Version]
- Wojtyla, A.; Gladych, M.; Rubis, B. Human telomerase activity regulation. Mol. Biol. Rep. 2011, 38, 3339–3349. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Podell, E.R.; Zaug, A.J.; Yang, Y.; Baciu, P.; Cech, T.R.; Lei, M. The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature 2007, 445, 506–510. [Google Scholar] [CrossRef]
- Masutomi, K.; Yu, E.Y.; Khurts, S.; Ben-Porath, I.; Currier, J.L.; Metz, G.B.; Brooks, M.W.; Kaneko, S.; Murakami, S.; DeCaprio, J.A.; et al. Telomerase maintains telomere structure in normal human cells. Cell 2003, 114, 241–253. [Google Scholar] [CrossRef] [Green Version]
- Shay, J.W.; Wright, W.E. Role of telomeres and telomerase in cancer. Semin. Cancer Biol. 2011, 21, 349–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackburn, E.H. Telomere states and cell fates. Nature 2000, 408, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Ramlee, M.K.; Wang, J.; Toh, W.X.; Li, S. Transcription regulation of the human telomerase reverse transcriptase (hTERT) gene. Genes 2016, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruden, M.; Puri, N. Novel anticancer therapeutics targeting telomerase. Cancer Treat. Rev. 2013, 39, 444–456. [Google Scholar] [CrossRef]
- Arndt, G.M.; MacKenzie, K. New prospects for targeting telomerase beyond the telomere. Nat. Rev. Cancer 2016, 16, 508–524. [Google Scholar] [CrossRef]
- Jäger, K.; Walter, M. Therapeutic Targeting of Telomerase. Genes 2016, 7, 39. [Google Scholar] [CrossRef] [Green Version]
- Neidle, S. Human telomeric G-quadruplex: The current status of telomeric G-quadruplexes as therapeutic targets in human cancer. FEBS J. 2009, 277, 1118–1125. [Google Scholar] [CrossRef]
- Islam, M.K.; Jackson, P.J.; Rahman, K.M.; Thurston, D.E. Recent advances in targeting the telomeric G-quadruplex DNA sequence with small molecules as a strategy for anticancer therapies. Futur. Med. Chem. 2016, 8, 1259–1290. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.M.; Tizkova, K.; Reszka, A.P.; Neidle, S.; Thurston, D.E. Identification of novel telomeric G-quadruplex-targeting chemical scaffolds through screening of three NCI libraries. Bioorg. Med. Chem. Lett. 2012, 22, 3006–3010. [Google Scholar] [CrossRef] [PubMed]
- Lauria, A.; Terenzi, A.; Bartolotta, R.; Bonsignore, R.; Perricone, U.; Tutone, M.; Martorana, A.; Barone, G.; Almerico, A.M. Does ligand symmetry play a role in the stabilization of DNA g-quadruplex host-guest complexes? Curr. Med. Chem. 2014, 21, 2665–2690. [Google Scholar] [CrossRef] [PubMed]
- Barata, P.; Sood, A.K.; Hong, D.S. RNA-targeted therapeutics in cancer clinical trials: Current status and future directions. Cancer Treat. Rev. 2016, 50, 35–47. [Google Scholar] [CrossRef]
- Asai, A.; Oshima, Y.; Yamamoto, Y.; Uochi, T.; Kusaka, H.; Akinaga, S.; Yamashita, Y.; Pongracz, K.; Pruzan, R.; Wunder, E.; et al. A novel telomerase template antagonist (GRN163) as a potential anticancer agent. Cancer Res. 2003, 63, 3931–3939. [Google Scholar]
- Damm, K.; Hemmann, U.; Garin-Chesa, P.; Hauel, N.; Kauffmann, I.; Priepke, H.; Niestroj, C.; Daiber, C.; Enenkel, B.; Guilliard, B.; et al. A highly selective telomerase inhibitor limiting human cancer cell proliferation. EMBO J. 2001, 20, 6958–6968. [Google Scholar] [CrossRef] [PubMed]
