
Molecular Modeling Insights into Upadacitinib Selectivity upon Binding to JAK Protein Family


Abstract
:1. Introduction
2. Results
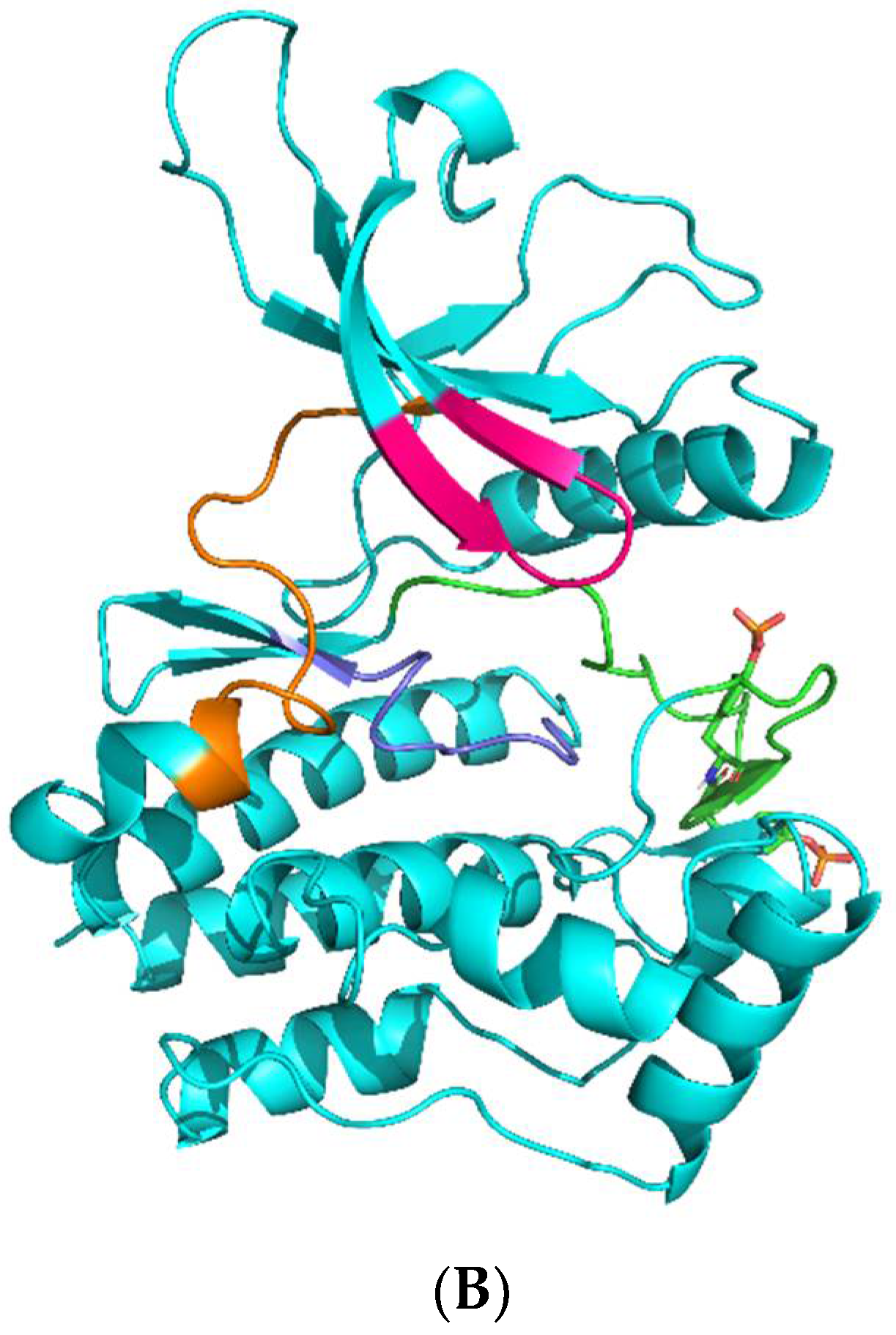
2.1. Molecular Docking
2.2. Molecular Dynamics
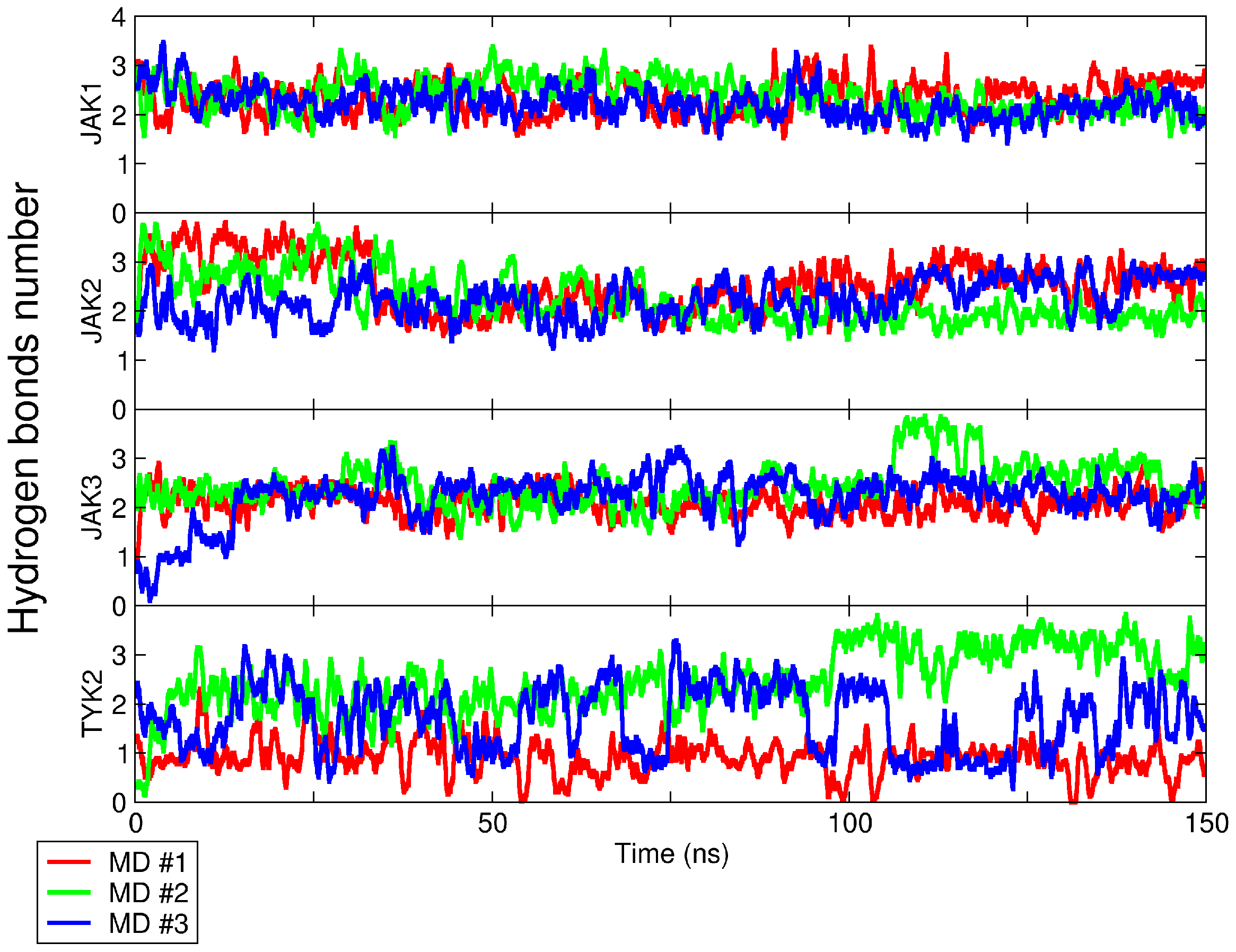
2.2.1. Number of Intermolecular Hydrogen Bonds
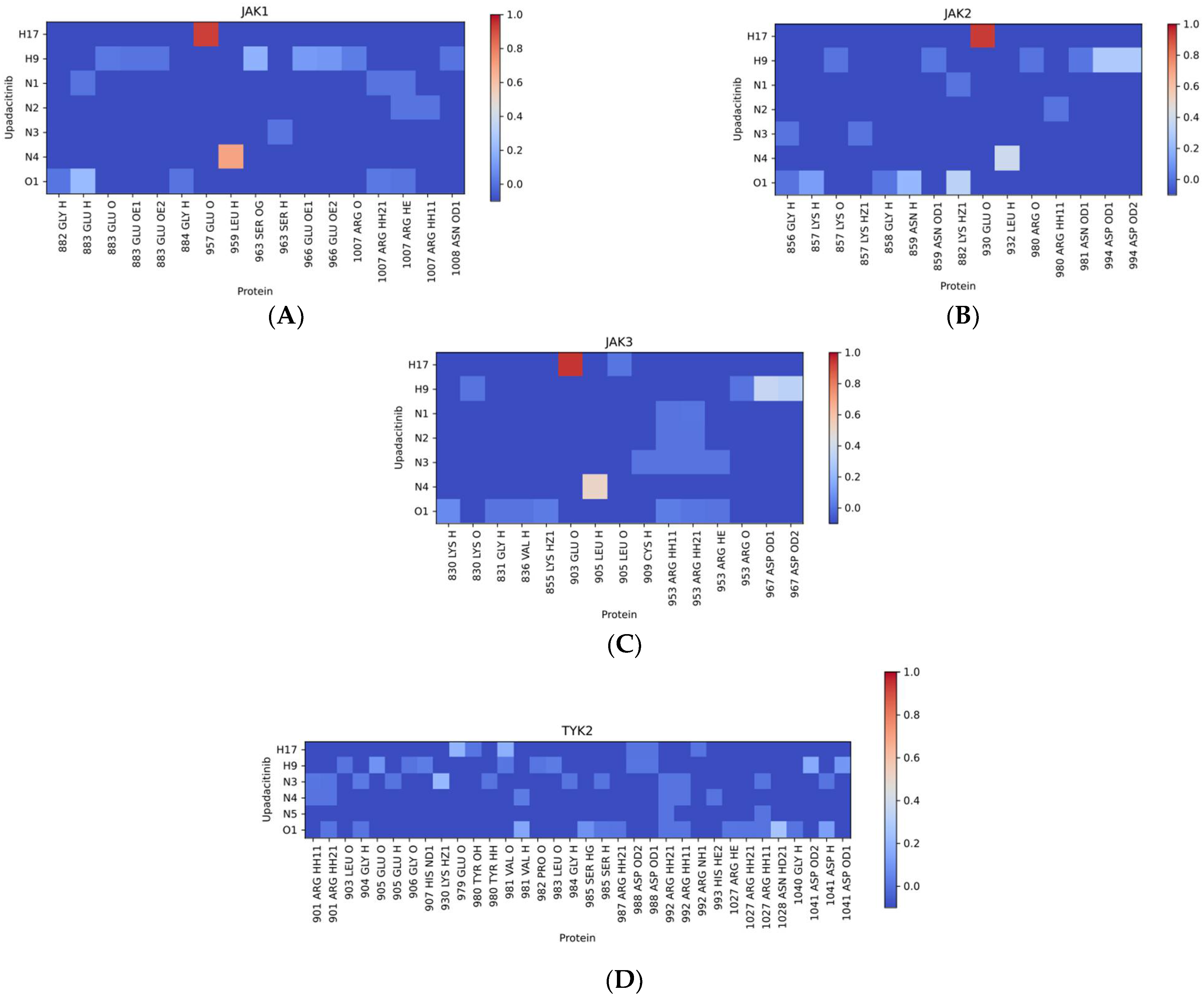
2.2.2. Distribution of Hydrogen Bonds
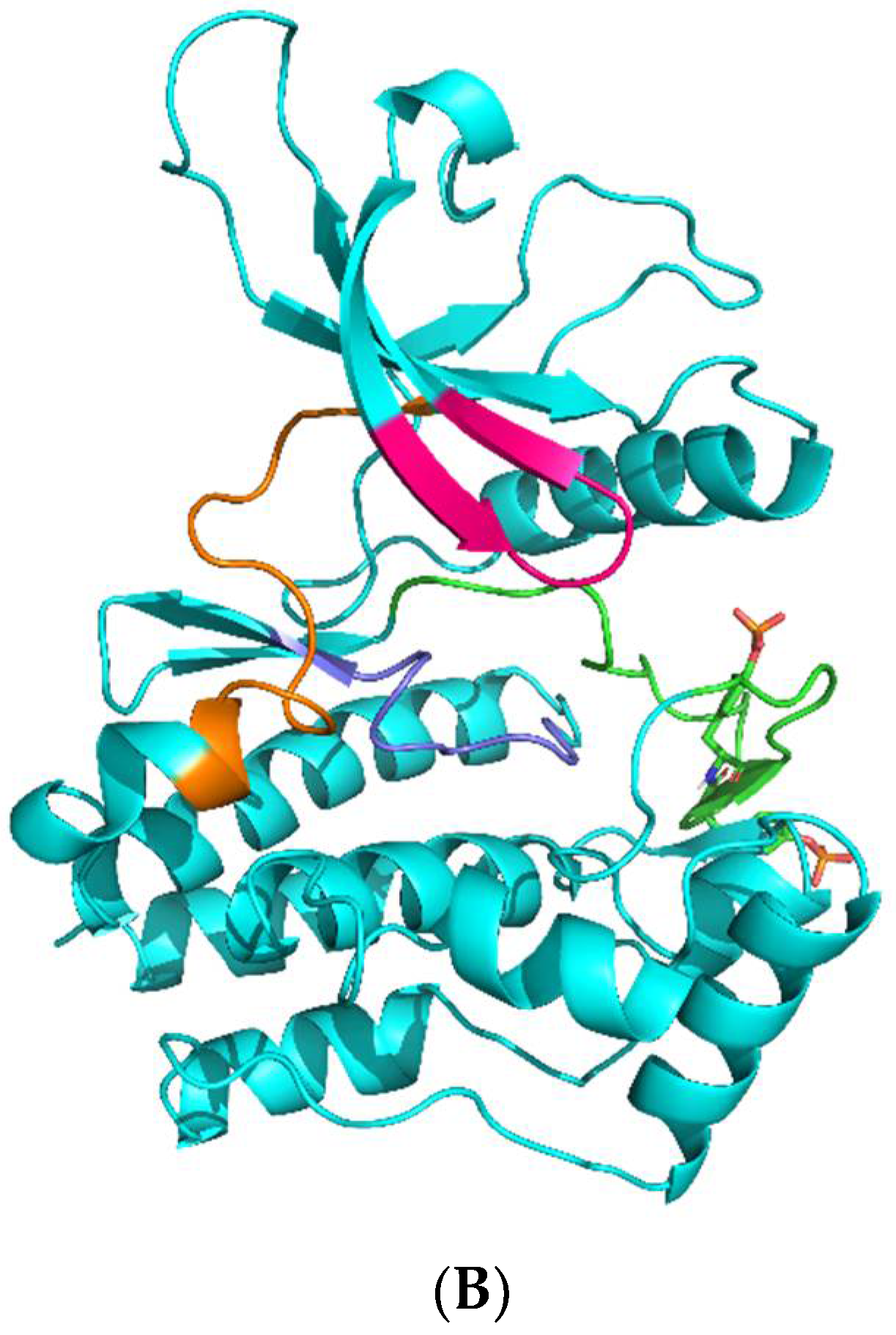
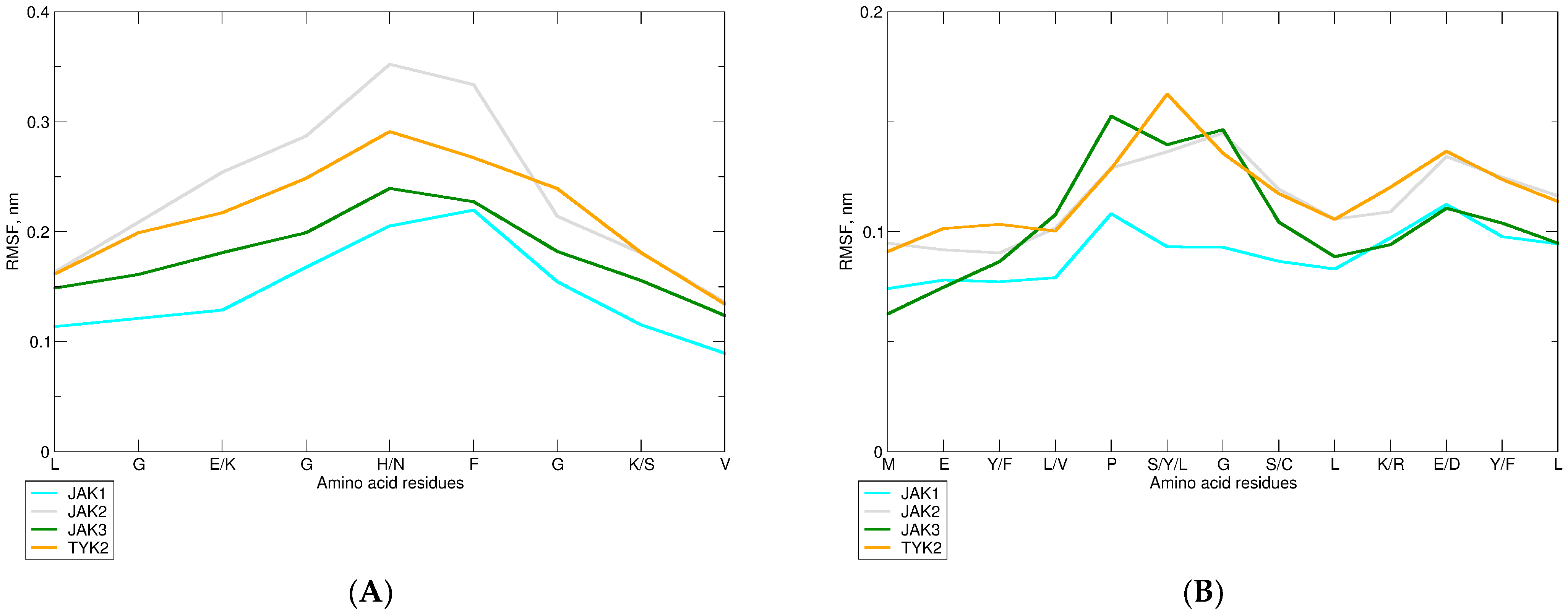
2.2.3. Changes in the JAK Structure upon Binding of Upadacitinib
2.2.4. Free Binding Energy Estimation via MM-PBSA Method
3. Discussion
4. Materials and Methods
4.1. Initial Molecular Structure Preparation
4.2. Molecular Docking
4.3. Molecular Dynamics
4.4. Molecular Dynamics Trajectories Analysis
4.5. Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) Free Binding Energy Estimation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parmentier, J.M.; Voss, J.; Graff, C.; Schwartz, A.; Argiriadi, M.; Friedman, M.; Camp, H.S.; Padley, R.J.; George, J.S.; Hyland, D.; et al. In Vitro and in Vivo Characterization of the JAK1 Selectivity of Upadacitinib (ABT-494). BMC Rheumatol. 