
The Role of LNK (SH2B3) in the Regulation of JAK-STAT Signalling in Haematopoiesis

 , ,
, ,  ,
, 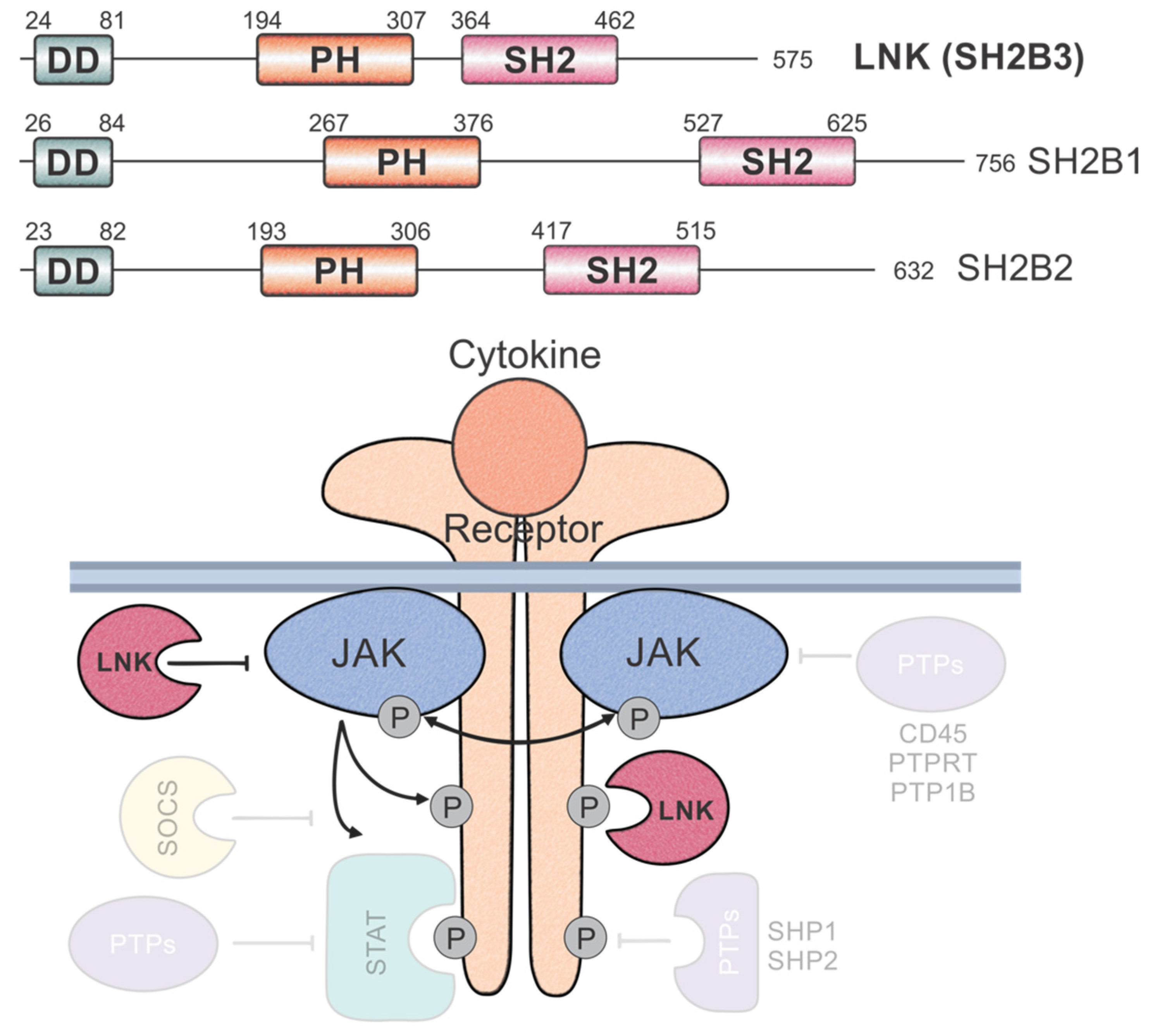{kind=link}
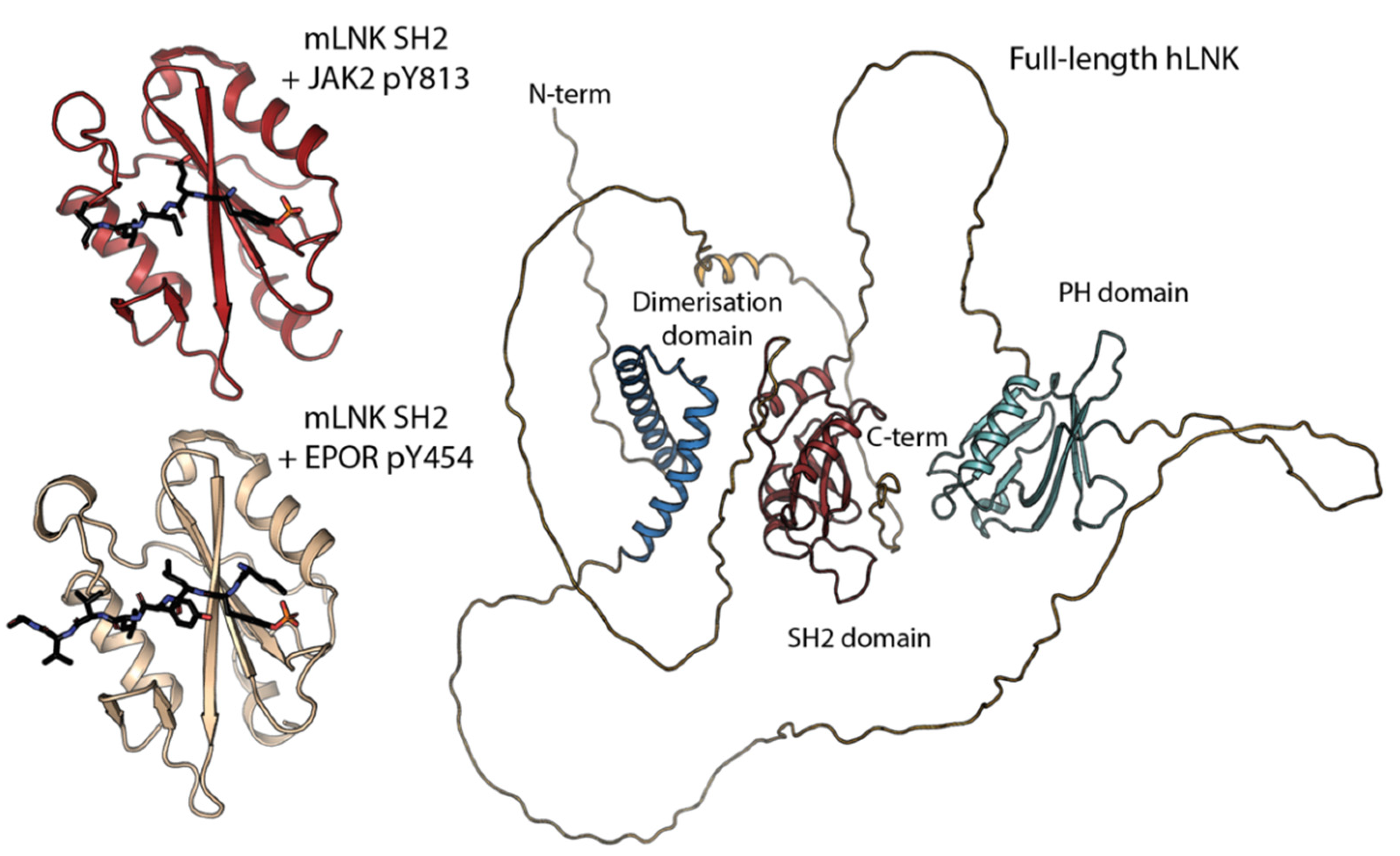{kind=link}
{kind=link}
Abstract
1. Introduction
2. Domain Architecture and Protein Structure
3. LNK as an Adaptor Protein
4. The Role of LNK in the Regulation of JAK-STAT Signalling
5. Other Potential Targets of LNK SH2 Domain
6. Regulation of LNK
7. LNK Mutations in Disease
8. LNK Mutations in Haematological Cancers
9. Mutations Identified in the LNK SH2 Domain
10. LNK as a Potential Therapeutic Target
11. Unanswered Questions and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, X.; Li, Y.; Tanaka, K.; Moore, K.G.; Hayashi, J.I. Cloning and characterization of Lnk, a signal transduction protein that links T-cell receptor activation signal to phospholipase C gamma 1, Grb2, and phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 1995, 92, 11618–11622. [Google Scholar] [CrossRef]
- Fitau, J.; Boulday, G.; Coulon, F.; Quillard, T.; Charreau, B. The adaptor molecule Lnk negatively regulates tumor necrosis factor-α-dependent VCAM-1 expression in endothelial cells through inhibition of the ERK1 and-2 pathways. J. Biol. Chem. 2006, 281, 20148–20159. [Google Scholar] [CrossRef]
- Wang, T.-C.; Chiu, H.; Chang, Y.-J.; Hsu, T.-Y.; Chiu, I.-M.; Chen, L. The adaptor protein SH2B3 (Lnk) negatively regulates neurite outgrowth of PC12 cells and cortical neurons. PLoS ONE 2011, 6, e26433. [Google Scholar] [CrossRef]
- Dhe-Paganon, S.; Werner, E.D.; Nishi, M.; Hansen, L.; Chi, Y.-I.; Shoelson, S.E. A phenylalanine zipper mediates APS dimerization. Nat. Struct. Mol. Biol. 2004, 11, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Harlan, J.E.; Hajduk, P.J.; Yoon, H.S.; Fesik, S.W. Pleckstrin homology domains bind to phosphatidylinositol-4,5-bisphosphate. Nature 1994, 371, 168–170. [Google Scholar] [CrossRef]
- Velazquez, L. The Lnk adaptor protein: A key regulator of normal and pathological hematopoiesis. Arch. Immunol. Ther. Exp. 2012, 60, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Takaki, S.; Watts, J.D.; Forbush, K.A.; Nguyen, N.T.; Hayashi, J.; Alberola-Ila, J.; Aebersold, R.; Perlmutter, R.M. Characterization of Lnk an adaptor protein expressed in lymphocytes. J. Biol. Chem. 1997, 272, 14562–14570. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; He, X.; Schembri-King, J.; Jakes, S.; Hayashi, J. Cloning and characterization of human Lnk, an adaptor protein with pleckstrin homology and Src homology 2 domains that can inhibit T cell activation. J. Immunol. 2000, 164, 5199–5206. [Google Scholar] [CrossRef]
