
Jakinibs of All Trades: Inhibiting Cytokine Signaling in Immune-Mediated Pathologies


Abstract
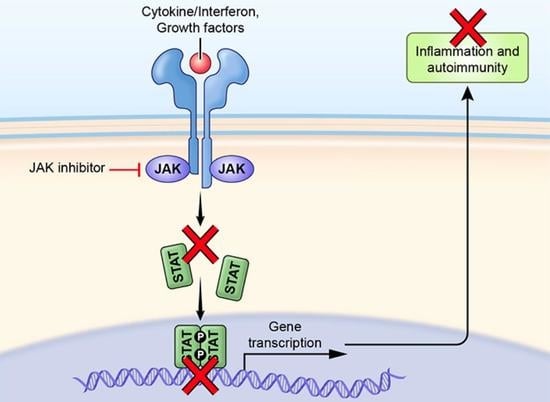
1. Introduction
Establishment of JAK Criticality
2. Jakinib Development and Approvals
3. Safety of Jakinibs
3.1. Infections
3.2. Hematologic Effects
3.3. Thromboembolic Risk
3.4. Impact on Lipids
3.5. Cardiovascular and Malignancy Risk
4. Selectivity and Pharmacokinetics of Jakinibs
5. Jakinib Discontinuation
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gadina, M.; Chisolm, D.A.; Philips, R.L.; McInness, I.B.; Changelian, P.S.; O’Shea, J.J. Translating JAKs to Jakinibs. J. Immunol. 2020, 204, 2011–2020. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, A.T.; Haikarainen, T.; Raivola, J.; Silvennoinen, O. Selective JAKinibs: Prospects in Inflammatory and Autoimmune Diseases. BioDrugs 2019, 33, 15–32. [Google Scholar] [CrossRef]
- Spagnuolo, R.; Dastoli, S.; Silvestri, M.; Cosco, C.; Garieri, P.; Bennardo, L.; Nistico, S.P. Anti-interleukin 12/23 in the treatment of erythema nodosum and Crohn disease: A case report. Dermatol. Ther. 2019, 32, e12811. [Google Scholar] [CrossRef]
- Burmester, G.R.; Bijlsma, J.W.J.; Cutolo, M.; McInnes, I.B. Managing rheumatic and musculoskeletal diseases—Past, present and future. Nat. Rev. Rheumatol. 2017, 13, 443–448. [Google Scholar] [CrossRef]
- Monaco, C.; Nanchahal, J.; Taylor, P.; Feldmann, M. Anti-TNF therapy: Past, present and future. Int. Immunol. 2015, 27, 55–62. [Google Scholar] [CrossRef]
- Gadina, M.; Le, M.T.; Schwartz, D.M.; Silvennoinen, O.; Nakayamada, S.; Yamaoka, K.; O’Shea, J.J. Janus kinases to jakinibs: From basic insights to clinical practice. Rheumatology 2019, 58, i4–i16. [Google Scholar] [CrossRef] [PubMed]
- Villarino, A.V.; Gadina, M.; O’Shea, J.J.; Kanno, Y. SnapShot: Jak-STAT Signaling II. Cell 2020, 181, 1696. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef]
- Russell, S.M.; Tayebi, N.; Nakajima, H.; Riedy, M.C.; Roberts, J.L.; Aman, M.J.; Migone, T.S.; Noguchi, M.; Markert, M.L.; Buckley, R.H.; et al. Mutation of Jak3 in a patient with SCID: Essential role of Jak3 in lymphoid development. Science 1995, 270, 797–800. [Google Scholar] [CrossRef]
- Eletto, D.; Burns, S.O.; Angulo, I.; Plagnol, V.; Gilmour, K.C.; Henriquez, F.; Curtis, J.; Gaspar, M.; Nowak, K.; Daza-Cajigal, V.; et al. Biallelic JAK1 mutations in immunodeficient patient with mycobacterial infection. Nat. Commun. 2016, 7, 13992. [Google Scholar] [CrossRef]
- Gruber, C.N.; Calis, J.J.A.; Buta, S.; Evrony, G.; Martin, J.C.; Uhl, S.A.; Caron, R.; Jarchin, L.; Dunkin, D.; Phelps, R.; et al. Complex Autoinflammatory Syndrome Unveils Fundamental Principles of JAK1 Kinase Transcriptional and Biochemical Function. Immunity 2020, 53, 672–684. [Google Scholar] [CrossRef]
- McIntosh, L.A.; Marion, M.C.; Sudman, M.; Comeau, M.E.; Becker, M.L.; Bohnsack, J.F.; Fingerlin, T.E.; Griffin, T.A.; Haas, J.P.; Lovell, D.J.; et al. Genome-Wide Association Meta-Analysis Reveals Novel Juvenile Idiopathic Arthritis Susceptibility Loci. Arthritis Rheumatol. 2017, 69, 2222–2232. [Google Scholar] [CrossRef]
- Witalisz-Siepracka, A.; Klein, K.; Prinz, D.; Leidenfrost, N.; Schabbauer, G.; Dohnal, A.; Sexl, V. Loss of JAK1 Drives Innate Immune Deficiency. Front. Immunol. 2018, 9, 3108. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef]
- Kreins, A.Y.; Ciancanelli, M.J.; Okada, S.; Kong, X.F.; Ramirez-Alejo, N.; Kilic, S.S.; El Baghdadi, J.; Nonoyama, S.; Mahdaviani, S.A.; Ailal, F.; et al. Human TYK2 deficiency: Mycobacterial and viral infections without hyper-IgE syndrome. J. Exp. Med. 2015, 212, 1641–1662. [Google Scholar] [CrossRef]
- Tao, J.H.; Zou, Y.F.; Feng, X.L.; Li, J.; Wang, F.; Pan, F.M.; Ye, D.Q. Meta-analysis of TYK2 gene polymorphisms association with susceptibility to autoimmune and inflammatory diseases. Mol. Biol. Rep. 2011, 38, 4663–4672. [Google Scholar] [CrossRef]
- Kleppe, M.; Spitzer, M.H.; Li, S.; Hill, C.E.; Dong, L.; Papalexi, E.; De Groote, S.; Bowman, R.L.; Keller, M.; Koppikar, P.; et al. Jak1 Integrates Cytokine Sensing to Regulate Hematopoietic Stem Cell Function and Stress Hematopoiesis. Cell Stem Cell 2017, 21, 489–501. [Google Scholar] [CrossRef]
- Hainzl, E.; Stockinger, S.; Rauch, I.; Heider, S.; Berry, D.; Lassnig, C.; Schwab, C.; Rosebrock, F.; Milinovich, G.; Schlederer, M.; et al. Intestinal Epithelial Cell Tyrosine Kinase 2 Transduces IL-22 Signals To Protect from Acute Colitis. J. Immunol. 2015, 195, 5011–5024. [Google Scholar] [CrossRef]
- Strobl, B.; Stoiber, D.; Sexl, V.; Mueller, M. Tyrosine kinase 2 (TYK2) in cytokine signalling and host immunity. Front. Biosci. (Landmark Ed.) 2011, 16, 3214–3232. [Google Scholar] [CrossRef]
- Ishizaki, M.; Muromoto, R.; Akimoto, T.; Ohshiro, Y.; Takahashi, M.; Sekine, Y.; Maeda, H.; Shimoda, K.; Oritani, K.; Matsuda, T. Tyk2 deficiency protects joints against destruction in anti-type II collagen antibody-induced arthritis in mice. Int. Immunol. 2011, 23, 575–582. [Google Scholar] [CrossRef][Green Version]
- Oyamada, A.; Ikebe, H.; Itsumi, M.; Saiwai, H.; Okada, S.; Shimoda, K.; Iwakura, Y.; Nakayama, K.I.; Iwamoto, Y.; Yoshikai, Y.; et al. Tyrosine kinase 2 plays critical roles in the pathogenic CD4 T cell responses for the development of experimental autoimmune encephalomyelitis. J. Immunol. 2009, 183, 7539–7546. [Google Scholar] [CrossRef]
- Changelian, P.S.; Flanagan, M.E.; Ball, D.J.; Kent, C.R.; Magnuson, K.S.; Martin, W.H.; Rizzuti, B.J.; Sawyer, P.S.; Perry, B.D.; Brissette, W.H.; et al. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science 2003, 302, 875–878. [Google Scholar] [CrossRef]
- Xeljanz® (Tofacitinib Citrate) Receives Marketing Authorization in the European Union for the Treatment of Active Polyarticular Juvenile Idiopathic Arthritis and Juvenile Psoriatic Arthritis. Available online: https://investors.pfizer.com/investor-news/press-release-details/2021/XELJANZ-tofacitinib-citrate-Receives-Marketing-Authorization-in-the-European-Union-for-the-Treatment-of-Active-Polyarticular-Juvenile-Idiopathic-Arthritis-and-Juvenile-Psoriatic-Arthritis/default.aspx (accessed on 30 August 2021).
