
Thienopyrimidine: A Promising Scaffold to Access Anti-Infective Agents



Abstract
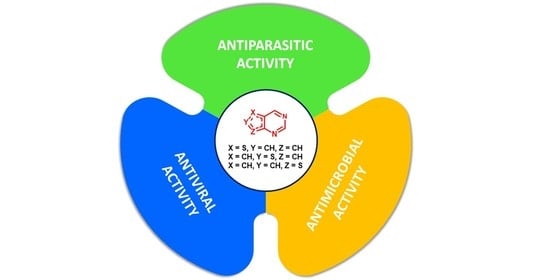
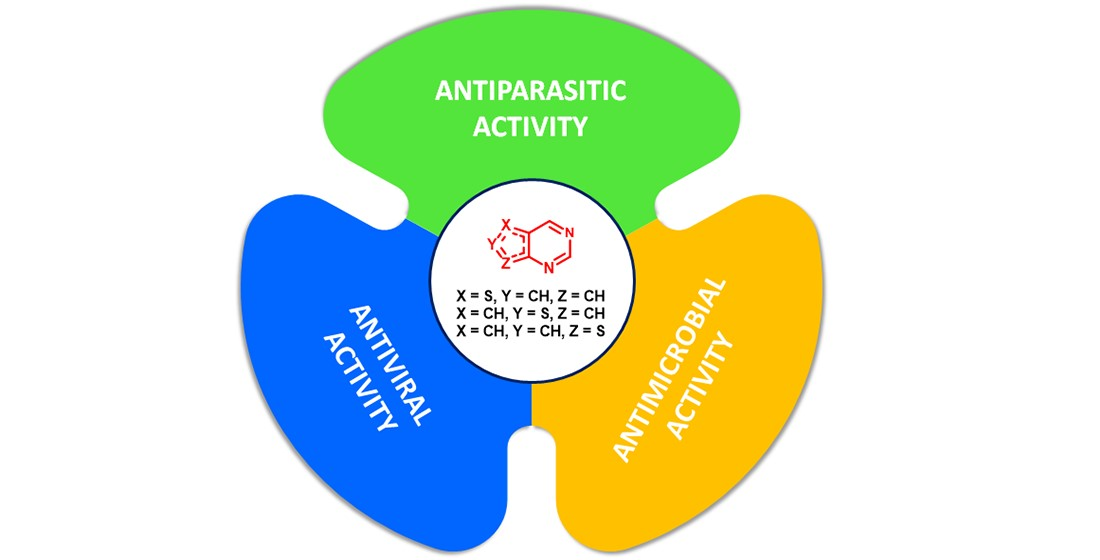
1. Introduction
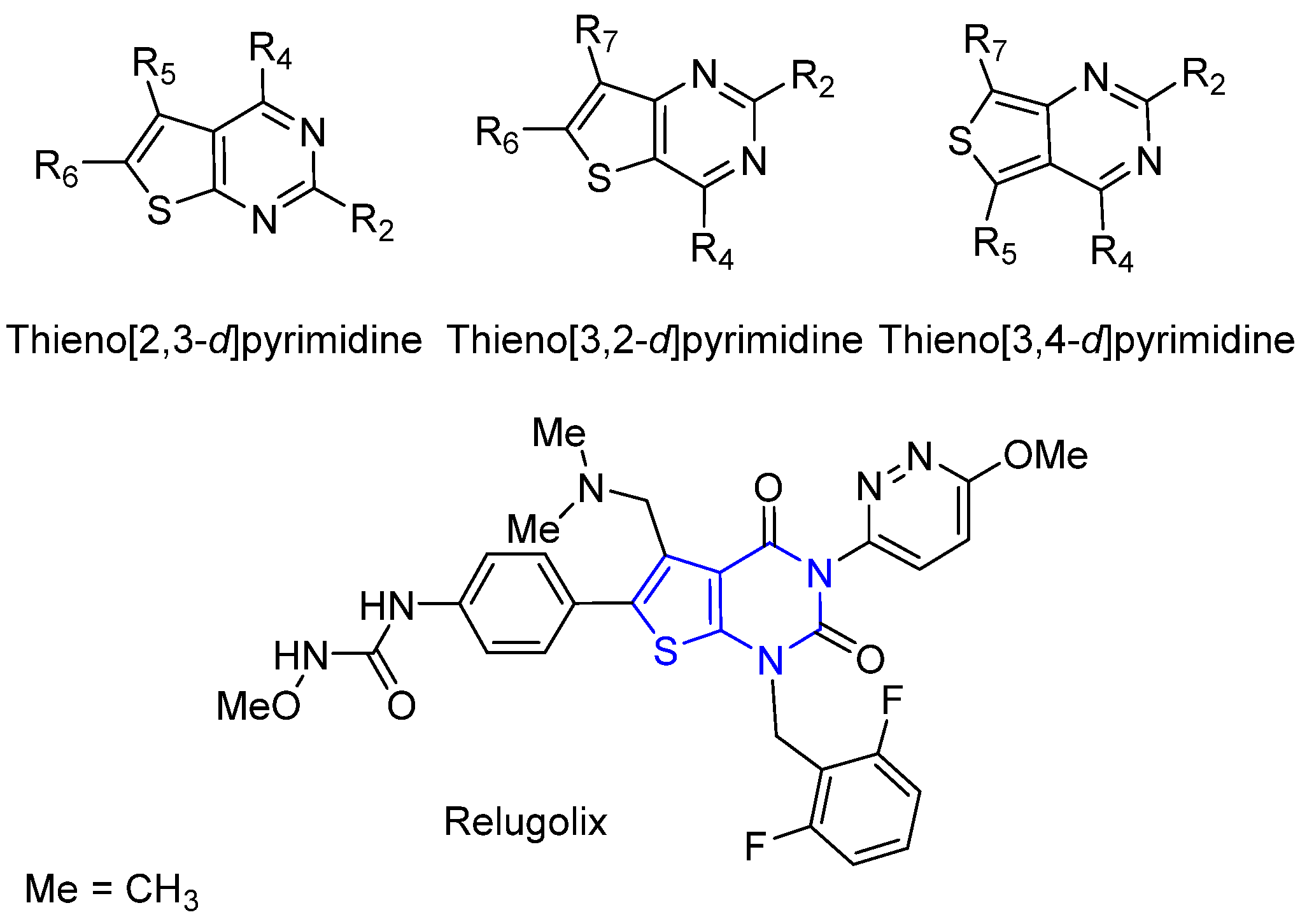
2. Synthesis of Thienopyrimidines
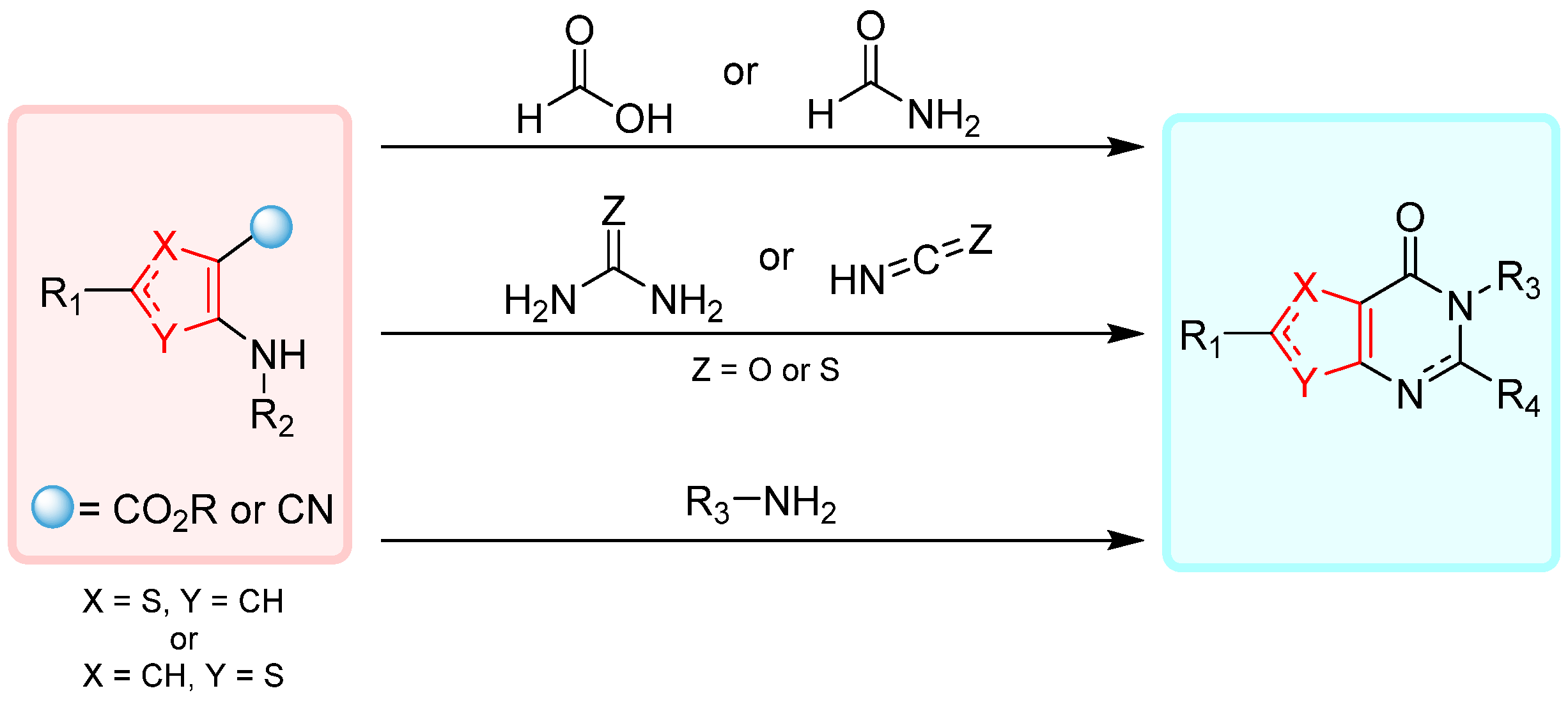
2.1. Synthesis from Thiophene Derivatives
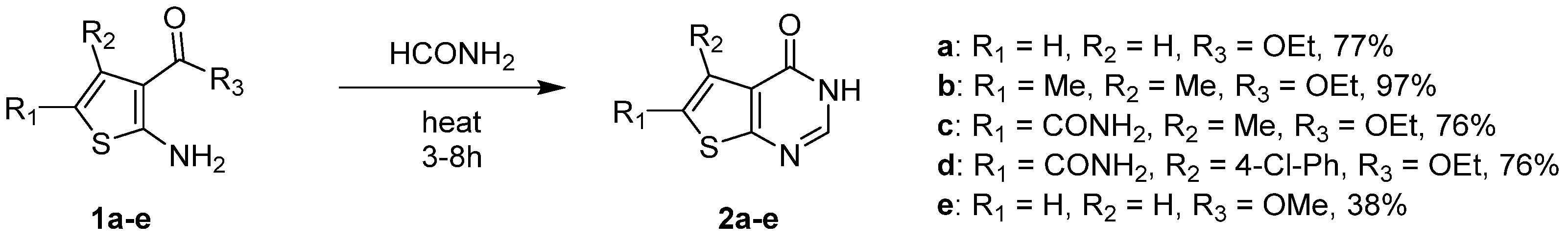
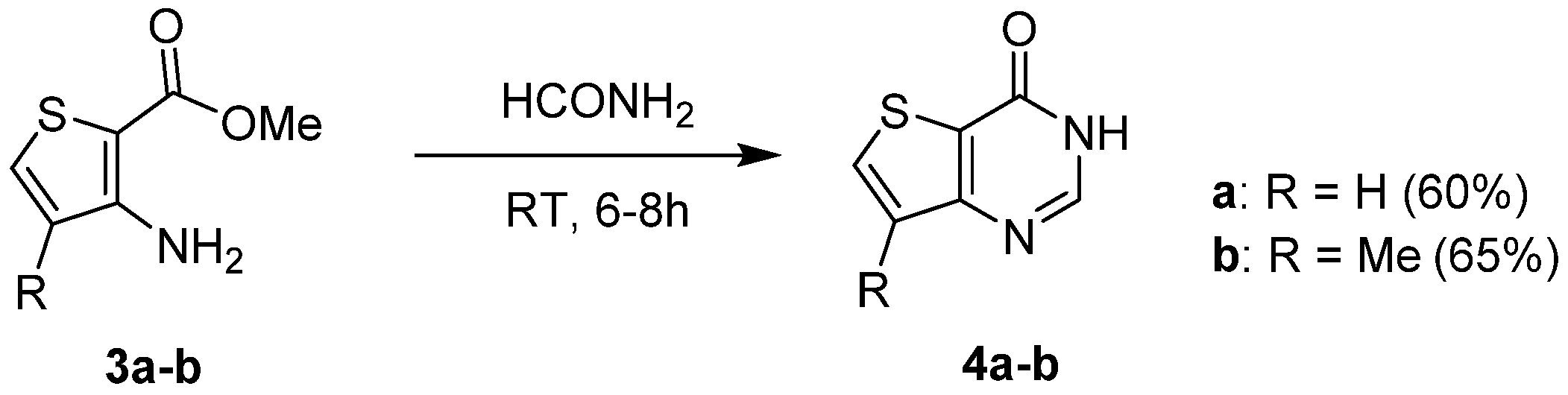
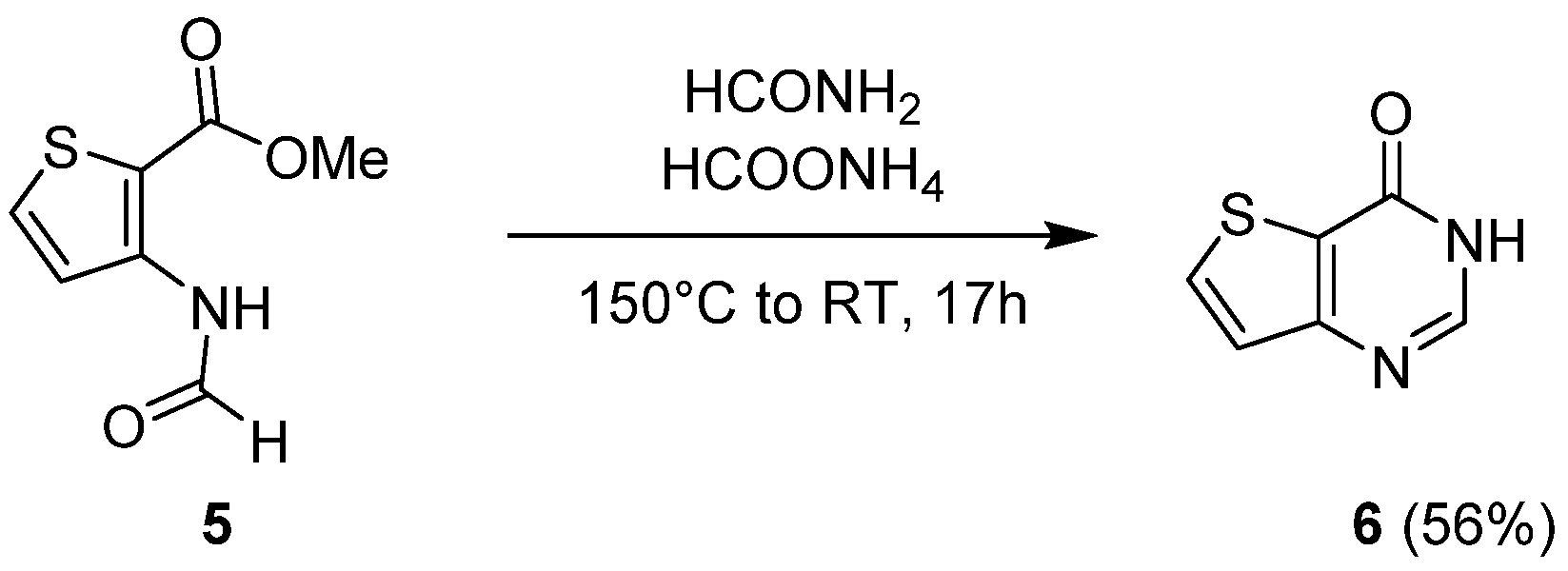
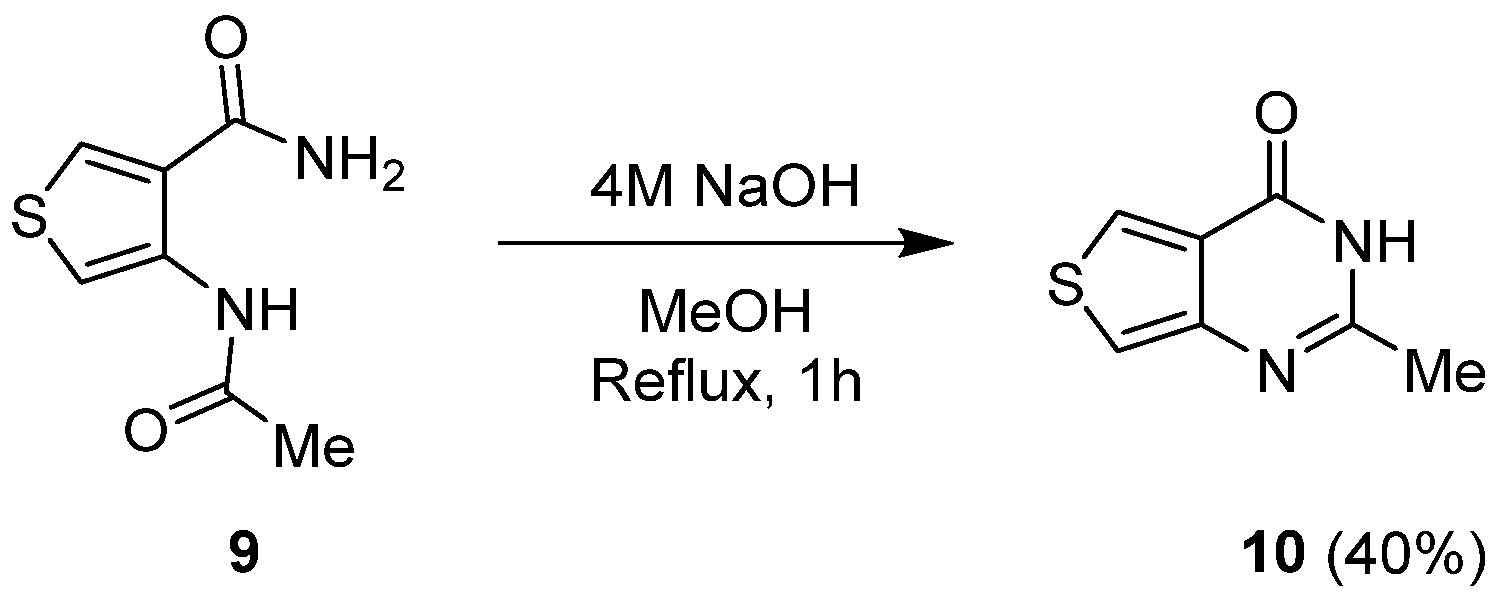
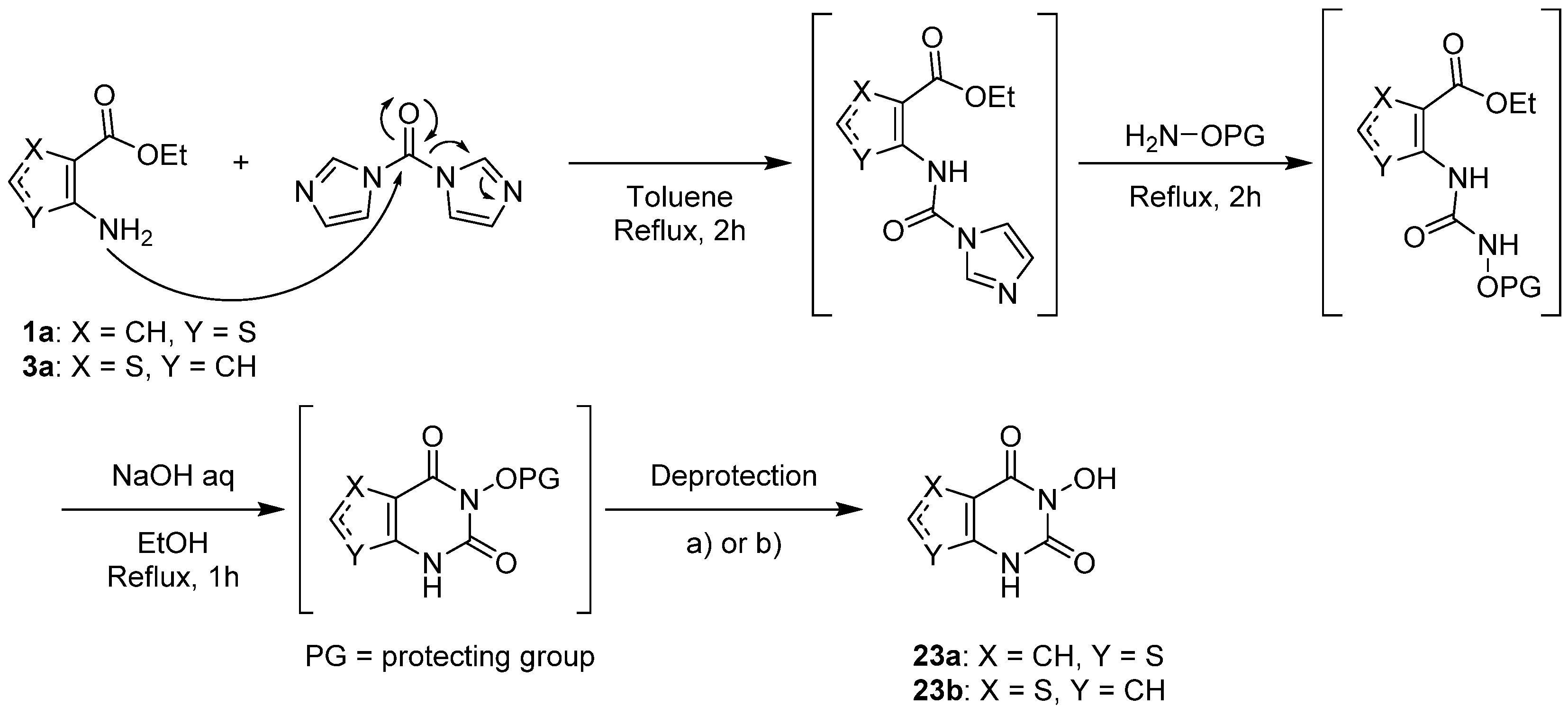
2.1.1. Cyclization with Carbonyl Reactants
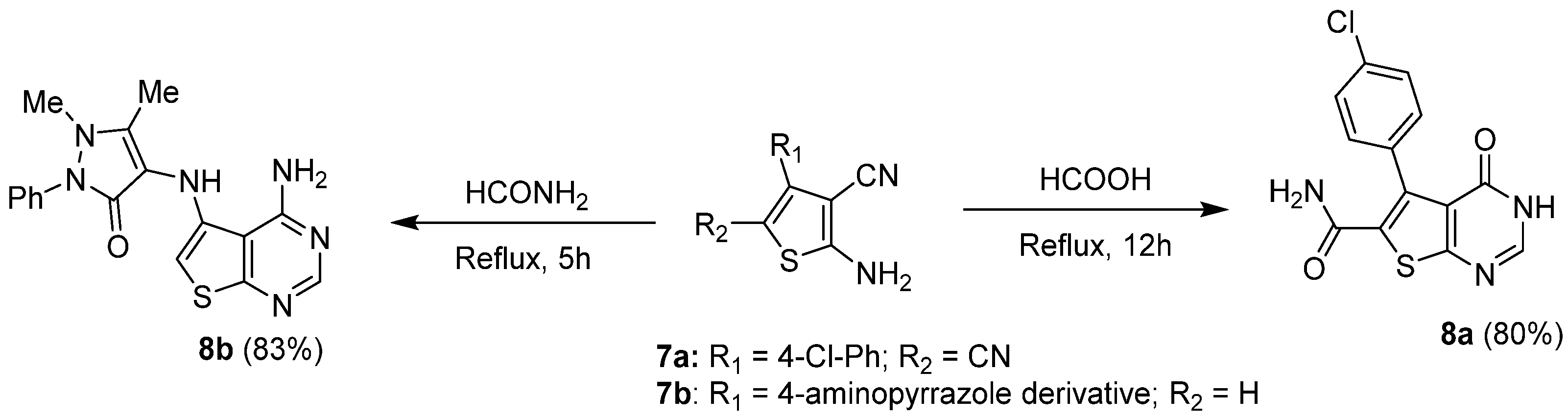
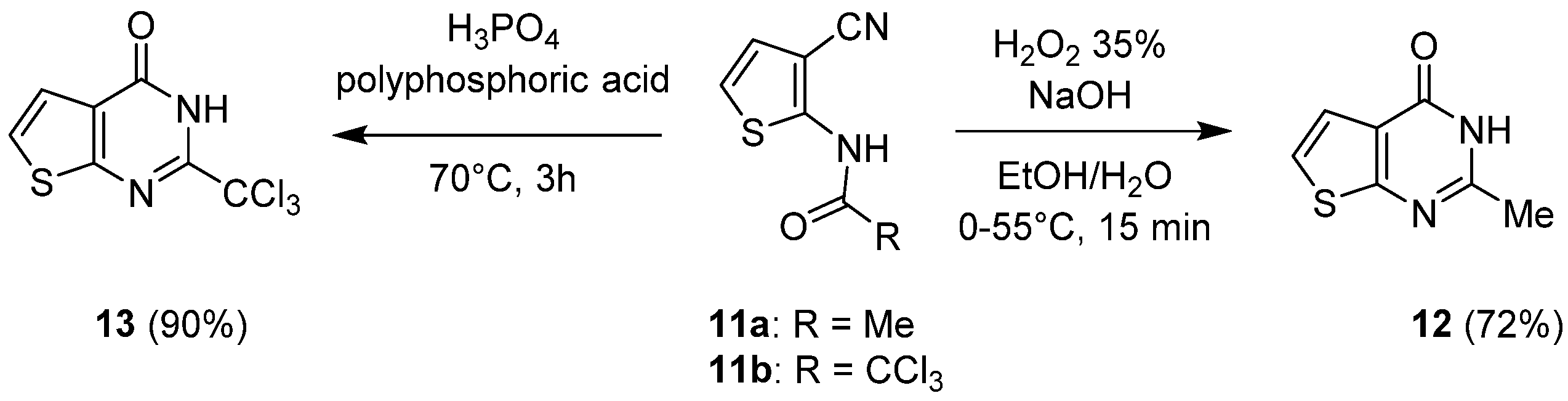
2.1.2. Cyclization with Nitrile Reactants
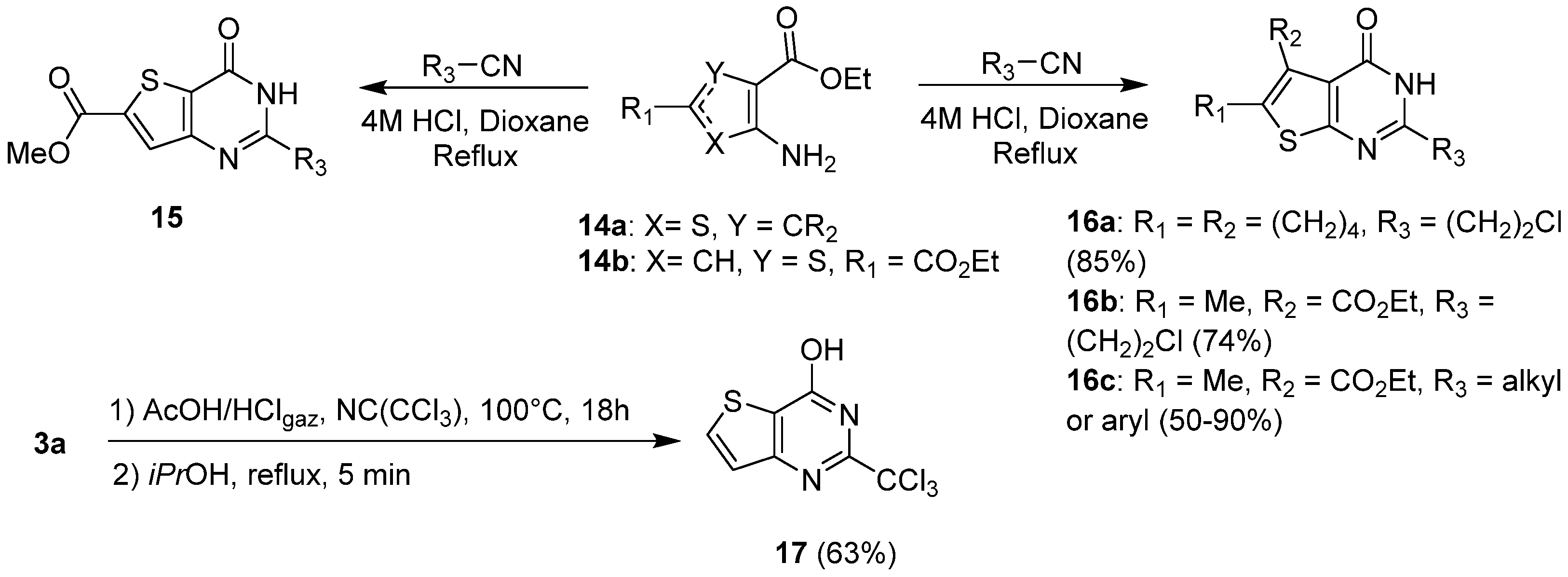
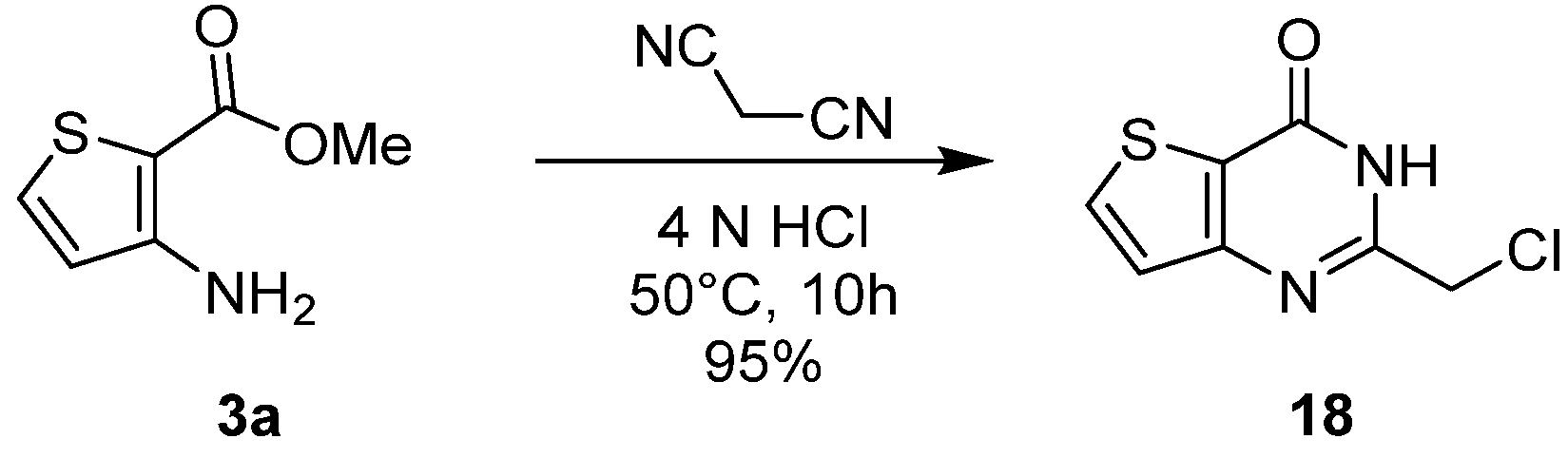
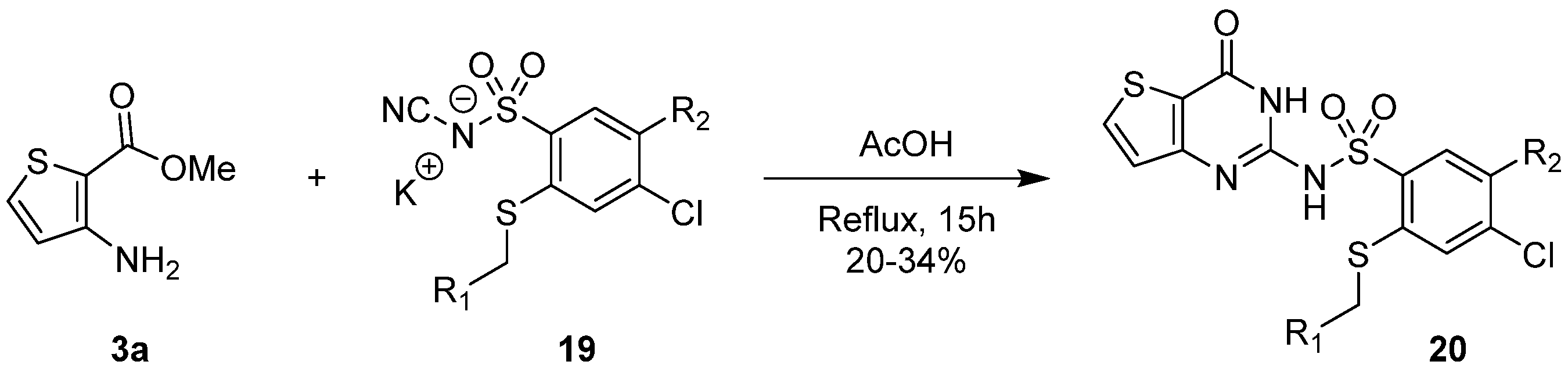
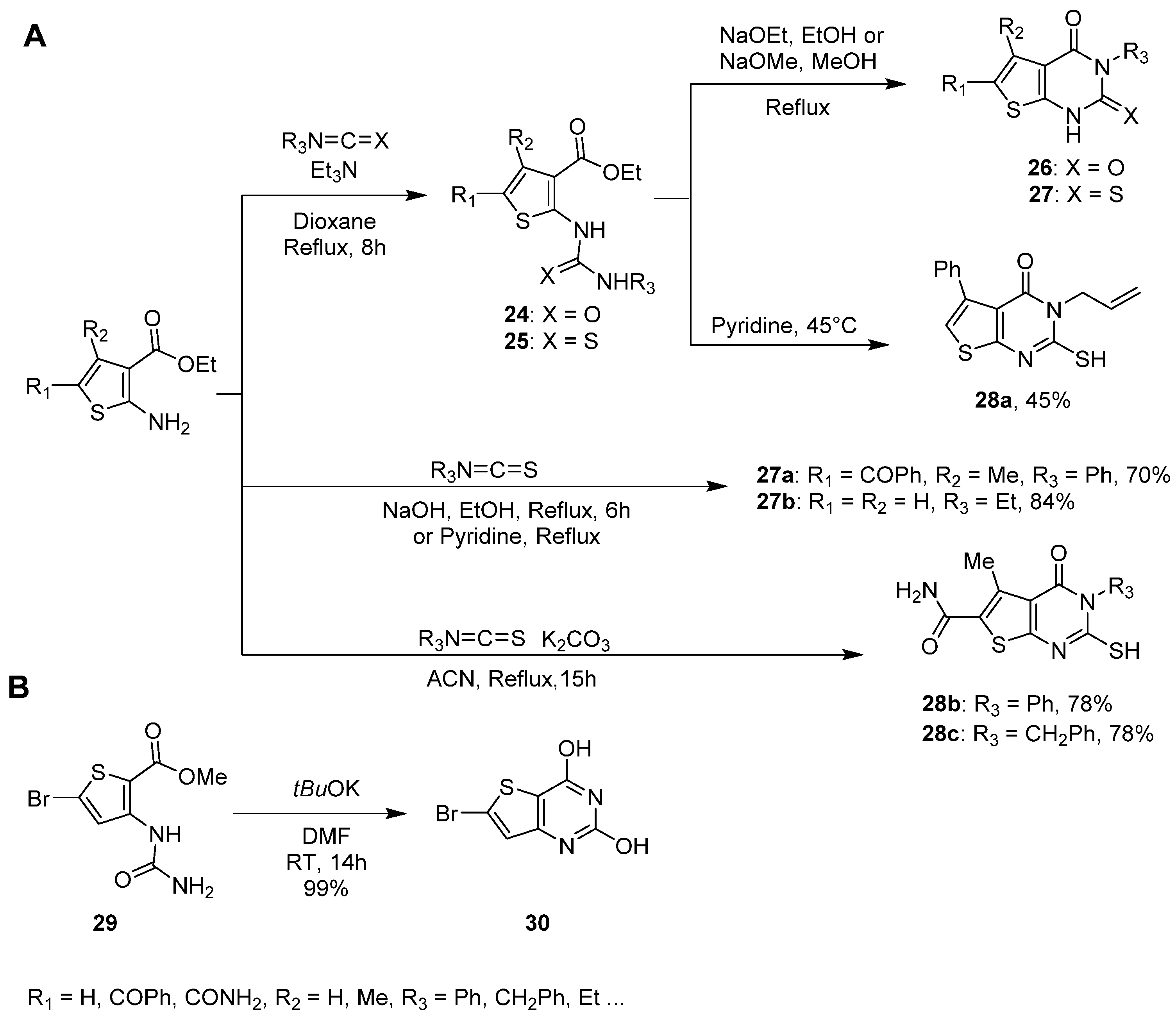
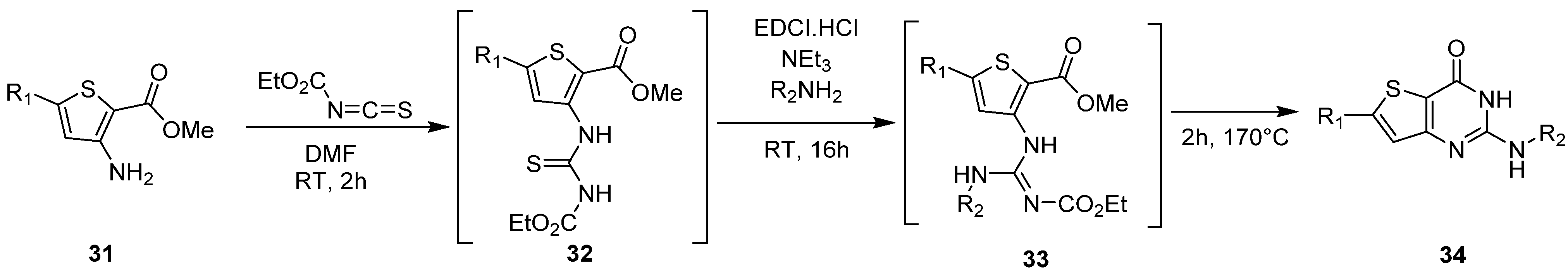
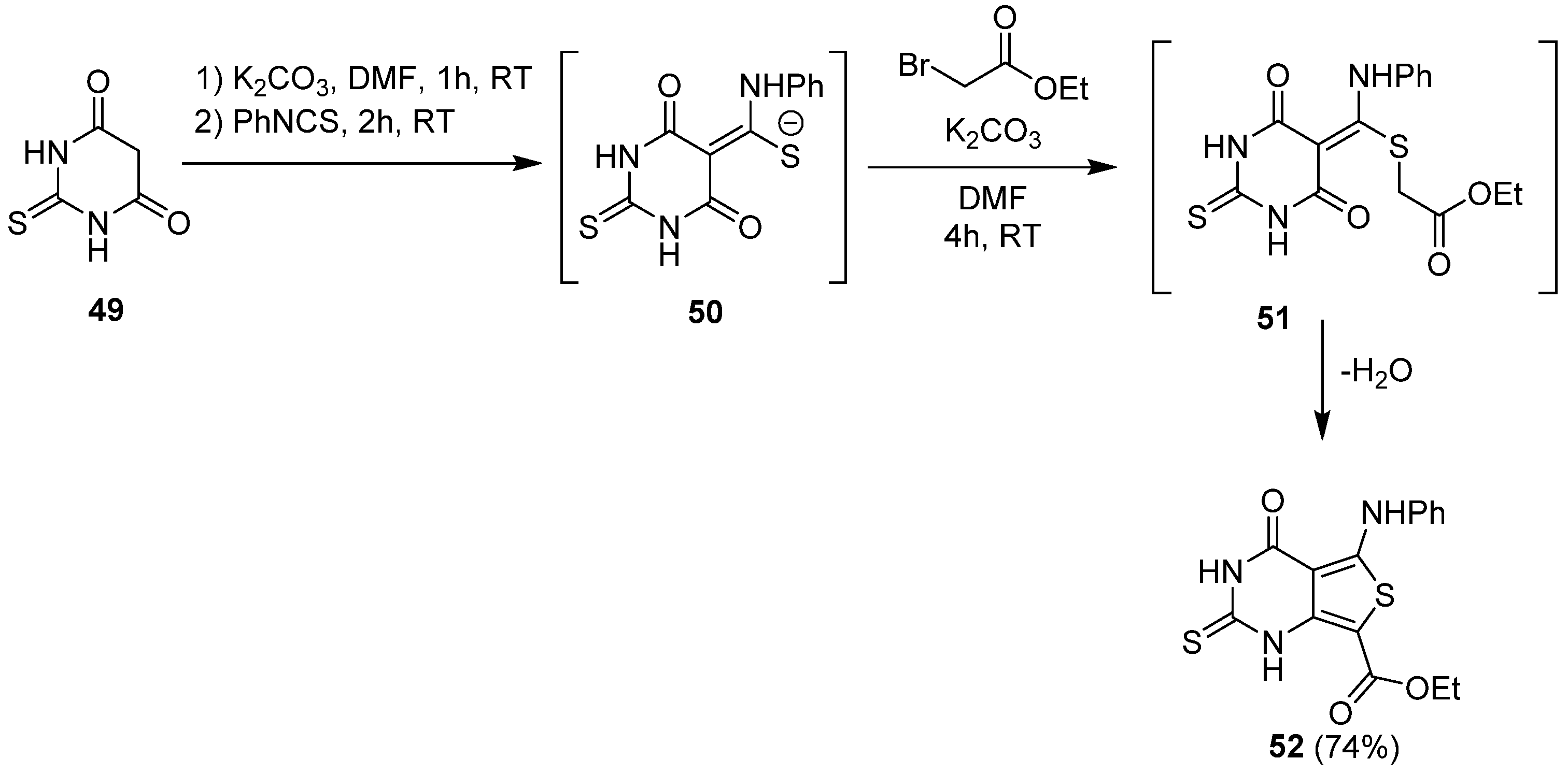
2.1.3. Synthesis from (Thio)urea Reagents, Iso(Thio)cyanate or (Thio)cyanate Derivatives
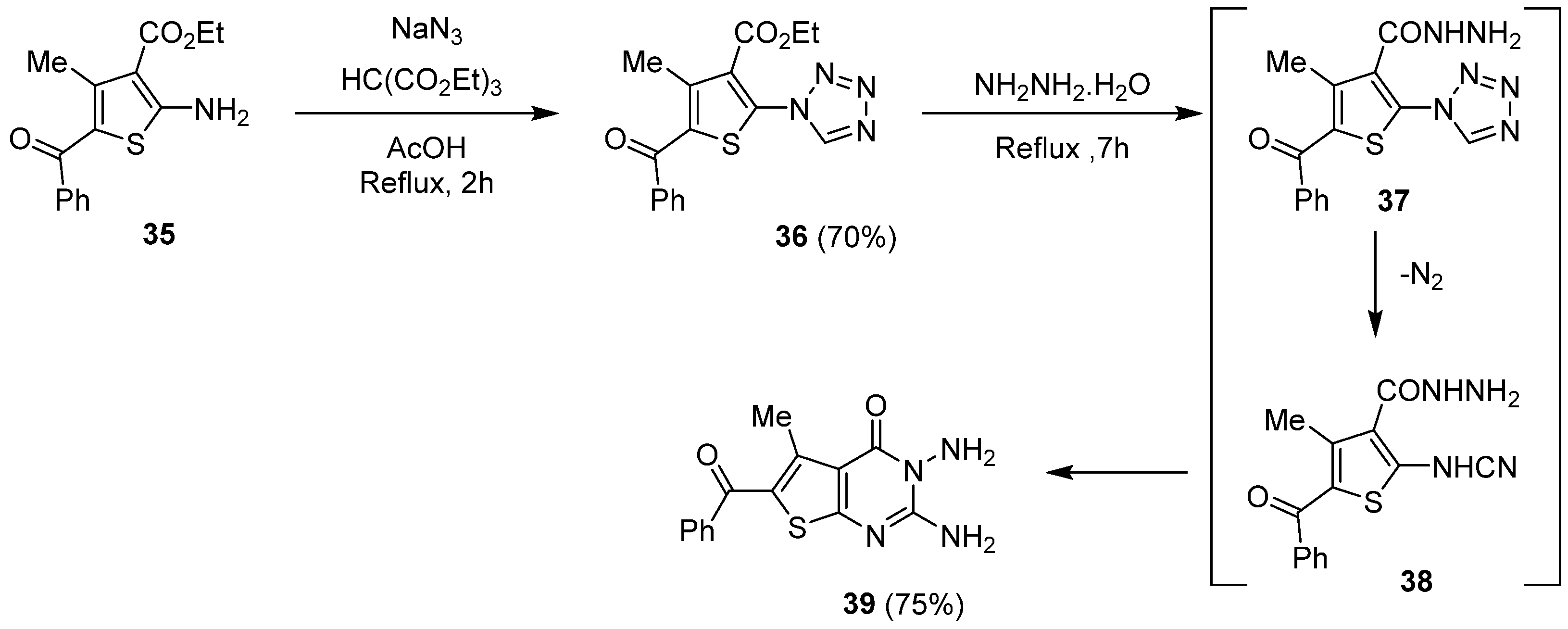
2.1.4. Synthesis via a Tetrazole Intermediate
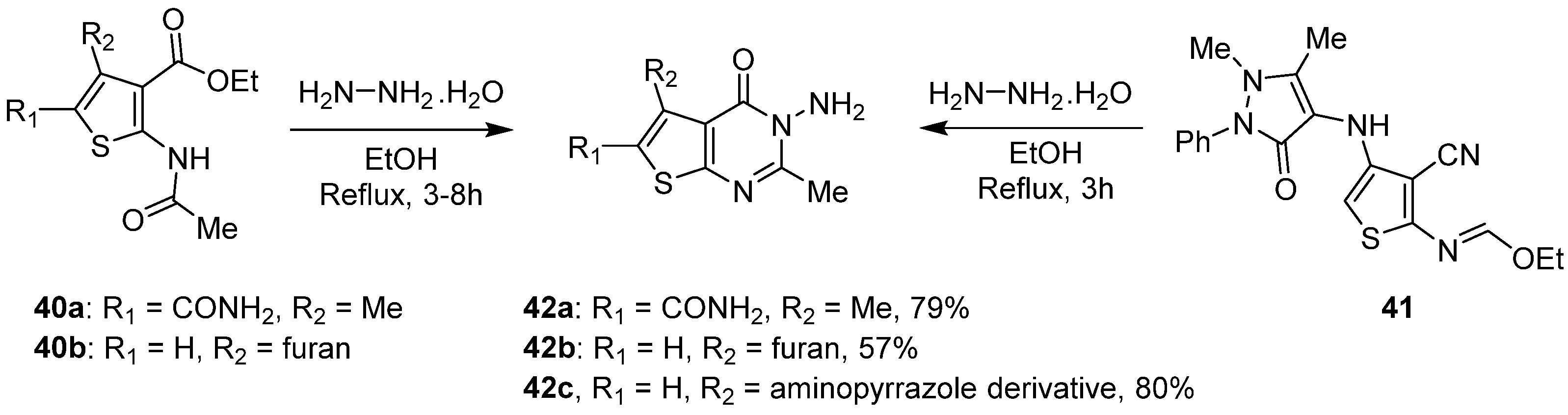
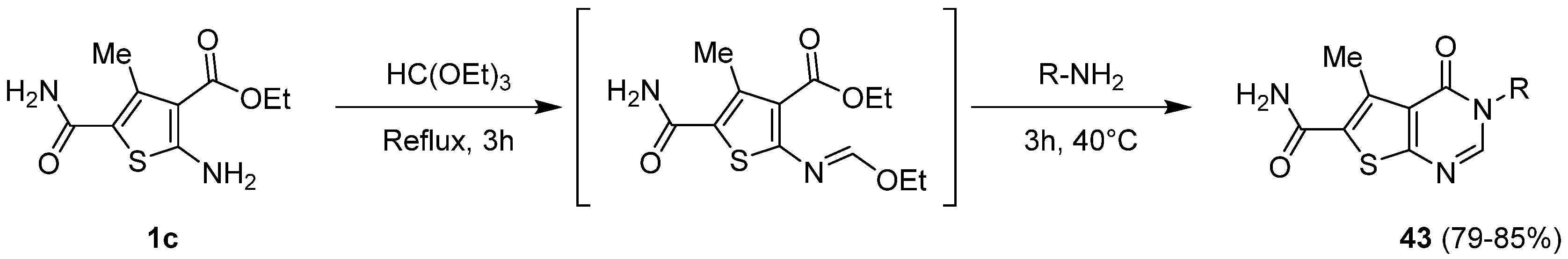
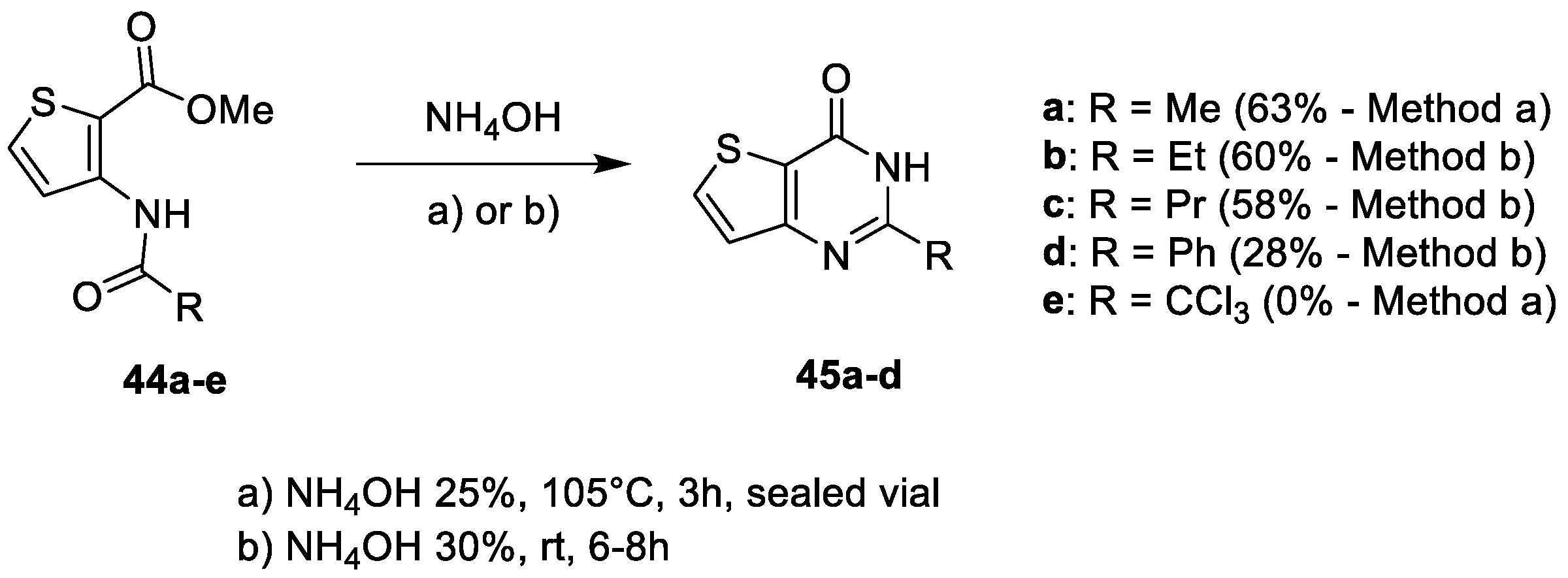
2.1.5. Cyclization with Amine/Hydrazine Derivatives
2.2. Synthesis of Thienopyrimidines from Pyrimidine Derivatives
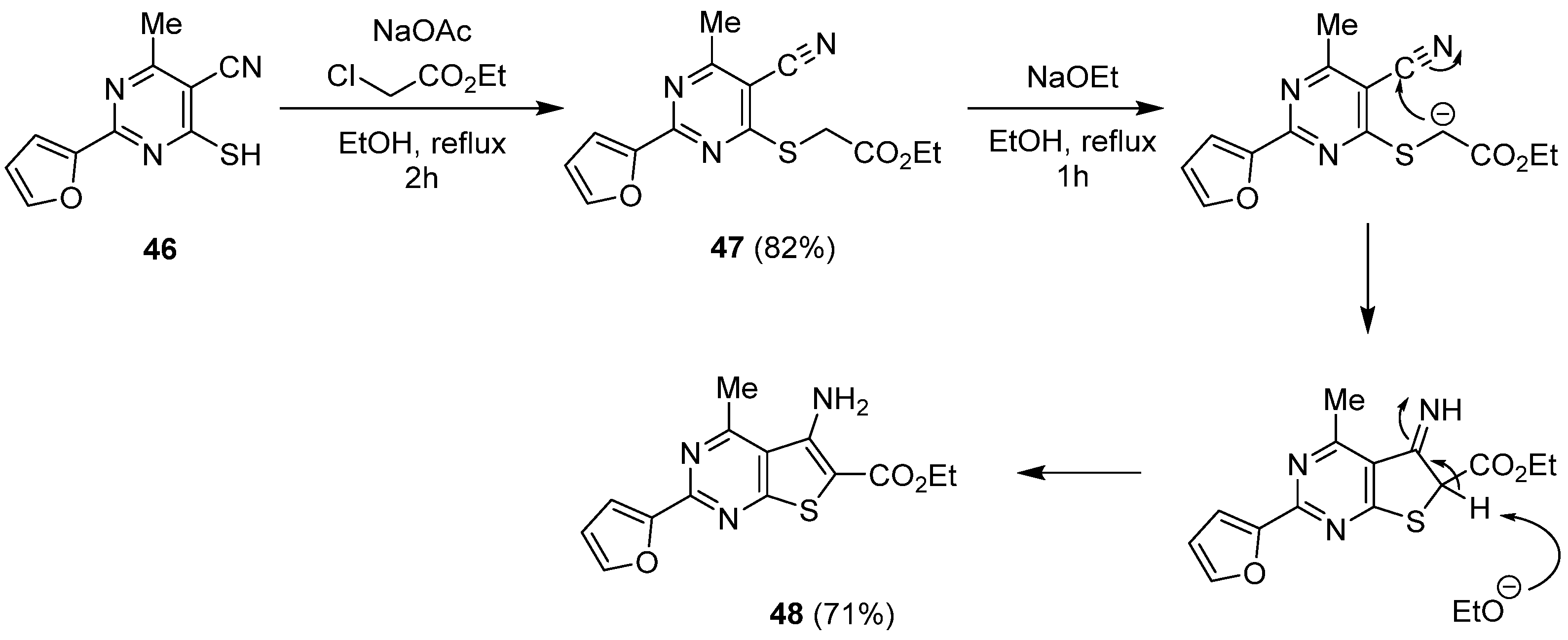
2.2.1. Synthesis from the Thorpe-Ziegler Reaction
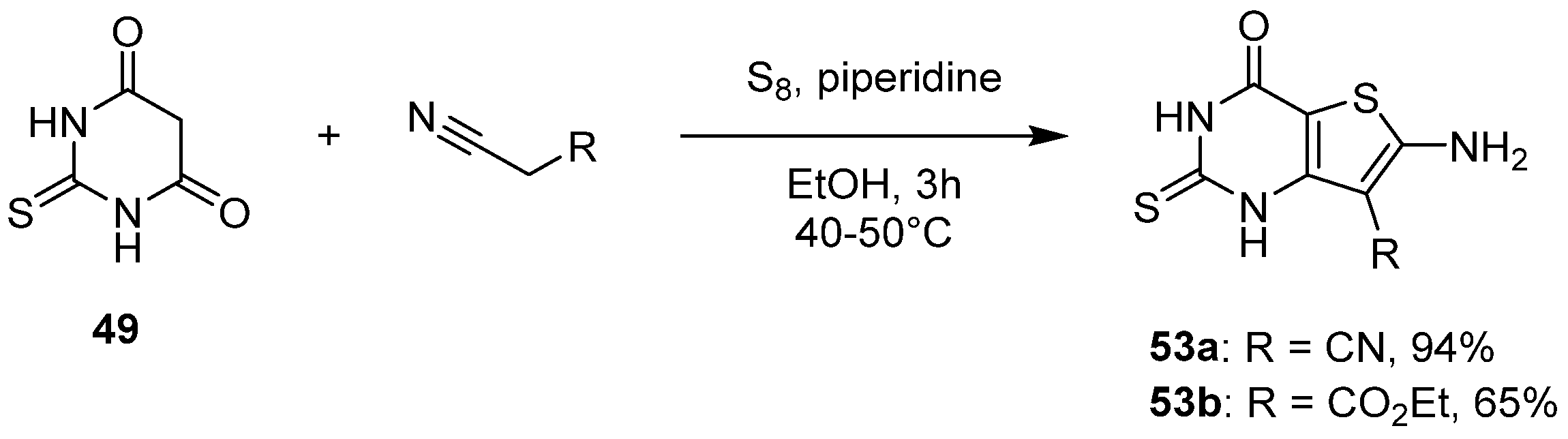
2.2.2. Synthesis from the Gewald Reaction
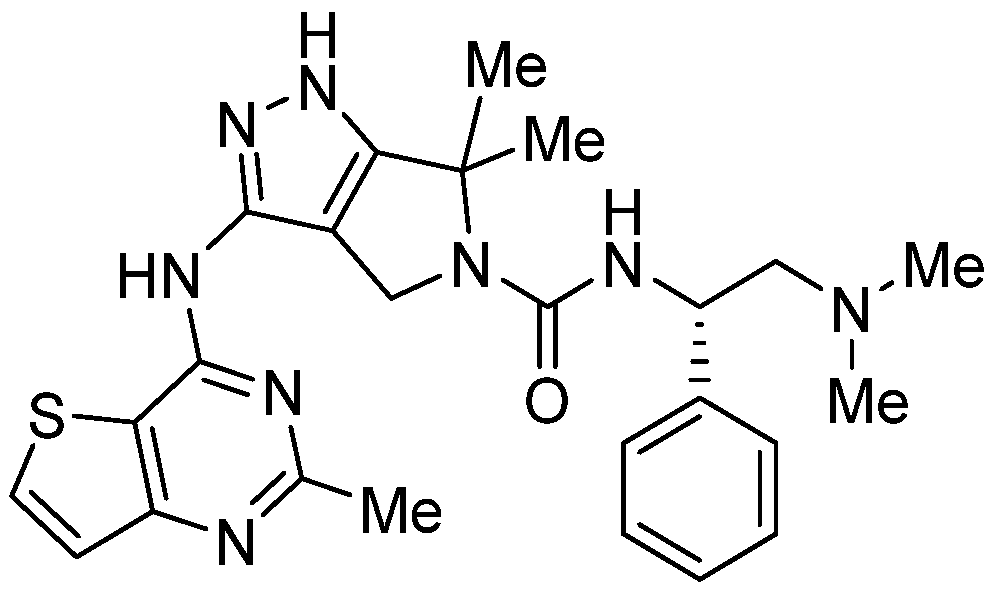
3. Antiparasitic Activity of Thienopyrimidines
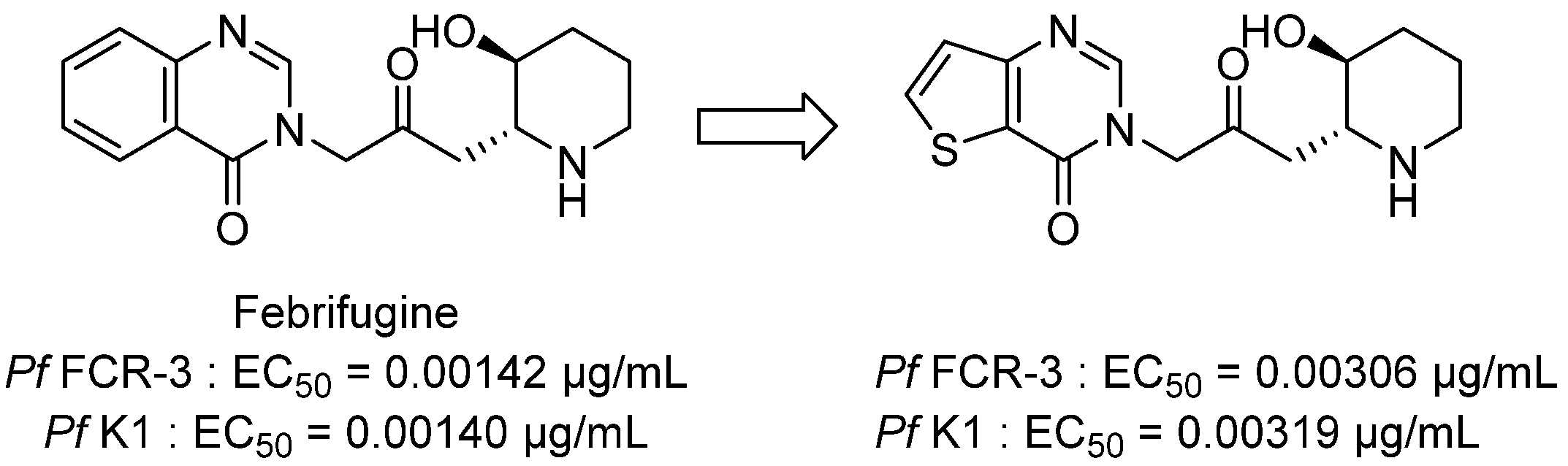
3.1. Antimalarial Activity
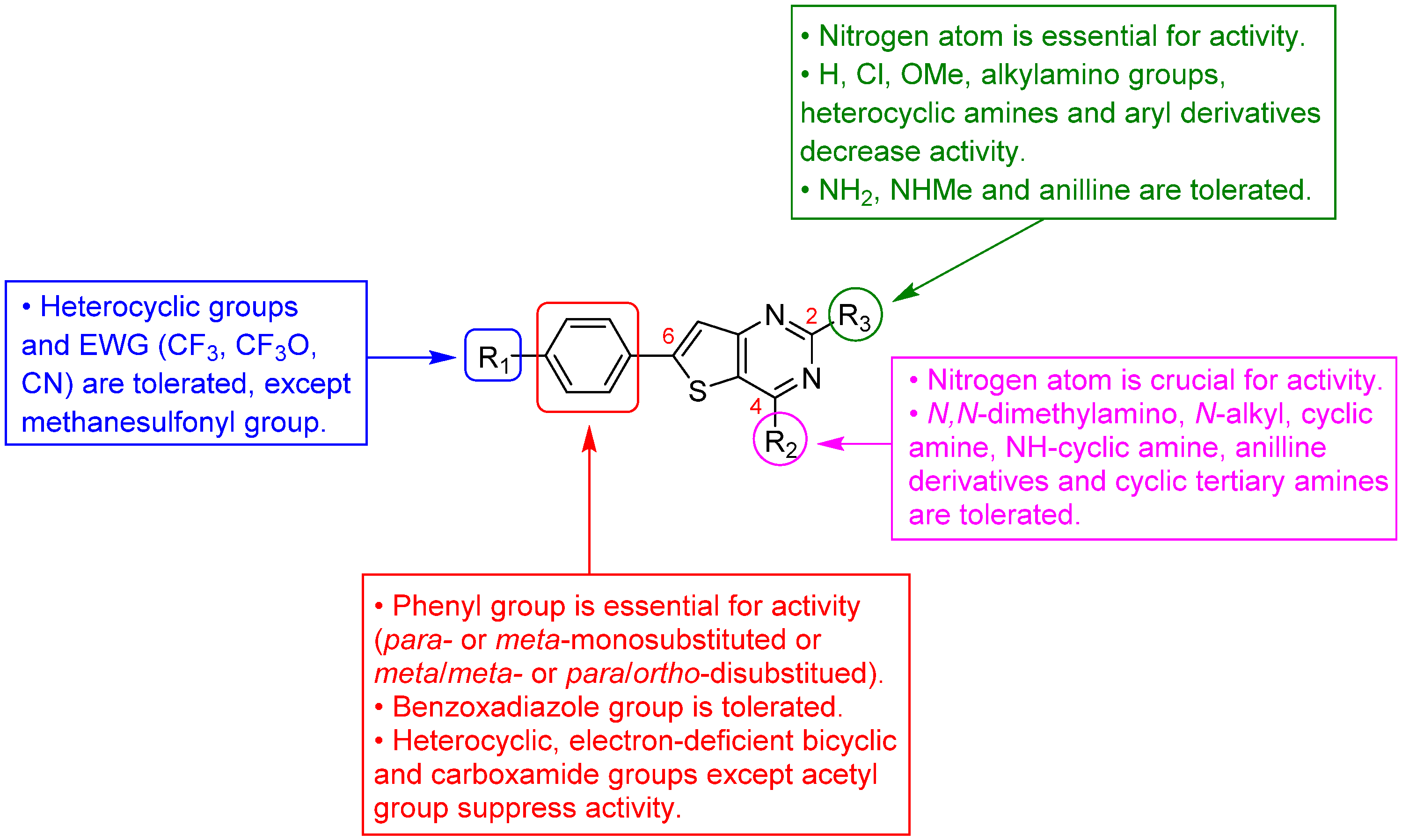
3.1.1. Thieno[2,3-d]pyrimidine Derivatives with Antiplasmodial Activity
3.1.2. Thieno[3,2-d]pyrimidine Derivatives with Antiplasmodial Activity
3.2. Anti-Trypanosomatid Activity
3.3. Antihelminthic Activity
4. Thienopyrimidines with Antituberculosis Activity
5. Thienopyrimidines with Antibacterial Activities Other than Tuberculosis
5.1. Inhibition of the Protein Glycosylation D (PglD) of Campylobacter Jejuni
5.2. Compounds wih Broad-Spectrum Antibacterial Activity
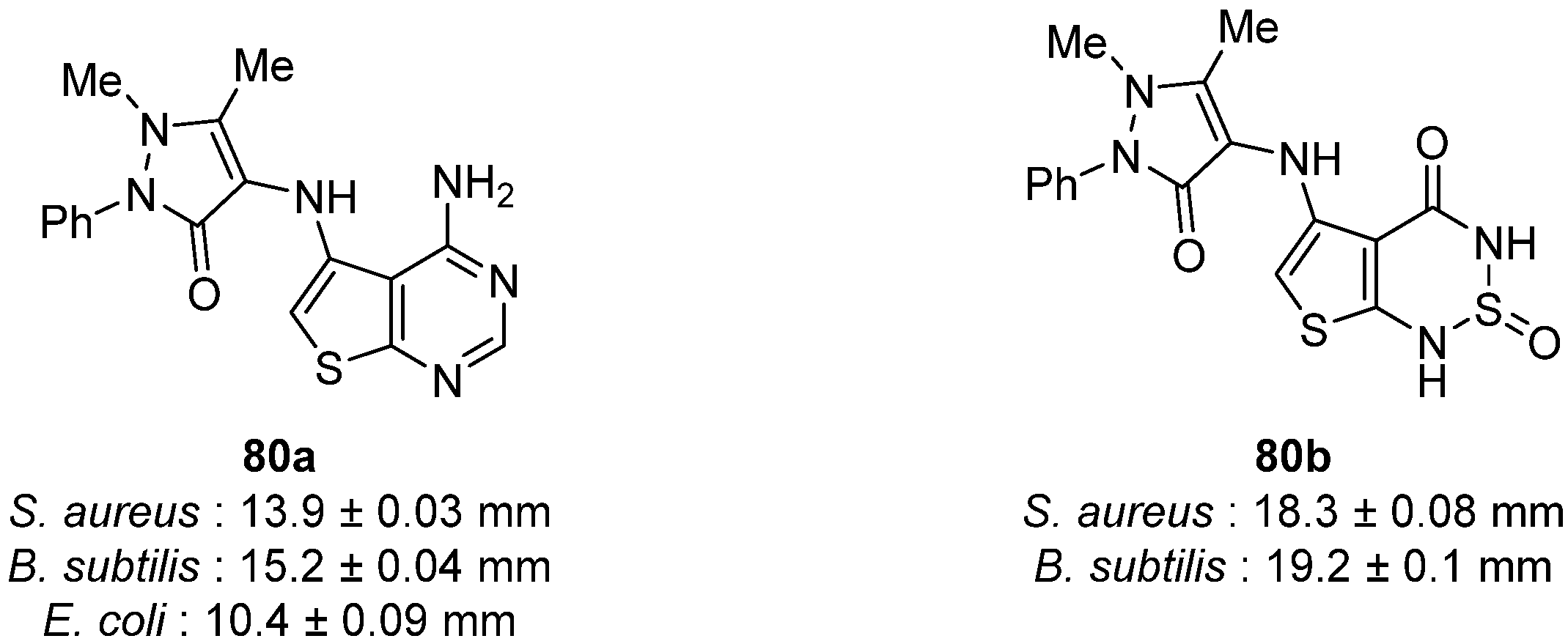
5.2.1. Thieno[2,3-d]pyrimidin-4-one and Pyrimidin-2,4-dione Derivatives
5.2.2. Other thieno[2,3-d]pyrimidine Derivatives
5.2.3. Thieno[3,2-d]pyrimidine Derivatives
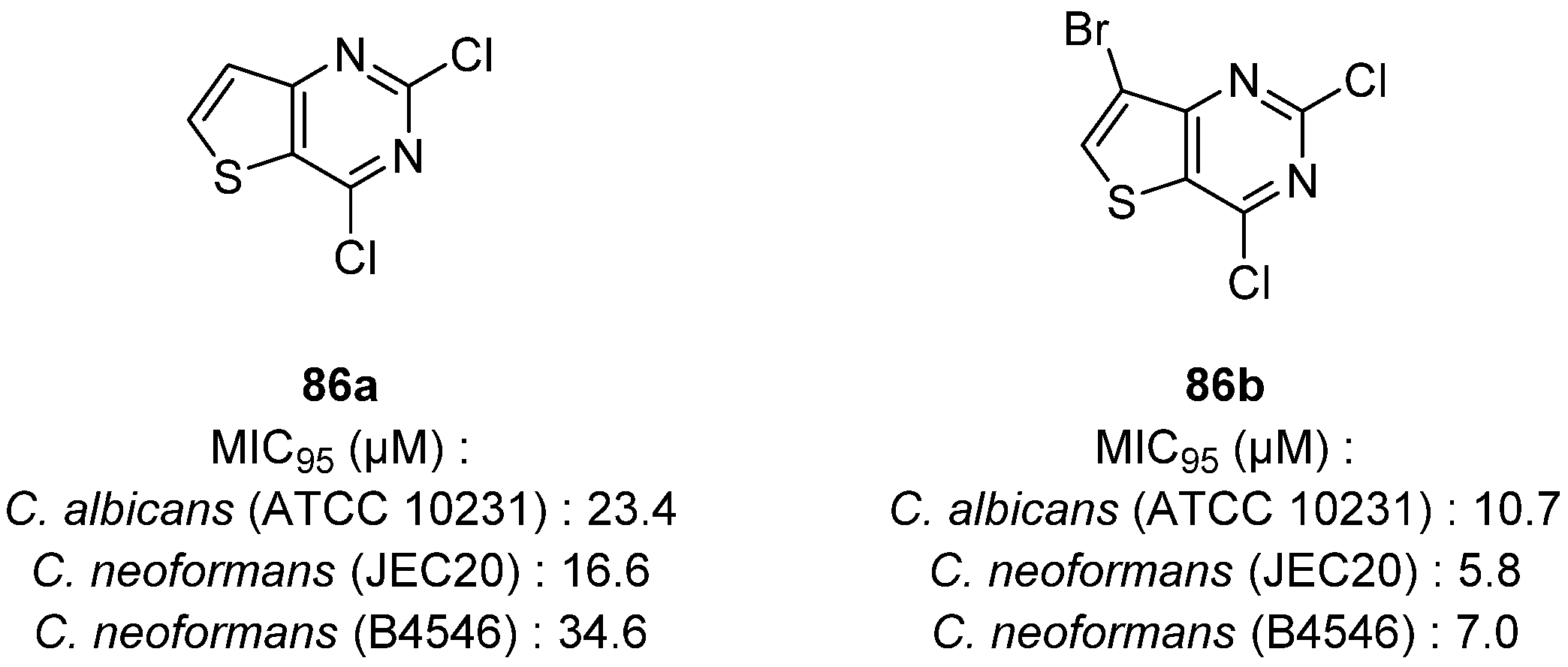
6. Thienopyrimidines with Antifungal Activity
7. Thienopyrimidines with Antiviral Activity
7.1. Activity against Influenza A Virus
7.2. Activity against Hepatitis B Virus
7.3. Activity against HIV-1
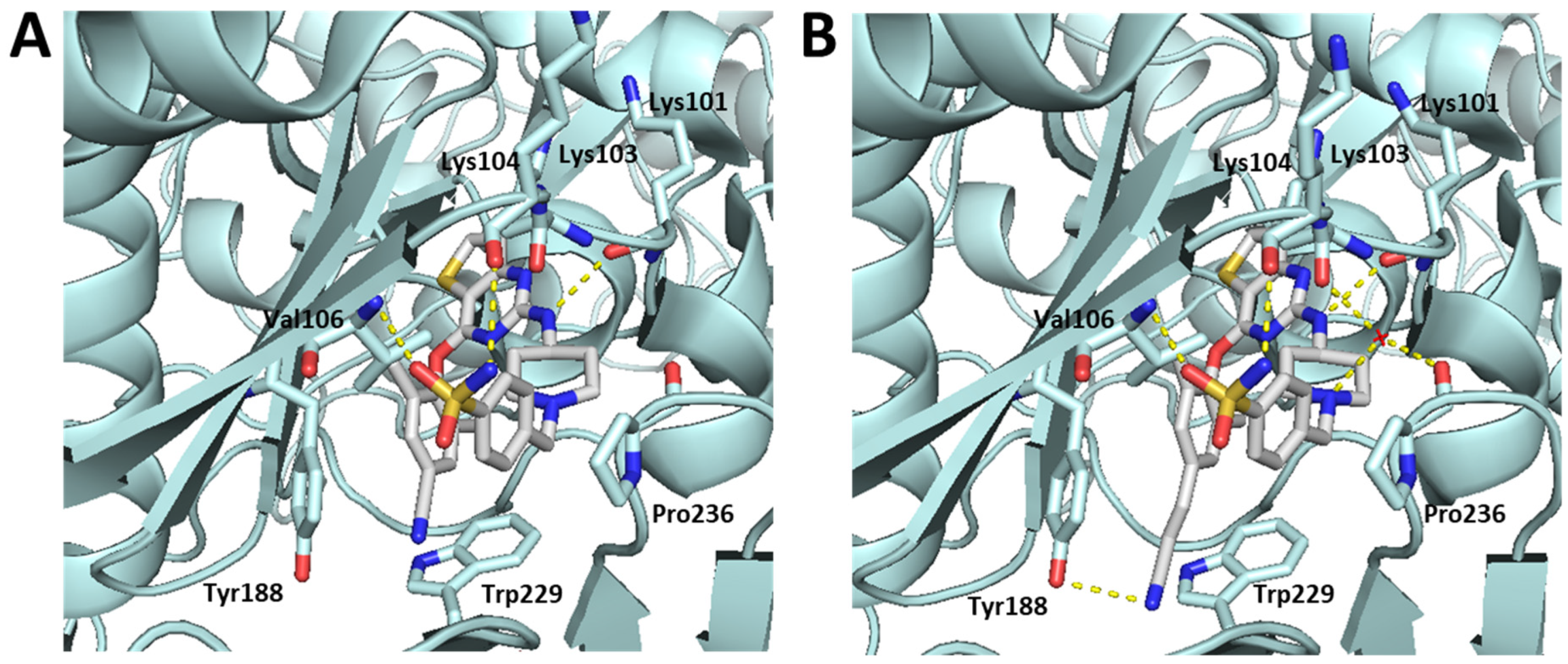
7.3.1. Thienopyrimidines as Reverse Transcriptase (RT) Inhibitors
7.3.2. Anti-HIV Thienopyrimidines with Other Mechanisms
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Litvinov, V.P.; Dotsenko, V.V.; Krivokolysko, S.G. Thienopyridines: Synthesis, Properties, and Biological Activity. Russ. Chem. Bull. 2005, 54, 864–904. [Google Scholar] [CrossRef]
- Ali, E.M.H.; Abdel-Maksoud, M.S.; Oh, C.-H. Thieno[2,3-d]Pyrimidine as a Promising Scaffold in Medicinal Chemistry: Recent Advances. Bioorganic Med. Chem. 2019, 27, 1159–1194. [Google Scholar] [CrossRef]
- Gramec, D.; Peterlin Mašič, L.; Sollner Dolenc, M. Bioactivation Potential of Thiophene-Containing Drugs. Chem. Res. Toxicol. 2014, 27, 1344–1358. [Google Scholar] [CrossRef]
- Maillard, L.T.; Bertout, S.; Quinonéro, O.; Akalin, G.; Turan-Zitouni, G.; Fulcrand, P.; Demirci, F.; Martinez, J.; Masurier, N. Synthesis and Anti-Candida Activity of Novel 2-Hydrazino-1,3-Thiazole Derivatives. Bioorganic Med. Chem. Lett. 2013, 23, 1803–1807. [Google Scholar] [CrossRef]