2018, 2, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guschin, D.; Rogers, N.; Briscoe, J.; Witthuhn, B.; Watling, D.; Horn, F.; Pellegrini, S.; Yasukawa, K.; Heinrich, P.; Stark, G.R. A Major Role for the Protein Tyrosine Kinase JAK1 in the JAK/STAT Signal Transduction Pathway in Response to Interleukin-6. EMBO J. 1995, 14, 1421. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Nakajima, H.; Saito, Y.; Saito, T.; Leonard, W.J.; Iwamoto, I. Janus Kinase 3 (Jak3) Is Essential for Common Cytokine Receptor Gamma Chain (gamma(c))-Dependent Signaling: Comparative Analysis of Gamma(c), Jak3, and Gamma(c) and Jak3 Double-Deficient Mice. Int. Immunol. 2000, 12, 123–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morinobu, A. JAK Inhibitors for the Treatment of Rheumatoid Arthritis. Immunol. Med. 2020, 43, 148–155. [Google Scholar] [CrossRef]
- Ptacek, J.; Hawtin, R.E.; Sun, D.; Louie, B.; Evensen, E.; Mittleman, B.B.; Cesano, A.; Cavet, G.; Bingham, C.O., 3rd; Cofield, S.S.; et al. Diminished Cytokine-Induced Jak/STAT Signaling Is Associated with Rheumatoid Arthritis and Disease Activity. PLoS ONE 2021, 16, e0244187. [Google Scholar] [CrossRef] [PubMed]