- Takizawa, H.; Kubo-Akashi, C.; Nobuhisa, I.; Kwon, S.-M.; Iseki, M.; Taga, T.; Takatsu, K.; Takaki, S. Enhanced engraftment of hematopoietic stem/progenitor cells by the transient inhibition of an adaptor protein, Lnk. Blood 2006, 107, 2968–2975. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.; Zhang, Y.; Ellyard, J.I.; Vinuesa, C.G.; Murphy, J.M.; Laktyushin, A.; Kershaw, N.J.; Babon, J.J. Structural and functional analysis of target recognition by the lymphocyte adaptor protein LNK. Nat. Commun. 2021, 12, 1–9. [Google Scholar] [CrossRef]
- Hu, J.; Liu, J.; Ghirlando, R.; Saltiel, A.R.; Hubbard, S.R. Structural basis for recruitment of the adaptor protein APS to the activated insulin receptor. Mol. Cell 2003, 12, 1379–1389. [Google Scholar] [CrossRef]
- Hu, J.; Hubbard, S.R. Structural basis for phosphotyrosine recognition by the Src homology-2 domains of the adapter proteins SH2-B and APS. J. Mol. Biol. 2006, 361, 69–79. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Lv, K.; Jiang, J.; Donaghy, R.; Riling, C.R.; Cheng, Y.; Chandra, V.; Rozenova, K.; An, W.; Mohapatra, B.C.; Goetz, B.T. CBL family E3 ubiquitin ligases control JAK2 ubiquitination and stability in hematopoietic stem cells and myeloid malignancies. Genes Dev. 2017, 31, 1007–1023. [Google Scholar] [CrossRef] [PubMed]
- Donaghy, R.; Han, X.; Rozenova, K.; Lv, K.; Jiang, Q.; Doepner, M.; Greenberg, R.A.; Tong, W. The BRISC deubiquitinating enzyme complex limits hematopoietic stem cell expansion by regulating JAK2 K63-ubiquitination. Blood J. Am. Soc. Hematol. 2019, 133, 1560–1571. [Google Scholar] [CrossRef]
- Rozenova, K.; Jiang, J.; Donaghy, R.; Aressy, B.; Greenberg, R.A.; Tong, W. MERIT40 deficiency expands hematopoietic stem cell pools by regulating thrombopoietin receptor signaling. Blood J. Am. Soc. Hematol. 2015, 125, 1730–1738. [Google Scholar] [CrossRef]
- Velazquez, L.; Cheng, A.M.; Fleming, H.E.; Furlonger, C.; Vesely, S.; Bernstein, A.; Paige, C.J.; Pawson, T. Cytokine signaling and hematopoietic homeostasis are disrupted in Lnk-deficient mice. J. Exp. Med. 2002, 195, 1599–1611. [Google Scholar] [CrossRef]