- Fersing, C.; Boudot, C.; Pedron, J.; Hutter, S.; Primas, N.; Caroline, D.; Bourgeade-Delmas, S.; Sournia-Saquet, A.; Moreau, A.; Cohen, A.; et al. 8-Aryl-6-Chloro-3-Nitro-2-(Phenylsulfonylmethyl)Imidazo[1,2-a]Pyridines as Potent Antitrypanosomatid Molecules Bioactivated by Type 1 Nitroreductases. Eur. J. Med. Chem. 2018, 157, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Fersing, C.; Basmaciyan, L.; Boudot, C.; Pedron, J.; Hutter, S.; Cohen, A.; Caroline, D.; Primas, N.; Laget, M.; Casanova, M.; et al. Nongenotoxic 3-Nitroimidazo[1,2-a]Pyridines Are NTR1 Substrates That Display Potent in Vitro Antileishmanial Activity. ACS Med. Chem. Lett. 2018, 10, 34–39. [Google Scholar] [CrossRef]
- Fersing, C.; Boudot, C.; Castera-Ducros, C.; Pinault, E.; Hutter, S.; Paoli-Lombardo, R.; Primas, N.; Pedron, J.; Seguy, L.; Bourgeade-Delmas, S.; et al. 8-Alkynyl-3-Nitroimidazopyridines Display Potent Antitrypanosomal Activity against Both T. b. Brucei and Cruzi. Eur. J. Med. Chem. 2020, 202, 112558. [Google Scholar] [CrossRef]
- Fersing, C.; Boudot, C.; Paoli-Lombardo, R.; Primas, N.; Pinault, E.; Hutter, S.; Castera-Ducros, C.; Kabri, Y.; Pedron, J.; Bourgeade-Delmas, S.; et al. Antikinetoplastid SAR Study in 3-Nitroimidazopyridine Series: Identification of a Novel Non-Genotoxic and Potent Anti-T. b. Brucei Hit-Compound with Improved Pharmacokinetic Properties. Eur. J. Med. Chem. 2020, 206, 112668. [Google Scholar] [CrossRef] [PubMed]
- Bosson-Vanga, H.; Primas, N.; Franetich, J.-F.; Lavazec, C.; Gomez, L.; Ashraf, K.; Tefit, M.; Soulard, V.; Dereuddre-Bosquet, N.; Le Grand, R.; et al. A New Thienopyrimidinone Chemotype Shows Multistage Activity against Plasmodium falciparum, Including Artemisinin-Resistant Parasites. Microbiol. Spectr. 2021, 9, e00274-21. [Google Scholar] [CrossRef]
- Pullarao, B.; Sharif, S.D.K.; Kumar, D.R.; Ramachandran, D. Design, Synthesis and Biological Evaluation of Thiophene Based Pyrimidin-4-One Derivatives as New Type of Antimicrobial Agents. Asian J. Chem. 2016, 28, 1997–2000. [Google Scholar] [CrossRef]
- Ortikov, I.S.; Turdibaev, Z.É.; Islamova, Z.I.; Élmuradov, B.Z.; Abdurazakov, A.S.; Bektemirov, A.M.; Osipova, S.O.; Khushbaktova, Z.A.; Syrov, V.N.; Shakhidoyatov, K.M. Search for Bactericides Among Derivatives of Deoxyvasicinone, Mackinazolinone, and Thienopyrimidinones. Pharm. Chem. J. 2017, 51, 456–464. [Google Scholar] [CrossRef]
- Habib, N.S.; Soliman, R.; El-Tombary, A.A.; El-Hawash, S.A.; Shaaban, O.G. Synthesis and Biological Evaluation of Novel Series of Thieno[2,3-d]Pyrimidine Derivatives as Anticancer and Antimicrobial Agents. Med. Chem. Res. 2013, 22, 3289–3308. [Google Scholar] [CrossRef]
- Kanawade, S.B.; Toche, R.B.; Rajani, D.P. Synthetic Tactics of New Class of 4-Aminothieno[2,3-d]Pyrimidine-6-Carbonitrile Derivatives Acting as Antimicrobial Agents. Eur. J. Med. Chem. 2013, 64, 314–320. [Google Scholar] [CrossRef]
- Woodring, J.L.; Patel, G.; Erath, J.; Behera, R.; Lee, P.J.; Leed, S.E.; Rodriguez, A.; Sciotti, R.J.; Mensa-Wilmot, K.; Pollastri, M.P. Evaluation of Aromatic 6-Substituted Thienopyrimidines as Scaffolds against Parasites That Cause Trypanosomiasis, Leishmaniasis, and Malaria. Med. Chem. Commun. 2015, 6, 339–346. [Google Scholar] [CrossRef] [PubMed]
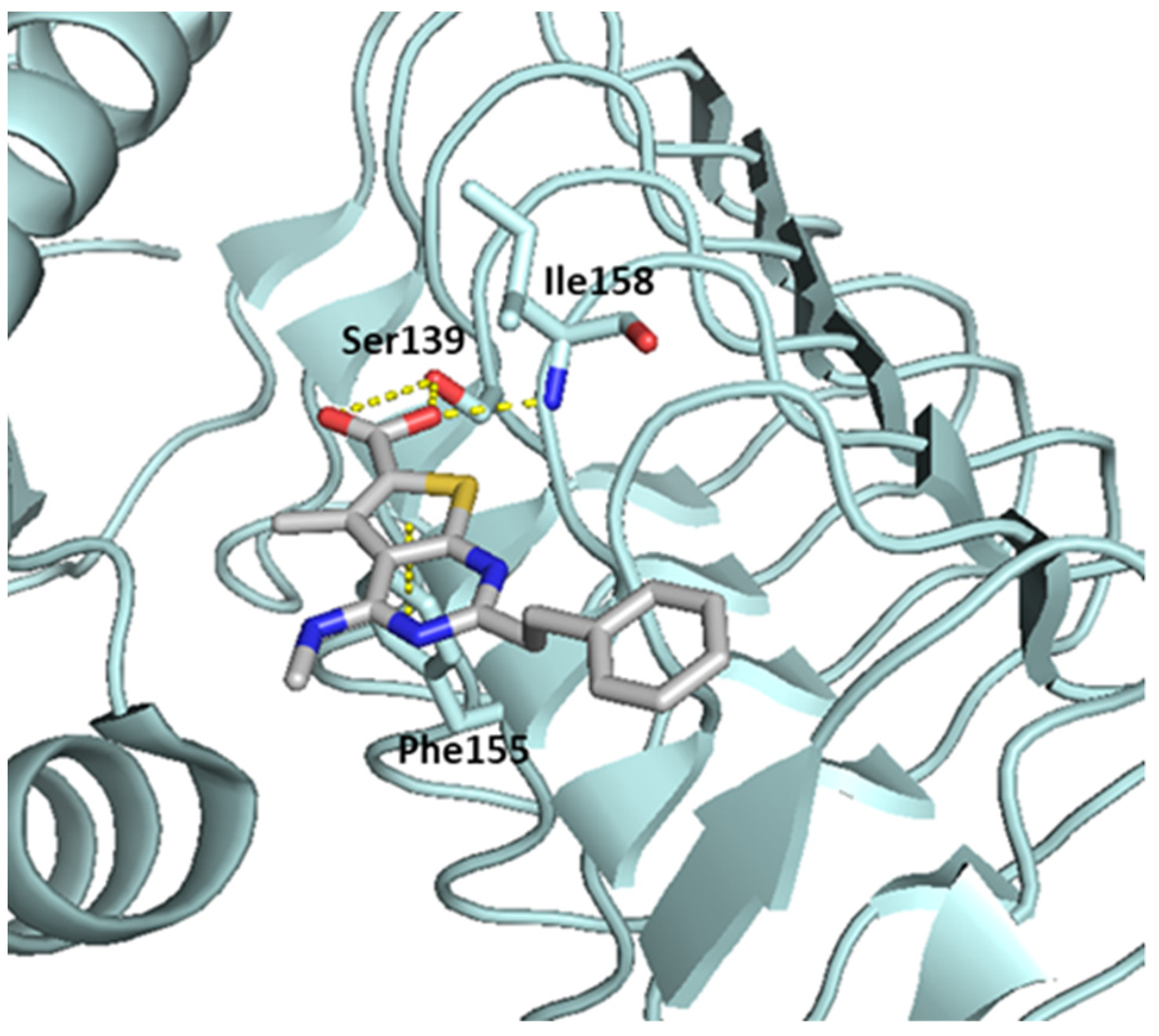
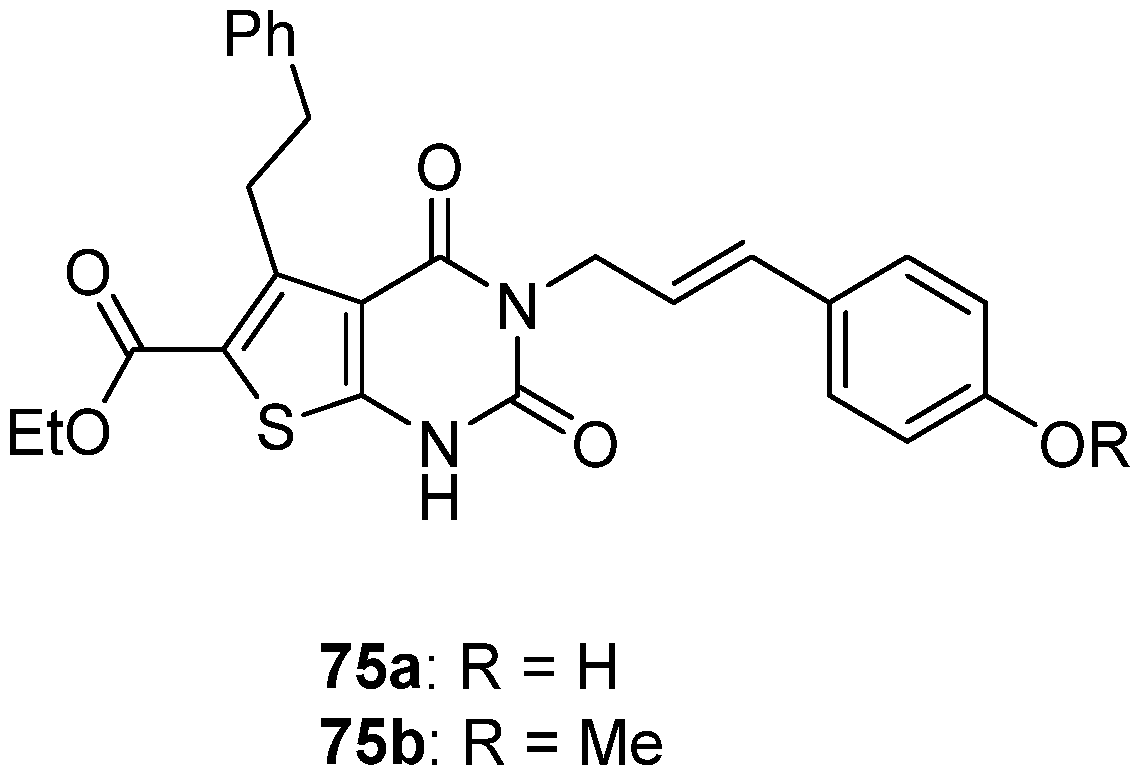
- Shao, X.; AbdelKhalek, A.; Abutaleb, N.S.; Velagapudi, U.K.; Yoganathan, S.; Seleem, M.N.; Talele, T.T. Chemical Space Exploration around Thieno[3,2-d]Pyrimidin-4(3H)-One Scaffold Led to a Novel Class of Highly Active Clostridium Difficile Inhibitors. J. Med. Chem. 2019, 62, 9772–9791. [Google Scholar] [CrossRef]
- Aly, H.M.; Saleh, N.M.; Elhady, H.A. Design and Synthesis of Some New Thiophene, Thienopyrimidine and Thienothiadiazine Derivatives of Antipyrine as Potential Antimicrobial Agents. Eur. J. Med. Chem. 2011, 46, 4566–4572. [Google Scholar] [CrossRef]