- Man, R.-J.; Chen, L.-W.; Zhu, H.-L. Telomerase inhibitors: A patent review (2010–2015). Expert Opin. Ther. Patents 2016, 26, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Baginski, M.; Serbakowska, K. In silico design of telomerase inhibitors. Drug Discov. Today 2020, 25, 1213–1222. [Google Scholar] [CrossRef]
- Bryan, C.; Rice, C.; Hoffman, H.; Harkisheimer, M.; Sweeney, M.; Skordalakes, E. Structural Basis of Telomerase Inhibition by the Highly Specific BIBR1532. Structure 2015, 23, 1934–1942. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.S.; Lee, D.-U. Synthesis, biological evaluation, drug-likeness, and in silico screening of novel benzylidene-hydrazone analogues as small molecule anticancer agents. Arch. Pharmacal Res. 2015, 39, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.-Y.; Yang, Y.; Liu, X.-H.; Qi, X.-B. Novel 3-(1-acetyl-5-(substituted-phenyl)-4,5-dihydro-1H-pyrazol-3-yl)-7-fluoro-2H-chromen-2-one Derivatives: Synthesis and Anticancer Activity. Lett. Drug Des. Discov. 2010, 7, 640–643. [Google Scholar] [CrossRef]
- Liu, X.-H.; Li, J.; Shi, J.B.; Song, B.-A.; Qi, X.-B. Design and synthesis of novel 5-phenyl-N-piperidine ethanone containing 4,5-dihydropyrazole derivatives as potential antitumor agents. Eur. J. Med. Chem. 2012, 51, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-H.; Liu, H.-F.; Chen, J.; Yang, Y.; Song, B.-A.; Bai, L.-S.; Liu, J.-X.; Zhu, H.-L.; Qi, X.-B. Synthesis and molecular docking study of novel coumarin derivatives containing 4,5-dihydropyrazole moiety as potential antitumor agents. Bioorg. Med. Chem. Lett. 2010, 20, 5705–5708. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zhang, S.; Qiu, K.-M.; Liu, Z.-J.; Yang, Y.-S.; Fu, J.; Zhong, W.-Q.; Zhu, H.-L. Synthesis, biological evaluation, 3D-QSAR studies of novel aryl-2H-pyrazole derivatives as telomerase inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 1091–1095. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zhou, Y.; Fu, J.; Zhu, H.-L. 4,5-Dihydropyrazole derivatives containing oxygen-bearing heterocycles as potential telomerase inhibitors with anticancer activity. RSC Adv. 2014, 4, 23904–23913. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Wu, X.Q.; Tang, W.J.; Shi, J.B.; Li, J.; Liu, X.H. Novel dihydropyrazole-chromen: Design and modulates hTERT inhibition proliferation of MGC-803. Eur. J. Med. Chem. 2016, 110, 65–75. [Google Scholar] [CrossRef]
- Xiao, X.; Ni, Y.; Jia, Y.-M.; Zheng, M.; Xu, H.-F.; Xu, J.; Liao, C. Identification of human telomerase inhibitors having the core of N -acyl-4,5-dihydropyrazole with anticancer effects. Bioorg. Med. Chem. Lett. 2016, 26, 1508–1511. [Google Scholar] [CrossRef] [PubMed]
- Kalathiya, U.; Padariya, M.; Baginski, M. Molecular Modeling and Evaluation of Novel Dibenzopyrrole Derivatives as Telomerase Inhibitors and Potential Drug for Cancer Therapy. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 11, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-H.; Liu, H.-F.; Shen, X.; Song, B.-A.; Bhadury, P.S.; Zhu, H.-L.; Liu, J.-X.; Qi, X.-B. Synthesis and molecular docking studies of novel 2-chloro-pyridine derivatives containing flavone moieties as potential antitumor agents. Bioorg. Med. Chem. Lett. 2010, 20, 4163–4167. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.-Z.; Zhang, X.-M.; Xu, Y.; Cheng, K.; Jiao, Q.-C.; Zhu, H.