- Firestein, G.S.; McInnes, I.B. Immunopathogenesis of Rheumatoid Arthritis. Immunity 2017, 46, 183–196. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.; Danila, M.I.; Cui, X.; Parks, L.; Baker, B.J.; Reynolds, R.J.; Raman, C.; Wanseck, K.C.; Redden, D.T.; Johnson, M.R.; et al. Expression of Interferon-γ Receptor Genes in Peripheral Blood Mononuclear Cells Is Associated With Rheumatoid Arthritis and Its Radiographic Severity in African Americans. Arthritis Rheumatol. 2015, 67, 1165–1170. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Dowty, M.E.; Lin, T.H.; Jesson, M.I.; Hegen, M.; Martin, D.A.; Katkade, V.; Menon, S.; Telliez, J.-B. Janus Kinase Inhibitors for the Treatment of Rheumatoid Arthritis Demonstrate Similar Profiles of in Vitro Cytokine Receptor Inhibition. Pharmacol. Res Perspect 2019, 7, e00537. [Google Scholar] [CrossRef] [Green Version]
- Friedman, M.; Frank, K.E.; Aguirre, A.; Argiriadi, M.A.; Davis, H.; Edmunds, J.J.; George, D.M.; George, J.S.; Goedken, E.; Fiamengo, B.; et al. Structure Activity Optimization of 6H-pyrrolo[2,3-e][1,2,4]triazolo[4,3-A]pyrazines as Jak1 Kinase Inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 4399–4404. [Google Scholar] [CrossRef]