- Desroches, J.; Kieffer, C.; Primas, N.; Hutter, S.; Gellis, A.; El-Kashef, H.; Rathelot, P.; Verhaeghe, P.; Azas, N.; Vanelle, P. Discovery of New Hit-Molecules Targeting Plasmodium Falciparum through a Global SAR Study of the 4-Substituted-2-Trichloromethylquinazoline Antiplasmodial Scaffold. Eur. J. Med. Chem. 2017, 125, 68–86. [Google Scholar] [CrossRef] [PubMed]
- De Schutter, J.W.; Morrison, J.P.; Morrison, M.J.; Ciulli, A.; Imperiali, B. Targeting Bacillosamine Biosynthesis in Bacterial Pathogens: Development of Inhibitors to a Bacterial Amino-Sugar Acetyltransferase from Campylobacter Jejuni. J. Med. Chem. 2017, 60, 2099–2118. [Google Scholar] [CrossRef]
- Mavrova, A.T.; Vuchev, D.; Anichina, K.; Vassilev, N. Synthesis, Antitrichinnellosis and Antiprotozoal Activity of Some Novel Thieno[2,3-d]Pyrimidin-4(3H)-Ones Containing Benzimidazole Ring. Eur. J. Med. Chem. 2010, 45, 5856–5861. [Google Scholar] [CrossRef]
- Kim, J.; Kwon, J.; Lee, D.; Jo, S.; Park, D.-S.; Choi, J.; Park, E.; Hwang, J.Y.; Ko, Y.; Choi, I.; et al. Serendipitous Discovery of 2-((Phenylsulfonyl)Methyl)-Thieno[3,2-d]Pyrimidine Derivatives as Novel HIV-1 Replication Inhibitors. Bioorganic Med. Chem. Lett. 2014, 24, 5473–5477. [Google Scholar] [CrossRef]
- Sławiński, J.; Żołnowska, B.; Pirska, D.; Kędzia, A.; Kwapisz, E. Synthesis and Antibacterial Activity of Novel 4-Chloro-2-Mercaptobenzenesulfonamide Derivatives. J. Enzym. Inhib. Med. Chem. 2013, 28, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Patel, A. Modi Synthesis and Biological Evaluation of Schiff Base Involving Thieno[2,3-d] Pyrimidine Moiety as Antimicrobial Agents. RJLBPCS 2019, 5, 31–41. [Google Scholar] [CrossRef]
- Giri, T.; Sailaja, G.; Laxminarayana, E.; Thirumala Chary, M.; Ramesh, M. Synthesis and Antibacterial Activity of Novel 4-{4-(Methylamino)Thieno[3,2-d]Pyrimidin-2-Yl}-Benzohydrazide Derivatives. Russ. J. Gen. Chem. 2017, 87, 1275–1280. [Google Scholar] [CrossRef]
- Tharikoppula, G.; Eppakayala, L.; Maringanti, T.C.; Kamalapuram, C.; Kudle, K.R. Synthesis and Antibacterial Activity of Thienopyrimidine Amide Derivatives. Asian J. Chem. 2017, 29, 1515–1521. [Google Scholar] [CrossRef]
- Temburnikar, K.W.; Zimmermann, S.C.; Kim, N.T.; Ross, C.R.; Gelbmann, C.; Salomon, C.E.; Wilson, G.M.; Balzarini, J.; Seley-Radtke, K.L. Antiproliferative Activities of Halogenated Thieno[3,2-d]pyrimidines. Bioorganic Med. Chem. 2014, 22, 2113–2122. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, V.; Babu Kondra, S.; Maddula, S.R. Synthesis, Structural Elucidation of Novel Thieno[2,3-d]Pyrimidine Core Unit Containing 1,2,4-Triazoles and Thiophenes as Potent Antimicrobial Activity. Org. Chem. Curr. Res. 2016, 5, 1000169. [Google Scholar] [CrossRef]
- Prabhakar, V.; Babu, S.J.; Siva Jyothi, S.V.L. Synthesis, Structural Elucidation and Anti-Bacterial Evaluation of Some Novel Heterocyclic Molecules Derived from Thieno[2,3-d]pyrimidine as a Core Unit. Org. Chem. Curr. Res. 2016, 5, 1000172. [Google Scholar] [CrossRef]
- Sang, Y.; Han, S.; Han, S.; Pannecouque, C.; De Clercq, E.; Zhuang, C.; Chen, F. Follow On-Based Optimization of the Biphenyl-DAPYs as HIV-1 Nonnucleoside Reverse Transcriptase Inhibitors against the Wild-Type and Mutant Strains. Bioorganic Chem. 2019, 89, 102974. [Google Scholar] [CrossRef]
- Al-Harbi, R.A.K.; Abdel-Rahman, A.A.H. Synthesis and Anti-HBV Activity of 2-(Methylthio)Thieno[3,2-d]Pyrimidin- 4(1H)-One Analogues of ACV. Der Pharma Chem. 2013, 5, 1–7. [Google Scholar]
- Kankanala, J.; Kirby, K.A.; Huber, A.D.; Casey, M.C.; Wilson, D.J.; Sarafianos, S.G.; Wang, Z. Design, Synthesis and Biological Evaluations of N-Hydroxy Thienopyrimidine-2,4-Diones as Inhibitors of HIV Reverse Transcriptase-Associated RNase H. Eur. J. Med. Chem. 2017, 141, 149–161. [Google Scholar] [CrossRef]