- Bersenev, A.; Wu, C.; Balcerek, J.; Tong, W. Lnk controls mouse hematopoietic stem cell self-renewal and quiescence through direct interactions with JAK2. J. Clin. Investig. 2008, 118, 2832. [Google Scholar] [CrossRef] [PubMed]
- Takaki, S.; Tezuka, Y.; Sauer, K.; Kubo, C.; Kwon, S.-M.; Armstead, E.; Nakao, K.; Katsuki, M.; Perlmutter, R.M.; Takatsu, K. Impaired lymphopoiesis and altered B cell subpopulations in mice overexpressing Lnk adaptor protein. J. Immunol. 2003, 170, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.; Lodish, H.F. Lnk inhibits Tpo–mpl signaling and Tpo-mediated megakaryocytopoiesis. J. Exp. Med. 2004, 200, 569–580. [Google Scholar] [CrossRef]
- Tong, W.; Zhang, J.; Lodish, H.F. Lnk inhibits erythropoiesis and Epo-dependent JAK2 activation and downstream signaling pathways. Blood 2005, 105, 4604–4612. [Google Scholar] [CrossRef]
- Seita, J.; Ema, H.; Ooehara, J.; Yamazaki, S.; Tadokoro, Y.; Yamasaki, A.; Eto, K.; Takaki, S.; Takatsu, K.; Nakauchi, H. Lnk negatively regulates self-renewal of hematopoietic stem cells by modifying thrombopoietin-mediated signal transduction. Proc. Nat. Acad. Sci. USA 2007, 104, 2349–2354. [Google Scholar] [CrossRef]
- Cheng, H.; Ross, J.A.; Frost, J.A.; Kirken, R.A. Phosphorylation of human Jak3 at tyrosines 904 and 939 positively regulates its activity. Mol. Cell. Biol. 2008, 28, 2271–2282. [Google Scholar] [CrossRef]
- Miyazaki, T.; Kawahara, A.; Fujii, H.; Nakagawa, Y.; Minami, Y.; Liu, Z.-J.; Oishi, I.; Silvennoinen, O.; Witthuhn, B.A.; Ihle, J.N. Functional activation of Jak1 and Jak3 by selective association with IL-2 receptor subunits. Science 1994, 266, 1045–1047. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.-W.; Sun, Q.-Y.; Edwards, J.J.; Fernández, L.T.; Ran, X.-B.; Zhou, S.-Q.; Scolyer, R.A.; Wilmott, J.S.; Thompson, J.F.; Doan, N. LNK suppresses interferon signaling in melanoma. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gueller, S.; Gery, S.; Nowak, V.; Liu, L.; Serve, H.; Koeffler, H.P. Adaptor protein Lnk associates with Tyr568 in c-Kit. Biochem. J. 2008, 415, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Gueller, S.; Goodridge, H.S.; Niebuhr, B.; Xing, H.; Koren-Michowitz, M.; Serve, H.; Underhill, D.M.; Brandts, C.H.; Koeffler, H.P. Adaptor protein Lnk inhibits c-Fms-mediated macrophage function. J. Leukoc. Biol. 2010, 88, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.-C.; Yin, T.; Koren-Michowitz, M.; Ding, L.-W.; Gueller, S.; Gery, S.; Tabayashi, T.; Bergholz, U.; Kazi, J.U.; Rönnstrand, L. Adaptor protein Lnk binds to and inhibits normal and leukemic FLT3. Blood 2012, 120, 3310–3317. [Google Scholar] [CrossRef] [PubMed]