- Chrencik, J.E.; Patny, A.; Leung, I.K.; Korniski, B.; Emmons, T.L.; Hall, T.; Weinberg, R.A.; Gormley, J.A.; Williams, J.M.; Day, J.E.; et al. Structural and Thermodynamic Characterization of the TYK2 and JAK3 Kinase Domains in Complex with CP-690550 and CMP-6. J. Mol. Biol. 2010, 400, 413–433. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xu, T.; Li, J.; Qiu, Y.; Rong, Y.; Gong, Z.; Cheng, X.; Dong, L.; Liu, W.; Li, J.; et al. A Novel Scalarized Scaffold Hopping Algorithm with Graph-Based Variational Autoencoder for Discovery of JAK1 Inhibitors. ACS Omega 2021, 6, 22945–22954. [Google Scholar] [CrossRef]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef]
- Stroet, M.; Caron, B.; Visscher, K.M.; Geerke, D.P.; Malde, A.K.; Mark, A.E. Automated Topology Builder Version 3.0: Prediction of Solvation Free Enthalpies in Water and Hexane. J. Chem. Theory Comput. 2018, 14, 5834–5845. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Zak, M.; Hanan, E.J.; Lupardus, P.; Brown, D.G.; Robinson, C.; Siu, M.; Lyssikatos, J.P.; Romero, F.A.; Zhao, G.; Kellar, T.; et al. Discovery of a Class of Highly Potent Janus Kinase 1/2 (JAK1/2) Inhibitors Demonstrating Effective Cell-Based Blockade of IL-13 Signaling. Bioorg. Med. Chem. Lett. 2019, 29, 1522–1531. [Google Scholar] [CrossRef] [PubMed]