- Abu-Hashem, A.A.; Abu-Zied, K.M.; El-Shehry, M.F. Synthetic Utility of Bifunctional Thiophene Derivatives and Antimicrobial Evaluation of the Newly Synthesized Agents. Mon. Chem. 2011, 142, 539–545. [Google Scholar] [CrossRef]
- Dewal, M.B.; Wani, A.S.; Vidaillac, C.; Oupický, D.; Rybak, M.J.; Firestine, S.M. Thieno[2,3-d]Pyrimidinedione Derivatives as Antibacterial Agents. Eur. J. Med. Chem. 2012, 51, 145–153. [Google Scholar] [CrossRef]
- Shaaban, O.G.; Issa, D.A.E.; El-Tombary, A.A.; Abd El Wahab, S.M.; Abdel Wahab, A.E.; Abdelwahab, I.A. Synthesis and Molecular Docking Study of Some 3,4-Dihydrothieno[2,3-d]Pyrimidine Derivatives as Potential Antimicrobial Agents. Bioorganic Chem. 2019, 88, 102934. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Kawai, K.; Asano, T.; Amano, S.; Asanuma, Y.; Sawada, K.; Onodera, Y.; Ueo, N.; Takahashi, N.; Sonoda, Y.; et al. 2-(Isopropylamino)Thieno[3,2-d]Pyrimidin-4(3H)-One Derivatives as Selective Phosphodiesterase 7 Inhibitors with Potent in Vivo Efficacy. Bioorganic Med. Chem. Lett. 2015, 25, 1910–1914. [Google Scholar] [CrossRef]
- González Cabrera, D.; Le Manach, C.; Douelle, F.; Younis, Y.; Feng, T.-S.; Paquet, T.; Nchinda, A.T.; Street, L.J.; Taylor, D.; de Kock, C.; et al. 2,4-Diaminothienopyrimidines as Orally Active Antimalarial Agents. J. Med. Chem. 2014, 57, 1014–1022. [Google Scholar] [CrossRef]
- Cohen, A.; Suzanne, P.; Lancelot, J.-C.; Verhaeghe, P.; Lesnard, A.; Basmaciyan, L.; Hutter, S.; Laget, M.; Dumètre, A.; Paloque, L.; et al. Discovery of New Thienopyrimidinone Derivatives Displaying Antimalarial Properties toward Both Erythrocytic and Hepatic Stages of Plasmodium. Eur. J. Med. Chem. 2015, 95, 16–28. [Google Scholar] [CrossRef]
- Chambhare, R.V.; Khadse, B.G.; Bobde, A.S.; Bahekar, R.H. Synthesis and Preliminary Evaluation of Some N-[5-(2-Furanyl)-2-Methyl-4-Oxo-4H-Thieno[2,3-d]Pyrimidin-3-Yl]-Carboxamide and 3-Substituted-5-(2-Furanyl)-2-Methyl-3H-Thieno[2,3-d]Pyrimidin-4-Ones as Antimicrobial Agents. Eur. J. Med. Chem. 2003, 38, 89–100. [Google Scholar] [CrossRef]
- Abdel Hamid, A.M.; Shehta, W. Synthesis of Some Novel Furan-Tagged Thienopyrimidine Derivatives as Antibacterial Agents: Synthesis of Some Novel Furan-Tagged Thienopyrimidine Derivatives as Antibacterial Agents. J. Heterocycl. Chem. 2019, 56, 485–492. [Google Scholar] [CrossRef]
- Aly, H.M.; Saleh, N.M. Utility of a Pyrimidine Thione Derivative in the Synthesis of New Fused Pyrimido[4,5-d]Pyrimidine, Pyrido[2,3-d]Pyrimidine and Different Types of Thienopyrimidine Derivatives. Int. J. Adv. Res. 2014, 2, 694–702. [Google Scholar]
- Malaria. Available online: https://www.who.int/news-room/fact-sheets/detail/malaria (accessed on 28 August 2020).
- World Health Organization. World Malaria Report 2020: 20 Years of Global Progress and Challenges; World Health Organization: Geneva, Switzerland, 2020; ISBN 978-92-4-001579-1.
- Ariey, F.; Witkowski, B.; Amaratunga, C.; Beghain, J.; Langlois, A.-C.; Khim, N.; Kim, S.; Duru, V.; Bouchier, C.; Ma, L.; et al. A Molecular Marker of Artemisinin-Resistant Plasmodium Falciparum Malaria. Nature 2014, 505, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Okombo, J.; Chibale, K. Recent Updates in the Discovery and Development of Novel Antimalarial Drug Candidates. Med. Chem. Comm. 2018, 9, 437–453. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, T.; Liu, J.; Ma, R.; Lu, W.; Huang, J.; Li, H.; Li, J.; Jiang, H. 2-(3,4-Dihydro-4-Oxothieno[2,3-d]Pyrimidin-2-Ylthio) Acetamides as a New Class of Falcipain-2 Inhibitors. 3. Design, Synthesis and Biological Evaluation. Molecules 2009, 14, 785–797. [Google Scholar] [CrossRef]