- Gueller, S.; Hehn, S.; Nowak, V.; Gery, S.; Serve, H.; Brandts, C.H.; Koeffler, H.P. Adaptor protein Lnk binds to PDGF receptor and inhibits PDGF-dependent signaling. Exp. Hematol. 2011, 39, 591–600. [Google Scholar] [CrossRef]
- Jiang, J.; Balcerek, J.; Rozenova, K.; Cheng, Y.; Bersenev, A.; Wu, C.; Song, Y.; Tong, W. 14-3-3 regulates the LNK/JAK2 pathway in mouse hematopoietic stem and progenitor cells. J. Clin. Investig. 2012, 122, 2079–2091. [Google Scholar] [CrossRef]
- Oh, S.T.; Simonds, E.F.; Jones, C.; Hale, M.B.; Goltsev, Y.; Gibbs, K.D.; Merker, J.D.; Zehnder, J.L.; Nolan, G.P.; Gotlib, J. Novel mutations in the inhibitory adaptor protein LNK drive JAK-STAT signaling in patients with myeloproliferative neoplasms. Blood 2010, 116, 988–992. [Google Scholar] [CrossRef]
- McMullin, M.F.; Cario, H. LNK mutations and myeloproliferative disorders. Am. J. Hematol. 2016, 91, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Lasho, T.; Finke, C.; Oh, S.; Gotlib, J.; Tefferi, A. LNK mutation studies in blast-phase myeloproliferative neoplasms, and in chronic-phase disease with TET2, IDH, JAK2 or MPL mutations. Leukemia 2010, 24, 1713. [Google Scholar] [CrossRef]
- Spolverini, A.; Pieri, L.; Guglielmelli, P.; Pancrazzi, A.; Fanelli, T.; Paoli, C.; Bosi, A.; Nichele, I.; Ruggeri, M.; Vannucchi, A.M. Infrequent occurrence of mutations in the PH domain of LNK in patients with JAK2 mutation-negative ‘idiopathic’ erythrocytosis. Haematologica 2013, 98, e101–e102. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.T.; Zahn, J.M.; Jones, C.D.; Zhang, B.; Loh, M.L.; Kantarjian, H.; Simonds, E.F.; Bruggner, R.V.; Abidi, P.; Natsoulis, G. Identification of novel LNK mutations in patients with chronic myeloproliferative neoplasms and related disorders. Am. Soc. Hematol. 2010, 116, 143–144. [Google Scholar] [CrossRef]
- Lasho, T.; Tefferi, A.; Finke, C.; Pardanani, A. Clonal hierarchy and allelic mutation segregation in a myelofibrosis patient with two distinct LNK mutations. Leukemia 2011, 25, 1056–1058. [Google Scholar] [CrossRef][Green Version]
- Dale, B.L.; Madhur, M.S. Linking inflammation and hypertension via LNK/SH2B3. Curr. Opin. Nephrol. Hypertens. 2016, 25, 87. [Google Scholar] [CrossRef]