- Zeiser, R.; von Bubnoff, N.; Butler, J.; Mohty, M.; Niederwieser, D.; Or, R.; Szer, J.; Wagner, E.M.; Zuckerman, T.; Mahuzier, B.; et al. Ruxolitinib for Glucocorticoid-Refractory Acute Graft-versus-Host Disease. N. Engl. J. Med. 2020, 382, 1800–1810. [Google Scholar] [CrossRef]
- Taylor, P.C.; Keystone, E.C.; van der Heijde, D.; Weinblatt, M.E.; Del Carmen Morales, L.; Reyes Gonzaga, J.; Yakushin, S.; Ishii, T.; Emoto, K.; Beattie, S.; et al. Baricitinib versus Placebo or Adalimumab in Rheumatoid Arthritis. N. Engl. J. Med. 2017, 376, 652–662. [Google Scholar] [CrossRef]
- Baricitinib EUA Letter of Authorization. Available online: https://www.fda.gov/media/143822/download (accessed on 27 April 2021).
- Fleischmann, R.; Pangan, A.L.; Song, I.H.; Mysler, E.; Bessette, L.; Peterfy, C.; Durez, P.; Ostor, A.J.; Li, Y.; Zhou, Y.; et al. Upadacitinib Versus Placebo or Adalimumab in Patients With Rheumatoid Arthritis and an Inadequate Response to Methotrexate: Results of a Phase III, Double-Blind, Randomized Controlled Trial. Arthritis Rheumatol. 2019, 71, 1788–1800. [Google Scholar] [CrossRef]
- European Commission Approves RINVOQ (upadacitinib) as First JAK Inhibitor in the European Union for the Treatment of Both Adults and Adolescents with Moderate to Severe Atopic Dermatitis. Available online: https://news.abbvie.com/news/press-releases/european-commission-approves-rinvoq-upadacitinib-as-first-jak-inhibitor-in-european-union-for-treatment-both-adults-and-adolescents-with-moderate-to-severe-atopic-dermatitis.htm (accessed on 27 August 2021).
- Jyseleca: Filgotinib. 2020. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/jyseleca (accessed on 9 December 2021).
- UK’s MHRA Grants Marketing Authorisation for Pfizer’s Cibinqo® (abrocitinib) for Adults and Adolescents with Moderate to Severe Atopic Dermatitis. 2021. Available online: https://www.pfizer.com/news/press-release/press-release-detail/uks-mhra-grants-marketing-authorisation-pfizers-cibinqor (accessed on 9 December 2021).
- FDA Approves Fedratinib For Myelofibrosis. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-fedratinib-myelofibrosis (accessed on 9 December 2021).
- Takeuchi, T.; Tanaka, Y.; Iwasaki, M.; Ishikura, H.; Saeki, S.; Kaneko, Y. Efficacy and safety of the oral Janus kinase inhibitor peficitinib (ASP015K) monotherapy in patients with moderate to severe rheumatoid arthritis in Japan: A 12-week, randomised, double-blind, placebo-controlled phase IIb study. Ann. Rheum. Dis. 2016, 75, 1057–1064. [Google Scholar] [CrossRef]
- Dhillon, S. Delgocitinib: First Approval. Drugs 2020, 80, 609–615. [Google Scholar] [CrossRef]
- Rynhoud, H.; Gibson, J.S.; Meler, E.; Soares Magalhaes, R.J. The Association between the Use of Oclacitinib and Antibacterial Therapy in Dogs With Allergic Dermatitis: A Retrospective Case-Control Study. Front. Vet. Sci. 2021, 8, 631443. [Google Scholar] [CrossRef] [PubMed]
- Zeiser, R.; Polverelli, N.; Ram, R.; Hashmi, S.K.; Chakraverty, R.; Middeke, J.M.; Musso, M.; Giebel, S.; Uzay, A.; Langmuir, P.; et al. Ruxolitinib for Glucocorticoid-Refractory Chronic Graft-versus-Host Disease. N. Engl. J. Med. 2021, 385, 228–238. [Google Scholar] [CrossRef]
- Zeiser, R.; Socie, G. The development of ruxolitinib for glucocorticoid-refractory acute graft-versus-host disease. Blood Adv. 2020, 4, 3789–3794. [Google Scholar] [CrossRef]
- Marconi, V.C.; Moser, C.; Gavegnano, C.; Deeks, S.G.; Lederman, M.M.; Overton, E.T.; Tsibris, A.; Hunt, P.W.; Kantor, A.; Sekaly, R.P.; et al. Randomized Trial of Ruxolitinib in Antiretroviral-Treated Adults with HIV. Clin. Infect. Dis. 2021. [Google Scholar] [CrossRef]
- Concert Pharmaceuticals Initiates THRIVE-AA2 Phase 3 Clinical Trial Evaluating CTP-543 for Alopecia Areata. Available online: https://ir.concertpharma.com/news-releases/news-release-details/concert-pharmaceuticals-initiates-thrive-aa2-phase-3-clinical (accessed on 15 November 2021).
- Webber, A.B.; Vincenti, F. An Update on Calcineurin Inhibitor-Free Regimens: The Need Persists, but the Landscape has Changed. Transplantation 2016, 100, 836–843. [Google Scholar] [CrossRef]
- Yan, Q.; Chen, W.; Song, H.; Long, X.; Zhang, Z.; Tang, X.; Chen, H.; Lin, H.; Sun, L. Tofacitinib Ameliorates Lupus through Suppression of T Cell Activation Mediated by TGF-Beta Type I Receptor. Front. Immunol. 2021, 12, 675542. [Google Scholar] [CrossRef]
- Hasni, S.A.; Gupta, S.; Davis, M.; Poncio, E.; Temesgen-Oyelakin, Y.; Carlucci, P.M.; Wang, X.; Naqi, M.; Playford, M.P.; Goel, R.R.; et al. Phase 1 double-blind randomized safety trial of the Janus kinase inhibitor tofacitinib in systemic lupus erythematosus. Nat. Commun. 2021, 12, 3391. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, P.O.; Quirk, D.; Furtado, R.H.; Maia, L.N.; Saraiva, J.F.; Antunes, M.O.; Kalil Filho, R.; Junior, V.M.; Soeiro, A.M.; Tognon, A.P.; et al. Tofacitinib in Patients Hospitalized with COVID-19 Pneumonia. N. Engl. J. Med. 2021. [Google Scholar] [CrossRef]
- Jenkins, M.K. The ups and downs of T cell costimulation. Immunity 1994, 1, 443–446. [Google Scholar] [CrossRef]
- Ford, M.L.; Adams, A.B.; Pearson, T.C. Targeting co-stimulatory pathways: Transplantation and autoimmunity. Nat. Rev. Nephrol 2014, 10, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Chong, A.S.; Alegre, M.L. The impact of infection and tissue damage in solid-organ transplantation. Nat. Rev. Immunol. 2012, 12, 459–471. [Google Scholar] [CrossRef]
- Braza, F.; Brouard, S.; Chadban, S.; Goldstein, D.R. Role of TLRs and DAMPs in allograft inflammation and transplant outcomes. Nat. Rev. Nephrol. 2016, 12, 281–290. [Google Scholar] [CrossRef]
- Todd, J.L.; Palmer, S.M. Danger signals in regulating the immune response to solid organ transplantation. J. Clin. Investig. 2017, 127, 2464–2472. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, M.; Khalifian, S.; Oh, B.C.; Zhang, Y.; Miller, D.; Beck, S.; Brandacher, G.; Raimondi, G. A short course of tofacitinib sustains the immunoregulatory effect of CTLA4-Ig in the presence of inflammatory cytokines and promotes long-term survival of murine cardiac allografts. Am. J. Transplant. 2020, 21, 2675–2687. [Google Scholar] [CrossRef]
- Majumder, P.; Zhang, Y.; Iglesias, M.; Fan, L.; Kelley, J.A.; Andrews, C.; Patel, N.; Stagno, J.R.; Oh, B.C.; Furtmuller, G.J.; et al. Multiphase Assembly of Small Molecule Microcrystalline Peptide Hydrogel Allows Immunomodulatory Combination Therapy for Long-Term Heart Transplant Survival. Small 2020, 16, e2002791. [Google Scholar] [CrossRef]
- Rovira, J.; Ramirez-Bajo, M.J.; Banon-Maneus, E.; Lazo-Rodriguez, M.; Moya-Rull, D.; Hierro-Garcia, N.; Tubita, V.; Pineiro, G.J.; Revuelta, I.; Ventura-Aguiar, P.; et al. Tofacitinib Halts Progression of Graft Dysfunction in a Rat Model of Mixed Cellular and Humoral Rejection. Transplantation 2018, 102, 1075–1084. [Google Scholar] [CrossRef]
- Assadiasl, S.; Fatahi, Y.; Mosharmovahed, B.; Mohebbi, B.; Nicknam, M.H. Baricitinib: From Rheumatoid Arthritis to COVID-19. J. Clin. Pharmacol. 2021, 61, 1274–1285. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.J.; Furie, R.A.; Tanaka, Y.; Kalunian, K.C.; Mosca, M.; Petri, M.A.; Dorner, T.; Cardiel, M.H.; Bruce, I.N.; Gomez, E.; et al. Baricitinib for systemic lupus erythematosus: A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2018, 392, 222–231. [Google Scholar] [CrossRef]
- Cantini, F.; Niccoli, L.; Nannini, C.; Matarrese, D.; Natale, M.E.D.; Lotti, P.; Aquilini, D.; Landini, G.; Cimolato, B.; Pietro, M.A.D.; et al. Beneficial impact of Baricitinib in COVID-19 moderate pneumonia; multicentre study. J. Infect. 2020, 81, 647–679. [Google Scholar] [CrossRef]
- Kalil, A.C.; Patterson, T.F.; Mehta, A.K.; Tomashek, K.M.; Wolfe, C.R.; Ghazaryan, V.; Marconi, V.C.; Ruiz-Palacios, G.M.; Hsieh, L.; Kline, S.; et al. Baricitinib plus Remdesivir for Hospitalized Adults with COVID-19. N. Engl. J. Med. 2021, 384, 795–807. [Google Scholar] [CrossRef]
- FDA News Release: Coronavirus (COVID-19) Update: FDA Authorizes Drug Combination for Treatment of COVID-19. 2020. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-drug-combination-treatment-covid-19 (accessed on 21 April 2021).