- Rosenthal, P.J. Falcipain Cysteine Proteases of Malaria Parasites: An Update. Biochim. Et Biophys. Acta (BBA)—Proteins Proteom. 2020, 1868, 140362. [Google Scholar] [CrossRef] [PubMed]
- Bekono, B.D.; Ntie-Kang, F.; Owono Owono, L.C.; Megnassan, E. Targeting Cysteine Proteases from Plasmodium Falciparum: A General Overview, Rational Drug Design and Computational Approaches for Drug Discovery. Curr. Drug Targets 2018, 19, 501–526. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.J.; Singh, A.; Chiang, P.; Kemp, S.J.; Goldman, E.A.; Weinhouse, M.I.; Vlasuk, G.P.; Rosenthal, P.J. Antimalarial Activities of Novel Synthetic Cysteine Protease Inhibitors. Antimicrob. Agents Chemother. 2003, 47, 3810–3814. [Google Scholar] [CrossRef]
- Edlin, C.D.; Morgans, G.; Winks, S.; Duffy, S.; Avery, V.M.; Wittlin, S.; Waterson, D.; Burrows, J.; Bryans, J. Identification and In-Vitro ADME Assessment of a Series of Novel Anti-Malarial Agents Suitable for Hit-to-Lead Chemistry. ACS Med. Chem. Lett. 2012, 3, 570–573. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Barrows, R.D.; Hammill, J.T.; Tran, M.C.; Falade, M.O.; Rice, A.L.; Davis, C.W.; Emge, T.J.; Rablen, P.R.; Kiplin Guy, R.; Knapp, S. Evaluation of 1,1-Cyclopropylidene as a Thioether Isostere in the 4-Thio-Thienopyrimidine (TTP) Series of Antimalarials. Bioorganic Med. Chem. 2020, 28, 115758. [Google Scholar] [CrossRef]
- Fletcher, S.; Avery, V.M. A Novel Approach for the Discovery of Chemically Diverse Anti-Malarial Compounds Targeting the Plasmodium Falciparum Coenzyme A Synthesis Pathway. Malar. J. 2014, 13, 343. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Lucantoni, L.; Sykes, M.L.; Jones, A.J.; Holleran, J.P.; Saliba, K.J.; Avery, V.M. Biological Characterization of Chemically Diverse Compounds Targeting the Plasmodium Falciparum Coenzyme A Synthesis Pathway. Parasites Vectors 2016, 9, 589. [Google Scholar] [CrossRef] [PubMed]
- Weidner, T.; Lucantoni, L.; Nasereddin, A.; Preu, L.; Jones, P.G.; Dzikowski, R.; Avery, V.M.; Kunick, C. Antiplasmodial Dihetarylthioethers Target the Coenzyme A Synthesis Pathway in Plasmodium Falciparum Erythrocytic Stages. Malar. J. 2017, 16, 192. [Google Scholar] [CrossRef]
- Kikuchi, H.; Yamamoto, K.; Horoiwa, S.; Hirai, S.; Kasahara, R.; Hariguchi, N.; Matsumoto, M.; Oshima, Y. Exploration of a New Type of Antimalarial Compounds Based on Febrifugine. J. Med. Chem. 2006, 49, 4698–4706. [Google Scholar] [CrossRef] [PubMed]
- Gonzàlez Cabrera, D.; Douelle, F.; Le Manach, C.; Han, Z.; Paquet, T.; Taylor, D.; Njoroge, M.; Lawrence, N.; Wiesner, L.; Waterson, D.; et al. Structure–Activity Relationship Studies of Orally Active Antimalarial 2,4-Diamino-Thienopyrimidines. J. Med. Chem. 2015, 58, 7572–7579. [Google Scholar] [CrossRef]
- Garrido, A.; Lepailleur, A.; Mignani, S.M.; Dallemagne, P.; Rochais, C. HERG Toxicity Assessment: Useful Guidelines for Drug Design. Eur. J. Med. Chem. 2020, 195, 112290. [Google Scholar] [CrossRef]
- van der Watt, M.E.; Reader, J.; Churchyard, A.; Nondaba, S.H.; Lauterbach, S.B.; Niemand, J.; Abayomi, S.; van Biljon, R.A.; Connacher, J.I.; van Wyk, R.D.J.; et al. Potent Plasmodium Falciparum Gametocytocidal Compounds Identified by Exploring the Kinase Inhibitor Chemical Space for Dual Active Antimalarials. J. Antimicrob. Chemother. 2018, 73, 1279–1290. [Google Scholar] [CrossRef]
- Woodring, J.L.; Behera, R.; Sharma, A.; Wiedeman, J.; Patel, G.; Singh, B.; Guyett, P.; Amata, E.; Erath, J.; Roncal, N.; et al. Series of Alkynyl-Substituted Thienopyrimidines as Inhibitors of Protozoan Parasite Proliferation. ACS Med. Chem. Lett. 2018, 9, 996–1001. [Google Scholar] [CrossRef]
- Trypanosomiasis, Human African (Sleeping Sickness). Available online: https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness) (accessed on 8 November 2021).