- Hong, L.; Jiang, Y.-F.; Chen, M.; Zhang, N.-N.; Yang, H.-J.; Rui, Q.; Zhou, Y.-F. Role of SH2B3 R262W gene polymorphism and risk of coronary heart disease: A PRISMA-compliant meta-analysis. Medicine 2018, 97, e13436. [Google Scholar] [CrossRef]
- Alcina, A.; Vandenbroeck, K.; Otaegui, D.; Sáiz, A.; Gonzalez, J.R.; Fernandez, O.; Cavanillas, M.; Cénit, M.; Arroyo, R.; Alloza, I. The autoimmune disease-associated KIF5A, CD226 and SH2B3 gene variants confer susceptibility for multiple sclerosis. Genes Immun. 2010, 11, 439–445. [Google Scholar] [CrossRef]
- Keefe, J.A.; Hwang, S.-J.; Huan, T.; Mendelson, M.; Yao, C.; Courchesne, P.; Saleh, M.A.; Madhur, M.S.; Levy, D. Evidence for a Causal Role of the SH2B3-β2M Axis in Blood Pressure Regulation: Framingham Heart Study. Hypertension 2019, 73, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Maslah, N.; Cassinat, B.; Verger, E.; Kiladjian, J.; Velazquez, L. The role of LNK/SH2B3 genetic alterations in myeloproliferative neoplasms and other hematological disorders. Leukemia 2017, 31, 1661–1670. [Google Scholar] [CrossRef]
- Roberts, K.G.; Morin, R.D.; Zhang, J.; Hirst, M.; Zhao, Y.; Su, X.; Chen, S.-C.; Payne-Turner, D.; Churchman, M.L.; Harvey, R.C. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell 2012, 22, 153–166. [Google Scholar] [CrossRef]
- Perez-Garcia, A.; Ambesi-Impiombato, A.; Hadler, M.; Rigo, I.; LeDuc, C.A.; Kelly, K.; Jalas, C.; Paietta, E.; Racevskis, J.; Rowe, J.M. Genetic loss of SH2B3 in acute lymphoblastic leukemia. Blood J. Am. Soc. Hematol. 2013, 122, 2425–2432. [Google Scholar] [CrossRef]
- Tan, M.; Rong, Y.; Tian, R.; Zhu, Y.; Zhu, P.; Chen, Y. Variation of LNK Gene in Chronic Myeloid Leukemia. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2017, 25, 706–710. [Google Scholar] [PubMed]
- Ge, Z.; Gu, Y.; Xiao, L.; Han, Q.; Li, J.; Chen, B.; Yu, J.; Kawasawa, Y.I.; Payne, K.J.; Dovat, S. Co-existence of IL7R high and SH2B3 low expression distinguishes a novel high-risk acute lymphoblastic leukemia with Ikaros dysfunction. Oncotarget 2016, 7, 46014. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.L.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163. [Google Scholar] [CrossRef]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.-L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef]