- NIH Closes Enrollment in Trial Comparing COVID-19 Treatment Regimens. Available online: https://www.nih.gov/news-events/news-releases/nih-closes-enrollment-trial-comparing-covid-19-treatment-regimens (accessed on 27 April 2021).
- Pérez-Alba, E.; Nuzzolo-Shihadeh, L.; Aguirre-Garcia, G.M.; Espinosa-Mora, J.; Lecona-Garcia, J.D.; Flores-Pérez, R.O.; Mendoza-Garza, M.; Camacho-Ortiz, A. Baricitinib plus dexamethasone compared to dexamethasone for the treatment of severe COVID-19 pneumonia: A retrospective analysis. J. Microbiol. Immunol. Infect. 2021, 54, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Lilly and Incyte Announce Results from the Phase 3 COV-BARRIER Study of Baricitinib in Hospitalized COVID-19 Patients. Available online: https://investor.lilly.com/news-releases/news-release-details/lilly-and-incyte-announce-results-phase-3-cov-barrier-study (accessed on 27 April 2021).
- Genovese, M.C.; Greenwald, M.; Codding, C.; Zubrzycka-Sienkiewicz, A.; Kivitz, A.J.; Wang, A.; Shay, K.; Wang, X.; Garg, J.P.; Cardiel, M.H. Peficitinib, a JAK Inhibitor, in Combination With Limited Conventional Synthetic Disease-Modifying Antirheumatic Drugs in the Treatment of Moderate-to-Severe Rheumatoid Arthritis. Arthritis Rheumatol. 2017, 69, 932–942. [Google Scholar] [CrossRef]
- Genovese, M.C.; Greenwald, M.W.; Gutierrez-Urena, S.R.; Cardiel, M.H.; Poiley, J.E.; Zubrzycka-Sienkiewicz, A.; Codding, C.E.; Wang, A.; He, W.; Amos, R.; et al. Two-Year Safety and Effectiveness of Peficitinib in Moderate-To-Severe Rheumatoid Arthritis: A Phase IIb, Open-Label Extension Study. Rheumatol. Ther. 2019, 6, 503–520. [Google Scholar] [CrossRef]
- Tanaka, Y.; Takeuchi, T.; Izutsu, H.; Kaneko, Y.; Kato, D.; Fukuda, M.; Rokuda, M.; Schultz, N.M. Patient- and physician-reported outcomes from two phase 3 randomized studies (RAJ3 and RAJ4) of peficitinib (ASP015K) in Asian patients with rheumatoid arthritis. Arthritis Res. Ther. 2021, 23, 221. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol. Res. 2016, 111, 784–803. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Iizuka, H.; Nemoto-Hasebe, I.; Nemoto, O.; Toyama, H.; Ohashi-Doi, K.; Kabashima, K. Clinical effect of delgocitinib 0.5% ointment on atopic dermatitis eczema intensity and skin barrier function. J. Cutan. Immunol. Allergy 2021, 1–9. [Google Scholar] [CrossRef]
- Nakagawa, H.; Nemoto, O.; Igarashi, A.; Saeki, H.; Kabashima, K.; Oda, M.; Nagata, T. Delgocitinib ointment in pediatric patients with atopic dermatitis: A phase 3, randomized, double-blind, vehicle-controlled study and a subsequent open-label, long-term study. J. Am. Acad. Dermatol. 2021, 85, 854–862. [Google Scholar] [CrossRef]
- Worm, M.; Bauer, A.; Elsner, P.; Mahler, V.; Molin, S.; Nielsen, T.S.S. Efficacy and safety of topical delgocitinib in patients with chronic hand eczema: Data from a randomized, double-blind, vehicle-controlled phase IIa study. Br. J. Dermatol. 2020, 182, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Ortalda, C.; Noli, C.; Colombo, S.; Borio, S. Oclacitinib in feline nonflea-, nonfood-induced hypersensitivity dermatitis: Results of a small prospective pilot study of client-owned cats. Vet. Dermatol. 2015, 26, 235-e52. [Google Scholar] [CrossRef] [PubMed]
- Drake, G.J.; Nuttall, T.; Lopez, J.; Magnone, W.; Leclerc, A.; Potier, R.; Lecu, A.; Guezenec, M.; Kolter, L.; Nicolau, A.; et al. Treatment Success in Three Andean Bears (Tremarctos Ornatus) with Alopecia Syndrome Using Oclacitinib Maleate (Apoquel(R)). J. Zoo Wildl. Med. 2017, 48, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Parmentier, J.M.; Voss, J.; Graff, C.; Schwartz, A.; Argiriadi, M.; Friedman, M.; Camp, H.S.; Padley, R.J.; George, J.S.; Hyland, D.; et al. In vitro and in vivo characterization of the JAK1 selectivity of upadacitinib (ABT-494). BMC Rheumatol. 2018, 2, 23. [Google Scholar] [CrossRef]
- Cai, F.; Sornasse, T.; Hong, F.; Camp, H.; Kato, K.; McInnes, I. Biomarker driven dissection of inflammation modulatory effect of upadacitinib versus abatacept in patients with active rheumatoid arthritis refractory to biologic DMARDs. In Proceedings of the ACR Convergence, Virtual, 5–9 November 2021. Abstract 1240. [Google Scholar]
- Bergman, M.; Buch, M.H.; Tanaka, Y.; Ditera, G.; Bahlas, S.; Wong, E.; Song, Y.; Tundia, N.; Suboticki, J.; Strand, V. Routine assessment of patient index data 3 (RAPID3) in patients with rheumatoid arthritis treated with long-term upadacitinib therapy. In Proceedings of the ACR Convergence, Virtual, 5–9 November 2021; Abstract POS0670. p. 579. [Google Scholar]
- Fleischmann, R.; Mysler, E.; Bessette, L.; Peterfy, C.; Durez, P.; Tanaka, Y.; Swierkot, J.; Khan, N.; Bu, X.; Li, Y.; et al. Long-term safety and efficacy of upadacitinib or adalimumab in patients with rheumatoid arthritis: Results at 3 years from the SELECT-COMPARE study. In Proceedings of the ACR Convergence, Virtual, 5–9 November 2021. Abstract 0828. [Google Scholar]
- RINVOQ® (upadacitinib) Receives U.S. FDA Approval for Active Psoriatic Arthritis. Available online: https://news.abbvie.com/news/press-releases/rinvoq-upadacitinib-receives-us-fda-approval-for-active-psoriatic-arthritis.htm (accessed on 15 December 2021).