-L. Synthesis, biological evaluation, and molecular docking studies of 2-chloropyridine derivatives possessing 1,3,4-oxadiazole moiety as potential antitumor agents. Bioorg. Med. Chem. 2010, 18, 7836–7841. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.M.; Qiu, M.; Sun, J.; Zhang, Y.B.; Yang, Y.S.; Wang, X.L.; Tang, J.F.; Zhu, H.L. Synthesis, biological evaluation, and molecular docking studies of 1,3,4-oxadiazole derivatives possessing 1,4-benzodioxan moiety as potential anticancer agents. Bioorg. Med. Chem. 2011, 19, 6518–6524. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, X.-L.; Shi, J.; Wang, S.-F.; Yin, Y.; Yang, Y.-S.; Zhang, W.-M.; Zhu, H.-L. Synthesis, molecular modeling, and biological evaluation of N-benzylidene-2-((5-(pyridin-4-yl)-1,3,4-oxadiazol-2-yl)thio)acetohydrazide derivatives as potential anticancer agents. Bioorg. Med. Chem. 2014, 22, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhu, H.; Yang, Z.M.; Zhu, H.L. Synthesis, molecular modeling and biological evaluation of 2-aminomethyl-5-(quinolin-2-yl)-1,3,4-oxadiazole-2(3H)-thione quinolone derivatives as novel anticancer agent. Eur. J. Med. Chem. 2013, 60, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Tutone, M.; Pecoraro, B.; Almerico, A.M. Investigation on Quantitative Structure-Activity Relationships of 1,3,4-Oxadiazole Derivatives as Potential Telomerase Inhibitors. Curr. Drug Discov. Technol. 2020, 17, 79–86. [Google Scholar] [CrossRef]
- Shi, J.B.; Tang, W.J.; Qi, X.B.; Li, R.; Liu, X.H. Novel pyrazole-5-carboxamide and pyrazole-pyrimidine derivatives: Synthesis and anticancer activity. Eur. J. Med. Chem. 2015, 90, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.J.; Wang, J.; Tong, X.; Shi, J.B.; Liu, X.H.; Li, J. Design and synthesis of celastrol derivatives as anticancer agents. Eur. J. Med. Chem. 2015, 95, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Song, B.A.; Zhao, H.J.; Qi, X.B.; Huang, Y.J.; Liu, X.H. Novel myricetin derivatives: Design, synthesis and anticancer activity. Eur. J. Med. Chem. 2015, 97, 155–163. [Google Scholar] [CrossRef]
- Hernandez-Sanchez, W.; Huang, W.; Plucinsky, B.; Garcia-Vazquez, N.; Robinson, N.J.; Schiemann, W.P.; Berdis, A.J.; Skordalakes, E.; Taylor, D.J. A non-natural nucleotide uses a specific pocket to selectively inhibit telomerase activity. PLoS Biol. 2019, 17, e3000204. [Google Scholar] [CrossRef]
- Fan, Z.F.; Ho, S.T.; Wen, R.; Fu, Y.; Zhang, L.; Wang, J.; Hu, C.; Shaw, P.C.; Liu, Y.; Cheng, M.S. Design, synthesis and molecular docking analysis of flavonoid derivatives as potential telomerase inhibitors. Molecules 2019, 24, 3180. [Google Scholar] [CrossRef] [Green Version]
- Betori, R.C.; Liu, Y.; Mishra, R.K.; Cohen, S.B.; Kron, S.J.; Scheidt, K.A. Targeted Covalent Inhibition of Telomerase. ACS Chem. Biol. 2020, 15, 706–717. [Google Scholar] [CrossRef]
- Saraswati, A.P.; Relitti, N.; Brindisi, M.; Gemma, S.; Zisterer, D.; Butini, S.; Campiani, G. Raising the bar in anticancer therapy: Recent advances in, and perspectives on, telomerase inhibitors. Drug Discov. Today 2019, 24, 1370–1388. [Google Scholar] [CrossRef]
- Chen, X.; Tang, W.-J.; Shi, J.B.; Liu, M.M.; Liu, X.-H. Therapeutic strategies for targeting telomerase in cancer. Med. Res. Rev. 2020, 40, 532–585. [Google Scholar] [CrossRef]