- Johnston, S.R.D.; Leary, A. Lapatinib: A Novel EGFR/HER2 Tyrosine Kinase Inhibitor for Cancer. Drugs Today 2006, 42, 441–453. [Google Scholar] [CrossRef]
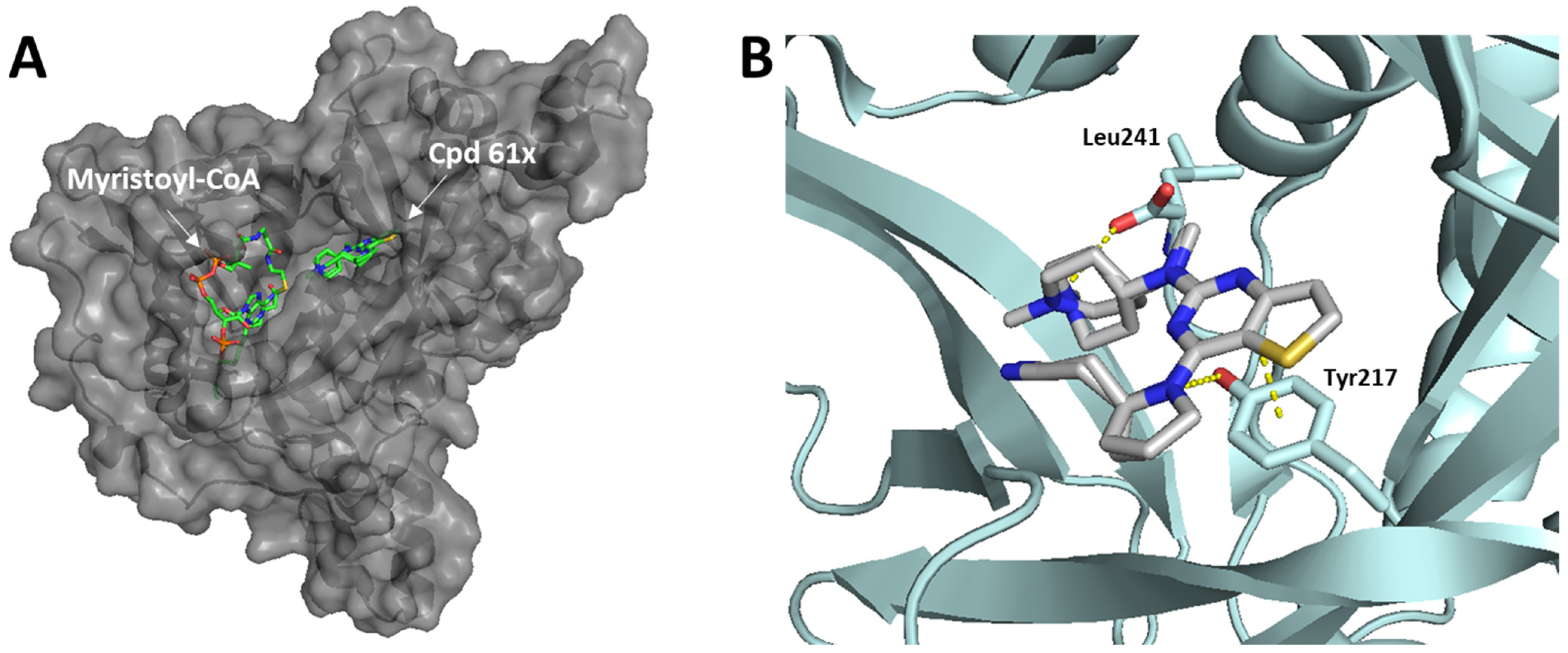
- Bell, A.S.; Yu, Z.; Hutton, J.A.; Wright, M.H.; Brannigan, J.A.; Paape, D.; Roberts, S.M.; Sutherell, C.L.; Ritzefeld, M.; Wilkinson, A.J.; et al. Novel Thienopyrimidine Inhibitors of Leishmania N -Myristoyltransferase with On-Target Activity in Intracellular Amastigotes. J. Med. Chem. 2020, 63, 7740–7765. [Google Scholar] [CrossRef] [PubMed]
- Tuberculosis (TB). Available online: https://www.who.int/news-room/fact-sheets/detail/tuberculosis (accessed on 6 August 2021).
- Rashmi, P.; Nargund, L.V.G.; Hazra, K.; Chandra, J.N.N.S. Thienopyrimidines as Novel Inhibitors of Mycobacterium Tuberclosis: Synthesis and In-Vitro Studies. Arch. Pharm. Pharm. Med. Chem. 2011, 344, 459–465. [Google Scholar] [CrossRef]
- Ananthan, S.; Faaleolea, E.R.; Goldman, R.C.; Hobrath, J.V.; Kwong, C.D.; Laughon, B.E.; Maddry, J.A.; Mehta, A.; Rasmussen, L.; Reynolds, R.C.; et al. High-Throughput Screening for Inhibitors of Mycobacterium Tuberculosis H37Rv. Tuberculosis 2009, 89, 334–353. [Google Scholar] [CrossRef] [PubMed]
- Harrison, G.A.; Mayer Bridwell, A.E.; Singh, M.; Jayaraman, K.; Weiss, L.A.; Kinsella, R.L.; Aneke, J.S.; Flentie, K.; Schene, M.E.; Gaggioli, M.; et al. Identification of 4-Amino-Thieno[2,3-d]Pyrimidines as QcrB Inhibitors in Mycobacterium Tuberculosis. mSphere 2019, 4, e00606-19. [Google Scholar] [CrossRef]
- Lu, X.; Williams, Z.; Hards, K.; Tang, J.; Cheung, C.-Y.; Aung, H.L.; Wang, B.; Liu, Z.; Hu, X.; Lenaerts, A.; et al. Pyrazolo[1,5-a]Pyridine Inhibitor of the Respiratory Cytochrome Bcc Complex for the Treatment of Drug-Resistant Tuberculosis. ACS Infect. Dis. 2019, 5, 239–249. [Google Scholar] [CrossRef]
- Antibiotic Resistance. Available online: https://www.who.int/news-room/fact-sheets/detail/antibiotic-resistance (accessed on 23 July 2021).
- Campylobacter. Available online: https://www.who.int/news-room/fact-sheets/detail/campylobacter (accessed on 30 August 2021).
- Luangtongkum, T.; Jeon, B.; Han, J.; Plummer, P.; Logue, C.M.; Zhang, Q. Antibiotic Resistance in Campylobacter: Emergence, Transmission and Persistence. Future Microbiol. 2009, 4, 189–200. [Google Scholar] [CrossRef] [PubMed]
- de Candia, M.; Altamura, C.; Denora, N.; Cellamare, S.; Nuzzolese, M.; De Vito, D.; Voskressensky, L.G.; Varlamov, A.V.; Altomare, C.D. Physicochemical Properties and Antimicrobial Activity of New Spirocyclic Thieno[2,3-d]Pyrimidin-4(3H)-One Derivatives. Chem. Heterocycl. Comp. 2017, 53, 357–363. [Google Scholar] [CrossRef]
- Tolba, M.S.; El-Dean, A.M.K.; Ahmed, M.; Hassanien, R.; Farouk, M. Synthesis and Antimicrobial Activity of Some New Thienopyrimidine Derivatives. Arkivoc 2017, 2017, 229–243. [Google Scholar] [CrossRef]
- Saddik, A.A.; Kamal El-Dean, A.M.; El-Said, W.A.; Hassan, K.M.; Abbady, M.S. Synthesis, Antimicrobial, and Anticancer Activities of a New Series of Thieno[2,3-d] Pyrimidine Derivatives: Synthesis of New Series from Thieno[2,3-d]Pyrimidine and Study the Antimicrobial and Anticancer Activities for Some Compounds. J. Heterocycl. Chem. 2018, 55, 2111–2122. [Google Scholar] [CrossRef]
- Bhagchand, J.; Santra, S. Prasanta Kumar Santra Synthesis and Evaluation of Antimicrobial Activity of Pyrimidine Derivatives. Asian J. Pharm. Clin. Res. 2019, 12, 156–163. [Google Scholar] [CrossRef]
- Hafez, H.N.; El-Gazzar, A.-R.B.A.; Zaki, M.E.A. Simple Approach to Thieno[3,2-d]Pyrimidines as New Scaffolds of Antimicrobial Activities. Acta Pharm. 2016, 66, 331–351. [Google Scholar] [CrossRef]
- Mathias, F.; Curti, C.; Montana, M.; Bornet, C.; Vanelle, P. Management of Adult Clostridium Difficile Digestive Contaminations: A Literature Review. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 209–231. [Google Scholar] [CrossRef] [PubMed]
- CDC Most Cases of C. Difficile Occur While Taking Antibiotics or Soon after. Available online: https://www.cdc.gov/cdiff/index.html (accessed on 31 August 2021).
- Antifungal Resistance|Fungal Diseases|CDC. Available online: https://www.cdc.gov/fungal/antifungal-resistance.html (accessed on 22 July 2021).
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Primers 2018, 4, 1–21. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, L.; Amichai, S.; Zandi, K.; Cox, B.; Schinazi, R.; Amblard, F. Novel Influenza Polymerase PB2 Inhibitors for the Treatment of Influenza A Infection. Bioorganic Med. Chem. Lett. 2019, 29, 126639. [Google Scholar] [CrossRef]
- Engelhardt, O.G.; Fodor, E. Functional Association between Viral and Cellular Transcription during Influenza Virus Infection. Rev. Med. Virol. 2006, 16, 329–345. [Google Scholar] [CrossRef] [PubMed]
- The Lancet Towards Elimination of Viral Hepatitis by 2030. Lancet 2016, 388, 308. [CrossRef]
- Nelson, N.P.; Easterbrook, P.J.; McMahon, B.J. Epidemiology of Hepatitis B Virus Infection and Impact of Vaccination on Disease. Clin. Liver Dis. 2016, 20, 607–628. [Google Scholar] [CrossRef] [PubMed]
- UNAIDS Data 2020. Available online: https://www.unaids.org/en/resources/documents/2020/unaids-data (accessed on 19 October 2020).
- HIV/AIDS. Available online: https://www.who.int/news-room/fact-sheets/detail/hiv-aids (accessed on 1 October 2020).
- Maartens, G.; Celum, C.; Lewin, S.R. HIV Infection: Epidemiology, Pathogenesis, Treatment, and Prevention. Lancet 2014, 384, 258–271. [Google Scholar] [CrossRef]
- WHO|HIV Drug Resistance Report 2019. Available online: http://www.who.int/hiv/pub/drugresistance/hivdr-report-2019/en/ (accessed on 19 October 2020).
- Kang, D.; Fang, Z.; Li, Z.; Huang, B.; Zhang, H.; Lu, X.; Xu, H.; Zhou, Z.; Ding, X.; Daelemans, D.; et al. Design, Synthesis, and Evaluation of Thiophene[3,2-d]Pyrimidine Derivatives as HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors with Significantly Improved Drug Resistance Profiles. J. Med. Chem. 2016, 59, 7991–8007. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Fang, Z.; Huang, B.; Lu, X.; Zhang, H.; Xu, H.; Huo, Z.; Zhou, Z.; Yu, Z.; Meng, Q.; et al. Structure-Based Optimization of Thiophene[3,2-d]Pyrimidine Derivatives as Potent HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors with Improved Potency against Resistance-Associated Variants. J. Med. Chem. 2017, 60, 4424–4443. [Google Scholar] [CrossRef]
- Kang, D.; Ding, X.; Wu, G.; Huo, Z.; Zhou, Z.; Zhao, T.; Feng, D.; Wang, Z.; Tian, Y.; Daelemans, D.; et al. Discovery of Thiophene[3,2-d]Pyrimidine Derivatives as Potent HIV-1 NNRTIs Targeting the Tolerant Region I of NNIBP. ACS Med. Chem. Lett. 2017, 8, 1188–1193. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Wang, Z.; Chen, M.; Feng, D.; Wu, G.; Zhou, Z.; Jing, L.; Zuo, X.; Jiang, X.; Daelemans, D.; et al. Discovery of Potent HIV -1 Non-nucleoside Reverse Transcriptase Inhibitors by Exploring the Structure–Activity Relationship of Solvent-exposed Regions I. Chem. Biol. Drug Des. 2019, 93, 430–437. [Google Scholar] [CrossRef]
- Kang, D.; Zhang, H.; Wang, Z.; Zhao, T.; Ginex, T.; Luque, F.J.; Yang, Y.; Wu, G.; Feng, D.; Wei, F.; et al. Identification of Dihydrofuro[3,4-d]Pyrimidine Derivatives as Novel HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors with Promising Antiviral Activities and Desirable Physicochemical Properties. J. Med. Chem. 2019, 62, 1484–1501. [Google Scholar] [CrossRef]
- Yang, Y.; Kang, D.; Nguyen, L.A.; Smithline, Z.B.; Pannecouque, C.; Zhan, P.; Liu, X.; Steitz, T.A. Structural Basis for Potent and Broad Inhibition of HIV-1 RT by Thiophene[3,2-d]Pyrimidine Non-Nucleoside Inhibitors. eLife 2018, 7, e36340. [Google Scholar] [CrossRef] [PubMed]
- Sang, Y.; Han, S.; Pannecouque, C.; De Clercq, E.; Zhuang, C.; Chen, F. Conformational Restriction Design of Thiophene-Biphenyl-DAPY HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors. Eur. J. Med. Chem. 2019, 182, 111603. [Google Scholar] [CrossRef] [PubMed]
- Richman, D.D.; Margolis, D.M.; Delaney, M.; Greene, W.C.; Hazuda, D.; Pomerantz, R.J. The Challenge of Finding a Cure for HIV Infection. Science 2009, 323, 1304–1307. [Google Scholar] [CrossRef] [PubMed]
- Vargas, B.; Giacobbi, N.S.; Sanyal, A.; Venkatachari, N.J.; Han, F.; Gupta, P.; Sluis-Cremer, N. Inhibitors of Signaling Pathways That Block Reversal of HIV-1 Latency. Antimicrob. Agents Chemother. 2018, 63, e01744-18. [Google Scholar] [CrossRef] [PubMed]


































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
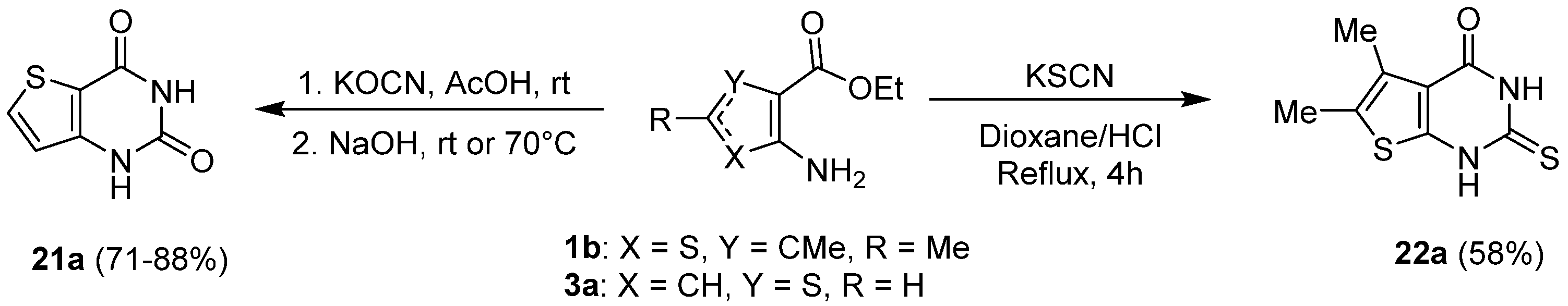{kind=link}
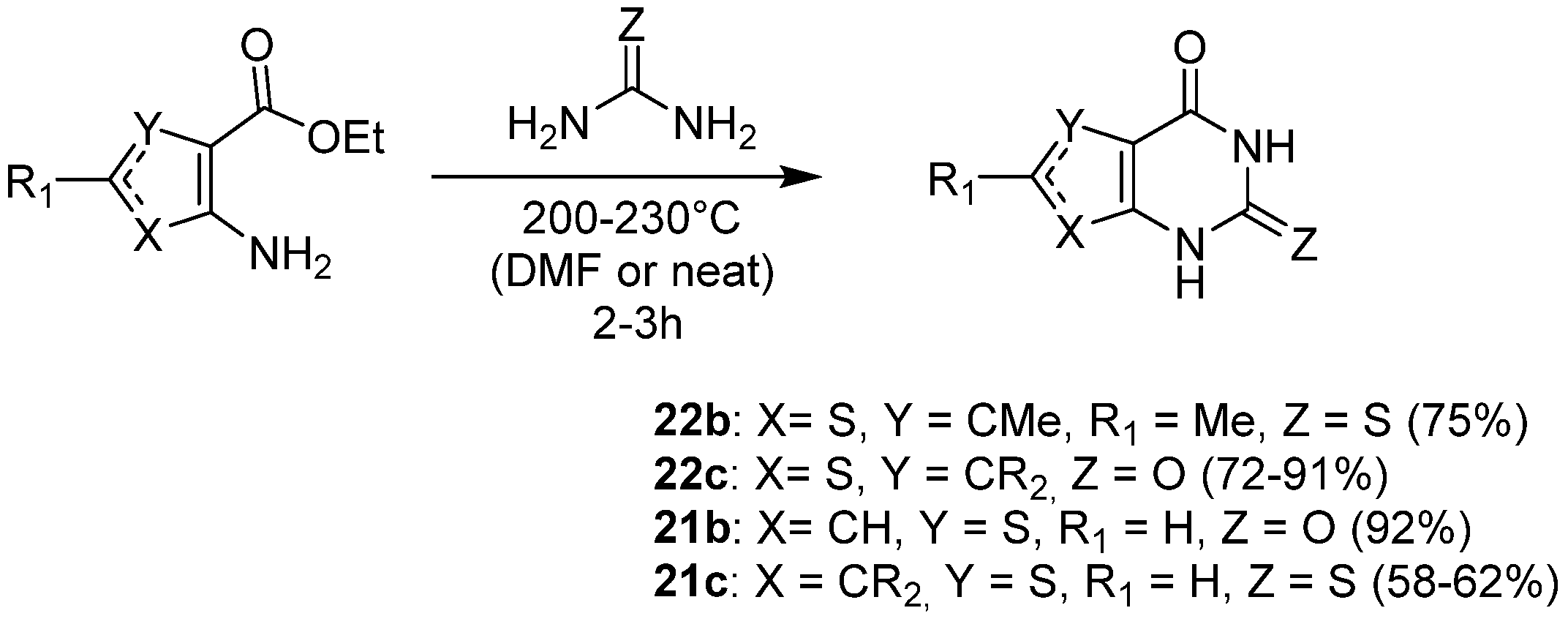{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Cpd | R1 | R2 | R3 | Inhibitory Activity Against FP-2 | |
| Inhibition Rate at 10 μM (%) | IC50 (μM) | ||||
| 54a | H | Allyl | - | 88.7 | 2.81 |
| 54b | H | Cyclohexyl | - | 92.7 | 1.46 |
| 54c | H | 3-F-Ph | - | 79.0 | 2.05 |
| 54d | H | 4-NO2-Ph | - | 85.4 | 2.77 |
| 54e | H | 4-Cl-Ph | - | 84.7 | 4.30 |
| 54f | H | Benzyl | - | 90.6 | 5.74 |
| 54g | 4-Cl | Allyl | - | 85.7 | 2.95 |
| 54h | 3-NO2 | Allyl | - | 53.0 | 11.8 |
| 55a | - | - |  | 93.3 | 6.63 |
| 55b | - | - |  | 94.3 | 5.70 |
| 55c | - | - |  | 90.3 | 3.31 |
| 55d | - | - |  | 93.2 | 2.49 |
| 55e | - | - |  | 72.0 | 5.58 |
| 55f | - | - |  | 92.0 | 5.43 |
 | ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | P. falciparum | Ref. | |
| NF54 EC50 (µM) | 3D7 EC50 (µM) | |||||
| 56a | H | H |  | 0.15 | - | [48] |
| 56b | H | H |  | 1.48 | - | [48] |
| 56c | H | H |  | 0.15 | - | [48] |
| 56d | Me | Me |  | 0.46 | - | [48] |
| 56e | H | H |  | - | 0.037 | [49] |
| 56f | H | H |  | - | 0.191 | [49] |
| 56g | H | H |  | - | >23 | [49] |
| 56h | H | H |  | - | >23 | [49] |
 | ||||||
|---|---|---|---|---|---|---|
| Compound | R | % Inhibition, PfNF54-Luc at 3 µM | Pf3D7 EC50 (µM) | Pf3D7 + 0.8 mM CoA EC50 (µM) | THP-1 EC50 (µM) | HEK-293 EC50 (µM) |
| 56i |  | 80.8 ± 0.5 | 0.283 ± 0.073 | 4.61 ± 0.15 | 6.13 | >10 [48] |
| 56j |  | 99.9 ± 0.0 | 0.0388 ± 0.0010 | 2.61 ± 0.42 | 27.5 | >10 |
| 56k |  | 99.5 ± 0.5 | 0.0747 ± 0.021 | 2.58 ± 0.42 | 33.6 | >10 |
| 56l |  | 99.7 ± 0.1 | 0.0958 ± 0.0063 | 11.2 ± 2.6 | 11 | >4 |
| 56m |  | 95.0 ± 1.1 | 0.0734 ± 0.0022 | 0.531 ± 0.007 | 16.5 | >10 |
| 56n |  | 98.0 ± 0.2 | 0.149 ± 0.021 | 1.06 ± 0.03 | 28 | >20 |
| 56o |  | - | 0.0370 ± 0.0017 | 1.51 ± 0.19 | - | >40 |
| Chloroquine | - | - | 0.0195 ± 0.0034 | 0.0271 ± 0.0094 | - | >40 |
| Artemisinin | - | - | 0.00337 ± 0.00065 | 0.00490 ± 0.0011 | - | - |
 | ||||
|---|---|---|---|---|
| Compound | R1 | R2 | Antiplasmodial Activity a on K1 EC50 (µM) | Cytotoxicity a on HepG2 CC50 (µM) |
| 57a | 4-Me-Ph | NH-tBu | 0.2 ± 0.02 | 25.6 ± 3.1 |
| 57b | 4-Me-Ph | NH-iPr | 0.8 ± 0.5 | 49.4 ± 1 |
| 57c | 4-Me-Ph | NH-nPr | >5 b | >62.5 b |
| 57d | Ph | NH-tBu | 1 | 12.5 ± 2.5 |
| 57e | H | NH-tBu | >5 b | 8.4 ± 3.5 |
| 57f | 3-Me-Ph | NH-tBu | 3.6 | 14.1 ± 1.4 |
| 57g | 2-Me-Ph | NH-tBu | 1.7 | 4.0 ± 1.1 |
| 57h | 4-Cl-Ph | NH-tBu | 0.8 | 15.0 ± 2.7 |