- Farmer, L.J.; Ledeboer, M.W.; Hoock, T.; Arnost, M.J.; Bethiel, R.S.; Bennani, Y.L.; Black, J.J.; Brummel, C.L.; Chakilam, A.; Dorsch, W.A.; et al. Discovery of VX-509 (Decernotinib): A Potent and Selective Janus Kinase 3 Inhibitor for the Treatment of Autoimmune Diseases. J. Med. Chem. 2015, 58, 7195–7216. [Google Scholar] [CrossRef]
- Forster, M.; Chaikuad, A.; Bauer, S.M.; Holstein, J.; Robers, M.B.; Corona, C.R.; Gehringer, M.; Pfaffenrot, E.; Ghoreschi, K.; Knapp, S.; et al. Selective JAK3 Inhibitors with a Covalent Reversible Binding Mode Targeting a New Induced Fit Binding Pocket. Cell Chem. Biol. 2016, 23, 1335–1340. [Google Scholar] [CrossRef] [Green Version]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational Protein-Ligand Docking and Virtual Drug Screening with the AutoDock Suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; van Gunsteren, W.F. Definition and Testing of the GROMOS Force-Field Versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef]
- Petrov, D.; Margreitter, C.; Grandits, M.; Oostenbrink, C.; Zagrovic, B. A Systematic Framework for Molecular Dynamics Simulations of Protein Post-Translational Modifications. PLoS Comput. Biol. 2013, 9, e1003154. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A.; Open Source Drug Discovery Consortium. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Modeling 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinase | Co-Crystallized Native Ligand Vina Score, kcal/mol 1 | RMSD between Re-Docked and Co-Crystallized Ligand, nm | IC50 of Upadacitinib, nM 2 | Upadacitinib Vina Score, kcal/mol 1 |
|---|---|---|---|---|
| JAK1 | −9.4 | 0.007 | 43 | −8.5 |
| JAK2 | −9.1 | 0.006 | 120 | −8.1 |
| JAK3 | −9.6 | 0.009 | 2300 | −7.9 |
| TYK2 | −10.9 | 0.000 | 4700 | −6.0 |
| Kinase | van der Waals Energy 1, kcal/mol | Electrostatic Energy 1, kcal/mol | Polar Solvation Energy 1, kcal/mol | SASA Energy 1,2, kcal/mol | Binding Energy 1, kcal/mol |
|---|---|---|---|---|---|
| JAK1 | −45.0 ± 3.5 | −12.6 ± 3.5 | 38.8 ± 6.7 | −4.4 ± 0.2 | −23.2 ± 4.2 |
| JAK2 | −39.3 ± 0.3 | −21.0 ± 0.4 | 48.6 ± 0.8 | −4.3 ± 0.0 | −16.0 ± 0.4 |
| JAK3 | −42.4 ± 0.3 | −15.7 ± 0.2 | 41.5 ± 2.8 | −4.4 ± 0.0 | −20.9 ± 0.3 |
| TYK2 | −35.8 ± 0.3 | −10.9 ± 0.3 | 38.2 ± 0.6 | −4.1 ± 0.0 | −12.6 ± 0.3 |