- Wong, L.H.; Unciti-Broceta, A.; Spitzer, M.; White, R.; Tyers, M.; Harrington, L. A Yeast Chemical Genetic Screen Identifies Inhibitors of Human Telomerase. Chem. Biol. 2013, 20, 333–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pibiri, I.; Lentini, L.; Melfi, R.; Tutone, M.; Baldassano, S.; Ricco Galluzzo, P.; Di Leonardo, A.; Pace, A. Rescuing the CFTR protein function: Introducing 1,3,4-oxadiazoles as translational readthrough inducing drugs. Eur. J. Med. Chem. 2018, 159, 126–142. [Google Scholar] [CrossRef] [PubMed]
- Almerico, A.M.; Tutone, M.; Lauria, A. Docking and multivariate methods to explore HIV-1 drug-resistance: A comparative analysis. J. Comput.-Aided Mol. Des. 2008, 22, 287–297. [Google Scholar] [CrossRef]
- Tutone, M.; Pibiri, I.; Lentini, L.; Pace, A.; Almerico, A.M. Deciphering the Nonsense Readthrough Mechanism of Action of Ataluren: An in Silico Compared Study. ACS Med. Chem. Lett. 2019, 10, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Tutone, M.; Virzì, A.; Almerico, A.M. Reverse screening on indicaxanthin from Opuntia ficus-indica as natural chemoactive and chemopreventive agent. J. Theor. Biol. 2018, 455, 147–160. [Google Scholar] [CrossRef]
- Tutone, M.; Pibiri, I.; Perriera, R.; Campofelice, A.; Culletta, G.; Melfi, R.; Pace, A.; Almerico, A.M.; Lentini, L. Pharmacophore-Based Design of New Chemical Scaffolds as Translational Readthrough-Inducing Drugs (TRIDs). ACS Med. Chem. Lett. 2020, 11, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Perricone, U.; Wieder, M.; Seidel, T.; Langer, T.; Padova, A.; Almerico, A.M.; Tutone, M. A Molecular Dynamics–Shared Pharmacophore Approach to Boost Early-Enrichment Virtual Screening: A Case Study on Peroxisome Proliferator-Activated Receptor α. ChemMedChem 2017, 12, 1399–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culletta, G.; Almerico, A.M.; Tutone, M. Comparing molecular dynamics-derived pharmacophore models with docking: A study on CDK-2 inhibitors. Chem. Data Collect. 2020, 28, 100485. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Doobary, S.; Sedikides, A.T.; Caldora, H.P.; Poole, D.L.; Lennox, A.J.J. Electrochemical Vicinal Difluorination of Alkenes: Scalable and Amenable to Electron-Rich Substrates. Angew. Chem. Int. Ed. 2019, 59, 1155–1160. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Meng, X.; Zhu, B.; Jia, Y.; Cao, X.; Huang, S. A Micellar Catalysis Strategy for Amidation of Alkynyl Bromides: Synthesis of Ynamides in Water. Eur. J. Org. Chem. 2019, 2019, 1166–1169. [Google Scholar] [CrossRef]
- Ge, S.; Zhong, H.; Ma, X.; Zheng, Y.; Zou, Y.; Wang, F.; Wang, Y.; Hu, Y.; Li, Y.; Liu, W.; et al. Discovery of secondary sulphonamides as IDO1 inhibitors with potent antitumour effects in vivo. J. Enzyme Inhib. Med. Chem. 2020, 35, 1240–1257. [Google Scholar] [CrossRef]
- Peng, H.; Ma, J.; Luo, W.; Zhang, G.; Yin, B. Methyl-triflate-mediated dearylmethylation of N -(arylmethyl)carboxamides via the retro-Mannich reaction induced by electrophilic dearomatization/rearomatization in an aqueous medium at room temperature. Green Chem. 2019, 21, 2252–2256. [Google Scholar] [CrossRef]
- Li, M.B.; Tang, X.L.; Tian, S.K. Cross-coupling of Grignard reagents with sulfonyl-activated sp3 carbon-nitrogen bonds. Adv. Synth. Catal. 2011, 353, 1980–1984. [Google Scholar] [CrossRef]