| 57i | 4-F-Ph | NH-tBu | >5 b | 5.1 ± 1.0 |
| 57j | 4-Me-Ph | NH-tBu·HCl | 0.045 | 24.0 |
| Chloroquine c | - | - | 0.5 | 30 |
| Compound |  | Activity on P. falciparum Strains EC50 (nM) a | Degradation Half-Life (min) | Ref. | |||
|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | K1 | NF54 | |||
| 58a | Ph |  | NHMe | 73 | 28 | 344.9 | [35] |
| 58b | Br |  | NHMe | 973 | 636 | - | [35] |
| 58c | 4-CN-Ph |  | NHMe | 19 | 9 | 238 | [35] |
| 58d | 4-CF3-Ph |  | NHMe | 32 | 29 | 124 | [35] |
| 58e | 4-CF3O-Ph |  | NHMe | 26 | 28 | >250 | [35] |
| 58f | 3-CN-Ph |  | NHMe | 17 | 4 | - | [35] |
| 58g | 3-CF3-Ph |  | NHMe | - | 13 | 31.1 | [35] |
| 58h | 2-Cl-4-CF3-Ph |  | NHMe | 47 | 16 | 130 | [35] |
| 58i |  |  | NHMe | 12 | 7 | 15 | [35] |
| 58j | 4-CF3-Ph |  | NHMe | >2436 | 1832 | - | [54] |
| 58k | 4-CF3-Ph |  | NHMe | 233 | 111 | - | [54] |
| 58l | 4-CF3-Ph |  | NHMe | >2608 | >2608 | - | [54] |
| 58m | 4-CF3-Ph | NH2 | NHMe | - | 1158 | - | [54] |
| 58n | 4-CF3-Ph |  | NHMe | - | 42 | - | [54] |
| 58o | 4-CF3-Ph |  | NHMe | 24 | 19 | - | [54] |
| 58p | 4-CF3-Ph |  | NHMe | - | 43 | 23.9 | [54] |
| 58q | 4-CF3-Ph |  | NH-benzyl | - | 42 | - | [54] |
| 58r | 4-CF3-Ph |  | NH2 | - | 58 | 104 | [54] |
| 58s | 4-CF3-Ph |  | NH2 | 24 | 25 | >150 | [54] |
| 58t | 4-CF3-Ph |  | NH2 | 33 | 20 | >150 | [54] |
| Chloroquine b | 194 | 16 | - | [54] | |||
| Artesunate b | 3 | 4 | - | [54] | |||
 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | P. falciparum D6 EC50 (µM) (r2) a | Ref. | |
| 59a | A |  | - | 0.027 (0.97) | [14] |
| 59b | B | 0.089 (0.81) | |||
| 59c | A | - |  | 0.64 (0.99) | [57] |
| 59d | B | 0.26 (0.94) | |||
| R |  |  | |
|---|---|---|---|
 | K1 P. falciparum IC50 (µM) | 0.6 | 0.5 |
| HepG2 CC50 (µM) | 3.2 | 6.2 | |
| SIa | 5.3 | 12.4 | |
 | K1 P. falciparum IC50 (µM) | 0.9 | 0.6 |
| HepG2 CC50 (µM) | 0.7 | 6.2 | |
| SI a | 0.8 | 6.7 | |
 | K1 P. falciparum IC50 (µM) | 0.6 | - |
| HepG2 CC50 (µM) | 4.3 | - | |
| SI a | 7.2 | - | |
 | K1 P. falciparum IC50 (µM) | 0.4 | 0.8 |
| HepG2 CC50 (µM) | 6.9 | 6.2 | |
| SI a | 17.2 | 16.6 | |
| Doxycycline b | K1 P. falciparum IC50 (µM) | 6.0 | |
| HepG2 CC50 (µM) | 20 | ||
| SI a | 3.3 | ||
| Atovaquone b | K1 P. falciparum IC50 (µM) | 0.001 | |
| HepG2 CC50 (µM) | >15.6 | ||
| SIa | 15,600 | ||
| Chloroquine b | K1 P. falciparum IC50 (µM) | 0.6 | |
| HepG2 CC50 (µM) | 30 | ||
| SI a | 50 | ||
| Doxorubicine | HepG2 CC50 (µM) | 0.2 | |
 | |||||
|---|---|---|---|---|---|
| Cpd | R | L. major Amastigotes EC50 (µM) (r2) a | L. major Promastigotes EC50 (µM) (r2) a | T. brucei brucei EC50 ± SEM (µM) | T. cruzi Amastigotes EC50 ± SEM (µM) |
| 59a (GW837016X) |  | - | - | 0.26 | - |
| 59b |  | >15 | 0.22 (0.84) | 1.1 ± 0.0 | >50.0 |
| 59c |  | >3 | >3 | 0.084 ± 0.0 | 3.3 ± 1.2 |
| 59d |  | 1.58 (0.83) | >20 | 2.2 ± 0.1 | 0.75 ± 0.02 |
| 59e |  | 0.38 (0.94) | - | 1.3 ± 0.3 | 12 ± 0.45 |
| 59f |  | 4.1 (0.77) | - | 0.28 ± 0.07 | 1.8 ± 0.17 |
| 59g |  | >15 | - | 0.76 ± 0.07 | >50 |
| 60a |  | 4.2 (0.94) | - | 0.22 ± 0.02 | 3.7 ± 0.23 |
| 60b |  | 9.1 (0.88) | - | 1.9 ± 0 | 0.61 ± 0.16 |
| Amphotericin B b | - | 0.035 (0.90) | - | - | - |
| Suramin b | - | - | - | 0.04 ± 0 | - |
| Benznidazole b | - | - | - | - | 0.79 ± 0.01 |
 | ||||||
|---|---|---|---|---|---|---|
| Cpd | R1 | R2 | R3 | CMP Assay IC50 (μM) | ||
| LdNMT | LmNMT | HsNMT1 | ||||
| 61a | H |  |  | 0.34 | 0.20 | 5.7 |
| 61b | H |  | 0.15 | 2.7 | >100 | |
| 61c | H |  | >100 | - | - | |
| 61d | H |  | >100 | - | - | |
| 61e | H |  | 28 | - | - | |
| 61f | H |  | 30 | - | - | |
| 61g | H |  | 7.7 | - | 46 | |
| 61h | H |  | >100 | - | - | |
| 61i | H |  | 0.5 | - | 11 | |
| 61j | H |  | 0.83 | 0.62 | 17 | |
| 61k | H |  | 4.0 | - | >100 | |
| 61l | H |  | 5.2 | - | - | |
| 61m | H |  | >100 | - | - | |
| 61n | H |  | >100 | - | - | |
| 61o | H | H |  | >100 | - | - |
| 61p | H |  | 44 | - | - | |
| 61q | H |  | 22 | 36 | - | |
| 61r | H |  | - | 13 | - | |
| 61s | H |  | >100 | - | >100 | |
| 61t | H |  | >100 | - | - | |
| 61u | H |  | 3.6 | - | 16 | |
| 61v | H |  | 0.15 | - | >100 | |
| 61w | t-Bu |  |  | 0.36 | 0.16 | - |
| 61x | t-Bu |  | 0.046 | 0.042 | 0.55 | |
 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Efficacy (%) a after 24 h b 5 μg/mL | Efficacy (%) after 48 h 5 μg/mL |
| 62a | H | - | - | 79.8 | 85.30 |
| 63a | - | Et | H | 39.07 | 59.75 |
| 63b | - | H | NO2 | 50.00 | 80.05 |
| 63c | - | H | Cl | 0.00 | 5.09 |
| Albendazole (20 μg/mL) | - | - | - | 10.8 | 14.8 |
 | |||
|---|---|---|---|
| Compound | R | M. tuberculosis H37Rv MIC (µM) | THP-1 IC50 (µM) |
| 64a | Ph | 320 | ND a |
| 64b | 4-((Me)2)N-Ph | 70 | 2492.90 |
| 64c | 2-Br-Ph | 64 | 2387.80 |
| 64d | 2-NO2-Ph | 70 | 2415.90 |
| 64e | 4-Cl-Ph | 71 | 2264.80 |
| 64f | 4-MeO-Ph | 66 | 2454.70 |
| 64g | 3,4,5-(MeO)3-Ph | 62 | 2019.90 |
| Pyrazinamide | - | 60.97 | ND |
 | ||||
|---|---|---|---|---|
| Compound | R1 | R2 | R3 | TB IC90 (µg·mL−1) |
| 65a | H | 4-Me-Ph | CH2Ph-Cl-4 | 1.0 |
| 65b | H | 3,4-(Me)2-Ph | Cyclohexyl | 1.7 |
| 65c | CH3 | Me | Cyclohexyl | 1.8 |
| 65d | H | 4-Br-Ph | CH2Ph | 2.2 |
| 65e | H | 4-Et-Ph | Et | 6.6 |
 | ||
|---|---|---|
| Compound | R | MABA in M. tuberculosis IC50 (µM) |
| 66a |  | 2.7 |
| 66b |  | 0.11 |
| 66c |  | 0.62 |
| 66d |  | 0.32 |
| 66e |  | 0.32 |
| 66f |  | 0.083 |
| 66g |  | 0.15 |
| Bedaquiline | - | <0.078 |
| Thioridazine | - | 11.2 |
 | |||
|---|---|---|---|
| Compound | R1 | R2 | C. jejuni PglD IC50 (µM) |
| 67a | Ph | Me | 2.2 ± 0.4 |
| 67b | Ph | PhCH2CH2 | 1.4 |
| 67c | Ph | 4-MeO-PhCH2CH2 | 0.54 |
| 67d | Ph | 4-F-PhCH2CH2 | 0.42 |
| 67e | Ph | 4-Me-PhCH2CH2 | 0.72 |
| 67f | Ph | 3,5-(MeO)2-PhCH2CH2 | 0.37 |
| 67g | Ph |  | 0.42 |
| 67h | Ph |  | 0.59 |
| 67i |  | Me | 0.46 ± 0.05 |
| 67j | 4-AcNH-Ph |  | 0.28 |
| 67k | 4-AcNH-Ph |  | 0.27 ± 0.09 |
| 67l | Benzyl |  | 0.87 |
| 67m | Benzyl |  | 0.42 |
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | X | Inhibition Zone Diameter (mm) | Ref. | |||
| S. aureus | E. coli | B. cereus | P. aeruginosa | ||||||
| 68 | COPh | Me | - | - | 26 | 24 | 27 | NT a | [31] |
| 69a | COPh | Me | Ph | S | 24 | 23 | 26 | NT | [31] |
| 69b | COPh | Me | Ph | O | 22 | 21 | 23 | NT | [31] |
| 69c | COPh | Me | H | S | 21 | 20 | 22 | NT | [31] |
| 71a | COPh | Me | COMe | O | 9 | 10 | 12 | NT | [31] |
| 71b | COPh | Me | CN | O | 11 | 12 | 14 | NT | [31] |
| 70a | Me | Me | - | - | 6 | 6 | 6 | 6 | [11] |
| 70b | Me | NO2 | - | - | 12 | 15 | 16 | 10 | [11] |
| 69d | Me | Me | H | O | 8 | 16 | 8 | 6 | [11] |
| 69e | Me | Me | H | S | 12 | 6 | 12 | 8 | [11] |
| Levofloxacin | - | - | - | - | 26 | 30 | 28 | NT | [31] |
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cpd | X | Y | R1 | R2 | R3 | Antibacterial Activity MIC (µM) | |||
| S. agalactiae (1) a | S. agalactiae (2) a | E. faecalisa | S. epidermidisa | ||||||
| 72a | H | CO2Me | Bn | H | CH2OMe | 25 | 25 | 25 | >250 |
| 72b | H | CO2Me | iPr | H | CH2Cl | 25 | 25 | 25 | >250 |
| 72c | H | CO2Me | Et | Ph | H | 25 | 25 | 25 | >250 |
| 72d | H | CO2Me | Et | 4-Cl-Ph | H | 125 | 25 | 25 | 25 |
| 72e | H | COMe | Et | 4-Cl-Ph | H | >250 | 250 | 250 | 250 |
| 72f | CO2Me | CO2Me | Et | 4-Cl-Ph | H | >250 | 250 | NT b | >250 |
| 72g | H | CO2Me | Et | 4-NO2-Ph | H | 250 | 250 | NT | >250 |
| 72h | H | CO2Me | Et | 4-MeO-Ph | H | 25 | 25 | 25 | 250 |
| Ampicillin | - | - | - | - | - | 1.5 | 1.5 | 25 | 25 |
| Compound | Cancer Cell Lines a | |||
|---|---|---|---|---|
| GI50 b (µM) | ||||
| MDA-MB-231 | OV2008 | HepG2 | C6 | |
| 72b | 8 | 7 | 21 | 23 |
| 72d | 44 | 16 | 40 | 48 |
 | ||||
|---|---|---|---|---|
| Compound | R | MIC (µg/mL) | ||
| Gram Positive | Gram Negative | |||
| B. subtitlis | P. aeruginosa | P. vulgaris | ||
| 73a |  | 50 | 50 | 25 |
| 73b |  | 25 | 25 | 100 |
| 73c |  | 50 | 25 | 100 |
| 73d |  | 100 | 100 | 25 |
| 73e |  | 25 | 25 | 100 |
| 73f |  | 100 | 100 | 25 |
| 73g |  | 25 | 25 | 100 |
| 73h |  | 25 | 25 | 100 |
| 73i |  | 100 | 100 | 25 |
| Ampicillin | - | 12.5 | 125 | - |
| Levofloxacin | - | - | 12.5 | 12.5 |
 | ||||||
|---|---|---|---|---|---|---|
| Compound | X | R2 | Antibacterial Activity MIC (µmol·L−1) | |||
| Gram-Positive Bacteria | Gram-Negative Bacteria | |||||
| S. aureus | B. subtilis | E. coli | S. typhi | |||
| 74a | NHCO | 4-F-Ph | 9 | 9 | 9 | 11 |
| 74b | NHCO | 4-MeO-Ph | 10 | 9 | 10 | 12 |
| 74c | NHCO | 4-NO2-Ph | 6 | 5 | 7 | 6 |
| 74d | NHCO | 2,4-F2-Ph | 4 | 5 | 4 | 5 |
| 74e | NCH | 4-F-Ph | 10 | 9 | 9 | 8 |
| 74f | NCH | 4-MeO-Ph | 11 | 11 | 12 | 12 |
| 74g | NCH | 4-NO2-Ph | 4 | 5 | 7 | 7 |
| 74h | NCH | 2,4-F2-Ph | 5 | 4 | 5 | 6 |
| Ampicillin | - | - | 50 | 4 | 4 | 49 |
| Penicillin-G | - | - | 6 | 22 | 5 | 5 |
| Chloramphenicol | - | - | 4 | 6 | 5 | 6 |
 | ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | Antibacterial Activity MIC (µg·mL−1) | |||
| Gram Positive | Gram Negative | |||||
| B. cereus | S. aureus | P. aeruginosa | E. coli | |||
| 76a | H |  | 5.0 | 5.0 | 4.0 | 5.0 |
| 76b | H |  | 4.0 | 5.0 | 5.0 | 4.0 |
| 76c | COCH2Cl |  | 4.0 | 5.0 | 4.0 | 5.0 |
| 76d | COCH2NHPh |  | 5.0 | 4.0 | 5.0 | 8.0 |
| Reference drugs | - | - | 5.0 Ofloxacin | 4.0 Levofloxacin | 4.0 Clindamycin | 5.0 Nitrofurantoin |
 | |||||
|---|---|---|---|---|---|
| Compound | R | Zone of Bacterial Inhibition at 10 mg/mL (in mm) | |||
| Gram Positive | Gram Negative | ||||
| S. aureus | B. subtilis | Salmonella sp. | E. coli | ||
| 77a | CN | 0 | 14 | 14 | 0 |
| 77b | CONH2 | 0 | 16 | 0 | 0 |
| 77c | CO2Et | 0 | 0 | 14 | 0 |
| 77d | COPh | 0 | 15 | 0 | 0 |
| 77e | COMe | 0 | 15 | 14 | 0 |
| 77f | CONHPh | 0 | 15 | 0 | 0 |
| 77g | CONHPh-Cl-4 | 15 | 0 | 0 | 0 |
| 77h | CONHPh-OMe-4 | 0 | 15 | 0 | 0 |
| 77i | CONHPh-Br-4 | 0 | 0 | 0 | 20 |
| Ampicillin | - | 23 | 32 | - | - |
| Gentamycin | - | - | - | 17 | 19 |
 | ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | Inhibition Zone Diameter (mm/mg Sample) | |||
| Gram Positive | Gram Negative | |||||
| B. subtilis | S. aureus | E. coli | P. aeruginosa | |||
| 78a | NH2 | CO2Et | 17 | 19 | 14 | 10 |
| 78b | NH2 | CONHNH2 | 18 | 18 | 10 | 14 |
| 78c | NH2 | CONHPh | 14 | 18 | 16 | 10 |
| 78d |  | CO2Et | 15 | 13 | 16 | 15 |
| 78e |  | CO2Et | 11 | 15 | 10 | 17 |
| 78f |  | CO2Et | 13 | 17 | 19 | 19 |
| 78g |  | CO2Et | 20 | 16 | 22 | 21 |
| Ampicillin | - | - | 26 | 21 | 25 | 26 |
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cpd | R1 | R2 | Inhibition Zone (mm) | Ref. | ||||
| E. coli | B. sphaericus | B. subtilis | S. aureus | K. pneumonia | ||||
| 79a | Cl |  | 16 a | 8 a | - | - | - | [72] |
| 79b | Cl |  | 17 a | 10 a | - | - | - | [72] |
| 79c | Cl |  | 11 a | 0 a | - | - | - | [72] |
| 79d | 4-CF3-Ph |  | 15.5 b | - | 11.5 b | 12.5 b | 14.5 b | [27] |
| 79e |  |  | 16.5 b | - | 12.5 b | 14.5 b | 15 b | [27] |
| 79f |  |  | 17 b | - | 13 b | 15 b | 16.5 b | [27] |
| 79g |  |  | 13 b | - | 11 b | 11.5 b | 12.5 b | [27] |
| Amoxicillin | - | - | 19.6 b | - | 15.7 b | 17.4 b | 18 b | [27] |
 | |||||
|---|---|---|---|---|---|