| Protein Molecule | UniProt ID | PDB ID | Native Ligand | Amino Acids | PTM (Phosphotyrosine) | Reference |
|---|---|---|---|---|---|---|
| JAK1 | P23458 | 6N7A | N-[3-(5-chloro-2-methoxyphenyl)-1-methyl-1H-pyrazol-4-yl]-2-methyl-2H-pyrazolo [4,3-c]pyridine-7-carboxamide | 865–1154 | 1034, 1035 | [16] |
| JAK2 | O60674 | 4YTH | N~2~-[2-(5-chloro-1H-pyrrolo[2,3-b]pyridin-3-yl)-5-fluoropyrimidin-4-yl]-2-methyl-N-(2,2,2-trifluoroethyl)-D-alaninamide | 842–1130 | 1007, 1008 | [17] |
| JAK3 | P52333 | 5LWN | (2~{S})-2-cyano-~{N},~{N}-dimethyl-3-[5-[3-[(1~{S},2~{R})-2-methylcyclohexyl]-3,5,8,10-tetrazatricyclo[7.3.0.0^{2,6}]dodeca-1,4,6,8,11-pentaen-4-yl]furan-2-yl]propanamide | 814–1103 | - | [18] |
| TYK2 | P29597 | 3LXP | 4-tert-butyl-15-fluoro-3,5,10-triazatetracyclo[11.4.0.02,6.07,12]heptadeca-1(13),2(6),4,7(12),8,14,16-heptaen-11-one | 888–1178 | 1054 | [11] |
| Simulated System Composition | MD Duration, ns | Clustering Cut-Off Values, nm |
|---|---|---|
| JAK1 | ||
| Protein(1)/Upadacitinib(1)/Water(11173)/Na+(4) | 3 × 150 | 0.10 |
| JAK2 | ||
| Protein(1)/Upadacitinib(1)/Water(10721)/Na+(4) | 3 × 150 | 0.12 |
| JAK3 | ||