- Kiruthika, S.E.; Nandakumar, A.; Perumal, P.T. Synthesis of pyrrolo-/indolo[1,2-a ]quinolines and naphtho[2,1- b ]thiophenes from gem -dibromovinyls and sulphonamides. Org. Lett. 2014, 16, 4424–4427. [Google Scholar] [CrossRef] [PubMed]
- Baraldi, P.G.; Moorman, A.R.; Borea, P.A. Novel Adenosine A3 Receptor Agonists. U.S. Patent 20090233878A1, 17 September 2009. [Google Scholar]
- Nardi, M.; Cano, N.H.; Costanzo, P.; Oliverio, M.; Sindona, G.; Procopio, A. Aqueous MW eco-friendly protocol for amino group protection. RSC Adv. 2015, 5, 18751–18760. [Google Scholar] [CrossRef]
- Huang, M.; Li, Y.; Liu, J.; Lan, X.B.; Liu, Y.; Zhao, C.; Ke, Z. A bifunctional strategy for N-heterocyclic carbene-stabilized iridium complex-catalyzed: N -alkylation of amines with alcohols in aqueous media. Green Chem. 2019, 21, 219–224. [Google Scholar] [CrossRef]
- Li, Q.Q.; Xiao, Z.F.; Yao, C.Z.; Zheng, H.X.; Kang, Y.B. Direct Alkylation of Amines with Alcohols Catalyzed by Base. Org. Lett. 2015, 17, 5328–5331. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, Y.; Yuan, G. Electrosynthesis of Arylsulfonamides from Amines and Sodium Sulfinates Using H2O-NaI as the Electrolyte Solution at Room Temperature. Chin. J. Chem. 2016, 34, 1277–1282. [Google Scholar] [CrossRef]
- Wang, H.; Wei, W.; Zhu, H.; Cui, H.; Wang, L. A Kind of Ethod of Sulfamide Compound. China Patent CN107033106A, 11 August 2017. [Google Scholar]
- Vicente, D.A.; Galdino, D.; Navarro, M.; Menezes, P.H. Electrochemical synthesis of sulfonamides in a graphite powder macroelectrode. Green Chem. 2020, 22, 5262–5266. [Google Scholar] [CrossRef]
- Deng, X.; Mani, N.S. A facile, environmentally benign sulfonamide synthesis in water. Green Chem. 2006, 8, 835–838. [Google Scholar] [CrossRef]
- Massah, A.; Kazemi, F.; Azadi, D.; Farzaneh, S.; Aliyan, H.; Naghash, H.; Momeni, A. A Mild and Chemoselective Solvent-Free Method for the Synthesis of N-Aryl and N-Alkylsulfonamides. Lett. Org. Chem. 2006, 3, 235–241. [Google Scholar] [CrossRef]
- Myung, K.; Ghosh, G.; Fattah, F.J.; Li, G.; Kim, H.; Dutia, A.; Pak, E.; Smith, S.; Hendrickson, E.A. Regulation of Telomere Length and Suppression of Genomic Instability in Human Somatic Cells by Ku86. Mol. Cell. Biol. 2004, 24, 5050–5059. [Google Scholar] [CrossRef] [Green Version]
- Raymond, E.; Sun, D.; Izbicka, E.; Mangold, G.; Silvas, E.; Windle, B.; Sharma, S.; Soda, H.; Laurence, R.; Davidson, K.; et al. A human breast cancer model for the study of telomerase inhibitors based on a new biotinylated-primer extension assay. Br. J. Cancer 1999, 80, 1332–1341. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, M.; Yamada, O.; Kanda, N.; Akita, S.; Kawano, T.; Ohno, T.; Mizoguchi, H.; Eto, Y.; Anderson, K.C.; Yamada, H. Telomerase overexpression in K562 leukemia cells protects against apoptosis by serum deprivation and double-stranded DNA break inducing agents, but not against DNA synthesis inhibitors. Cancer Lett. 2002, 178, 187–197. [Google Scholar] [CrossRef]
- Badisa, R.B.; Darling-Reed, S.F.; Joseph, P.; Cooperwood, J.S.; Latinwo, L.M.; Goodman, C.B. Selective cytotoxic activities of two novel synthetic drugs on human breast carcinoma MCF-7 cells. Anticancer Res. 2009, 29, 2993–2996. [Google Scholar]