- Cheng, Y.; Chikwava, K.; Wu, C.; Zhang, H.; Bhagat, A.; Pei, D.; Choi, J.K.; Tong, W. LNK/SH2B3 regulates IL-7 receptor signaling in normal and malignant B-progenitors. J. Clin. Investig. 2016, 126, 1267–1281. [Google Scholar] [CrossRef] [PubMed]
- Loh, P.-R.; Genovese, G.; McCarroll, S.A. Monogenic and polygenic inheritance become instruments for clonal selection. Nature 2020, 584, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Prchal, J. Bone-marrow responses in polycythemia vera. N. Engl. J. Med. 1974, 290, 1382. [Google Scholar] [PubMed]
- Tefferi, A.; Pardanani, A. Essential Thrombocythemia. N. Engl. J. Med. 2019, 381, 2135–2144. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M. Management of myelofibrosis. Hematology 2011, 2011, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.V.; Kreil, S.; Zoi, K.; Waghorn, K.; Curtis, C.; Zhang, L.; Score, J.; Seear, R.; Chase, A.J.; Grand, F.H. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood 2005, 106, 2162–2168. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.M.; Campbell, P.J.; Baxter, E.J.; Todd, T.; Stephens, P.; Edkins, S.; Wooster, R.; Stratton, M.R.; Futreal, P.A.; Green, A.R. The V617F JAK2 mutation is uncommon in cancers and in myeloid malignancies other than the classic myeloproliferative disorders. Blood 2005, 106, 2920–2921. [Google Scholar] [CrossRef]
- Carpinelli, M.R.; Hilton, D.J.; Metcalf, D.; Antonchuk, J.L.; Hyland, C.D.; Mifsud, S.L.; Di Rago, L.; Hilton, A.A.; Willson, T.A.; Roberts, A.W.; et al. Suppressor screen in Mpl−/− mice: C-Myb mutation causes supraphysiological production of platelets in the absence of thrombopoietin signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 6553–6558. [Google Scholar] [CrossRef]
- Pardanani, A.D.; Levine, R.L.; Lasho, T.; Pikman, Y.; Mesa, R.A.; Wadleigh, M.; Steensma, D.P.; Elliott, M.A.; Wolanskyj, A.P.; Hogan, W.J. MPL515 mutations in myeloproliferative and other myeloid disorders: A study of 1182 patients. Blood 2006, 108, 3472–3476. [Google Scholar] [CrossRef]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.; Berg, T.; Gisslinger, B.; Pietra, D. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef]
- Tefferi, A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia 2010, 24, 1128. [Google Scholar] [CrossRef]
- McMullin, M.F.; Wu, C.; Percy, M.J.; Tong, W. A nonsynonymous LNK polymorphism associated with idiopathic erythrocytosis. Am. J. Hematol. 2011, 86, 962–964. [Google Scholar] [CrossRef]