- Nash, P.; Richette, P.; Gossec, L.; Marchesoni, A.; Kato, K.; Blondell, E.; Lesser, E.; McCaskill, R.M.; Feng, D.; Anderson, J.K.; et al. Upadacitinib as monotherapy and in combination with non-biologic DMARDs for the treatment of psoriatic arthritis: Subgroup analysis from two phase 3 trials. In Proceedings of the ACR Convergence, Virtual, 5–9 November 2020. Abstract 1345. [Google Scholar]
- McInnes, I.B.; Anderson, J.K.; Magrey, M.; Merola, J.F.; Liu, Y.; Kishimoto, M.; Jeka, S.; Pacheco-Tena, C.; Wang, X.; Chen, L.; et al. Trial of Upadacitinib and Adalimumab for Psoriatic Arthritis. N. Engl. J. Med. 2021, 384, 1227–1239. [Google Scholar] [CrossRef]
- Mease, P.J.; Lertratanakul, A.; Anderson, J.K.; Papp, K.; Van den Bosch, F.; Tsuji, S.; Dokoupilova, E.; Keiserman, M.; Wang, X.; Zhong, S.; et al. Upadacitinib for psoriatic arthritis refractory to biologics: SELECT-PsA 2. Ann. Rheum. Dis. 2020, 80, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J.; Lertratanakul, A.; Strober, B.; Tsuji, S.; Richetee, P.; Lovan, C.; Feng, D.; Anderson, J.; Van den Bosch, F. Efficacy of upadacitinib in patients with psoriatic arthritis stratified by number of prior biologic disease-modifying anti-rheumatic drugs. In Proceedings of the ACR Convergence, Virtual, 5–9 November 2021. Abstract 1356. [Google Scholar]
- Merola, J.F.; Richette, P.; Lubrano, E.; Drescher, E.; Soto, L.; Lovan, C.; Kato, K.; Lippe, R.; Lane., M.; Kishimoto, M. Efficacy of upadacitinib in patients with psoriatic arthritis stratified by baseline skin severity: A subgroup analysis of two phase III trials. In Proceedings of the 2021 EULAR, Virtual, 2–5 June 2021; Abstract POS1030. p. 786. [Google Scholar]
- Deodhar, A.; Baraliakos, X.; Mcinnes, I.; De Vlam, K.; Bessette, L.; Maniccia, A.; Lippe, R.; Saffore, C.; Gao, T.; Song, I.H.; et al. Effect of upadacitinib on reducing pain in patients with active ankylosing spondylitis and inadequate response to nonsteroidal anti-inflammatory drugs. In Proceedings of the 2021 the EULAR, Virtual, 2–5 June 2021. Abstract POS0907. [Google Scholar]
- Reich, K.; Teixeira, H.D.; de Bruin-Weller, M.; Bieber, T.; Soong, W.; Kabashima, K.; Werfel, T.; Zeng, J.; Huang, X.; Hu, X.; et al. Safety and efficacy of upadacitinib in combination with topical corticosteroids in adolescents and adults with moderate-to-severe atopic dermatitis (AD Up): Results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2021, 397, 2169–2181. [Google Scholar] [CrossRef]
- Orbai, A.M.; Ogdie, A.; Gossec, L.; Tillett, W.; Leung, Y.Y.; Gao, J.; Trivedi, M.; Tasset, C.; Meuleners, L.; Besuyen, R.; et al. Effect of filgotinib on health-related quality of life in active psoriatic arthritis: A randomized phase 2 trial (EQUATOR). Rheumatology 2020, 59, 1495–1504. [Google Scholar] [CrossRef]
- McInnes, I.B.; Szekanecz, Z.; McGonagle, D.; Maksymowych, W.P.; Pfeil, A.; Lippe, R.; Song, I.H.; Lertratanakul, A.; Sornasse, T.; Biljan, A.; et al. A review of JAK-STAT signalling in the pathogenesis of spondyloarthritis and the role of JAK inhibition. Rheumatology 2021, keab740. [Google Scholar] [CrossRef]
- Van der Heijde, D.; Baraliakos, X.; Gensler, L.S.; Maksymowych, W.P.; Tseluyko, V.; Nadashkevich, O.; Abi-Saab, W.; Tasset, C.; Meuleners, L.; Besuyen, R.; et al. Efficacy and safety of filgotinib, a selective Janus kinase 1 inhibitor, in patients with active ankylosing spondylitis (TORTUGA): Results from a randomised, placebo-controlled, phase 2 trial. Lancet 2018, 392, 2378–2387. [Google Scholar] [CrossRef]
- Bieber, T.; Simpson, E.L.; Silverberg, J.I.; Thaci, D.; Paul, C.; Pink, A.E.; Kataoka, Y.; Chu, C.Y.; DiBonaventura, M.; Rojo, R.; et al. Abrocitinib versus Placebo or Dupilumab for Atopic Dermatitis. N. Engl. J. Med. 2021, 384, 1101–1112. [Google Scholar] [CrossRef]
- Sun, X.; He, Q.; Yang, J.; Wang, A.; Zhang, F.; Qiu, H.; Zhou, K.; Wang, P.; Ding, X.; Yuan, X.; et al. Preventive and Therapeutic Effects of a Novel JAK Inhibitor SHR0302 in Acute Graft-Versus-Host Disease. Cell Transplant. 2021, 30, 9636897211033778. [Google Scholar] [CrossRef] [PubMed]
- Reistone Announces Positive Topline Phase 2 Results for SHR0302, a Selective JAK1 Inhibitor, for Treatment of Patients with Alopecia Areata. Available online: http://www.reistonebio.com/view/id/124.html (accessed on 30 August 2021).
- Reistone Announces Positive Results from a Phase II Study Evaluating SHR0302 Ointment for Patients with Mild-to-Moderate Atopic Dermatitis. Available online: http://www.reistonebio.com/view/id/126.html (accessed on 30 August 2021).
- Reistone Announces First Patient Dosed in Phase III Global Study in Atopic Dermatitis. Available online: http://www.reistonebio.com/view/id/116.html (accessed on 30 August 2021).
- Krueger, J.G.; McInnes, I.B.; Blauvelt, A. Tyrosine kinase 2 and Janus kinase signal transducer and activator of transcription signaling and inhibition in plaque psoriasis. J. Am. Acad. Dermatol. 2021, 86, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Bristol Myers Squibb Announces Deucravacitinib (BMS-986165) Demonstrated Superiority to Placebo and Otezla (Apremilast) in Pivotal Phase 3 Psoriasis Study. Available online: https://news.bms.com/news/corporate-financial/2020/Bristol-Myers-Squibb-Announces-Deucravacitinib-BMS-986165-Demonstrated-Superiority-to-Placebo-and-Otezla-apremilast-in-Pivotal-Phase-3-Psoriasis-Study/default.aspx (accessed on 4 May 2021).
- Bristol Myers Squibb Announces Positive Topline Results from Second Pivotal Phase 3 Psoriasis Study Showing Superiority of Deucravacitinib Compared to Placebo and Otezla (Apremilast). Available online: https://news.bms.com/news/details/2021/Bristol-Myers-Squibb-Announces-Positive-Topline-Results-from-Second-Pivotal-Phase-3-Psoriasis-Study-Showing-Superiority-of-Deucravacitinib-Compared-to-Placebo-and-Otezla-apremilast/default.aspx (accessed on 4 May 2021).
- Mease, P.J.; Deodhar, A.; Van den Heijde, D.; Behrens, F.; Kivitz, A.; Lehman, T.; Wei, L.; Nys, M.; Banerjee, S.; Nowark, M. Efficacy of Deucravacitinib, An Oral, Selective Tyrosine Kinase 2 Inhibitor, In Musculoskeletal Manifestations Of Active Psoriatic Arthritis In A Phase 2, Randomized, Double-Blind, Placebo-Controlled Trial. In Proceedings of the 2021 EULAR, Virtual, 2–5 June 2021. Abstract OP0227. [Google Scholar]
- Bristol Myers Squibb Presents Late-Breaking Phase 2 Data Demonstrating the Safety and Efficacy of Deucravacitinib (BMS-986165) in Patients with Psoriatic Arthritis. Available online: https://news.bms.com/news/details/2020/Bristol-Myers-Squibb-Presents-Late-Breaking-Phase-2-Data-Demonstrating-the-Safety-and-Efficacy-of-Deucravacitinib-BMS-986165-in-Patients-with-Psoriatic-Arthritis/default.aspx (accessed on 30 August 2021).
- King, B.; Guttman-Yassky, E.; Peeva, E.; Banerjee, A.; Sinclair, R.; Pavel, A.B.; Zhu, L.; Cox, L.A.; Craiglow, B.; Chen, L.; et al. A phase 2a randomized, placebo-controlled study to evaluate the efficacy and safety of the oral Janus kinase inhibitors ritlecitinib and brepocitinib in alopecia areata: 24-week results. J. Am. Acad. Dermatol. 2021, 85, 379–387. [Google Scholar] [CrossRef]
- Coffey, G.; Betz, A.; DeGuzman, F.; Pak, Y.; Inagaki, M.; Baker, D.C.; Hollenbach, S.J.; Pandey, A.; Sinha, U. The novel kinase inhibitor PRT062070 (Cerdulatinib) demonstrates efficacy in models of autoimmunity and B-cell cancer. J. Pharmacol. Exp Ther. 2014, 351, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.D.; Munoz, J.; Stevens, D.; Smith, S.M.; Feldman, T.A.; Ye, J.C.; de Vos, S.; Hess, B.T.; Miller, C.B.; Khatcheressian, J.L.; et al. Rapid and Durable Responses with the SYK/JAK Inhibitor Cerdulatinib in a Phase 2 Study in Relapsed/Refractory Follicular Lymphoma-Alone or in Combination with Rituximab. Blood 2019, 134, 3981. [Google Scholar] [CrossRef]
- Horwitz, S.M.; Feldman, T.A.; Brian, T.H.; Khodadoust, M.S.; Kim, Y.H.; Munoz, J.; Patel, M.R.; Phillips, T.J.; Smith, S.D.; Smith, S.M.; et al. A Phase 2 Study of the Dual SYK/JAK Inhibitor Cedulatinib Demonstrates Good Tolerability and Clinical Response in Relapsed/Refractory Peripheral T-Cell Lymphoma and Cutaneous T-Cell Lymphoma. Blood 2019, 134, 466. [Google Scholar] [CrossRef]
- Pardanani, A.; Lasho, T.; Smith, G.; Burns, C.J.; Fantino, E.; Tefferi, A. CYT387, a selective JAK1/JAK2 inhibitor: In vitro assessment of kinase selectivity and preclinical studies using cell lines and primary cells from polycythemia vera patients. Leukemia 2009, 23, 1441–1445. [Google Scholar] [CrossRef]
- Duenas-Perez, A.B.; Mead, A.J. Clinical potential of pacritinib in the treatment of myelofibrosis. Ther. Adv. Hematol. 2015, 6, 186–201. [Google Scholar] [CrossRef]
- Hart, S.; Goh, K.C.; Novotny-Diermayr, V.; Hu, C.Y.; Hentze, H.; Tan, Y.C.; Madan, B.; Amalini, C.; Loh, Y.K.; Ong, L.C.; et al. SB1518, a novel macrocyclic pyrimidine-based JAK2 inhibitor for the treatment of myeloid and lymphoid malignancies. Leukemia 2011, 25, 1751–1759. [Google Scholar] [CrossRef]
- Mesa, R.A.; Vannucchi, A.M.; Mead, A.; Egyed, M.; Szoke, A.; Suvorov, A.; Jakucs, J.; Perkins, A.; Prasad, R.; Mayer, J.; et al. Pacritinib versus best available therapy for the treatment of myelofibrosis irrespective of baseline cytopenias (PERSIST-1): An international, randomised, phase 3 trial. Lancet Haematol. 2017, 4, e225–e236. [Google Scholar] [CrossRef]
- Pfizer Announces Positive Top-Line Results from Phase 2b/3 Trial of Ritlecitinib in Alopecia Areata. 2021. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-announces-positive-top-line-results-phase-2b3-trial (accessed on 30 August 2021).