- Le, M.-T.; Mai, T.T.; Huynh, P.N.H.; Tran, T.-D.; Thai, K.-M.; Nguyen, Q.-T. Structure-based discovery of interleukin-33 inhibitors: A pharmacophore modelling, molecular docking, and molecular dynamics simulation approach. SAR QSAR Environ. Res. 2020, 31, 883–904. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Azam, F.; Madi, A.M.; Ali, H.I. Molecular Docking and Prediction of Pharmacokinetic Properties of Dual Mechanism Drugs that Block MAO-B and Adenosine A2A Receptors for the Treatment of Parkinson’s Disease. J. Young Pharm. 2012, 4, 184–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.H.; Abraham, M.H.; Le, J.; Hersey, A.; Luscombe, C.N.; Beck, G.; Sherborne, B.; Cooper, I. Rate-limited steps of human oral absorption and QSAR studies. Pharm. Res. 2002, 19, 1446–1457. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving force field accuracy on challenging regimes of chemical space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Allegra, M.; Tutone, M.; Tesoriere, L.; Attanzio, A.; Culletta, G.; Almerico, A.M. Evaluation of the IKKβ Binding of Indicaxanthin by Induced-Fit Docking, Binding Pose Metadynamics, and Molecular Dynamics. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, T.; Yu, R. Molecular dynamics and free energy studies on the wild-type and double mutant HIV-1 protease complexed with amprenavir and two amprenavir-related inhibitors: Mechanism for binding and drug resistance. J. Med. Chem. 2007, 50, 1177–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massova, I.; Kollman, P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. Des. 2000, 18, 113–135. [Google Scholar] [CrossRef]
- Culletta, G.; Gulotta, M.R.; Perricone, U.; Zappalà, M.; Almerico, A.M.; Tutone, M. Exploring the SARS-CoV-2 Proteome in the Search of Potential Inhibitors via Structure-Based Pharmacophore Modeling/Docking Approach. Computation 2020, 8, 77. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, Q.Q.; Liang, S.; Hu, L.-M.; Little, R.D.; Zeng, C.-C. Electrochemical Oxidative Amination of Sodium Sulfinates: Synthesis of Sulfonamides Mediated by NH4I as a Redox Catalyst. J. Org. Chem. 2016, 81, 4713–4719. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Kin Tse, M.; Zhou, S.; Pohl, M.-M.; Radnik, J.; Hübner, S.; Jähnisch, K.; Brückner, A.; Beller, M. Green and Efficient Synthesis of Sulfonamides Catalyzed by Nano-Ru/Fe3O4. J. Am. Chem. Soc. 2009, 131, 1775–1779. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Ge, S.; Zheng, Y.; Zou, Y.; Wang, F.; Xu, Q.; Guo, W.; Wang, Y.; Hu, Y. Benzene sulfonamide IDO1 inhibitor, its preparation method and application. China Patent CN106928101A, 7 July 2017. [Google Scholar]
- Molander, G.A.; Fleury-Brégeot, N.; Hiebel, M.-A. Synthesis and Cross-Coupling of Sulfonamidomethyltrifluoroborates. Org. Lett. 2011, 13, 1694–1697. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Asencio, A.; Ramón, D.J.; Yus, M. N-Alkylation of poor nucleophilic amines and derivatives with alcohols by a hydrogen autotransfer process catalyzed by copper(II) acetate: Scope and mechanistic considerations. Tetrahedron 2011, 67, 3140–3149. [Google Scholar] [CrossRef]
- Burness, D.M. β-Keto Acetals. I. Synthesis of Pyrazoles and Pyrimidines and the Steric Inhibition of Resonance in 5-Alkyl-1-p-nitrophenylpyrazoles. J. Org. Chem. 1956, 21, 97–101. [Google Scholar] [CrossRef]
- Bouchet, P. Bulletin de la Societe Chimique de France; Société chimique de France: Paris, France, 1976; Volume 5–6, pp. 839–844. [Google Scholar]