| Protein(1)/Upadacitinib(1)/Water(10919) | 3 × 150 | 0.12 |
| TYK2 | ||
| Protein(1)/Upadacitinib(1)/Water(11456)/Na+(7) | 3 × 150 | 0.10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taldaev, A.; Rudnev, V.R.; Nikolsky, K.S.; Kulikova, L.I.; Kaysheva, A.L. Molecular Modeling Insights into Upadacitinib Selectivity upon Binding to JAK Protein Family. Pharmaceuticals 2022, 15, 30. https://doi.org/10.3390/ph15010030
Taldaev A, Rudnev VR, Nikolsky KS, Kulikova LI, Kaysheva AL. Molecular Modeling Insights into Upadacitinib Selectivity upon Binding to JAK Protein Family. Pharmaceuticals. 2022; 15(1):30. https://doi.org/10.3390/ph15010030
Chicago/Turabian StyleTaldaev, Amir, Vladimir R. Rudnev, Kirill S. Nikolsky, Liudmila I. Kulikova, and Anna L. Kaysheva. 2022. "Molecular Modeling Insights into Upadacitinib Selectivity upon Binding to JAK Protein Family" Pharmaceuticals 15, no. 1: 30. https://doi.org/10.3390/ph15010030
APA StyleTaldaev, A., Rudnev, V. R., Nikolsky, K. S., Kulikova, L. I., & Kaysheva, A. L. (2022). Molecular Modeling Insights into Upadacitinib Selectivity upon Binding to JAK Protein Family. Pharmaceuticals, 15(1), 30. https://doi.org/10.3390/ph15010030