| Compound | R | Antibacterial Activity MIC (µg/mL) | |||
| Gram-Negative Bacteria | Gram-Positive Bacteria | ||||
| E. coli | P. aeruginosa | S. aureus | S. pyogenes | ||
| 81 | - | 100 | 62.5 | 200 | 250 |
| 82a | Piperazinyl | 125 | 250 | 62.5 | 100 |
| 82b | Piperazinyl-carboxylate | 62.5 | 100 | 250 | 250 |
| 82c | 4-Cl-Ph | 125 | 62.5 | 200 | 100 |
| Ampicillin | - | 100 | NA a | 250 | 100 |
| Chloramphenicol | - | 50 | 50 | 50 | 50 |
| Ciprofloxacin | - | 25 | 25 | 50 | 50 |
 | |||||
|---|---|---|---|---|---|
| Compound | R | Zone of Inhibition of the Bacteria (mm, C = 30 µL) | |||
| E. coli | Pseudomonas sp. | S. aureus | Bacillus sp. | ||
| 83a |  | 13 | 17 | 25 | 17 |
| 83b |  | 14 | 5 | 14 | NA a |
| 83c |  | NA a | NA a | 17 | NA a |
| 83d |  | 15 | 14 | 16 | 15 |
| 83e |  | 15 | 14 | 19 | NA a |
| 83f |  | NA a | 14 | NA a | NA a |
| Streptomycin | - | 35 | 32 | 34 | 37 |
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cpd | X | R | MIC (µmol·L−1) | |||||
| Gram-Negative Bacteria | Gram-Positive Bacteria | |||||||
| E. coli | K. pneumoniae | P. aeruginosa | S. lactis | S. aureus | E. faecalis | |||
| 84a | O | - | 8 | 8 | 7 | 6 | 5 | 8 |
| 84b | N |  | 2 | 3 | 2 | 2 | 2 | 4 |
| 84c | N |  | 7 | 7 | 5 | 5 | 4 | 7 |
| 84d | N |  | 6 | 5 | 5 | 5 | 4 | 6 |
| 84e | N |  | 4 | 3 | 3 | 2 | 2 | 4 |
| 84f | N |  | 9 | 7 | 6 | 5 | 5 | 7 |
| 84g | N |  | 7 | 8 | 7 | 8 | 6 | 8 |
| 84h | N |  | 3 | 2 | 3 | 2 | 1 | 2 |
| 84i | N |  | 3 | 3 | 2 | 3 | 2 | 4 |
| 84j | N |  | 2 | 1 | 4 | 1 | 2 | 3 |
| Ciprofloxacin | - | - | 5 | 4 | 4 | 2 | 2 | 4 |
 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | C. difficile MIC (µg·mL−1) | |
| ATCC BAA 1870 | ATCC 43255 | ||||
| 85a | H | H |  | 4 | 2 |
| 85b | Me | H |  | 4 | 8 |
| 85c |  | H |  | 4 | 16 |
| 85d |  | H |  | 4 | 4 |
| 85e |  | H |  | 1 | 2 |
| 85f |  | H |  | 2 | 4 |
| 85g |  | H |  | 4 | 4 |
| 85h |  | H |  | 2 | 2 |
| 85i |  | H |  | 2 | 4 |
| 85j | Me | Benzyl |  | 2 | 16 |
| 85k | Me | 4-NO2-PhCH2 |  | 2 | 4 |
| 85l | Me | H | Cl | 4 | 8 |
| Vancomycin | - | - | - | 1 | 0.5 |
| Metronidazole | - | - | - | 0.125 | 0.25 |
| Fidaxomicin | - | - | - | 0.0625 | 0.0625 |
 | |||
|---|---|---|---|
| Compounds | R | Inhibition Zone Diameter (mm) | |
| P. aeruginosa | S. aureus | ||
| 52 | - | 13 | 12 |
| 53a | CN | 14 | 14 |
| 53b | CO2Et | 13 | 14 |
| Tetracyclin | - | 28 | 26 |
| Compound | Inhibition Zone Diameter (mm) a | |
|---|---|---|
| C. albicans | A. niger | |
| 68 | 26 | 30 |
| 69a | 24 | 28 |
| 69b | 22 | 26 |
| 69d | 23 | 25 |
| 71a | 11 | 15 |
| 71b | 13 | 17 |
| Nystatin | 27 | 26 |
| Compound | Inhibition Zone Diameter (mm/g of Samples) | |
|---|---|---|
| A. flavus | C. albicans | |
| 52 | 0 | 10 |
| 53a | 13 | 14 |
| 53b | 14 | 13 |
| Amphotericin B | 16 | 19 |
 | ||||
|---|---|---|---|---|
| Compound | R | MIC (µmol·mL−1) | ||
| C. albicans | A. flavus | G. lucidum | ||
| 84k |  | 4 | 5 | 4 |
| 84l |  | 2 | 2 | 1 |
| 84m |  | 3 | 2 | 2 |
| 84n |  | 7 | 4 | 6 |
| 84o |  | 5 | 4 | 4 |
| 84p |  | 4 | 3 | 3 |
| 84q |  | 4 | 4 | 5 |
| Ketoconazole | - | 3 | 2 | 3 |
 | ||||
|---|---|---|---|---|
| Compound | R | MIC (µg/mL) | ||
| C. albicans | A. fumigatus | R. oryzae | ||
| 73j |  | 100 | 50 | 50 |
| 73k |  | 25 | 100 | 50 |
| 73l |  | 25 | 50 | 100 |
| 73m |  | 100 | 50 | 50 |
| 73n |  | 25 | 50 | 50 |
| 73o | OEt | 50 | 50 | 50 |
| 73p | OH | 100 | 100 | 50 |
| Clotrimazole | - | 12.5 | 100 | 100 |
| Compound | Antifungal Activity MIC (µg·mL−1) | |||
|---|---|---|---|---|
| G. candidum | C. albicans | T. rubrum | A. flavus | |
| 76a | 4.0 | 5.0 | 4.0 | 5.0 |
| 76b | 4.0 | 5.0 | 4.0 | 5.0 |
| 76c | 4.0 | 5.0 | 4.0 | 5.0 |
| 76d | 4.0 | - | 4.0 | 5.0 |
| Clotrimazole | 4.0 | 5.0 | 4.0 | 5.0 |
| Compound | Inhibition Zone Diameter a (mm) | |||
|---|---|---|---|---|
| A. fumigatus | G. candidum | C. albicans | S. racemosum | |
| 80a | 12.5 ± 0.09 | 12.8 ± 0.1 | 11.0 ± 0.05 | 13.4 ± 0.08 |
| 80b | 15.1 ± 0.01 | 14.4 ± 0.1 | 13.4 ± 0.4 | 10.6 ± 0.2 |
| Itraconazole | 28 ± 0.05 | 27 ± 0.1 | 26 ± 0.02 | 22 ± 0.09 |
| Clotrimazole | 26 ± 0.1 | 23 ± 0.03 | 18 ± 0.1 | 20 ± 0.2 |
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Structure | Anti-Influenza A Activity in A549 Cells EC50 (nM ± SD) | Cytotoxicity CC50 (µM) | |||||
| X | A | PBM | CEM | Vero | Huh7 | A549 | ||
| 87a | C |  | 6.5 ± 1.1 | 9.3 | 4.2 | 65.9 | 24.0 | 14.5 |
| 87b | C |  | 17 ± 10 | 64.6 | 31.6 | 74.9 | 68.7 | 14.7 |
| 87c | C |  | 29 ± 6 | >100 | >100 | >100 | >100 | >100 |
| 87d | C |  | 27 ± 14 | 78.4 | 12.9 | 90.3 | 31.2 | 41.7 |
| 87e | N |  | 12 ± 1 | >100 | 13.7 | >100 | 62.7 | >100 |
| 87f | N |  | 42 ± 7 | >100 | 67.0 | 52.5 | 70.7 | 32.5 |
| Pimodivir | - | - | 4 ± 2 | >100 | 48.9 | >100 | 95.8 | 20.0 |
 | ||
|---|---|---|
| Compound | R | HBV DNA EC50 (µM) |
| 88 | 4-Me | 1.7 |
| 89 | 4-Me | 1.6 |
| 90a | 4-Me | 0.2 |
| 90b | H | 0.7 |
| 90c | 2-Me | 0.6 |
| 90d | 4-MeO | 0.6 |
| Lamivudine a | - | 0.1 |
 | ||||||
|---|---|---|---|---|---|---|
| Cpd | R1 | R2 | EC50 a (nM) | Ref. | ||
| IIIB | ROD | NL4-3 | ||||
| 91a | CN |  | 1.4 ± 0.4 | >227,890 | 1.16 ± 0.43 | [86,90] |
| 91b | Me |  | 3552 ± 848 | >16,460 | - | [86] |
| 91c |  |  | 1.22 ± 0.26 | >2.30 | - | [87] |
| 91d | CN |  | >1.17 × 103 | >1.17 | - | [87] |
| 91e | CN |  | 7.1 ± 0.5 | >9.287 | - | [88] |
| 91f | CN |  | 10 ± 8 | >3734 | - | [88] |
| 91g | CN |  | 58 ± 29 | >19,390 | - | [89] |
| 91h | CN |  | - | - | >217 | [90] |
| 91i | CN |  | - | - | 2.20 ± 0.67 | [90] |
| 91j | CN |  | - | - | 8.69 ± 2.74 | [90] |
| 91k | CN |  | - | - | 104 ± 36.8 | [90] |
| 91l | CN |  | - | - | 4.53 ± 1.30 | [90] |
| ETV | - | - | 5.8 ± 4.0 | - | - | [86] |
| Cpd | EC50 a (nM) | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|
| L100I | K103N | Y181C | Y188L | E138K | F227L + V106A | RES056 (K103N + Y181C) | ||
| 91a | 3.4 ± 0.6 | 2.9 | 3.2 ± 0.4 | 3.0 ± 0.1 | 2.9 | 4.2 ± 1.2 | 30.6 ± 12 | [86] |
| 91b | 4519 ± 158 | 4937 ± 407 | 4845 ± 118 | 8471 ± 197 | 5505 ± 315 | 4547 ± 13 | >16,462 | [86] |
| 91c | 1.34 ± 0.5 | 0.958 ± 0.07 | 5.00 ± 0.1 | 5.45 ± 0.2 | 4.74 ± 0.2 | 2.70 ± 1.74 | 5.50 ± 0.811 | [87] |
| 91d | >1.17 × 103 | >1.17 × 103 | >1.17 × 103 | >1.17 × 103 | >1.17 × 103 | >1.17 × 103 | >1.17 × 103 | [87] |
| 91e | 424 ± 361 | 70 ± 25 | 428 ± 294 | 675 ± 91 | 45 ± 1 | 3583 ± 241 | >9280 | [88] |
| 91f | 562 ± 487 | 32 ± 2 | 513 ± 415 | 903 ± 248 | 35 ± 1 | 1208 ± 333 | >3727 | [88] |
| 91g | 280 ± 61 | 14 ± 1 | 780 ± 9 | 790 ± 43 | 31 ± 4 | 770 ± 110 | >19,390 | [89] |
| ETV | 5.4 ± 2.1 | 2.4 ± 0.6 | 15.8 ± 2.1 | 20.5 ± 2.9 | 14.4 ± 2.2 | 29.4 ± 7.7 | 17 ± 1.8 | [86] |
 | ||||||
|---|---|---|---|---|---|---|
| Compound | R | R5 | R6 | EC50 a | Ref. | |
| HIV-1 (IIIB) (nM) | ROD (µM) | |||||
| 92a | 2′-NO2 | - | - | 7.8 ± 3.7 | >107.2 | [28] |
| 92b | 3′-NO2 | - | - | 87.7 ± 30.6 | >34.4 | [28] |
| 92c | 2′,3′-(Me)2 | - | - | 38 ± 12.7 | 1.4 ± 0.3 | [28] |
| 92d | 2′,3′-F2 | - | - | 13.5 ± 5.6 | >104.3 | [28] |
| 92e | 2′,6′-F2 | - | - | 12.3 ± 2.1 | >37.8 | [28] |
| 92f | 2′-Me-6′-Cl | - | - | 34.5 ± 16.2 | >70.3 | [28] |
| 92g | - | H | 2-F | 11 ± 8 | - | [92] |
| 92h | - | 3-Me | 2-F | 29 ± 5 | - | [92] |
| 92i | - | 3-Cl | 3-Cl | 21 ± 10 | - | [92] |
| 92j | - | 2-Me | 2-Me | 14 ± 5 | - | [92] |
| 92k | - | 2-Me | 3-Me | 17 ± 12 | - | [92] |
| NVP | - | - | - | 309.4 ± 57.7 | >4.0 | [28] |
| ETV | - | - | - | 5.5 ± 4.1 | >2.0 | [28] |
| Cpd | EC50 a (nM) | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|
| L100I | K103N | Y181C | Y188L | E138K | F227L + V106A | K103N + Y181C | ||
| 92a | 18.2 ± 3.9 | 5.5 ± 0.6 | 55.1 ± 0.6 | >15,140 | 6.5 ± 0.5 | ≥40,000 | >20,000 | [28] |
| 92c | 23.2 ± 4.2 | 33.8 ± 4.2 | 57.1 ± 10.6 | 50.7 ± 17.0 | 42.3 ± 4.2 | 253.6 ± 8.5 | 152.2 ± 42.3 | [28] |
| 92d | 52.0 ± 8.3 | 9.4 ± 2.1 | 58.2 ± 33.3 | 228 ± 41.6 | 17.0 ± 0.2 | 603 ± 21 | >2037.1 | [28] |
| 92e | 47.8 ± 8.3 | 8.7 ± 2.7 | 120.6 ± 18.7 | >20,308 | 16.6 ± 2.1 | ≥7233.70 | >78,552.4 | [28] |
| 92f | 28.4 ± 16.2 | 30.4 ± 6.1 | 48.7 ± 14.2 | 107.5 ± 42 | 52.7 ± 6.1 | 405.6 ± 6.1 | 148.0 ± 20.3 | [28] |
| 92g | 610 ± 270 | 90 ± 10 | 630 ± 490 | - | 130 ± 110 | - | - | [92] |
| 92h | 440 ± 450 | 210 ± 130 | 710 ± 10 | - | 400 ± 70 | - | - | [92] |
| 92i | 940 ± 50 | >20,000 | 660 ± 90 | - | 40 ± 30 | - | - | [92] |
| 92j | 130 ± 80 | 20 ± 10 | 30 ± 10 | - | 40 ± 10 | - | - | [92] |
| 92k | 1970 ± 660 | 130 ± 10 | >19000 | - | 160 ± 120 | - | - | [92] |
| NVP | 2102 ± 751 | >10,075.0 | >15,866.7 | >15,866.7 | 210 ± 26 | >15,866.7 | >15,866.7 | [28] |
| ETV | 7.1 ± 2.8 | 3.2 ± 0.5 | 12.0 ± 1.4 | 20.0 ± 7.6 | 6.5 ± 5.8 | 15.2 ± 16.1 | 55.3 ± 9.2 | [28] |
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compounds | R1 | R2 | R3 | RT IC50 a (μM) | INST IC50 a (μM) | MAGI Antiviral | ||
| RNase H | Pol | EC50 b (μM) | CC50 c (μM) | |||||
| 93a | Ph | - | - | 0.10 ± 0.06 | >10 | 4.5 ± 0.8 | 11 ± 2 | 28 ± 0.2 |
| 93b | 4-F-Ph | - | - | 0.20 ± 0.1 | >10 | 17 ± 4 | >20 | >100 |
| 93c | 4-Cl-Ph | - | - | 0.070 ± 0.05 | >10 | 23 ± 6 | >20 | >100 |
| 94a | - | Ph | H | 0.084 ± 0.006 | >10 | 2.2 ± 0.4 | >20 | >100 |
| 94b | - | Benzyl | H | 0.043 ± 0.008 | >10 | 5.0 ± 1 | >20 | >100 |
| 94c | - | H | Ph | 0.10 ± 0.03 | >10 | 1.3 ± 0.1 | 14 ± 1 | >100 |
| 94d | - | H | 4-Cl-Ph | 0.040 ± 0.02 | >10 | 2.1 ± 0.3 | 7.4 ± 0.3 | >100 |
| 94e | - | H | H | 0.20 ± 0.03 | >10 | 35 ± 6 | 18 ± 1 | 54 ± 6 |
| 94f | - | H | Me | 0.10 ± 0.02 | >10 | 12 ± 2 | >20 | 81 ± 6 |
| 94g | - | Me | Me | 0.10 ± 0.03 | >10 | 8.4 ± 1 | 8.9 ± 1 | 62 ± 1 |
| 94h | - | CO2H | Me | 0.10 ± 0.02 | >10 | 7.9 ± 1 | >20 | >100 |
| Raltegravir | - | - | - | >10 | ND d | 0.65 | 0.030 ± 0.005 | ND d |
 | ||||
|---|---|---|---|---|
| Compound | R1 | X | R2 | EC50 a (µM) |
| 95a |  | SO2 | H | 0.025 |
| 95b |  | SO2 | H | 3.2 |
| 95c |  | SO2 | H | 0.915 |
| 95d |  | SO2 | H | 0.485 |
| 95e |  | SO2 | H | 0.59 |
| 95f |  | SO2 | H | 0.140 |
| 95g |  | SO2 | H | 0.775 |
| 95h |  | SO2 | H | 0.103 |
| 95i |  | S | H | 0.014 |
| 95j |  | CH2 | H | 1.6 |
| 95k |  | CO | H | 0.20 |
| 95l |  | S | Cl | 0.728 |
| Nevirapine b | - | - | 0.150 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lagardère, P.; Fersing, C.; Masurier, N.; Lisowski, V. Thienopyrimidine: A Promising Scaffold to Access Anti-Infective Agents. Pharmaceuticals 2022, 15, 35. https://doi.org/10.3390/ph15010035
Lagardère P, Fersing C, Masurier N, Lisowski V. Thienopyrimidine: A Promising Scaffold to Access Anti-Infective Agents. Pharmaceuticals. 2022; 15(1):35. https://doi.org/10.3390/ph15010035
Chicago/Turabian StyleLagardère, Prisca, Cyril Fersing, Nicolas Masurier, and Vincent Lisowski. 2022. "Thienopyrimidine: A Promising Scaffold to Access Anti-Infective Agents" Pharmaceuticals 15, no. 1: 35. https://doi.org/10.3390/ph15010035
APA StyleLagardère, P., Fersing, C., Masurier, N., & Lisowski, V. (2022). Thienopyrimidine: A Promising Scaffold to Access Anti-Infective Agents. Pharmaceuticals, 15(1), 35. https://doi.org/10.3390/ph15010035