- Allegra, M.; De Cicco, P.; Ercolano, G.; Attanzio, A.; Busà, R.; Cirino, G.; Tesoriere, L.; Livrea, M.A.; Ianaro, A. Indicaxanthin from Opuntia Ficus Indica (L. Mill) impairs melanoma cell proliferation, invasiveness, and tumor progression. Phytomedicine 2018, 50, 19–24. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | R | R’ | Method X | No Solvent * |
|---|---|---|---|---|
| C | NO2 | 4-Cl | 78 | 100 |
| D | Me | 4-OMe | 100 | 60 |
| E | NHAc | 4-OMe | 80 | 70 |
| Compound | % Inhibition Cell Proliferation | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HCT-116 | MCF-7 | K-562 | |||||||||||||
| Compound Concentration (µm) | |||||||||||||||
| 10 | 75 | 150 | 300 | 400 | 10 | 75 | 150 | 300 | 400 | 10 | 75 | 150 | 300 | 400 | |
| 1A | 0 | 0 | 19 ± 2 | 29 ± 3 | 40 ± 2 | 0 | 0 | 0 | 12 ± 1 | 13 ± 1 | 0 | 0 | 0 | 33 ± 2 | 38 ± 2 |
| 1B | 0 | 5 ± 0.2 | 23 ± 2 | * Sol | * Sol | 0 | 15 ± 1 | 21 ± 2 | * Sol | * Sol | 0 | 5 ± 0.3 | 21 ± 1 | * Sol | * Sol |
| 1C | 0 | 0 | 2 ± 0.1 | 8 ± 0.2 | 15 ± 2 | 0 | 5 ± 0.1 | 15 ± 1 | 19 ± 1 | 30 ± 2 | 0 | 12 ± 1 | 13 ± 1 | 16 ± 1 | 25 ± 1 |
| 1D | 0 | 0 | 0 | * Sol | * Sol | 0 | 0 | 0 | * Sol | * Sol | 0 | 0 | 0 | * Sol | * Sol |
| 1E | 0 | 5 ± 0.1 | * Sol | * Sol | * Sol | 0 | 20 ± 2 | * Sol | * Sol | * Sol | 0 | 16 ± 1 | * Sol | * Sol | * Sol |
| 1F | 0 | 0 | 6 ± 0.3 | * Sol | * Sol | 0 | 0 | 0 | * Sol | * Sol | 0 | 0 | 0 | * Sol | * Sol |
| 1G | 0 | 5 ± 0.2 | 28 ± 2 | * Sol | * Sol | 0 | 12 ± 1 | 35 ± 2 | * Sol | * Sol | 0 | 2 ± 0.1 | 37 ± 3 | * Sol | * Sol |
| 2A | 0 | 5 ± 0.1 | 30 ± 1 | 44 ± 2 | 55 ± 3 | 0 | 0 | 0 | 0 | 0 | 0 | 13 ± 1 | 42 ± 2 | 55 ± 3 | 60 ± 3 |
| 2B | 0 | 0 | 37 ± 2 | * Sol | * Sol | 0 | 17 ± 1 | 34 ± 2 | * Sol | * Sol | 0 | 19 ± 1 | 23 ± 2 | * Sol | * Sol |
| 2C | 15 ± 2 | 31 ± 2 | 56 ± 2 | 75 ± 2 | 91 ± 4 | 10 ± 1 | 19 ± 3 | 59 ± 2 | 76 ± 2 | 85 ± 2 | 35 ± 2 | 70 ± 3 | 76 ± 1 | 80 ± 3 | 85 ± 2 |
| BIBR-1532 | 25 ± 2 | 47 ± 3 | 56 ± 3 | 89 ± 2 | 95 ± 5 | 12 ± 2 | 23 ± 2 | 57 ± 3 | 74 ± 2 | 91 ± 3 | 10 ± 2 | 19 ± 3 | 32 ± 1 | 62 ± 3 | 79 ± 2 |
| Compound | IC50 (µm) | |||
|---|---|---|---|---|
| HCT-116 | MCF-7 | K-562 | BALB/3-T3 | |
| BIBR-1532 | 70 ± 4 | 120 ± 8 | 208 ± 11 | N/A |
| 2C | 109 ± 8 | 127 ± 9 | 33 ± 4 | 324 ± 15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Culletta, G.; Allegra, M.; Almerico, A.M.; Restivo, I.; Tutone, M. In Silico Design, Synthesis, and Biological Evaluation of Anticancer Arylsulfonamide Endowed with Anti-Telomerase Activity. Pharmaceuticals 2022, 15, 82. https://doi.org/10.3390/ph15010082
Culletta G, Allegra M, Almerico AM, Restivo I, Tutone M. In Silico Design, Synthesis, and Biological Evaluation of Anticancer Arylsulfonamide Endowed with Anti-Telomerase Activity. Pharmaceuticals. 2022; 15(1):82. https://doi.org/10.3390/ph15010082
Chicago/Turabian StyleCulletta, Giulia, Mario Allegra, Anna Maria Almerico, Ignazio Restivo, and Marco Tutone. 2022. "In Silico Design, Synthesis, and Biological Evaluation of Anticancer Arylsulfonamide Endowed with Anti-Telomerase Activity" Pharmaceuticals 15, no. 1: 82. https://doi.org/10.3390/ph15010082
APA StyleCulletta, G., Allegra, M., Almerico, A. M., Restivo, I., & Tutone, M. (2022). In Silico Design, Synthesis, and Biological Evaluation of Anticancer Arylsulfonamide Endowed with Anti-Telomerase Activity. Pharmaceuticals, 15(1), 82. https://doi.org/10.3390/ph15010082