- Sandborn, W.J.; Nguyen, D.D.; Beattie, D.T.; Brassil, P.; Krey, W.; Woo, J.; Situ, E.; Sana, R.; Sandvik, E.; Pulido-Rios, M.T.; et al. Development of Gut-Selective Pan-Janus Kinase Inhibitor TD-1473 for Ulcerative Colitis: A Translational Medicine Programme. J. Crohns Colitis 2020, 14, 1202–1213. [Google Scholar] [CrossRef] [PubMed]
- Theravance Biopharma, Inc. Reports Third Quarter 2020 Financial Results and Provides Business Update. 2020. Available online: https://investor.theravance.com/news-releases/news-release-details/theravance-biopharma-inc-reports-third-quarter-2020-financial (accessed on 15 July 2021).
- Singh, D.; Bogus, M.; Moskalenko, V.; Lord, R.; Moran, E.J.; Crater, G.D.; Bourdet, D.L.; Pfeifer, N.D.; Woo, J.; Kaufman, E.; et al. A phase 2 multiple ascending dose study of the inhaled pan-JAK inhibitor nezulcitinib (TD-0903) in severe COVID-19. Eur. Respir J. 2021, 58, 2100673. [Google Scholar] [CrossRef]
- Bechman, K.; Subesinghe, S.; Norton, S.; Atzeni, F.; Galli, M.; Cope, A.P.; Winthrop, K.L.; Galloway, J.B. A systematic review and meta-analysis of infection risk with small molecule JAK inhibitors in rheumatoid arthritis. Rheumatology 2019, 58, 1755–1766. [Google Scholar] [CrossRef]
- Harigai, M. Growing evidence of the safety of JAK inhibitors in patients with rheumatoid arthritis. Rheumatology 2019, 58, i34–i42. [Google Scholar] [CrossRef]
- van Gurp, E.; Weimar, W.; Gaston, R.; Brennan, D.; Mendez, R.; Pirsch, J.; Swan, S.; Pescovitz, M.D.; Ni, G.; Wang, C.; et al. Phase 1 dose-escalation study of CP-690 550 in stable renal allograft recipients: Preliminary findings of safety, tolerability, effects on lymphocyte subsets and pharmacokinetics. Am. J. Transplant. 2008, 8, 1711–1718. [Google Scholar] [CrossRef]
- Busque, S.; Vincenti, F.G.; Tedesco Silva, H.; O’Connell, P.J.; Yoshida, A.; Friedewald, J.J.; Steinberg, S.M.; Budde, K.; Broeders, E.N.; Kim, Y.S.; et al. Efficacy and Safety of a Tofacitinib-based Immunosuppressive Regimen After Kidney Transplantation: Results From a Long-term Extension Trial. Transplant. Direct 2018, 4, e380. [Google Scholar] [CrossRef]
- Galloway, J.; Buch, M.H.; Yamaoka, K.; Leatherwood, C.; Pechonkina, A.; Tiamiyu, I.; Jiang, D.; Ye, L.; Besuyen, R.; Aletaha, D. Infections and serious infections in the filgotinib rheumatoid arthritis program. Ann. Rheum. Dis. 2021, 80, 70–71. [Google Scholar] [CrossRef]
- Cohen, S.B.; van Vollenhoven, R.F.; Winthrop, K.L.; Zerbini, C.A.F.; Tanaka, Y.; Bessette, L.; Zhang, Y.; Khan, N.; Hendrickson, B.; Enejosa, J.V.; et al. Safety profile of upadacitinib in rheumatoid arthritis: Integrated analysis from the SELECT phase III clinical programme. Ann. Rheum. Dis. 2020, 21, 9004. [Google Scholar] [CrossRef]
- Blauvelt, A.; Teixeira, H.D.; Simpson, E.L.; Costanzo, A.; De Bruin-Weller, M.; Barbarot, S.; Prajapati, V.H.; Lio, P.; Hu, X.; Wu, T.; et al. Efficacy and Safety of Upadacitinib vs Dupilumab in Adults With Moderate-to-Severe Atopic Dermatitis: A Randomized Clinical Trial. JAMA Dermatol. 2021, 157, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.; Schwartz, D.M. JAK-STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs 2017, 77, 521–546. [Google Scholar] [CrossRef]
- Lee, Y.H.; Song, G.G. Comparative efficacy and safety of tofacitinib, baricitinib, upadacitinib, and filgotinib in active rheumatoid arthritis refractory to biologic disease-modifying antirheumatic drugs. Z. Rheumatol. 2021, 80, 379–392. [Google Scholar] [CrossRef]
- Papp, K.; Gordon, K.; Thaci, D.; Morita, A.; Gooderham, M.; Foley, P.; Girgis, I.G.; Kundu, S.; Banerjee, S. Phase 2 Trial of Selective Tyrosine Kinase 2 Inhibition in Psoriasis. N. Engl. J. Med. 2018, 379, 1313–1321. [Google Scholar] [CrossRef]
- Dowty, M.E.; Jesson, M.I.; Ghosh, S.; Lee, J.; Meyer, D.M.; Krishnaswami, S.; Kishore, N. Preclinical to clinical translation of tofacitinib, a Janus kinase inhibitor, in rheumatoid arthritis. J. Pharmacol. Exp. Ther. 2014, 348, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Kremer, J.M.; Bloom, B.J.; Breedveld, F.C.; Coombs, J.H.; Fletcher, M.P.; Gruben, D.; Krishnaswami, S.; Burgos-Vargas, R.; Wilkinson, B.; Zerbini, C.A.; et al. The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: Results of a double-blind, placebo-controlled phase IIa trial of three dosage levels of CP-690,550 versus placebo. Arthritis Rheum. 2009, 60, 1895–1905. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.; Cutolo, M.; Genovese, M.C.; Lee, E.B.; Kanik, K.S.; Sadis, S.; Connell, C.A.; Gruben, D.; Krishnaswami, S.; Wallenstein, G.; et al. Phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) or adalimumab monotherapy versus placebo in patients with active rheumatoid arthritis with an inadequate response to disease-modifying antirheumatic drugs. Arthritis Rheum. 2012, 64, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Koops, H.; Strand, V.; Nduaka, C.; DeMasi, R.; Wallenstein, G.; Kwok, K.; Wang, L. Analysis of haematological changes in tofacitinib-treated patients with rheumatoid arthritis across phase 3 and long-term extension studies. Rheumatology 2017, 56, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Genovese, M.C.; Takeuchi, T.; Hyslop, D.L.; Macias, W.L.; Rooney, T.; Chen, L.; Dickson, C.L.; Riddle Camp, J.; Cardillo, T.E.; et al. Safety Profile of Baricitinib in Patients with Active Rheumatoid Arthritis with over 2 Years Median Time in Treatment. J. Rheumatol. 2019, 46, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Kay, J.; Harigai, M.; Rancourt, J.; Dickson, C.; Melby, T.; Issa, M.; de la Torre, I.; Isaka, Y.; Cardoso, A.; Saifan, C.; et al. Changes in selected haematological parameters associated with JAK1/JAK2 inhibition observed in patients with rheumatoid arthritis treated with baricitinib. RMD Open 2020, 6, e001370. [Google Scholar] [CrossRef] [PubMed]