- Gery, S.; Gueller, S.; Chumakova, K.; Kawamata, N.; Liu, L.; Koeffler, H.P. Adaptor protein Lnk negatively regulates the mutant MPL, MPLW515L associated with myeloproliferative disorders. Blood 2007, 110, 3360–3364. [Google Scholar] [CrossRef][Green Version]
- Gery, S.; Cao, Q.; Gueller, S.; Xing, H.; Tefferi, A.; Koeffler, H.P. Lnk inhibits myeloproliferative disorder-associated JAK2 mutant, JAK2V617F. J. Leukoc. Biol. 2009, 85, 957–965. [Google Scholar] [CrossRef]
- Camps, C.; Petousi, N.; Bento, C.; Cario, H.; Copley, R.R.; McMullin, M.F.; van Wijk, R.; Ratcliffe, P.J.; Robbins, P.A.; Taylor, J.C. Gene panel sequencing improves the diagnostic work-up of patients with idiopathic erythrocytosis and identifies new mutations. Haematologica 2016, 101, 1306–1318. [Google Scholar] [CrossRef]
- Chen, Y.; Fang, F.; Hu, Y.; Liu, Q.; Bu, D.; Tan, M.; Wu, L.; Zhu, P. The polymorphisms in LNK gene correlated to the clinical type of myeloproliferative neoplasms. PLoS ONE 2016, 11, e0154183. [Google Scholar] [CrossRef]
- Koren-Michowitz, M.; Gery, S.; Tabayashi, T.; Lin, D.; Alvarez, R.; Nagler, A.; Koeffler, H.P. SH 2B3 (LNK) mutations from myeloproliferative neoplasms patients have mild loss of function against wild type JAK 2 and JAK 2 V617F. Br. J. Haematol. 2013, 161, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Coltro, G.; Lasho, T.L.; Finke, C.M.; Gangat, N.; Pardanani, A.; Tefferi, A.; Jevremovic, D.; Altman, J.K.; Patnaik, M.M. Germline SH2B3 pathogenic variant associated with myelodysplastic syndrome/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis. Am. J. Hematol. 2019, 94, E231–E234. [Google Scholar] [CrossRef] [PubMed]
- Angona, A.; Fernández-Rodríguez, C.; Alvarez-Larrán, A.; Camacho, L.; Longarón, R.; Torres, E.; Pairet, S.; Besses, C.; Bellosillo, B. Molecular characterisation of triple negative essential thrombocythaemia patients by platelet analysis and targeted sequencing. Blood Cancer J. 2016, 6, e463. [Google Scholar] [CrossRef] [PubMed]
- Giani, F.C.; Fiorini, C.; Wakabayashi, A.; Ludwig, L.S.; Salem, R.M.; Jobaliya, C.D.; Regan, S.N.; Ulirsch, J.C.; Liang, G.; Steinberg-Shemer, O. Targeted application of human genetic variation can improve red blood cell production from stem cells. Cell Stem Cell 2016, 18, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Olsson, L.; Albitar, F.; Castor, A.; Behrendtz, M.; Biloglav, A.; Paulsson, K.; Johansson, B. Cooperative