- Kameda, H.; Takeuchi, T.; Yamaoka, K.; Oribe, M.; Kawano, M.; Yokoyama, M.; Pangan, A.L.; Konishi, Y.; Meerwein, S.; Tanaka, Y. Efficacy and safety of upadacitinib over 84 weeks in Japanese patients with rheumatoid arthritis (SELECT-SUNRISE). Arthritis Res. Ther. 2021, 23, 9. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Ghosh, S.; Panes, J.; Schreiber, S.; D’Haens, G.; Tanida, S.; Siffledeen, J.; Enejosa, J.; Zhou, W.; Othman, A.A.; et al. Efficacy of Upadacitinib in a Randomized Trial of Patients With Active Ulcerative Colitis. Gastroenterology 2020, 158, 2139–2149.e2114. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.D.; Zuckerman, A.; Krishnaswami, S.; Nduaka, C.; Lan, S.; Hutmacher, M.M.; Boy, M.G.; Kowalski, K.; Menon, S.; Riese, R. Changes in serum creatinine in patients with active rheumatoid arthritis treated with tofacitinib: Results from clinical trials. Arthritis Res. Ther. 2014, 16, R158. [Google Scholar] [CrossRef]
- Strober, B.; Buonanno, M.; Clark, J.D.; Kawabata, T.; Tan, H.; Wolk, R.; Valdez, H.; Langley, R.G.; Harness, J.; Menter, A.; et al. Effect of tofacitinib, a Janus kinase inhibitor, on haematological parameters during 12 weeks of psoriasis treatment. Br. J. Dermatol. 2013, 169, 992–999. [Google Scholar] [CrossRef]
- Reddy, V.; Cohen, S. JAK Inhibitors: What Is New? Curr. Rheumatol. Rep. 2020, 22, 50. [Google Scholar] [CrossRef]
- Xie, F.; Yun, H.; Bernatsky, S.; Curtis, J.R. Brief Report: Risk of Gastrointestinal Perforation among Rheumatoid Arthritis Patients Receiving Tofacitinib, Tocilizumab, or Other Biologic Treatments. Arthritis Rheumatol. 2016, 68, 2612–2617. [Google Scholar] [CrossRef] [PubMed]
- Winthrop, K.L. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat. Rev. Rheumatol. 2017, 13, 234–243. [Google Scholar] [CrossRef] [PubMed]
- FitzGerald, O.; Gladman, D.; Mease, P.; Ritchlin, C.; Smolen, J.; Gao, L.; Hu, S.; Nowak, M.; Banerjee, S.; Catless, I.; et al. Biomarker changes with selective tyrosine kinase 2 inhibitor, deucravacitinib, in PsA: Effects on disease markers and tyrosine kinase 2- versus janus kinase 1/2/3-mediated pathways. In Proceedings of the ACR Convergence, Virtual, 5–9 November 2021. Abstract 0490. [Google Scholar]
- Stebbing, J.; Sanchez Nievas, G.; Falcone, M.; Youhanna, S.; Richardson, P.; Ottaviani, S.; Shen, J.X.; Sommerauer, C.; Tiseo, G.; Ghiadoni, L.; et al. JAK inhibition reduces SARS-CoV-2 liver infectivity and modulates inflammatory responses to reduce morbidity and mortality. Sci. Adv. 2021, 7, eabe4724. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Ugel, S.; Tinazzi, E.; Vella, A.; De Sanctis, F.; Cane, S.; Batani, V.; Trovato, R.; Fiore, A.; Petrova, V.; et al. Baricitinib restrains the immune dysregulation in patients with severe COVID-19. J. Clin. Investig. 2020, 130, 6409–6416. [Google Scholar] [CrossRef]
- Cantini, F.; Niccoli, L.; Matarrese, D.; Nicastri, E.; Stobbione, P.; Goletti, D. Baricitinib therapy in COVID-19: A pilot study on safety and clinical impact. J. Infect. 2020, 81, 318–356. [Google Scholar] [CrossRef]
- Baricitinib improves respiratory function in patients treated with corticosteroids for SARS-CoV-2 pneumonia: An observational cohort study. Rheumatology 2021, 60, 399–407. [CrossRef]
- Sands, B.E.; Colombel, J.F.; Ha, C.; Farnier, M.; Armuzzi, A.; Quirk, D.; Friedman, G.S.; Kwok, K.; Salese, L.; Su, C.; et al. Lipid Profiles in Patients With Ulcerative Colitis Receiving Tofacitinib-Implications for Cardiovascular Risk and Patient Management. Inflamm. Bowel Dis. 2021, 27, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Wolk, R.; Armstrong, E.J.; Hansen, P.R.; Thiers, B.; Lan, S.; Tallman, A.M.; Kaur, M.; Tatulych, S. Effect of tofacitinib on lipid levels and lipid-related parameters in patients with moderate to severe psoriasis. J. Clin. Lipidol. 2017, 11, 1243–1256. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Zhao, X.; She, L.; Shi, Z.; Deng, Z.; Tan, L.; Tu, X.; Jiang, S.; Tang, B. Baricitinib induces LDL-C and HDL-C increases in rheumatoid arthritis: A meta-analysis of randomized controlled trials. Lipids Health Dis. 2019, 18, 54. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves Boxed Warning about Increased Risk of Blood Clots and Death with Higher Dose of Arthritis and Ulcerative Colitis Medicine Tofacitinib (Xeljanz, Xeljanz XR). 2021. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-approves-boxed-warning-about-increased-risk-blood-clots-and-death-higher-dose-arthritis-and (accessed on 4 May 2021).
- FDA Requires Warnings about Increased Risk of Serious Heart-Related Events, Cancer, Blood Clots, and Death for JAK Inhibitors That Treat Certain Chronic Inflammatory Conditions. 2021. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-requires-warnings-about-increased-risk-serious-heart-related-events-cancer-blood-clots-and-death (accessed on 21 September 2021).
- Pfizer Shares Co-Primary Endpoint Results from Post-Marketing Required Safety Study of Xeljanz® (tofacitinib) in Subjects with Rheumatoid Arthritis (RA). 2021. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-shares-co-primary-endpoint-results-post-marketing (accessed on 24 September 2021).
- Bilal, J.; Riaz, I.B.; Naqvi, S.A.A.; Bhattacharjee, S.; Obert, M.R.; Sadiq, M.; Abd El Aziz, M.A.; Nooman, Y.; Prokop, L.J.; Ge, L.; et al. Janus Kinase Inhibitors and Risk of Venous Thromboembolism: A Systematic Review and Meta-analysis. Mayo Clin. Proc 2021, 96, 1861–1873. [Google Scholar] [CrossRef]
- Kremer, J.M.; Bingham, C.O., 3rd; Cappelli, L.C.; Greenberg, J.D.; Madsen, A.M.; Geier, J.; Rivas, J.L.; Onofrei, A.M.; Barr, C.J.; Pappas, D.A.; et al. Postapproval Comparative Safety Study of Tofacitinib and Biological Disease-Modifying Antirheumatic Drugs: 5-Year Results from a United States-Based Rheumatoid Arthritis Registry. ACR Open Rheumatol. 2021, 3, 173–184. [Google Scholar] [CrossRef]
- Roubille, C.; Richer, V.; Starnino, T.; McCourt, C.; McFarlane, A.; Fleming, P.; Siu, S.; Kraft, J.; Lynde, C.; Pope, J.; et al. The effects of tumour necrosis factor inhibitors, methotrexate, non-steroidal anti-inflammatory drugs and corticosteroids on cardiovascular events in rheumatoid arthritis, psoriasis and psoriatic arthritis: A systematic review and meta-analysis. Ann. Rheum. Dis. 2015, 74, 480–489. [Google Scholar] [CrossRef]
- Greenberg, J.D.; Kremer, J.M.; Curtis, J.R.; Hochberg, M.C.; Reed, G.; Tsao, P.; Farkouh, M.E.; Nasir, A.; Setoguchi, S.; Solomon, D.H.; et al. Tumour necrosis factor antagonist use and associated risk reduction of cardiovascular events among patients with rheumatoid arthritis. Ann. Rheum. Dis. 2011, 70, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Khorrow-Khavar, F.; Kim, S.; Lee, H.; Been Lee, S.; and Desai, R. Risk of cardiovascular outcomes in patients treated with tofacitinib: First results from the safety of tofacitinib in routine care patients with rheumatoid arthritis (STAR-RA) study. In Proceedings of the ACR Convergence, Virtual, 5–9 November 2021. Abstract 1939. [Google Scholar]
- Charles-Schoeman, C.; Buch, M.; Dougados, M.; Bhatt, D.L.; Giles, J.; Vranic, I.; Wu, J.; Wang, C.; Menon, S.; Rivas, J.L.; et al. Risk factors for major adverse cardiovascular events in patients aged ≥ 50 years with RA and ≥1 additional cardiovascular risk factor: Results from a phase 3b/4 randomized safety study of tofacitinib vs TNF inhibitors. In Proceedings of the ACR Convergence, Virtual, 5–9 November 2021. Abstract 0958. [Google Scholar]
- Taylor, P.C.; Takeuchi, T.; Burmester, G.R.; Durez, P.; Smolen, J.S.; Deberdt, W.; Issa, M.; Terres, J.R.; Bello, N.; Winthrop, K.L. Safety of baricitinib for the treatment of rheumatoid arthritis over a median of 4.6 and up to 9.3 years of treatment: Final results from long-term extension study and integrated database. Ann. Rheum. Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Byers, N.L.; Higgs, R.E.; Lee, J.; Macias, W.L.; Na, S.; Ortmann, R.A.; Rocha, G.; Rooney, T.P.; Wehrman, T.; et al. Comparison of baricitinib, upadacitinib, and tofacitinib mediated regulation of cytokine signaling in human leukocyte subpopulations. Arthritis Res. Ther. 2019, 21, 183. [Google Scholar] [CrossRef]
- Traves, P.G.; Murray, B.; Campigotto, F.; Galien, R.; Meng, A.; Di Paolo, J.A. JAK selectivity and the implications for clinical inhibition of pharmacodynamic cytokine signalling by filgotinib, upadacitinib, tofacitinib and baricitinib. Ann. Rheum. Dis. 2021, 80, 865–875. [Google Scholar] [CrossRef]
- Dowty, M.E.; Lin, T.H.; Jesson, M.I.; Hegen, M.; Martin, D.A.; Katkade, V.; Menon, S.; Telliez, J.B. Janus kinase inhibitors for the treatment of rheumatoid arthritis demonstrate similar profiles of in vitro cytokine receptor inhibition. Pharmacol. Res. Perspect. 2019, 7, e00537. [Google Scholar] [CrossRef] [PubMed]
- Chimalakonda, A.; Burke, J.; Cheng, L.; Catlett, I.; Tagen, M.; Zhao, Q.; Patel, A.; Shen, J.; Girgis, I.G.; Banerjee, S.; et al. Selectivity Profile of the Tyrosine Kinase 2 Inhibitor Deucravacitinib Compared with Janus Kinase 1/2/3 Inhibitors. Dermatol. Ther. 2021, 11, 1763–1776. [Google Scholar] [CrossRef]
- Shi, J.G.; Chen, X.; Emm, T.; Scherle, P.A.; McGee, R.F.; Lo, Y.; Landman, R.R.; McKeever, E.G., Jr.; Punwani, N.G.; Williams, W.V.; et al. The effect of CYP3A4 inhibition or induction on the pharmacokinetics and pharmacodynamics of orally administered ruxolitinib (INCB018424 phosphate) in healthy volunteers. J. Clin. Pharmacol. 2012, 52, 809–818. [Google Scholar] [CrossRef]
- Arana Yi, C.; Tam, C.S.; Verstovsek, S. Efficacy and safety of ruxolitinib in the treatment of patients with myelofibrosis. Future Oncol. 2015, 11, 719–733. [Google Scholar] [CrossRef]
- Krishnaswami, S.; Chow, V.; Boy, M.; Wang, C.; Chan, G. Pharmacokinetics of tofacitinib, a janus kinase inhibitor, in patients with impaired renal function and end-stage renal disease. J. Clin. Pharmacol. 2014, 54, 46–52. [Google Scholar] [CrossRef]
- Zhao, X.; Sheng, X.Y.; Payne, C.D.; Zhang, X.; Wang, F.; Cui, Y.M. Pharmacokinetics, Safety, and Tolerability of Single- and Multiple-Dose Once-Daily Baricitinib in Healthy Chinese Subjects: A Randomized Placebo-Controlled Study. Clin. Pharmacol. Drug Dev. 2020, 9, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.F.; Zeng, J.; Marroum, P.J.; Song, I.H.; Othman, A.A. Pharmacokinetics of Upadacitinib With the Clinical Regimens of the Extended-Release Formulation Utilized in Rheumatoid Arthritis Phase 3 Trials. Clin. Pharmacol. Drug Dev. 2019, 8, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Klunder, B.; Mohamed, M.F.; Othman, A.A. Population Pharmacokinetics of Upadacitinib in Healthy Subjects and Subjects with Rheumatoid Arthritis: Analyses of Phase I and II Clinical Trials. Clin. Pharmacokinet. 2018, 57, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Namour, F.; Diderichsen, P.M.; Cox, E.; Vayssiere, B.; Van der Aa, A.; Tasset, C.; Van’t Klooster, G. Pharmacokinetics and Pharmacokinetic/Pharmacodynamic Modeling of Filgotinib (GLPG0634), a Selective JAK1 Inhibitor, in Support of Phase IIB Dose Selection. Clin. Pharmacokinet 2015, 54, 859–874. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.G.; Fraczkiewicz, G.; Williams, W.V.; Yeleswaram, S. Predicting drug-drug interactions involving multiple mechanisms using physiologically based pharmacokinetic modeling: A case study with ruxolitinib. Clin. Pharmacol. Ther. 2015, 97, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shi, J.G.; Emm, T.; Scherle, P.A.; McGee, R.F.; Lo, Y.; Landman, R.R.; Punwani, N.G.; Williams, W.V.; Yeleswaram, S. Pharmacokinetics and pharmacodynamics of orally administered ruxolitinib (INCB018424 phosphate) in renal and hepatic impairment patients. Clin. Pharmacol. Drug Dev. 2014, 3, 34–42. [Google Scholar] [CrossRef]
- Gong, X.; Chen, X.; Kuligowski, M.E.; Liu, X.; Liu, X.; Cimino, E.; McGee, R.; Yeleswaram, S. Pharmacokinetics of Ruxolitinib in Patients with Atopic Dermatitis Treated With Ruxolitinib Cream: Data from Phase II and III Studies. Am. J. Clin. Dermatol. 2021, 22, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Lefevre, P.L.C.; Vande Casteele, N. Clinical Pharmacology of Janus Kinase Inhibitors in Inflammatory Bowel Disease. J. Crohns Colitis 2020, 14, S725–S736. [Google Scholar] [CrossRef]
- Posada, M.M.; Cannady, E.A.; Payne, C.D.; Zhang, X.; Bacon, J.A.; Pak, Y.A.; Higgins, J.W.; Shahri, N.; Hall, S.D.; Hillgren, K.M. Prediction of Transporter-Mediated Drug-Drug Interactions for Baricitinib. Clin. Transl. Sci. 2017, 10, 509–519. [Google Scholar] [CrossRef]
- Kim, H.; Brooks, K.M.; Tang, C.C.; Wakim, P.; Blake, M.; Brooks, S.R.; Montealegre Sanchez, G.A.; de Jesus, A.A.; Huang, Y.; Tsai, W.L.; et al. Pharmacokinetics, Pharmacodynamics, and Proposed Dosing of the Oral JAK1 and JAK2 Inhibitor Baricitinib in Pediatric and Young Adult CANDLE and SAVI Patients. Clin. Pharmacol. Ther. 2018, 104, 364–373. [Google Scholar] [CrossRef] [PubMed]
- FDA Olumiant Label, Highlights of Prescribing Information. 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/207924s000lbl.pdf (accessed on 20 August 2021).