genetic changes in pediatric B-cell precursor acute lymphoblastic leukemia with deletions or mutations of IKZF1. Genes Chromosomes Cancer 2015, 54, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, P.B.; Ryan, S.; Bashton, M.; Hollern, S.; Hanna, R.; Case, M.; Schwalbe, E.C.; Schwab, C.J.; Cranston, R.E.; Young, B.D. SH2B3 inactivation through CN-LOH 12q is uniquely associated with B-cell precursor ALL with iAMP21 or other chromosome 21 gain. Leukemia 2019, 33, 1881–1894. [Google Scholar] [CrossRef]
- Balcerek, J.; Jiang, J.; Li, Y.; Jiang, Q.; Holdreith, N.; Singh, B.; Chandra, V.; Lv, K.; Ren, J.-G.; Rozenova, K. Lnk/Sh2b3 deficiency restores hematopoietic stem cell function and genome integrity in Fancd 2 deficient Fanconi anemia. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Looi, C.Y.; Imanishi, M.; Takaki, S.; Sato, M.; Chiba, N.; Sasahara, Y.; Futaki, S.; Tsuchiya, S.; Kumaki, S. Octa-arginine mediated delivery of wild-type Lnk protein inhibits TPO-induced M-MOK megakaryoblastic leukemic cell growth by promoting apoptosis. PLoS ONE 2011, 6, e23640. [Google Scholar] [CrossRef] [PubMed]
- Baran-Marszak, F.; Magdoud, H.; Desterke, C.; Alvarado, A.; Roger, C.; Harel, S.; Mazoyer, E.; Cassinat, B.; Chevret, S.; Tonetti, C. Expression level and differential JAK2-V617F–binding of the adaptor protein Lnk regulates JAK2-mediated signals in myeloproliferative neoplasms. Blood J. Am. Soc. Hematol. 2010, 116, 5961–5971. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morris, R.; Butler, L.; Perkins, A.; Kershaw, N.J.; Babon, J.J. The Role of LNK (SH2B3) in the Regulation of JAK-STAT Signalling in Haematopoiesis. Pharmaceuticals 2022, 15, 24. https://doi.org/10.3390/ph15010024
Morris R, Butler L, Perkins A, Kershaw NJ, Babon JJ. The Role of LNK (SH2B3) in the Regulation of JAK-STAT Signalling in Haematopoiesis. Pharmaceuticals. 2022; 15(1):24. https://doi.org/10.3390/ph15010024
Chicago/Turabian StyleMorris, Rhiannon, Liesl Butler, Andrew Perkins, Nadia J. Kershaw, and Jeffrey J. Babon. 2022. "The Role of LNK (SH2B3) in the Regulation of JAK-STAT Signalling in Haematopoiesis" Pharmaceuticals 15, no. 1: 24. https://doi.org/10.3390/ph15010024
APA StyleMorris, R., Butler, L., Perkins, A., Kershaw, N. J., & Babon, J. J. (2022). The Role of LNK (SH2B3) in the Regulation of JAK-STAT Signalling in Haematopoiesis. Pharmaceuticals, 15(1), 24. https://doi.org/10.3390/ph15010024