- Namour, F.; Fagard, L.; Van der Aa, A.; Harrison, P.; Xin, Y.; Tasset, C. Influence of age and renal impairment on the steady state pharmacokinetics of filgotinib, a selective JAK1 inhibitor. Br. J. Clin. Pharmacol. 2018, 84, 2779–2789. [Google Scholar] [CrossRef]
- Muensterman, E.; Engelhardt, B.; Gopalakrishnan, S.; Anderson, J.; Mohamed, M.E. Upadacitinib pharmacokinetics and ex-posure-response relationships for efficacy and safety in psoriatic arthritis—Analysis of the phase 3 SELECT-PsA studies. In Proceedings of the ACR Convergence, Virtual, 5–9 November 2021; Abstract POS1054. pp. 805–806. [Google Scholar]
- Fleischmann, R.; Wollenhaupt, J.; Takiya, L.; Maniccia, A.; Kwok, K.; Wang, L.; van Vollenhoven, R.F. Safety and maintenance of response for tofacitinib monotherapy and combination therapy in rheumatoid arthritis: An analysis of pooled data from open-label long-term extension studies. RMD Open 2017, 3, e000491. [Google Scholar] [CrossRef] [PubMed]
- Ebina, K.; Hirano, T.; Maeda, Y.; Yamamoto, W.; Hashimoto, M.; Murata, K.; Onishi, A.; Jinno, S.; Hara, R.; Son, Y.; et al. Drug retention of sarilumab, baricitinib, and tofacitinib in patients with rheumatoid arthritis: The ANSWER cohort study. Clin. Rheumatol. 2021, 40, 2673–2680. [Google Scholar] [CrossRef]
- Lwin, M.N.; Holroyd, C.; Edwards, C.J. O10 Characteristics of patients who discontinued baricitinib treatment within 12 months and reasons for discontinuation: Real-world data. Rheumatology 2021, 60, keab246.009. [Google Scholar] [CrossRef]
- Alunno, A.; Najm, A.; Machado, P.M.; Bertheussen, H.; Burmester, G.R.; Carubbi, F.; De Marco, G.; Giacomelli, R.; Hermine, O.; Isaacs, J.D.; et al. 2021 update of the EULAR points to consider on the use of immunomodulatory therapies in COVID-19. Ann. Rheum. Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- Seror, R.; Camus, M.; Salmon, J.H.; Roux, C.; Dernis, E.; Basch, A.; Germain, V.; Leske, C.; Brousseau, S.; Truchetet, M.E.; et al. Do JAK inhibitors affect immune response to COVID-19 vaccination? Data from the MAJIK-SFR Registry. Lancet Rheumatol. 2021, 4, E8–E11. [Google Scholar] [CrossRef]
- Le, R.Q.; Li, L.; Yuan, W.; Shord, S.S.; Nie, L.; Habtemariam, B.A.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Tocilizumab for Treatment of Chimeric Antigen Receptor T Cell-Induced Severe or Life-Threatening Cytokine Release Syndrome. Oncologist 2018, 23, 943–947. [Google Scholar] [CrossRef]
- Quatrini, L.; Ugolini, S. New insights into the cell- and tissue-specificity of glucocorticoid actions. Cell Mol Immunol. 2021, 18, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Siegel, A.; Kreider, K. Physiologic Steroid Tapering. J. Nurse Pract. 2019, 15, 463–464. [Google Scholar] [CrossRef]
- Abebe, R.; Hatiya, H.; Abera, M.; Megersa, B.; Asmare, K. Bovine mastitis: Prevalence, risk factors and isolation of Staphylococcus aureus in dairy herds at Hawassa milk shed, South Ethiopia. BMC Vet. Res. 2016, 12, 270. [Google Scholar] [CrossRef] [PubMed]
- Citera, G.; Jain, R.; Irazoque-Palazuelos, F.; Guzman, R.; Madariaga, H.; Gruben, D.C.; Wang, L.; Stockert, L.; Hsu, M.A.; San-tana, K.; et al. Tofacitinib in patients with rheumatoid arthritis and indicative of depression and/or anxiety: A post hoc analysis of phase 3 and phase 3B/4 clinical trials. In Proceedings of the 2020 EULAR, Virtual, 3–6 June 2020. Abstract THU0196. [Google Scholar]
- Khandaker, G.M.; Oltean, B.P.; Kaser, M.; Dibben, C.R.M.; Ramana, R.; Jadon, D.R.; Dantzer, R.; Coles, A.J.; Lewis, G.; Jones, P.B. Protocol for the insight study: A randomised controlled trial of single-dose tocilizumab in patients with depression and low-grade inflammation. BMJ Open 2018, 8, e025333. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| JAK Inhibitor | Target | Indication | References |
|---|---|---|---|
| tofacitinib | JAK1, JAK3 | RA, PsA, UC, adult and juvenile PsA (patients > 2) | [23] |
| JAK2 | juvenile idiopathic arthritis (patients > 2 years old) | ||
| ruxolitinib | JAK1 | PV, intermediate-high myelofibrosis, SR-GVHD | [22,24] |
| JAK2 | AD (topical cream) | ||
| baricitinib | JAK1 | RA 1, AD 2 | [25,26] |
| JAK2 | COVID-19 3 | ||
| upadacitinib | JAK1 | RA, PsA, AS 4, moderate-severe AD 4 | [27,28] |
| filgotinib | JAK1 | RA 4, UC 2 | [29] |
| abrocitinib | JAK1 | AD 5 | [30] |
| fedratinib | JAK2 | intermediate-high risk | [31] |
| FLT3 | primary or secondary myelofibrosis | ||
| peficitinib | pan-JAK | RA 6 | [32] |
| delgocitinib | pan-JAK | AD 7 | [33] |
| ocalacitinib | JAK1, JAK2 | canine allergic dermatitis | [34] |
| JAK3 |
| JAK Inhibitor | Indication under Investigation | Trial |
|---|---|---|
| ruxolitinib | pediatric moderate-severe chronic GVHD | NCT03774082 |
| hemophagocytic lymphohistiocytosis | NCT04551131 | |
| hypereosinophilic syndrome/primary eosinophilic disorders | NCT03801434 | |
| inflammation in HIV patients | NCT02475655 | |
| severe AA 1 | NCT04518995, NCT04797650 | |
| tofacitinib | AS | NCT03502616 |
| SLE | NCT02535689 | |
| cutaneous lupus | NCT03288324 | |
| Sjogren’s syndrome | NCT04496960 | |
| inflammatory eye disease | NCT03580343 | |
| COVID-19 | NCT04469114 | |
| baricitinib | SLE | NCT02708095, NCT03616912, NCT03616964, NCT03843125 |
| COVID-19 | NCT04401579, NCT04640168, NCT04421027 | |
| peficitinib | RA | NCT01649999, NCT01711814, NCT01565655, NCT02308163, NCT02305849 |
| psoriasis | NCT01096862 | |
| UC | NCT01959282 | |
| impaired renal function | NCT02603497 | |
| delgocitinib | chronic hand eczema | NCT04871711, NCT04872101, NCT04949841 |
| inverse psoriasis | NCT02695940 | |
| AD | NCT03725722, NCT03826901 | |
| upadacitinib | PsA | NCT03104374, NCT03104400, NCT03104374 |
| AS | NCT03178487 | |
| AD 2 | NCT03569293, NCT03607422, NCT03568318 | |
| giant cell arteritis | NCT03725202 | |
| Takayasu arteritis | NCT04161898 | |
| filgotinib | CD | NCT03077412, NCT02914561, NCT02914600 |
| PsA | NCT03101670 | |
| AS | NCT03117270 | |
| abrocitinib | AD | NCT03349060, NCT03575871, NCT03720470 |
| itacitinib | acute GVHD 3 | NCT03139604 |
| asthma | NCT04129931 | |
| bronchiolitis obliterans syndrome | NCT03978637 | |
| MDS/MPN overlap syndrome | NCT04061421 | |
| SHR0302 | CD | NCT03677648 |
| RA | NCT02892370, NCT02665910, NCT02423538 | |
| AS | NCT04481139 | |
| AA | NCT04346316 | |
| AD | NCT04717310, NCT04162899 | |
| UC | NCT03675477 | |
| vitiligo | NCT04774809 | |
| deucravacitinib | psoriasis | NCT03624127, NCT03611751, NCT04167462, NCT03924427, NCT04036435 |
| PsA | NCT03881059 | |
| CD | NCT03599622 | |
| UC | NCT03934216 | |
| SLE | NCT03252587 | |
| brepocitinib | psoriasis 4 | NCT03850483 |
| AD 1 | NCT03903822 | |
| PsA | NCT03963401 | |
| UC | NCT02958865 | |
| CD | NCT03395184 | |
| vitiligo | NCT03715829 | |
| SLE | NCT03845517 | |
| cicatricial alopecia | NCT05076006 | |
| hidradenitis suppurativa | NCT04092452 | |
| AA | NCT02974868 | |
| gusacitinib | AD | NCT03531957 |
| chronic hand eczema | NCT03728504 | |
| cerdulatinib | follicular lymphoma | NCT04021082 |
| T-cell lymphoma | NCT01994382 | |
| vitiligo 4 | NCT04103060 | |
| momelotinib | myelofibrosis | NCT01969838 |
| primary myelofibrosis, post-PV, post-ET myelofibrosis | NCT02101268 | |
| pacritinib | severe thrombocytopenia in myelofibrosis patients | NCT01773187 |
| ritlecitinib | AA | NCT03732807 |
| vitiligo | NCT03715829 | |
| UC | NCT02958865 | |
| CD | NCT03395184 | |
| AA | NCT04006457 | |
| izencitinib | UC | NCT03758443 |
| CD | NCT03635112 | |
| OST-122 | UC | NCT04353791 |
| TD-8236 | asthma | NCT03652038, NCT04150341 5 |
| nezulcitinib | COVID-19-associated pulmonary disease | NCT04402866 |
| JAK Inhibitor | Time to Peak Concentration | Half-Life (Hours) | References |
|---|---|---|---|
| ruxolitinib | <2 h | approximately 3 h | [151] |
| 5 most common ruxolitinib metabolites | 1.5–2 h | 4.3–5.7 h | [150] |
| tofacitinib | 0.5–1 h | 3 h | [152] |
| baricitinib | 0.75–1 h | 5.9–7.4 h | [153] |
| upadacitinib | within 1 h 1; 2–3 h 2 | 4 h | [154,155] |
| filgotinib | 0.5–5 h | 5–6 h | [152] |
| GS829845 | 3–5 h | 20–23 h | [156] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alexander, M.; Luo, Y.; Raimondi, G.; O’Shea, J.J.; Gadina, M. Jakinibs of All Trades: Inhibiting Cytokine Signaling in Immune-Mediated Pathologies. Pharmaceuticals 2022, 15, 48. https://doi.org/10.3390/ph15010048
Alexander M, Luo Y, Raimondi G, O’Shea JJ, Gadina M. Jakinibs of All Trades: Inhibiting Cytokine Signaling in Immune-Mediated Pathologies. Pharmaceuticals. 2022; 15(1):48. https://doi.org/10.3390/ph15010048
Chicago/Turabian StyleAlexander, Madison, Yiming Luo, Giorgio Raimondi, John J. O’Shea, and Massimo Gadina. 2022. "Jakinibs of All Trades: Inhibiting Cytokine Signaling in Immune-Mediated Pathologies" Pharmaceuticals 15, no. 1: 48. https://doi.org/10.3390/ph15010048
APA StyleAlexander, M., Luo, Y., Raimondi, G., O’Shea, J. J., & Gadina, M. (2022). Jakinibs of All Trades: Inhibiting Cytokine Signaling in Immune-Mediated Pathologies. Pharmaceuticals, 15(1), 48. https://doi.org/10.3390/ph15010048